Real-time probing of β-amyloid self-assembly and inhibition using fluorescence self-quenching between neighbouring dyes†
Steven D.
Quinn
a,
Paul A.
Dalgarno‡
a,
Ryan T.
Cameron
b,
Gordon J.
Hedley
a,
Christian
Hacker
c,
John M.
Lucocq
c,
George S.
Baillie
b,
Ifor D. W.
Samuel
*a and
J. Carlos
Penedo
*ad
aSUPA School of Physics and Astronomy, University of St Andrews, North Haugh, Fife, Scotland, KY16 9SS, UK. E-mail: jcp10@st-andrews.ac.uk, idws@st-andrews.ac.uk; Fax: +44 (0)1334 463104; Tel: +44 (0)1334 463106
bInstitute of Cardiovascular and Medical Sciences, CMVLS, Glasgow University, Glasgow, G12 8QQ, UK
cSchool of Medicine, University of St Andrews, North Haugh, Fife, Scotland, KY16 9SS, UK
dBiomedical Science Research Complex, School of Biology, University of St Andrews, North Haugh, Fife, Scotland, KY16 9SS, UK
First published on 22nd October 2013
Abstract
The fluorescence response of the Thioflavin-T (ThT) dye and derivatives has become the standard tool for detecting β-amyloid aggregates (Aβ) in solution. However, it is accepted that ThT-based methods suffer from important drawbacks. Some of these are due to the cationic structure of ThT, which limits its application at slightly acidic conditions; whereas some limitations are related to the general use of an extrinsic-dye sensing strategy and its intrinsic requirement for the formation of a sensor-binding site during the aggregation process. Here, we introduce fluorescence-self-quenching (FSQ) between N-terminally tagged peptides as a strategy to overcome some of these limitations. Using a combination of steady-state, picosecond time-resolved fluorescence and transmission electron microscopy, we characterize the fluorescence response of HiLyte fluor 555-labelled Aβ peptides and demonstrate that Aβ self-assembly organizes the covalently attached probes in close proximity to trigger the self-quenching sensing process over a broad range of conditions. Importantly, we prove that N-terminal tagging of β-amyloid peptides does not alter the self-assembly kinetics or the resulting aggregated structures. We also tested the ability of FSQ-based methods to monitor the inhibition of Aβ1–42 aggregation using the small heat-shock protein Hsp20 as a model system. Overall, FSQ-based strategies for amyloid-sensing fill the gap between current morphology-specific protocols using extrinsic dyes, and highly-specialized single-molecule techniques that are difficult to implement in high-throughput analytical determinations. When performed in Förster resonance energy transfer (FRET) format, the method becomes a ratiometric platform to gain insights into amyloid structure and for standardizing in vitro studies of amyloid aggregation.
Introduction
A major hallmark of Alzheimer's disease (AD) is the formation of extracellular β-amyloid (Aβ) peptides which aggregate into neurologically toxic structures.1 Aβ1–42 and Aβ1–40 are the two amyloid allomorphs generated by β-secretase cleavage of the amyloid precursor protein (APP).2 It is known that the less abundant Aβ1–42 has a higher propensity to aggregate and affects neuronal cells more easily under physiological conditions than Aβ1–40.1,2 It is also well established that the self-assembly of both allomorphs is highly dependent on the environmental conditions (pH, temperature, ionic strength, metal ions) and results in a variety of morphologies ranging from small oligomeric intermediates,3,4 to the fibrils and plaques observed in advanced AD.5,6 Although the β-amyloid peptide has served as a paradigm for studies of amyloid assembly for many years,7 and it is generally believed that Aβ oligomerization leads to the neuron-cell death;8 to date, the mechanism through which amyloid peptides assemble into higher order structures and how this leads to cytotoxicity is still not entirely understood.3–10 Although a variety of structural techniques including solid-state NMR,11 electron-paramagnetic resonance (EPR),12 cryo-electron microscopy (cryo-EM),13 atomic force microscopy (AFM)14 and X-ray diffraction15 have provided insights into the molecular basis of amyloid aggregation, optical techniques based on absorption and/or fluorescence detection have been the most widely employed methods to monitor in real-time the kinetics of the aggregation process in solution.16,17 In this context, the green birefringence of Congo Red18 and the fluorescence enhancement of Thioflavin T (ThT) upon binding to amyloid aggregates19 have been essential tools for detecting Aβ(1–42) and Aβ(1–40) aggregates for many years. Unfortunately, the interaction between both molecules and the amyloid structures remains controversial, thus making unclear what the dye binding probes.16,20,21 It is also known that because of its positive charge, the affinity of ThT towards amyloid aggregates shows a marked pH-dependence that limits its application at slightly acidic conditions. Moreover, ThT-based methods require the formation of a binding site in the aggregated structure, a feature that may be absent at early stages of the aggregation process or only be formed in certain types of aggregates.1,2,20–25 Furthermore, when screening for amyloid inhibitors, it has been found that ThT and certain inhibitor compounds compete for amyloid binding, thus leading to false positive results.22–25 To overcome these limitations, substantial efforts have been directed to develop fluorescent probes specifically tailored to sense particular morphologies using extrinsic probes.24–27 Although, it is clear that existing and newly designed extrinsic probes will continue to provide very valuable information about the presence and structure of specific amyloid structures; the development of robust sensing platforms applicable across a wide range of conditions remains highly desirable.16,17,24,25,28In this work, we demonstrate that fluorescence self-quenching (FSQ) between identically HiLyte Fluor 555 labelled Aβ peptides (Aβ555) at the N-terminal position constitutes a powerful and easy-to-implement single-color method to monitor the self-assembly process in real-time across a wide range of aggregation conditions. We demonstrate that the self-assembly of dye-labelled Aβ peptides organizes the covalently attached fluorescence probes in close proximity to trigger the self-quenching process without altering the aggregation mechanism or the resulting structures. Using steady-state and picosecond time-resolved fluorescence, we further characterize the proximity-dependent self-quenching between identical dyes as the underlying sensing mechanism. The possibility of monitoring the interaction between different amyloid structures and inhibitor agents by FSQ was also tested using the small heat shock protein Hsp20 as a model.29,30 Our findings demonstrate that Hsp20's inhibitory efficiency strongly depends on the amyloid-growing conditions and suggests some degree of specificity of Hsp20 towards certain amyloid morphologies. Lastly, we also introduce a dual-color ratiometric modification based on Förster resonance energy transfer (FRET) that provides a quantifiable metric to ensure that samples of starting Aβ material are identical. This is a crucial feature to avoid common discrepancies observed with in vitro studies of Aβ caused by the presence of pre-aggregated peptides in freshly prepared samples.10,17 Overall, our results indicate that fluorescence self-quenching between neighbouring peptides may represent a complementary strategy to extrinsic fluorophores for real-time in vitro studies of amyloid self-assembly mechanisms, and an attractive alternative to ThT-staining when searching for inhibitor compounds.
