 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanocrystal synthesis in microfluidic reactors: where next?
Thomas W.
Phillips†
a,
Ioannis G.
Lignos†
b,
Richard M.
Maceiczyk†
b,
Andrew J.
deMello
*b and
John C.
deMello
*a
aCentre for Plastic Electronics and Department of Chemistry, Imperial College London, Exhibition Road, South Kensington, London, SW7 2AZ, UK. E-mail: j.demello@imperial.ac.uk
bDepartment of Chemistry and Applied Biosciences, ETH Zürich, Vladimir Prelog Weg 1, 8093 Zürich, Switzerland. E-mail: andrew.demello@chem.ethz.ch
First published on 9th June 2014
Abstract
The past decade has seen a steady rise in the use of microfluidic reactors for nanocrystal synthesis, with numerous studies reporting improved reaction control relative to conventional batch chemistry. However, flow synthesis procedures continue to lag behind batch methods in terms of chemical sophistication and the range of accessible materials, with most reports having involved simple one- or two-step chemical procedures directly adapted from proven batch protocols. Here we examine the current status of microscale methods for nanocrystal synthesis, and consider what role microreactors might ultimately play in laboratory-scale research and industrial production.
 Thomas W. Phillips | Thomas Phillips is a PhD student in the Plastic Electronics Centre for Doctoral Training at Imperial College London. He received a first class MSci degree in Chemistry from Imperial in 2011, followed by a Research Masters in Plastic Electronics in 2012. He works on the synthesis of metal nanoparticles using droplet flow reactors. |
 Ioannis G. Lignos | Ioannis Lignos received his diploma in Chemical Engineering from NTUA, Greece in 2009 and his MSc in Chemical and Bioengineering from EPFL, Switzerland in 2011. He is now a PhD student at ETH Zürich, specialising in the microfluidic synthesis of semiconductor nanomaterials for solar cell applications. |
 Richard M. Maceiczyk | Richard Maceiczyk received his bachelor degree in Chemistry from Heidelberg University and his master degree from ETH Zürich, conducting his masters research project at Nagoya University. He is now a PhD student at ETH Zürich working on the synthesis of nanoparticles in microfluidic systems. |
 Andrew J. deMello | Andrew deMello is Professor of Biochemical Engineering in the Department of Chemistry and Applied Biosciences at ETH Zürich. Between 1997 and 2011 Andrew was Professor of Chemical Nanosciences in the Chemistry Department at Imperial College London. He has given over 250 invited lectures worldwide, published over 230 papers in the areas of microfluidics and nanoscale science, and co-authored two books. |
 John C. deMello | John deMello is Professor of Nanomaterials in the Department of Chemistry at Imperial College London, and a Royal Society Industrial Fellow. His research is focused on the flow synthesis of electronic materials, and their applications to electronic devices and sensors. He has authored more than 100 papers in the fields of microfluidics and electronic materials. |
1. Beyond batch
The availability of high quality nanometre-sized colloidal crystals is of critical importance in many fields of applied science, including optoelectronics,1 catalysis,2 and nanomedicine.3 In each of these fields, the size-controlled physical and chemical properties of nanocrystals can be exploited to improve on conventional bulk materials in terms of chemical reactivity, bioavailability, or magnetic, optical, or electronic behaviour. In all cases, this requires tightly specified nanocrystals of well-defined size, shape, composition and crystallinity.Over the years a multitude of synthetic strategies have been reported for producing high quality nanocrystals. Many of these methods, however, suffer from significant batch-to-batch variability arising from inadequate reaction control. In 2002, some of the authors of this article proposed the use of microscale reactors for nanocrystal synthesis, reasoning that a reduction in reaction volume would ensure better uniformity in the thermal and chemical environment, which in turn would lead to improved product control.4 A simple demonstration of the idea was provided, using the direct reaction of cadmium nitrate and sodium sulphide (with sodium polyphosphate as a capping agent) to form cadmium sulphide nanocrystals of tuneable band gap. While this and related work demonstrated the feasibility of adapting a conventional batch synthesis route to flow – and in particular demonstrated the ease with which reaction conditions could be systematically varied in a flow environment – it did not at the time improve upon the quality of nanocrystals achievable in batch.