Results and discussion
A single-color assay to monitor amyloid aggregation in real-time using N-terminally tagged Aβ peptides
To investigate the potential use of Aβ peptides covalently tagged with fluorescence probes to detect and monitor amyloid aggregation in real time, we explored the fluorescence response of Aβ peptides labeled at the N-terminal position with HiLyte Fluor 555 (Aβ555), a member of the cyanine family of fluorescence compounds (Fig. S1, ESI†), at experimental conditions known to promote different morphologies. For instance, it has been recently shown that low concentrations of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) (1–4% v/v) drastically promote the formation of ring-like and globular structures of Aβ1–42 and Aβ1–40 (ref. 31) via a mechanism involving the formation of HFIP micro-droplets in the aqueous solvent that act as interfaces to promote Aβ1–42 aggregation. In our case, the addition of 1.5% (v/v) of HFIP to a fresh non-aggregated solution of Aβ555 in aqueous buffer (pH 7.9) induced a 61% decrease in the fluorescence intensity over a 30 min time window (Fig. 1a and b) and a 16% decrease in the absorbance value (Fig. 1b). The reproducibility of the assay was confirmed by performing four replicates using different amyloid preparations and we obtained values of (60 ± 2)% and (16 ± 2)% for the HFIP-induced variation in fluorescence and absorbance signals, respectively. | ||
| Fig. 1 Representative aggregation time course and relative fluorescence quenching during the HFIP-induced aggregation of Aβ1–42. (a) Normalized variation in fluorescence intensity of a 7 μM freshly prepared non-aggregated sample of Aβ555 as a function of time at 4 °C after injection of 1.5% (v/v) HFIP. The solid line is a fit to a biexponential decay function. Inset: normalized fluorescence enhancement of ThT as a function of time after injection of 1.5% (v/v) compared to that obtained for Aβ555 at identical experimental conditions. (b) Relative variation in absorption and fluorescence emission observed during the HFIP-induced aggregation of 100% labelled Aβ555 peptides (blue). Solid black lines represent the initial stage (t = 0) before injection of 1.5% (v/v) HIFP and dash blue lines correspond to spectra obtained at ∼30 min. Inset: normalized fluorescence spectra obtained at the initial (black) and final aggregation stage (blue). (c) Transmission electron micrograph (TEM) of negatively stained Aβ555 for aggregates obtained the addition of 1.5% (v/v) of HFIP. (d) Unlabeled Aβ1–42 and Aβ555 show an identical distribution of amyloid morphologies. Amyloid aggregates generated by injection of 1.5% (v/v) HFIP were imaged and quantified using TEM as described in the methods section. | ||
Additional time courses at increasing concentrations of HFIP were also in agreement with previous analysis on the effect of HFIP on the aggregation process of Aβ1–42 peptides31 (Fig. S2, ESI†). The time-dependent decrease of the fluorescence intensity with 1.5% (v/v) added HFIP showed a bi-exponential decay with kinetic rates very similar to those obtained for the aggregation of unlabelled Aβ1–42 using the established ThT method (Fig. 1a inset and Table S1, ESI†). Indeed, a global fit of the time courses obtained for Aβ555 and unlabeled amyloid reproduced both decays with similar rate constants and amplitudes (Table S1, ESI†). This confirms that the observed reduction in fluorescence signal is due to Aβ555 aggregation and that covalent labeling of Aβ1–42 at the N-terminal position does not alter the aggregation kinetics, in agreement with previous studies.28,31,32 Although a biexponential model is clearly an oversimplification of the underlying aggregation kinetics, for the purpose of this work, it provides a useful way to quantitatively compare both assays. We also tested whether FSQ could be used to monitor Aβ aggregation at more physiologically-relevant peptide concentrations. The real-time assembly of a 23-fold lower concentration of Aβ555 (300 nM) was monitored over a 40 minute time window. The fluorescence time-course revealed a similar decrease in fluorescence emission to that observed previously at 7 μM Aβ555 (Fig. S3, ESI†).
To further evaluate whether the morphology of the aggregates was conserved between unlabeled Aβ1–42 and Aβ555, transmission electron microscopy images (TEM) of both Aβ aggregates generated at 1.5% HFIP were obtained as described in the methods section.
TEM images of freshly formed HFIP-induced aggregates of Aβ555 (Fig. 1c) and unlabeled Aβ1–42 (Fig. S4, ESI† and Fig. 1d) were identical and the images revealed the presence of globular structures as the predominant morphologies over the 30 min time window of our fluorescence assay (Fig. 1c and d). Indeed, a quantitative analysis of the Aβ555 globules revealed an average diameter of 22.32 nm (standard deviation = 5.55 nm; standard error of the mean = 0.74 nm; n = 56), which was comparable to the 23.94 nm obtained for the unlabelled species (standard deviation = 6.92 nm; standard error of the mean = 0.93 nm; n = 56) (Fig. S5, ESI†). This average diameter is in good agreement with that observed by Nichols et al.31 after 1 h incubation of Aβ1–42 with 2% HFIP. Although it is not clear whether the globule-like structures obtained under HFIP-growing conditions represent or not a biologically relevant polymorph in the context of amyloid cytotoxicity, it is worth to mention that acceleration of amyloid aggregation has already been observed at the interface of biomimetic membranes including ganglioside micelles and lipid-rafts.33 Also recently, it has been suggested that highly fluorinated anesthetic compounds, similar to HFIP, promote Aβ aggregation and that the cytotoxicity of these aggregates may be related to the appearance of cognitive problems.34 In this context, amyloid-β derived diffusible ligands (ADDLs), which are soluble aggregates of the Aβ1–42 peptide ranging in size from trimers to 12 mer and higher, may constitute a more toxic and physiologically relevant oligomeric species.9,35 However, the in vitro characterization of the structure and kinetics of these ADDLs in solution has been challenging because the propensity of these species to evolve into larger structures when the concentration of amyloid peptide is too high. We have applied our FSQ-based assay at conditions reported to promote the formation of ADDLs (100 μM Aβ1–42, phenol red free F-12 cell culture media, pH 7.3, 4 °C)36–38 but using a much lower concentration of Aβ1–42 peptide (0.5 μM). Under these conditions, the fluorescence time-course of Aβ555 self-assembly displayed a self-quenching magnitude of 77 ± 3% and a decrease in absorbance of 67 ± 4% (Fig. S6, ESI†). The time-dependent variation of the FSQ efficiency reached a plateau at ∼24 h which is consistent with previous reports on the kinetics of ADDLs formation.36–38
Fluorescence self-quenching between neighboring dyes as the underlying sensing mechanism
Having confirmed that Aβ555 forms identical aggregated structures to the unlabeled peptide and in a comparable timescale, we next explored the nature of the quenching mechanism by repeating the aggregation process at 1.5% (v/v) of HFIP maintaining the total concentration of Aβ peptide but using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of Aβ555 and unlabeled Aβ1–42. Under these conditions, the fluorescence decreased to <20% (Fig. 2a and b), whilst the relative variation in absorbance value remained similar to that obtained when using only Aβ555 (Fig. 2b). The observed dependence of the relative variation in fluorescence intensity with the ratio between Aβ555 and unlabeled Aβ1–42 strongly suggests a self-quenching mechanism in which increasing the distance between fluorophores, by inserting unlabeled amyloid, decreases the inter-dye quenching interaction and thus increases their fluorescence intensity. Although it has been known for decades that when identical fluorophores are placed in close proximity their fluorescence intensity decreases due to intermolecular interactions,39 the exact underlying physical mechanism is still not entirely understood and it may involve a range of dye-specific photophysical processes.40 Nevertheless, fluorescence self-quenching between identical dyes has already been used to study the folding pathway of multi-labeled proteins at the single-molecule level,39 to monitor the avidin–biotin interaction using BODIPY-labeled biotin,41 and more recently, the early kinetic steps of amyloid aggregation into fibrillar structures.28
:1 ratio of Aβ555 and unlabeled Aβ1–42. Under these conditions, the fluorescence decreased to <20% (Fig. 2a and b), whilst the relative variation in absorbance value remained similar to that obtained when using only Aβ555 (Fig. 2b). The observed dependence of the relative variation in fluorescence intensity with the ratio between Aβ555 and unlabeled Aβ1–42 strongly suggests a self-quenching mechanism in which increasing the distance between fluorophores, by inserting unlabeled amyloid, decreases the inter-dye quenching interaction and thus increases their fluorescence intensity. Although it has been known for decades that when identical fluorophores are placed in close proximity their fluorescence intensity decreases due to intermolecular interactions,39 the exact underlying physical mechanism is still not entirely understood and it may involve a range of dye-specific photophysical processes.40 Nevertheless, fluorescence self-quenching between identical dyes has already been used to study the folding pathway of multi-labeled proteins at the single-molecule level,39 to monitor the avidin–biotin interaction using BODIPY-labeled biotin,41 and more recently, the early kinetic steps of amyloid aggregation into fibrillar structures.28
 | ||
| Fig. 2 Fluorescence self-quenching between neighboring dyes as the underlying sensing mechanism. (a) Normalized variation in steady-state fluorescence emission as a function of time for a sample containing a 1:1 mixture of Aβ555 and unlabelled Aβ1–42 at a total peptide concentration of 7 μM. The solid line represents the best fit to a biexponential function. (b) Relative variation in absorption and fluorescence emission observed during the HFIP-induced aggregation of a 1:1 mixture of unlabeled and labelled Aβ555 peptides. Solid black lines represent the initial stage (t = 0) before injection of 1.5% (v/v) HIFP and dash magenta lines correspond to spectra obtained at the plateau region of the time-dependent aggregation process. Inset: normalized fluorescence spectra obtained at the initial (black) and final aggregation stage (blue). (c) Time-resolved fluorescence decays obtained for Aβ555 in the monomeric state (black), and at the final stage of the aggregation process induced by injection of 1.5% (v/v) of HFIP using 100% Aβ555 (blue) or a sample containing a 1:1 mixture of Aβ555 and unlabeled Aβ1–42 (magenta). Solid lines represent the fit to mono-exponential (black and magenta) or biexponential decay functions (blue). Residuals obtained for each decay after fitting to a mono- or biexponential function are also shown. (d) Schematic showing the principle of the fluorescence self-quenching assay. N-terminally attached HiLyte Fluor 555 is progressively quenched as monomers aggregate, resulting in a combination of partially quenched and completely non-emissive fluorophores. The range and form of the morphologies shown are illustrative, and not to scale. | ||
A self-quenching process taking place between neighbouring fluorophores is further supported from the fluorescence recovery observed for freshly prepared HFIP-induced globular aggregates following dilution in aqueous buffer (Fig. S7, ESI†). These rapidly formed aggregates have shown to be initially unstable against dilution and progressively evolved into mature and stable fibril-like structures.31 In fact, the fluorescence recovery of HFIP-induced aggregates monitored after dilution in aqueous buffer was found to be reduced approximately 4-fold after 18 h maturation, confirming the slow conversion of freshly prepared HFIP-induced aggregates into more stable structures as previously reported31 (Fig. S7, ESI†).
Additional evidence for the decrease in Aβ555 being caused by a self-quenching mechanism was also obtained from picosecond time-resolved fluorescence measurements (Fig. 2c and Table S2, ESI†). The average fluorescence lifetime obtained at the end point of the aggregation time course (∼20 min) showed a smaller decrease, relative to the monomeric state (1.24 ± 0.04 ns), for a 1:1 ratio of Aβ555 and unlabeled Aβ1–42 (0.71 ± 0.01 ns) than when using only labeled peptide (0.52 ± 0.03 ns). This result confirms that the variation in the excited-state properties of the fluorophore relates to the probability of having identically labeled peptides in close proximity as shown schematically in Fig. 2d. Importantly, for both ratios of labeled:unlabeled peptide investigated, 50% Aβ555 and 100% Aβ555, the fluorescence decay did not show any component associated to the lifetime of the monomeric species (1.24 ± 0.04 ns) (Table S2, ESI†), thus ruling out that the observed variations in fluorescence intensity were due to changes in the relative population of monomer and aggregated species.
Wide-range probing of amyloid self-assembly using fluorescence self-quenching
Amyloid self-assembly is known to be extremely sensitive to small variations in the environmental conditions,7–10 therefore aggregation protocols and initial solution conditions are critical to consistently obtain reproducible results.9,10,17 Using a combination of atomic force microscopy (AFM) and biochemical assays, Stine et al.36–38 have demonstrated that the aggregation behavior of Aβ1–42 is strongly dependent on initial solution conditions and that purified dry stocks of synthetic amyloid subjected to a strong solvent monomerize the peptide (i.e., 100% HFIP), thus erasing any pre-existing morphology.37 Using this initial approach and the optimized methods developed by Stine et al.,36,37 we tested the possibility of using FSQ to monitor in real-time amyloid self-assembly at conditions known to promote the formation of different biologically-relevant morphologies. We started by carrying out aggregation experiments at conditions known to promote fibril-like structures36–38 (pH 7.9, 150 mM NaCl and 37 °C). The different morphology obtained for these aggregates compared to the globular shape observed under HFIP-growing conditions was confirmed by TEM as shown in Fig. 3a. A quantitative analysis of the Aβ555 aggregates produced revealed the presence of fibrils with an average diameter of 4.25 nm (standard deviation = 1.18 nm; standard error of the mean = 0.17 nm; n = 50). This was comparable to the 4.15 nm found for fibrillar aggregates formed via self-assembly of unlabelled Aβ (standard deviation = 0.86 nm; standard error of the mean = 0.12 nm; n = 50) (Fig. S8, ESI†). The width obtained for both fibrils, with and without dye, is also in excellent agreement with previous studies in which values ranging from 4 (short fibrils) to 7 nm (longer fibrils) were observed under similar growing conditions.42,43 The fluorescence time-course of 7 μM Aβ555 assembly also showed evidence of self-quenching, however its relative magnitude was now only (27 ± 2)% (Fig. 3b and c) and the absorbance decreased by a similar percentage (Fig. 3c). A representative fluorescence time-course for Aβ555 aggregation (Fig. 3b) also followed a biphasic decay (Table S3, ESI†) with rates similar to that obtained using unlabeled Aβ1–42 and ThT emission as the aggregation assay (Fig. 3b, inset). A smaller effect on the fluorescence output of HiLyte Fluor 555 under these aggregation conditions, compared to HFIP-induced self-assembly, was also confirmed by evaluating the changes in its fluorescence lifetime (Fig. 3d and Table S4, ESI†). Incubation of Aβ555 under fibril forming conditions resulted in a decrease in the fluorescence lifetime from 1.24 ± 0.04 ns (monomeric state) to a value of 0.90 ± 0.02 ns for the aggregated state. This value represents a ∼28% decrease in the fluorescence lifetime and is in excellent agreement with the self-quenching magnitude reported using steady-state fluorescence (Fig. 3b). To explore the possibility of using a lower peptide concentration at these aggregation conditions, we also monitored the assembly of 1 μM Aβ555 in real-time (Fig. S9, ESI†). The fluorescence time-course revealed a similar quenching magnitude (∼25%) to that observed when using a 7-fold higher concentration, thus confirming that the relative magnitude of FSQ efficiency in 100% fluorescently labelled samples is independent of the total concentration of amyloid peptide used in the assay. | ||
| Fig. 3 Representative aggregation time course and relative fluorescence quenching during the aggregation of Aβ1–42 under fibril-forming conditions. (a) Transmission electron micrograph of negatively stained Aβ555 obtained by incubation at pH 7.9 and 150 mM NaCl at 37 °C. (b) Normalized variation in steady-state fluorescence emission as a function of time obtained during the aggregation of a 7 μM sample of Aβ555 at 37 °C (pH 7.9, 150 mM NaCl). The solid line represents the non-linear squares fitting to a biexponential decay function. Inset: normalized variation in the fluorescence enhancement of ThT observed during the aggregation of a 7 μM sample of unlabeled Aβ1–42 under the same conditions used for Aβ555. The solid line is the non-linear squares fitting to a biexponential function showing identical kinetic components as those obtained for Aβ555 and the dotted lines shows the result from a global fit of both ThT and Aβ555 aggregation time courses. (c) Relative variation in the absorption and fluorescence spectra between Aβ555 monomers (solid lines) and Aβ555 aggregates (dash lines) grown at 37 °C, pH 7.9, 150 mM NaCl. Inset: normalized fluorescence spectra obtained at the initial (black) and final (red) stages of aggregation. (d) Time-resolved fluorescence decays obtained for Aβ555 in the monomeric state (black), and at the final stage of the aggregation process (red). Solid lines represent the fitting to a single exponential decay function. Residuals for each fit are also shown. | ||
Given that Aβ is generated within intracellular compartments (i.e., endocytic vesicles), we next investigated the performance of FSQ under slightly acidic conditions (pH 6) mimicking those present in the endosome.44 It has been proposed that exposure of Aβ to these endosomal conditions accelerates amyloid self-assembly into high order structures.44
It is important to mention that acidic conditions are known to significantly affect the binding affinity of ThT, thus compromising quantitative mechanistic studies of amyloid polymorphism and its dependence on experimental conditions.21,24,45–47 For example, it has been reported a 30-fold variation in KD between ThT and Aβ1–40 aggregates generated at pH 8.5 and pH 6.0,24 and more than two orders of magnitude variation for insulin aggregates produced at pH 7.5 (KD ∼ 0.5 μM) compared to pH 1.6 (KD ∼ 64 μM).24 Because ThT-based methods have to be used with caution under low pH conditions, we decided to also explore the potential of FSQ to monitor the aggregation process at more acidic conditions (pH 4). Although in the context of Aβ cytotoxicity, low pH conditions may not represent a relevant amyloidogenic pathway, understanding amyloid polymorphism and its dependence with pH and ionic strength has attracted considerable interest.9,44,48 At pH 6 we obtained a (47 ± 3)% fluorescence quenching (Fig. 4a and c) that increased to (67 ± 3)% at pH 4 (Fig. 4b and d), respectively, whilst the absorbance decreased by 26 and 28%, respectively (Fig. 4c and d). TEM images of Aβ555 incubated at pH 6 (Fig. 4e) and pH 4 (Fig. 4f) revealed the presence of aggregates of different morphology to those obtained under HFIP and at pH 7.9. We observed plaque-like aggregates at pH 6 (Fig. 4e); whilst a mixture of fibrils and globular structures was present at pH 4 (Fig. 4f). The heterogeneity of the aggregates obtained at these conditions was further explored using fluorescence lifetime measurements. At both pH conditions, the fluorescence lifetime of the aggregated sample required fitting to a biexponential function with broadly similar amplitudes but very different lifetimes (Fig. 4a inset, Fig. 4b inset and Table S5, ESI†), therefore suggesting the presence of at least two different organizations between neighbouring dyes. The calculated average lifetimes showed values of 0.47 ± 0.02 ns and 0.36 ± 0.01 ns at pH 6 and pH 4, respectively. A more pronounced effect on the fluorescence lifetime of HiLyte Fluor 555 at pH 4 compared to pH 6 is in agreement with the higher self-quenching efficiency observed previously by steady-state fluorescence.