In the ensuing twelve years, there have been numerous reports of nanocrystal syntheses in microfluidic reactors, ranging widely in sophistication and complexity with regards to the microfluidic network employed, the synthetic strategy, and the nature of the particles produced. There have been many articles by ourselves and others reviewing the application of microfluidic reactors to nanocrystal synthesis, and it is not our intention to add to that list here.5–11 Rather, our aim is to identify areas where microfluidic methods can offer advantages over batch procedures, and to draw attention to a number of challenges that must now be addressed to realize their full potential for nanocrystal synthesis.
The usual motivation for using microfluidic reactors to synthesise nanocrystals is a desire to attain improved sample-to-sample consistency relative to batch methods. In conventional batch processes it can be difficult to ensure consistent reaction conditions from one time to the next, especially when reactions are carried out manually. Minor variations in the times, locations and rates of precursor addition or the way in which the reaction is eventually quenched can have a strong influence on the final product, meaning the quality of the nanocrystals obtained can vary greatly from one batch to the next. Furthermore, inhomogeneities in the temperature or chemical composition of the reaction mixture due to uneven heating or inadequate mixing can be an unwanted source of polydispersity. In moving to a microfluidic format improved sample consistency can be achieved by virtue of the reduced reaction volumes, which ensure a more uniform reaction environment. Nanocrystal properties can be readily tuned by varying the volumetric flow rates of the injected reagents or the temperature distribution along the flow profile. Finally, once the desired conditions have been determined, an arbitrary amount of optimised, highly consistent material can be obtained by operating the reactor continuously until production requirements have been met. This at least is the goal. To what extent can it be achieved?
2. Single-phase versus two-phase reactors
Reactors for nanocrystal synthesis fall into two broad categories: capillary- and chip-based systems. Capillary reactors are generally simpler in structure and more easily fabricated, using simple fluidic components joined by appropriate lengths of tubing. Chips are typically fabricated from a plastic, glass or silicon substrate, using soft-lithography, wet etching or micromachining techniques, and can be precisely tailored to the individual requirements of the reaction; multiple chemical processes (e.g. heating, mixing, cooling, reagent addition) can be integrated into a single, small-footprint device. Both types of reactor have been applied to good effect in nanocrystal synthesis.12–15 Which kind you should select is largely a matter of taste: do you prefer the convenience of capillaries or the highly integrated nature of chips?On a different level, microfluidic reactors can be categorised as single-phase or two-phase reactors. Most research to date has focused on single-phase reactors, in which miscible streams of reagents are injected into a channel or capillary where they mix and react (Fig. 1(a)). Single-phase reactors offer a high degree of synthetic flexibility, tolerating a wide range of flow rates and solvents. In addition, they facilitate the easy injection of additional reagents in a controlled manner through the use of downstream inlets in the reaction channel, making it straightforward to conduct multistep reactions and produce more complex structures.16 Two issues, however, limit their performance. Firstly, the flowing liquid drags against the channel walls inducing a parabolic velocity profile across the flow path, with the fluid moving fastest at the channel centre; this in turn generates a distribution of residence times within the reactor, leading to undesirable dispersion in nanoparticle properties.17 Secondly, precursor and/or product deposition on the walls can lead to fouling, which affects the flow and ages the reactor. Left unchecked this can, and frequently does, lead to eventual failure through blockage.