 | ||
| Fig. 4 Real-time probing of Aβ1–42 aggregation at endosomal pH and at pH 4 using fluorescence self-quenching. (a, b) Normalized variation in steady-state fluorescence emission as a function of time obtained during the aggregation of a 7 μM sample of Aβ555 at endosomal pH 6 (a) and at pH 4 (b) in the presence of 150 mM NaCl at 37 °C. The solid lines represent the non-linear squares fitting to a biexponential decay function. Insets: time-resolved fluorescence decays obtained for Aβ555 at the final stage of the aggregation process at pH 6 (a) and pH 4 (b). Solid line represents the fitting to a biexponential decay function. Comparison between residuals obtained from mono- and biexponential fits at each aggregation condition are also shown. (c, d) Relative variation in the absorption and fluorescence spectra between Aβ555 monomers (black solid lines) and Aβ555 aggregates (dash lines) grown at pH 6 (c) and pH 4 (d). Inset: normalized fluorescence spectra obtained at the initial (black) and final stages of aggregation at pH 6 (green) and pH 4 (grey). (e, f) Transmission electron micrograph of negatively stained Aβ555 obtained by incubation at pH 6 (e) and pH 4 (f) at 37 °C. | ||
N-terminal labelling as a multi-parameter optical fingerprint of amyloid self-assembly
To quantitatively estimate if the mean values of self-quenching efficiency were significantly different between the four experimental conditions used to induce aggregation, we applied one-way ANOVA. We found that the self-quenching efficiencies differed significantly between aggregation conditions at a simultaneous significance level of p < 0.001. To get a clearer picture of which pairs of aggregation conditions were the source of variation, we performed a post-ANOVA Tukey test including the four groups of aggregation conditions. This analysis revealed that all groups exhibit significant differences between each other (p < 0.05), with the exception of the comparison of FSQ efficiencies in 1.5% HIFP and pH 4. This is expected taking into account that the mean value difference between both self-quenching efficiencies is only 60% versus 67%, respectively. From this analysis, we concluded that the FSQ efficiency obtained from repeated experiments consistently showed a statistically significant difference for amyloid aggregates in the order (globules, pH 4 > pH 6 > pH 7.4) (Fig. 5), whilst the decrease in absorption remained at a constant 30% value with the exception of globular structures, where it showed a 16% decrease (Fig. 5). Although the exact nature of these variations in absorbance and FSQ efficiency needs further investigation, our data suggest that the N-terminal fluorophore is sensitive to the tridimensional structure of the aggregate and that the quenching of the fluorescence signal may arise from a combination of static and dynamic contributions. For instance at pH 7.4, the decrease in absorbance accounts almost entirely for the quenching observed in steady-state fluorescence (27%), suggesting that under these conditions, static quenching may be the dominant process. This is further supported by the small variation in fluorescence lifetime, from 1.24 ns for the monomer peptide to 0.9 ns for the fibril structure. In contrast, for HFIP-induced globular aggregates, the decrease in absorbance was much smaller (16%) than the observed emission quenching (61%) and the fluorescence lifetime decreased to a much shorter value (0.52 ns). A similar interpretation can be done at pH 6 and pH 4, and we reasoned that under these aggregation conditions, the decrease in emission intensity results mostly from a dynamic quenching process. Overall, HiLyte Fluor 555 covalently attached to Aβ1–42 exhibits optical signatures (absorption variation, FSQ efficiency and changes in the fluorescence lifetime) that depend on the aggregation conditions and thus the resulting structure. | ||
| Fig. 5 Comparative bar plot summarizing the relative variation in fluorescence quenching (filled box) and absorbance (pattern-filled box) observed for Aβ555 self-assembly as a function of aggregation conditions (blue: 1.5% v/v HFIP; red: pH 7.9 and 37 °C; green: pH 6 and 37 °C; grey: pH 4 and 37 °C). | ||
This may be a useful feature to spectroscopically compare aggregates of a given protein at different conditions or aggregated structures from different amyloidogenic proteins grown at similar conditions.