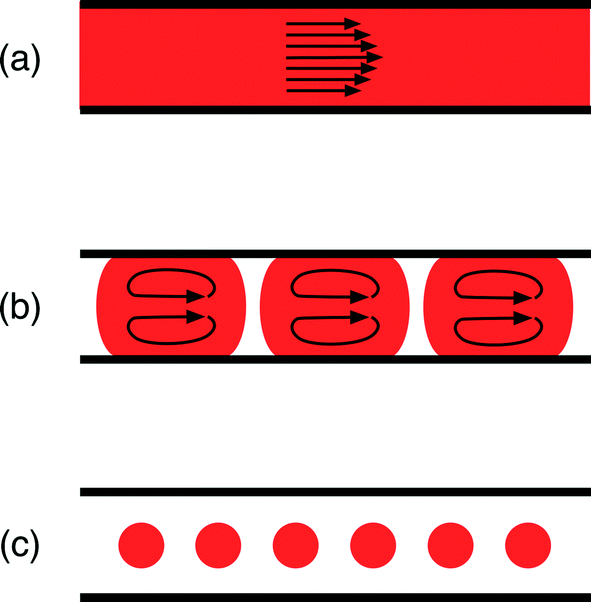 | ||
| Fig. 1 Diagram showing three types of fluid flow in a microfluidic channel: (a) single-phase continuous flow, where contact with the channel walls leads to a parabolic flow profile; (b) segmented or “slug” flow where the reaction mixture is divided up into discrete units by an immiscible fluid; (c) droplet flow where the reaction mixture is completely isolated from the channel walls by the immiscible fluid. | ||
Two-phase reactors elegantly overcome the drawbacks of single-phase reactors. Injection of an additional immiscible fluid (which can be a gas or a liquid) into the channel divides the reaction mixture into a succession of discrete “slugs” or “droplets” (Fig. 1(b) and (c)) that pass through the reactor at a common speed, eliminating velocity dispersion. In the case of slug flow, the reaction mixture still makes contact with the channel wall, but abrasion from the second phase can help to reduce or even eliminate fouling, prolonging reactor lifetimes. Yen et al. were the first to use gas–liquid segmented flow reactors for high temperature nanocrystal synthesis, reporting the synthesis of CdSe from cadmium 2,4-pentanedionate and selenium at 260 °C.18 They achieved good size control by tuning the injection rates of the cadmium and selenium precursor solutions and reported significantly narrower emission spectra than an equivalent single-phase synthesis, consistent with the non-dispersive motion of the reagent slugs.
In the case of droplet flow, the reaction mixture is fully isolated from the wall by the immiscible liquid, thereby preventing reagent deposition from happening in the first place. Furthermore, if the droplets are large enough to fill the cross-section of the channel, they are forced to move in a direction parallel to the channel and cannot take unguided off-axis routes that would induce velocity dispersion. The use of droplet reactors for nanocrystal synthesis was first demonstrated by Shestopalov et al.19 They used chip-based oil–water droplet reactors operating at room temperature to prepare cadmium sulphide quantum dots from ionic cadmium and sulphur precursor solutions. Chan and co-workers subsequently developed a high temperature chip-based droplet reactor in glass, using octadecene as the droplet phase, a high boiling point perfluorinated polyether as the carrier fluid, and a surface coating of perfluoroalkylsilane to ensure preferential wetting of the channel walls by the carrier phase.20 They were able to controllably synthesise high quality CdSe by reacting cadmium oxide and elemental selenium in the presence of oleic acid and trioctylphosphine at 300 °C, achieving good stable operation for up to five hours of continuous use.
Nightingale et al. later developed a capillary-based droplet reactor, using polytetrafluoroethylene tubing, capable of operating at temperatures of up to 250 °C.21 Using the same carrier fluid and reagents as Chan et al. they synthesized CdSe quantum dots continuously over a 24 hour period, obtaining consistent product over the full duration of the synthesis run, with no drift in the spectral properties of the resultant particles. They applied the same reactor to the synthesis of nanocrystalline silver, titania and (in later work22) superparamagnetic iron oxide, reporting good operational stability in all cases. Indeed, the titania synthesis involved the formation of insoluble titanium oxide hydrate intermediates, clearly visible as blue clouds inside the droplets, yet did not cause reactor fouling. The authors noted anecdotally that during six months of testing with a wide variety of materials systems and synthesis routes they had yet to experience a single instance of reactor fouling, confirming the effectiveness of the droplet regime in preventing reactor fouling. Capillary reactors have most recently been applied by Lignos et al. to the high temperature synthesis of near-infra-red-emitting PbS and PbSe nanocrystals, with excellent operational stability again being reported over prolonged (three hour) operation.23
The best flow regime to use is dependent on the chemistry at hand. When single-phase flow can be used, it is clearly the simplest method to implement. However, there are few (if any) situations where single-phase flow can be expected to offer a clear performance advantage over two-phase flow, and the circumstances in which it can be reliably applied are limited. In our experience single-phase reactors are suited to a narrow range of low temperature chemistries in aqueous solutions. High temperature reactions in organic solvents inevitably cause single-phase reactors to foul, and should instead be carried out in two-phase reactors. For research purposes, slug-flow and droplet-flow may often be used interchangeably. In our own research, however, we work mainly with droplet-based systems, due to their increased resilience to reactor fouling. (We prefer methods that could in principle be transferred directly from the laboratory to a production environment without alteration).