Monitoring inhibition of Aβ1–42 aggregation using fluorescence self-quenching
To test the ability of FSQ to report the interaction between amyloid and inhibitory agents, we chose to investigate the effect of a small heat shock protein (Hsp20) as a model system. Small heat shock proteins (Hsps) constitute a family of proteins whose primary function is to act as folding chaperones to ensure that polypeptides and proteins adopt their functional structures.29,30 Although the exact mechanism by which these proteins act is not entirely understood, it has been shown that certain Hsps including Hsp27, Hsp17.7, Hsp16.2 and Hsp20 attenuate the oligomerization process of Aβ1–40 and Aβ1–42.29,30,49,50 We have chosen the Hsp20 protein as a model because it has been shown to be the most efficient Hsp at decreasing Aβ1–42 toxicity in human brain pericytes (HBP) and effectively reduces β-sheet formation when incubated with Aβ1–40 carrying the ‘Dutch’ variant.30 Hsp20 and other α-crystallins have also shown propensity to form multimeric species;29,50 however, to date, there is still no clear evidences regarding whether the inhibitory effect observed for Hsp20 against amyloid aggregation is due to its monomer form or whether higher order oligomers are the active species. Additionally, the equilibrium distribution of these Hsp20 oligomers is likely to be influenced by the morphological nature of the amyloid species present in solution.29To take into account these aspects of Hsp20 structure, we decided to investigate the interaction between Hsp20 and Aβ1–42 at two molar ratios 4:1 and 1:2 (Aβ:Hsp20) under globular- and fibril-forming conditions. We observed a pronounced difference in the inhibitory effect of human Hsp20 to the amyloid aggregation process under both experimental conditions. The fluorescence self-quenching magnitude observed when Hsp20 was co-incubated with Aβ555 in the presence of 1.5% (v/v) HFIP (Fig. 6) remained similar, within the error, to that observed in the absence of Hsp20 (62 ± 3%) reaching values of 56 ± 7% and 63 ± 6 at 4:1 and 1:2 (Aβ:Hsp20) molar ratios, respectively. In contrast, when the aggregation experiments were performed under fibril-forming conditions in the presence of Hsp20 at 1:2 (Aβ:Hsp20) molar ratio, the magnitude of the fluorescence quenching decreased from 25 ± 5% (no Hsp20) to practically undetectable levels (4 ± 2%) (Fig. 6). However, at 4:1 (Aβ:Hsp20) molar ratio, no effect on amyloid aggregation was observed. Although our current data do not allow us to confirm whether or not oligomeric forms of the heat-shock protein are the inhibitory species, the complete inhibition of Aβ1–42 aggregation at high Hsp20 to Aβ1–42 molar ratios is in good agreement with previous studies showing no Aβ1–42 aggregates formed after 3 h of co-incubating Hsp20 and Aβ1–42 at 1:1 molar ratio.30 Similar evidences for a concentration-dependence on the effect of Hsp20 on Aβ-induced cytotoxicity were obtained from human brain pericytes (HBPs). From this analysis it was found that molar ratios Aβ:sHsp20 of 25:1 and lower failed to protect HBPs against Aβ-mediated toxicity; whilst molar ratios near 1:1 resulted in complete inhibition of cell death.30
 | ||
| Fig. 6 The interaction between the small heat-shock protein Hsp20 and Aβ1–42 labelled at the N-terminus with HiLyte Fluor 555 (Aβ555) was monitored using fluorescence self-quenching under globular (blue) and fibrillar (red) growing conditions. No variation in self-quenching efficiency (∼60%) at 4:1 and 1:2 (Aβ:Hsp20) molar ratios (blue) was observed under HFIP-induced growing conditions. Under fibril growing conditions (37 °C, pH 7.9, 150 mM NaCl), Hsp20 induced a decrease from 25% to 4% in fluorescence quenching at 1:2 (Aβ:Hsp20) molar ratio, whilst no effect was observed at 4:1 (Aβ:Hsp20) molar ratio (red). *Statistical analysis was performed using Student's t test. The level of significance of the difference between both (Aβ:Hsp20) molar ratios under fibril growing conditions was P < 0.05. | ||
Towards a ratiometric FRET-based assay for amyloid aggregation
The fact that fluorescence self-quenching intrinsically depends on the close positioning of the dyes along the aggregated structure prompted us to explore whether the assay could be transformed into a ratiometric method by using Förster resonance energy transfer (FRET) between a mixture of Aβ555 and Aβ647 (Aβ1–42 N-terminally labelled with HiLyte Fluor 647, a fluorescence acceptor for HiLyte Fluor 555). The chemical structure of cyanine derivative HiLyte Fluor 647 is shown in Fig. S1 (ESI†). We measured the efficiency of energy transfer for a sample aggregated under HFIP conditions using the RatioA method.51 The RatioA method provides a value proportional to the FRET efficiency by comparing the amount of acceptor fluorescence obtained by energy transfer from the donor (sensitized acceptor emission) with that obtained by direct excitation of the acceptor.51 We found that optimal conditions for minimizing self-quenching, in favour of FRET as the main route to deactivate the donor from its excited state, as schematically shown in Fig. 7a, could be obtained by using a molar ratio of ∼0.6 (unlabelled Aβ1–42):0.4 (labelled Aβ1–42), where the labelled amyloid contained a 1 to 1 ratio of Aβ555 and Aβ647 (Fig. 7b). Using this combination, the time trajectory of RatioA from a sample induced to aggregate by injection of 1.5% (v/v) of HFIP (Fig. 7c) or under fibril-like forming conditions (pH 7.9, 150 mM NaCl, 37 °C) (Fig. 7d) showed an increase in RatioA from a similar initial value of ∼0.034 to values of 0.09 and 0.037, respectively. It is important to emphasize that the reproducibility of the RatioA value for the non-aggregated state provides a quantifiable metric to ensure that samples of starting Aβ material are identical, thus avoiding common inconsistencies with in vitro studies of Aβ caused by variations in the aggregation baseline. At all the aggregation conditions investigated using the FRET assay, the time-dependent variation of RatioA displayed a biphasic behavior with kinetic parameters (Table S6, ESI†) similar to those obtained using FSQ (Fig. 1a and 3b). When similar FRET-based experiments were carried out at ADDL-forming conditions (phenol red free F-12 cell culture media, pH 7.3, 4 °C) an increase in RatioA, again from an initial value of ∼0.034, to a value of 0.044 was observed (Fig. S10, ESI†). The time scale of the aggregation process monitored by FRET (Fig. S10, ESI†) followed a similar profile to that previously observed for ADDL-growing conditions using FSQ (Fig. S6, ESI†).
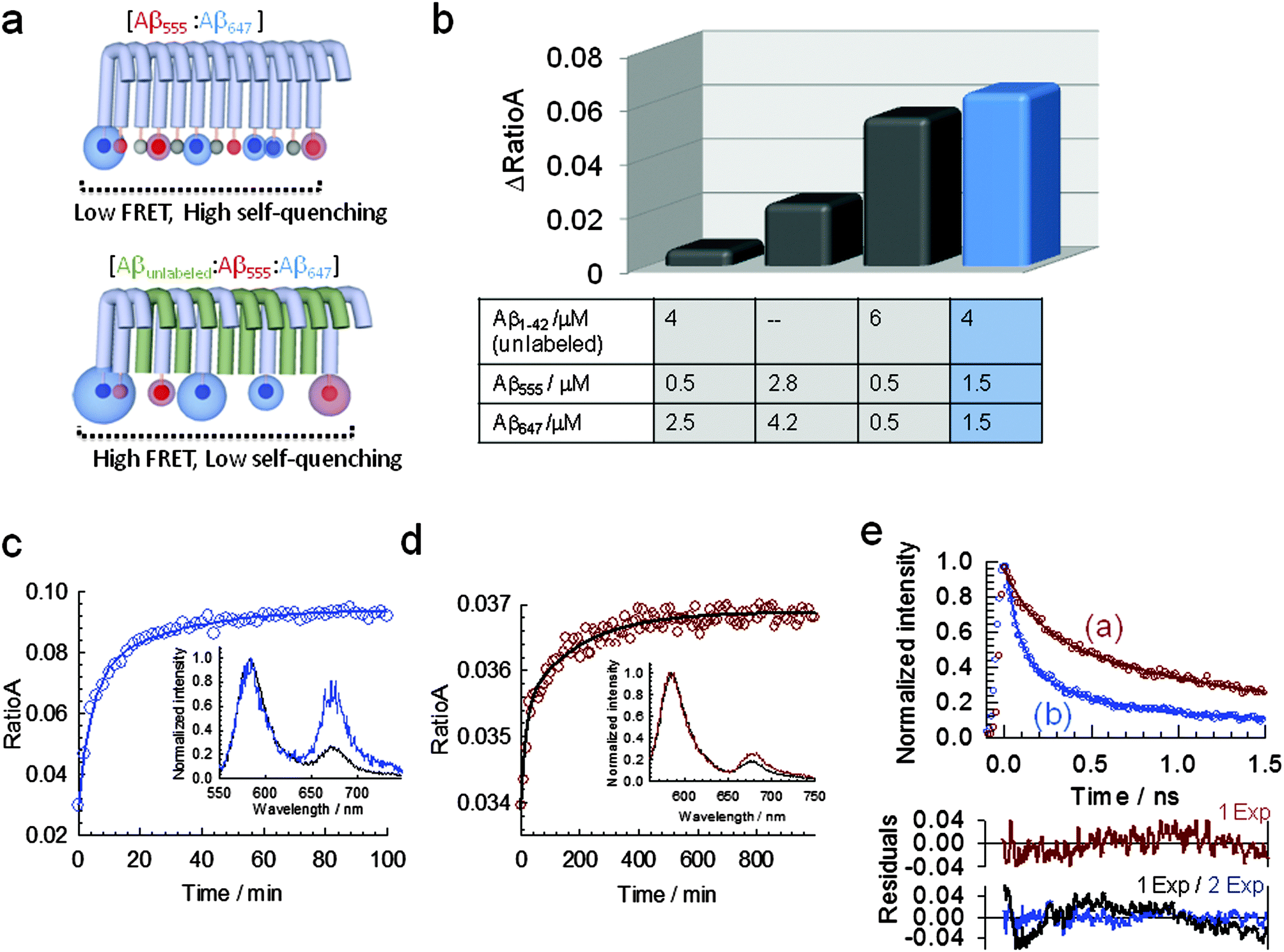 | ||