One issue with two phase systems that requires further attention is the potential influence of the second (supposedly inert) phase on the nucleation and growth kinetics of the nanocrystals (and hence on their final crystal structure, defect levels and polydispersity). While the second phase is typically chosen to be unreactive, some degree of interaction with the solvent phase is inevitable due to the high interfacial contact areas. If any reagents or reaction products are soluble in the carrier liquid, they will partition between the two phases on the basis of relative solubility. Fluorous carrier liquids for instance can support high levels of dissolved gases, allowing gaseous side products from a reaction to leech from the solvent into the carrier liquid. Conversely, pre-dissolved gases in the carrier liquid (originating for example from imperfect degassing or deliberate loading) may pass into the solvent phase. The effects of solute exchange between the two phases has been largely overlooked in the microreactor literature, but can clearly be expected to have a significant influence on the final particle properties. Indeed, at an extreme level, it forms the basis of multiphase synthesis routes where precursors are dissolved in separate phases and meet and react at the phase boundaries. Multiphase chemistries have been widely exploited in batch nanocrystal synthesis but, with a few notable exceptions (see e.g.ref. 24 and 25), have rarely been applied on the microscale. This is somewhat surprising since the large interfacial contact areas and short diffusion distances in microreactors should enhance reaction rates relative to batch reactions, leading to shorter nucleation times and better controlled product.
3. Where next?
3.1. Plug-and-play droplet manipulation
From a stability perspective, droplet reactors represent a near ideal environment in which to perform nanocrystal synthesis, but they are not without their own limitations. To date they have been largely restricted to simple one-step procedures, in which all reagents are loaded into the droplets at the outset. There are very few reports of multistep nanocrystal synthesis in droplet reactors due to the difficulty of performing discrete chemical operations on individual droplets in a moving droplet stream.In the past decade a wide range of creative solutions have been devised for manipulating droplets, including synchronization, loading, merging, splitting, trapping, dilution and concentration.26 Using an appropriate series of these operations, it is possible to envisage sophisticated multistep synthesis procedures being carried out in a single uninterrupted production line. Many of these techniques, however, have been developed for analytical applications where reliable operation is required only over a narrow set of operating conditions. In particular many droplet procedures have been specifically developed for the manipulation of aqueous droplets, limiting the range of chemistries and materials to which they can be applied. What is required for droplet chemistry to become a standard tool for nanocrystal (and other) synthesis is a series of robust, modular components that can be easily inserted into a droplet-based fluidic network and readily applied to a broad range of solvents and reaction conditions – a flow-based equivalent of Quickfit glassware in batch chemistry. Until such tools are routinely available, droplet reactors will continue to lag behind single-phase reactors in terms of the range of accessible chemistries and materials.
The most important operation for multistep chemistry is reagent addition, which requires the ability to deliver precise quantities of reagents to the droplets as they move. One way to do this is via droplet fusion, introducing the new reagents into the flow reactor as a separate droplet stream which is then merged with the original droplet stream on a pairwise basis. This can be achieved using special channel architectures to bring the droplets together, sometimes assisted by the application of an electric field to lower the interfacial tension between droplets. Hung and co-workers used this approach in a polydimethylsiloxane (PDMS) chip-based device to generate CdS nanocrystals from an alternating train of Cd2+- and S2−-containing aqueous droplets in silicone oil, using a tapered chamber to induce pairwise droplet merging.27 They identified a narrow range of flow conditions over which reliable pairwise merging could be achieved, with conditions outside this range leading to random coalescence of multiple droplets or no merging at all. Frenz et al. later reported an improved chip-based procedure, using an AC electric field to induce pairwise fusion of two water-in-oil droplet streams at flow-rate ratios of up to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5.28 The reactor was successfully applied to the synthesis of superparamagnetic iron oxide nanoparticles by incorporating an iron salt precursor and ammonium hydroxide into the two droplet streams, with co-precipitation of the iron oxide particles occurring after droplet merging due to the increased pH.
:5.28 The reactor was successfully applied to the synthesis of superparamagnetic iron oxide nanoparticles by incorporating an iron salt precursor and ammonium hydroxide into the two droplet streams, with co-precipitation of the iron oxide particles occurring after droplet merging due to the increased pH.