| Fig. 7 A ratiometric assay based on Förster resonance energy transfer (FRET) between N-terminally labelled peptides to monitor Aβ1–42 self-assembly. (a) Schematic showing the competition between self-quenching and FRET (top panel) and the optimization of the FRET efficiency by inserting unlabeled Aβ1–42 (bottom panel). The morphology shown is only for displaying purposes and is not drawn to scale. (b) Variation in the efficiency of resonance energy transfer, measured as RatioA value, at different molar ratios of labeled and unlabeled peptides. Relative concentrations of Aβ1–42:Aβ555:Aβ647 at a molar ratio of 0.6:0.2:0.2 maximize the efficiency of the FRET process (blue bar). (c) Real-time variation in RatioA during the aggregation of 7 μM Aβ1–42 (molar ratios of Aβ1–42:Aβ555:Aβ647 as described in (b)) induced by injection of 1.5% (v/v) (pH 7.9, 4 °C). Inset: normalized fluorescence spectra at the donor emission maximum corresponding to the initial (pre-injection of HFIP, black line) and at the final stage of the aggregation process (>60 min, blue line) showing the relative increase in acceptor fluorescence. (d) Real-time variation in RatioA during the aggregation of 7 μM Aβ1–42 (molar ratios of Aβ1–42:Aβ555:Aβ647 as described in (b)) induced by incubation at 37 °C (pH 7.9, 150 mM NaCl). Inset: normalized fluorescence spectra at the donor emission maximum corresponding to the initial (black line) and at the final stage of the aggregation process (>600 min, red line). (e) Time-resolved fluorescence decay of the FRET donor HiLyte Fluor 555 in presence of the FRET acceptor HiLyte Fluor 647 in a mixture containing a 0.6:0.2:0.2 molar ratio of Aβ1–42 (unlabeled), Aβ555 and Aβ647, respectively. Data were taken at the final stage of the aggregation process induced by incubation at pH 7.9 and 37 °C (a) as shown in Fig. 7d and at, the corresponding final stage of the HFIP-induced aggregation process (b) as shown in Fig. 7c. Solid lines represent the fit to a monoexponential (a) and biexponential function (b), respectively. Residuals obtained from the fitting to each function are also shown. | ||
Further evidence confirming the presence of resonance energy transfer between HiLyte Fluor 555 and HiLyte Fluor 647 peptides were obtained from the decrease in the average fluorescence lifetime of the donor in the presence of acceptor, which changed for HFIP-induced aggregation from 0.71 ± 0.01 ns in the absence of acceptor to τav = 0.36 ± 0.02 ns (Fig. 7e and Table S7, ESI†) when half of the Aβ555 concentration was replaced by Aβ647. When similar experiments were repeated under fibril-growing conditions, the average lifetime of the donor decreased to τav = 0.48 ± 0.01 (Fig. 7e and Table S7, ESI†), thus confirming a higher FRET efficiency for HFIP-growing conditions. A closer inter-dye distance and therefore a higher FRET efficiency in globular structures (ΔRatioA ∼ 0.06) (Fig. 7c) compared to fibrillar morphologies (ΔRatioA ∼ 0.003) (Fig. 7d) agrees well with the results obtained from fluorescence self-quenching and emphasizes the potential of N-terminally labelled peptides as a versatile single- and dual-colour platform to monitor amyloid assembly in real-time and to provide structural insights into the aggregation state.
Conclusions
We demonstrate a robust and easy-to-implement multi-parameter method for real-time probing of amyloid self-assembly across a wide range of aggregation conditions based on fluorescence self-quenching (FSQ) between identical N-terminal tagged peptides. Using a combination of steady-state and picosecond time-resolved fluorescence spectroscopy in mixtures of labelled and unlabelled amyloid, we prove that the observed quenching is due to the close positioning of the dyes during the aggregation process. N-terminal tagging of amyloid peptides is complementary to the use of extrinsic dyes, with identical kinetic features for amyloid aggregation to those reported using Thioflavin T. In principle, FSQ should enable inhibitor screening without interfering or competing with inhibitor binding. Moreover, because the fluorescence reporter is covalently attached to the peptide monomer, monitoring amyloid aggregation is not limited by the formation of probe-specific binding sites along the aggregation pathway or affected by variations in the affinity constant of the extrinsic fluorophore for amyloid aggregates at different experimental conditions. However, as with any fluorescence-based assay, care should be taken to ensure that the variation in fluorescence output is not due to non-specific interactions between the reporter probe and any potential inhibitor compound. In this context, it is worth to notice that FSQ-based assays may offer the possibility of assessing the presence of these artifacts by changing the ratio of labelled to unlabelled amyloid peptide. Here, any dependence of the inhibitory effect with the degree of amyloid labelling should be indicative of some interaction between the fluorophore and the tested compound. Importantly, we also demonstrate that the excited-state fluorescence lifetime of the covalently attached dye in the monomer and aggregated forms can easily be determined, thus providing a means to quantify variations in their relative populations as a function of time or aggregation conditions. It is interesting to note that such discrimination between monomer and aggregated populations is not possible using exclusively steady-state fluorescence intensity measurements or extrinsic probes that do not interact with monomeric Aβ. This may be an important feature if we take into account that several lines of evidence have suggested that amyloid toxicity may not be directly related to the presence of specific and discrete morphologies, but rather to the ability of these species to grow and dynamically exchange in the presence of monomeric Aβ1–42.52 We envisage that such combination of time-resolved, steady-state fluorescence and absorption could be insightful for distinguishing between the formation of different types of aggregates.N-terminal tagging of amyloidogenic peptides with small molecules shares some common features with recent approaches using fluorescence proteins fused to α-synuclein,53,54 Aβ peptides55 and prion sequences.56 In both cases, perturbation of the photophysical properties is explored to monitor the aggregation process. It has been found that aggregation promotes misfolding of the fused protein, and thus a decrease in the fluorescence output similar to that observed by us using small-molecule tagging. However, it has also been indicated that certain peptide sequences aggregate without affecting the native function of the fluorescent protein,55 therefore compromising the use of fused proteins as a general strategy to investigate peptide aggregation. Lastly, we show that dual-colour N-terminal tagging with a FRET pair constitutes a platform not only for monitoring aggregation and provide insights into the structure of the aggregates, but also to ensure identical pre-aggregation conditions. This is crucial for in vitro studies of amyloid aggregation, where the presence of pre-aggregated amyloid structures in stock solutions of amyloid has led to significant discrepancies in the literature. We believe this FRET assay could be used for standardizing measurements across laboratories given that it is concentration-independent and provides a reproducible magnitude to which samples can be compared to.
There are also obvious limitations to the use of N-terminal tagging for certain applications. For instance, the requirement to employ synthetically labelled peptides makes difficult its implementation as a diagnostic tool. In summary, in vitro probing of amyloid aggregation using FSQ fills the gap between current amyloid-sensing methods using non-covalent probes and highly specialized single-molecule techniques.57,58 It offers the advantage that it can be easily performed in any laboratory using a conventional fluorimeter and can be adapted for continuous large-scale analytical determinations. Although we demonstrated the potential of FSQ using exclusively Aβ1–42 as a model for protein aggregation, it is likely that it could be applied to investigate the aggregation process of several misfolded proteins and peptides implicated in various pathologies, including the tau protein in Alzheimer's disease, prion in spongiform encephalopathy and α-synuclein in Parkinson disease.
Experimental section
Monomer preparation
Synthetic unlabelled and dye labelled Aβ1–42 peptides incorporating either HiLyte Fluor 555 or HiLyte 647 were purchased from Anaspec Inc. (USA) and used with no additional purification. HiLyte Fluor 555 and HiLyte Fluor 647 are derivatives of the widely used Cy3 and Cy5 dyes, respectively, in which a long chain linking both aromatic groups has been incorporated to decrease the flexibility of the molecule and improve the photophysical behaviour (Fig. S1, ESI†). Fluorophore attachment to the N-terminal position of the Aβ peptide was performed by the manufacturer and peptides were characterized by the manufacturer using mass spectrometry and high-performance liquid chromatography (HPLC). Peptides were suspended in 100% 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) at 5 mg mL−1 and incubated for complete solubilization at room temperature for 1.5 h. HFIP was subsequently removed by evaporation under vacuum for 4 h and stored at −20 °C.17,36–39,44,49Protein expression and purification
The full length Hsp20 sequence was cloned into a pET28 vector (Novagen) in order to express an N-terminal His-tag and then transformed into competent BL21 cells (Invitrogen, Paisley). Cells were grown until OD600 = 1, 1 M of IPTG was then added and cells were grown for a further 24 hours at 37 °C. The protein was then purified using nickel affinity chromatography. The resulting protein product was then checked for impurities on a 4% gel and then verified through western blotting techniques.Aggregation protocols
To obtain HFIP-induced aggregates, pre-treated Aβ1–42 monomers were resuspended in dimethylsulfoxide (DMSO) to a concentration of 2.5 mg mL−1. Aβ1–42 peptides were subsequently diluted in Tris-HCl buffer solution (50 mM, pH 7.9) to the final desired concentration and ≤4% HFIP was added to induce aggregation.31 All aqueous solutions were prepared with deionized water (Millipore, USA). Incubation of the peptides for 1 hour at 4 °C with vigorous agitation by continued vortexing results in the progressive formation of Aβ1–42 globular structures.To obtain ADDLs, Aβ1–42 monomers were resuspended in dimethylsulfoxide (DMSO) to a concentration of 2.5 mg mL−1. Aβ1–42 peptides were subsequently diluted in phenol red free HAM's F-12 media (pH 7.3) to the final desired concentration to induce aggregation.35–39 Incubation of the peptides for 24 hours at 4 °C results in the progressive formation of Aβ1–42 ADDL structures. Fluorescence scans were recorded at discrete time points along the time course of the aggregation process.
For oligomeric and fibril-like aggregates formed at pH 7.9, HFIP-pre-treated Aβ1–42 monomers were resuspended in DMSO to a concentration of 2.5 mg mL−1 and subsequently diluted to the desired concentration in 50 mM Tris-HCl buffer solution containing 150 mM NaCl and incubated at 37 °C for 24 hours as reported previously.17,35–39 pH 6-induced aggregates were created by re-suspending HFIP-pre-treated Aβ1–42 monomers in DMSO to a concentration of 2.5 mg mL−1 and subsequently diluting to the final concentration in 50 mM 2-(N-morpholino)ethanesulfonic acid (MES) buffer at pH 6.0. Samples were incubated without agitation at 37 °C for 5 hours.44 Finally, pH 4-induced aggregates were created by re-suspending HFIP-pre-treated Aβ1–42 in DMSO to a concentration of 2.5 mg mL−1 and subsequently diluting in Tris-HCl buffer solution (50 mM, pH 7.9). 0.1% (v/v) of C2H4O2 was subsequently added to switch the pH from 7.9 to 4.0. After an incubation period of 1.5 hours (no agitation), pH 4-induced aggregates were produced.48 Final Aβ concentrations were determined by absorbance spectroscopy using extinction coefficients of 150000 cm−1 M−1 at 560 nm for Aβ555 and 250000 cm−1 M−1 at 653 nm for Aβ647. Final concentrations of ThT were determined using an extinction coefficient of 36000 cm−1 M−1 at 412 nm.24
Fluorescence spectroscopy of Aβ1–42 aggregates
Fluorescence emission spectra from N-terminally labelled Aβ1–42 aggregates were obtained using a Peltier-cooled Varian Eclipse fluorescence spectrophotometer during incubation. Cuvettes with 1 cm path length were used and agitation was achieved with the insertion of magnetic stirring bars. Spectra from Aβ555 and Aβ647 were recorded using excitation wavelengths of 547 nm and 647 nm respectively. Fluorescence spectra of ThT in the presence of unlabelled Aβ were recorded using an excitation wavelength of 440 nm.Fluorescence lifetime measurements
Fluorescence lifetime measurements were performed with a Hamamatsu C6860 Synchroscan streak camera. The 80 MHz, 100 fs (full width half maximum) 800 nm output of a Ti:sapphire oscillator was frequency doubled with a beta barium borate crystal, giving 400 nm excitation pulses. The 400 nm light, with an average power of less than 1 mW, was subsequently focussed through the optical path length (1 cm) of the solution cuvette. The photoluminescence from the sample was then collected and collimated with a lens before being focussed onto the entrance slit of a Chromex 250 is imaging spectrograph. Excitation light was removed with a yellow Schott glass filter that cuts all light below 420 nm. Spectral windows of 540–680 nm (single-colour quenching assay) and 540–590 nm (dual-colour FRET assay) were selected with the spectrograph before the light was directed into the streak camera. Time resolved photoluminescence dynamics were then recorded over a number of time ranges, giving an ultimate resolution with deconvolution with the instrument response function of ∼2 ps.Transmission electron microscopy
The structures of Aβ1–42 amyloid aggregates were analysed by negative staining for transmission electron microscopy. Pioloform and carbon-coated 100-mesh copper grids (Agar Scientific, UK) were placed face down on droplets containing Aβ aggregates and incubated for 2 min at room temperature to allow binding of the protein structures to the grids. The grids were subsequently washed and stained twice on droplets of 3% aqueous uranyl acetate for 2 min each followed by removal of excess staining solution by gently blotting the side of the grid with filter paper. The grids were then air dried and analysed in the electron microscope. Aβ amyloid structures were sampled by taking 5 micrographs per sample with a JEOL 1200 transmission electron microscope on Ditabis imaging plates (DITABIS Digital Biomedical Imaging Systems AG, Germany). Micrographs were selected to represent the average distribution, density and size of the Aβ aggregates.59 Globular structures were sampled using the forbidden line unbiased counting rule applied to quadrats positioned systematic uniform random (approximately 6 quadrats per micrograph, 0.01148 μm2 per quadrat) on micrographs which were displayed in Photoshop CS6. The mean calliper diameter was measured both horizontally and vertically and the average of both measurements calculated for each globular structure. Fibrillar structures were sampled using a systematic uniform random scanning method in which micrographs were taken at nominal magnification of 30 K each time a fibrillar structure was encountered. The fibril diameters were measured in Photoshop CS6 on fibrils selected by line intersections with a grid applied to the micrographs (grid spacing = 103.63 nm). Each time a fibril intersected with the grid, a diameter was acquired. The mean diameters of globular and fibrillar structures were subsequently calculated, along with the standard deviation and standard error of the mean.Statistical analysis
Data are expressed as the means ± SEM from 3–5 separated experiments using different amyloid preparations and stocks. Statistical comparison of the results obtained between the different experimental conditions tested was carried out using one-way analysis of variance (ANOVA). Post hoc ANOVA tests for pairwise multiple comparisons between experimental conditions were performed using the Tukey method. A P value of <0.05 was considered statistically significant.Acknowledgements
We are grateful to Arvydas Ruseckas for his help with time resolved fluorescence measurements and Stefan Heucke for performing initial fluorescence studies.Notes and references
- Y. Huang and L. Mucke, Cell, 2012, 148, 1204–1222 CrossRef CAS PubMed.