The above methods were applied in aqueous systems, although it is possible the method of Hung et al. could be generalized to organic droplets. (The second method relies on electrofusion and cannot therefore be applied to non-polar organic solvents). The two techniques, moreover, were applied to a single droplet-fusion step and it is not clear whether they could be applied repeatedly to carry out multistep chemical operations. As an alternative to droplet fusion, direct injection can be used to add reagents.29 In this approach, the new reagent is injected into the flowing droplet stream as a continuous laminar stream that spontaneously adds to the droplets as they pass. In principle direct injection is the easier of the two methods to implement since it does not rely on special channel structures or extraneous equipment, making it the more practical approach for repeated reagent injection. In practice, however, it is typically rather unreliable, with the injected reagent adding inconsistently to the flowing droplets and frequently forming new droplets in preference to adding to the existing ones.
In recent work Nightingale and co-workers reported an improvement to the direct injection method, in which a third inert gaseous phase was injected alongside the solvent and the immiscible fluid.30 The gas maintained an even separation of solvent droplets, ensuring they received equal amounts of the injected reagent, and also suppressed the unwanted formation of new droplets. Using this approach they carried out a five-step quantum dot synthesis, in which feedstock was repeatedly added to the growing nanocrystals to sustain particle growth. To our knowledge, this is the first example of a multistep dosing procedure being applied in the field of nanocrystal synthesis. Techniques like this one, which offer a means of repeatedly dosing droplets, can be expected to play a critical role in extending the reach of microfluidics in nanocrystal synthesis.
Beyond synthesis of the nanocrystal itself, additional steps such as surface modification, purification and phase transfer are typically required to obtain a usable material. In virtually all cases, these procedures are carried out using standard batch techniques and little if any work has been reported on in-line post-synthesis processing. Such processing is particularly important for a production environment where the ability to combine synthesis and purification/treatment in a single continuous process would significantly enhance efficiency, reliability and control. The need for robust post-processing methodologies is set to grow steadily as quantum dots and other precision engineered nanocrystals gain deeper industrial traction, and the need to purify, functionalise or otherwise modify large (kilogram) quantities arises. Batch purification procedures, which typically rely on repeated cycles of washing and centrifugation, are ill-suited to large volumes. A fully integrated flow-based procedure capable of converting injected reagents to fully processed ready-to-use nanocrystals would consequently have significant industrial benefit.
3.2. Inline analysis for product optimisation
Full exploitation of microfluidic synthesis procedures requires real-time information about the progression of the reaction, allowing appropriate changes to be made to the reaction conditions to optimise yield, size-distributions or physico-chemical properties. Optical techniques are especially easy to integrate with microfluidic devices due to their non-invasive nature, and can generate useful and immediate information about the nanocrystal properties.31 Absorption and fluorescence methods are routinely used to extract information about size distributions and surface uniformity. However there is a much broader range of optical methods that could in principle be employed, with light-scattering and time-dependent fluorescence spectroscopy being particularly interesting in this regard due to their respective abilities to probe particle size and fluorescence quantum efficiency. Beyond optical methods there is a range of other techniques that can provide important information about crystal structure, crystallite shape or reaction yield. Size exclusion chromatography,32,33 gel electrophoresis34 and particle tracking can provide information about size distributions, while nuclear magnetic resonance spectroscopy35 can characterise nanocrystal surface chemistry. Traditionally these analysis techniques have been considered off-line techniques but, as new miniaturised systems are developed and equipment costs fall, it is becoming possible to integrate dedicated analysis systems into flow reactors, greatly increasing the depth of information available at the time of particle production.In the simplest case, this information can be used to manually tweak the reaction conditions to obtain a better product. However, by coupling both the sensor(s) and reactor to real-time control algorithms, the entire process of nanocrystal synthesis can be automated, with the control algorithm repeatedly and systematically updating the reaction conditions until a desired end-point is achieved. This approach was used by Krishnadasan et al. to develop a fully automated system for synthesizing CdSe quantum dots of a desired emission wavelength and optimized fluorescence quantum yield.36 Toyota et al. later reported a rapid screening system for CdSe quantum dots, using five parallel microreactors and inline fluorescence spectroscopy to rapidly trawl through multiple reaction conditions with a view to obtaining improved particle characteristics and uncovering new mechanistic information (Fig. 2).37 Taking the idea of self-optimisation one step further, instead of designing specific reactors for a particular chemical procedure (as is done now), one might consider the development of generic multipurpose reactors whose architectures can autonomously adapt to a new set of reagents so as to achieve a given objective. (The software controlling the reactors might even suggest changes or extensions to the reactor if the objective proves unachievable with the original reactor configuration). This “open-ended” approach to reactor design could potentially drive chemical exploration in highly productive directions that a human chemist would never consider taking in advance.