- N. De Strooper, R. Vassar and T. Golde, Nat. Rev. Neurol., 2010, 6, 99–107 CrossRef PubMed.
- D. M. Walsh and D. J. Selkoe, J. Neurochem., 2007, 101, 1172–1184 CrossRef CAS PubMed.
- C. G. Glabe, J. Biol. Chem., 2008, 283, 29639–29643 CrossRef CAS PubMed.
- L. Kreplak and U. Aebi, Adv. Protein. Chem., 2006, 73, 217–233 CrossRef CAS.
- R. P. Friedrich, et al. , Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1942–1947 CrossRef CAS PubMed.
- J. Greenwald and R. Riek, Structure, 2010, 18, 1244–1260 CrossRef CAS PubMed.
- D. Eisenberg and M. Jucker, Cell, 2012, 148, 1188–1203 CrossRef CAS PubMed.
- Y. Miller, B. Ma and R. Nussinov, Chem. Rev., 2010, 110, 4820–4838 CrossRef CAS PubMed.
- W. Hamley, Chem. Rev., 2012, 112, 5147–5192 CrossRef PubMed.
- K. Iwata, et al. , Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18119–18124 CrossRef CAS PubMed.
- M. Török, et al. , J. Biol. Chem., 2002, 277, 40810–40815 CrossRef PubMed.
- R. Zhang, et al. , Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4653–4658 CrossRef CAS PubMed.
- A. Mastrangelo, J. Mol. Biol., 2006, 358, 106–119 CrossRef PubMed.
- M. Sunde, et al. , J. Mol. Biol., 1997, 273, 729–739 CrossRef CAS PubMed.
- A. Reinke and J. E. Gestwicki, Chem. Biol. Drug Des., 2011, 77, 399–411 CAS.
- M. Lindgren, K. Sörgjerd and P. Hammarström, Biophys. J., 2005, 88, 4200–4212 CrossRef CAS PubMed.
- J. Howie and D. B. Brewer, Micron, 2009, 40, 285–301 CrossRef PubMed.
- H. Levine 3rd, Protein Sci., 1993, 2, 404–410 CrossRef CAS PubMed.
- E. Brandenburg, H. v. Berlepsch and B. Koksch, Mol. BioSyst., 2012, 8, 557–564 RSC.
- R. Mishra, D. Sjölander and P. Hammarström, Mol. BioSyst., 2011, 7, 1232–1240 RSC.
- S. A. Hudson, H. Ecroyd, T. W. Kee and J. A. Carver, FEBS J., 2009, 276, 5960–5972 CrossRef CAS PubMed.
- F. Meng, P. Marek, K. J. Potter, C. B. Verchere and D. P. Raleigh, Biochemistry, 2008, 47, 6016–6024 CrossRef CAS PubMed.
- M. J. Groenning, J. Chem. Biol., 2010, 3, 1–18 CrossRef PubMed.
- C. W. Bertoncini and M. S. Celej, Curr. Op. Prot. Pep. Sci., 2011, 12, 206–221 CrossRef CAS.
- L. Qin, J. Vastl and J. Gao, Mol. BioSyst., 2010, 6, 1791–1795 RSC.
- P. Marek, R. Gupta and D. P. Raleigh, ChemBioChem, 2008, 9, 1372–1374 CrossRef CAS PubMed.
- K. Garai K and C. Frieden, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3321–3326 CrossRef PubMed.
- H. W. Edwards, R. T. Cameron and G. S. Baillie, Cell. Signalling, 2011, 23, 1447–1454 CrossRef CAS PubMed.
- M. M. Wilhelmus, et al. , Brain Res., 2006, 1089, 67–78 CrossRef CAS PubMed.
- M. R. Nichols, et al. , J. Biol. Chem., 2005, 280, 2471–2480 CrossRef CAS PubMed.
- L. M. Jungbauer, C. Yu, K. J. Laxton and M. J. LaDu, J. Mol. Recognit., 2009, 22, 403–413 CrossRef CAS PubMed.
- E. Chi, S. L. Frey and K. Y. C Lee, Biochemistry, 2007, 46, 1913–1924 CrossRef CAS PubMed.
- R. G. Eckenhoff, et al. , Anesthesiology, 2004, 3, 703–709 CrossRef PubMed.
- W. L. Klein, Neurochem. Int., 2002, 41, 345–352 CrossRef CAS.
- W. B. Stine, L. Jungbauer, C. Yu and M. J. LaDu, Methods Mol. Biol., 2011, 670, 13–32 CAS.
- W. B. Stine, et al. , J. Biol. Chem., 2003, 278, 11612–11622 CrossRef CAS PubMed.
- A. Jan, D. M. Hartley and H. A. Lashuel, Nat. Protocols, 2010, 5, 1186–1209 CAS.
- X. Zhuang, T. Ha, D. K. Harold, T. Centner, S. Labeit and S. Chu, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 14241–14244 CrossRef CAS PubMed.
- V. M. Agranovich and M. D. Galanin, North-Holland, Amsterdam.
- X. Song and B. I. Swanson, Anal. Chem. Acta, 2001, 442, 78–87 CrossRef.
- G. Thankur, et al. , Int. J. Alzheimer's Dis., 2011, 502386 Search PubMed.
- M. Schmidt, et al. , Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 19813–19818 CrossRef CAS PubMed.
- P. M. Gorman, C. M. Yip, P. E. Fraser and A. Chakrabartty, J. Mol. Biol., 2003, 325, 743–757 CrossRef CAS.
- R. Khurana, et al. , J. Struct. Biol., 2005, 151, 229–238 CrossRef CAS PubMed.
- W. E. Klunk, et al. , Life Sci., 2001, 69, 1471–1484 CrossRef CAS.
- N. Amdursky, Y. Erez and D. Huppert, Acc. Chem. Res., 2012, 45, 1548–1557 CrossRef CAS PubMed.
- P. E. Fraser, J. T. Nguyen, W. T. Surewicz and D. A. Kirschner, Biophys. J., 2001, 60, 1190–1201 CrossRef.
- S. Lee, K. Carson, A. Rice-Ficht and T. Good, Protein Sci., 2005, 14, 593–601 CrossRef CAS PubMed.
- S. Lee, K. Carson, A. Rice-Ficht and T. Good, Biochem. Biophys. Res. Commun., 2006, 347, 527–533 CrossRef CAS PubMed.
- R. M. Clegg, Methods Enzymol., 1992, 211, 353–388 CAS.
- A. Jan, O. Adolfsson, I. Allaman, A. L. Buccarello, P. J. Magistretti, A. Pfeifer, A. Muhs and H. A. Lashuel, J. Biol. Chem., 2011, 286, 8585–8596 CrossRef CAS PubMed.
- T. J. Van Ham, et al. , J. Mol. Biol., 2010, 395, 627–642 CrossRef CAS PubMed.
- G. S. Schierle, et al. , ChemPhysChem, 2011, 12, 673–680 CrossRef PubMed.
- C. Wurth, N. K. Guimard and M. H. Hecht, J. Mol. Biol., 2002, 319, 1279–1290 CrossRef CAS.
- D. Hamada, et al. , Proteins, 2008, 72, 811–821 CrossRef CAS PubMed.
- N. Cremades, et al. , Cell, 2012, 149, 1048–1059 CrossRef CAS PubMed.
- P. Narayan, et al. , Nat. Struct. Mol. Biol., 2012, 19, 79–83 CAS.
- C. Goldsbury, J. Struct. Biol., 2011, 173, 1–13 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of aggregation protocols, steady-state and time-resolved measurements, transmission electron microscopy characterization and Fig. S1–S10 and Tables S1–S6. See DOI: 10.1039/c3mb70272c |
| ‡ Current address: Institute of Biological Chemistry, Biophysics and Bioengineering, School of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, UK. |
| This journal is © The Royal Society of Chemistry 2014 |