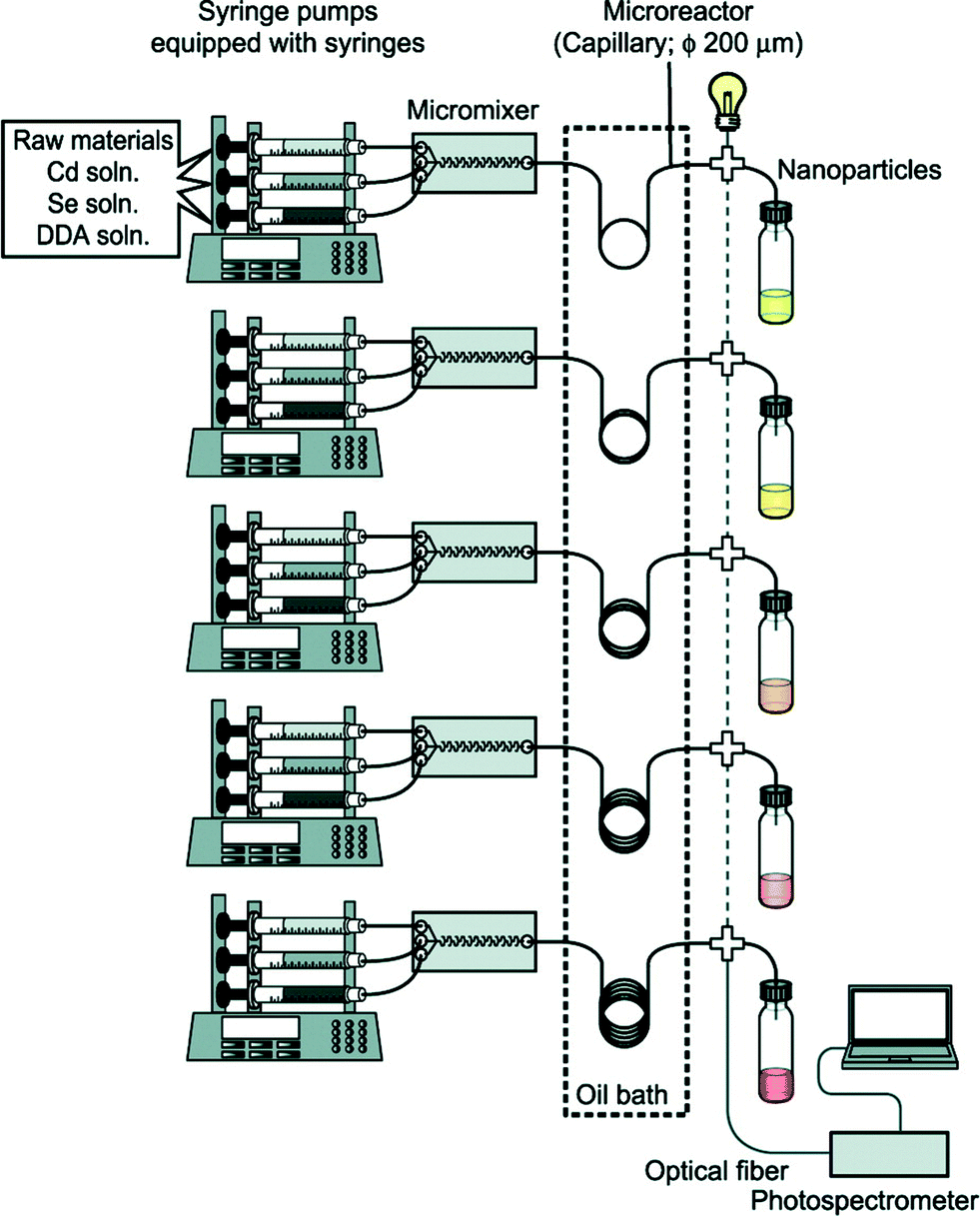 | ||
| Fig. 2 Schematic of a microfluidic system developed by Toyota et al. for the rapid combinatorial synthesis of CdSe quantum dots. Reprinted with permission from ref. 37. Copyright 2010 American Chemical Society. | ||
The use of autonomous control and rapid screening methodologies could transform nanocrystal science, enabling the discovery of superior-performing or novel particles that form under conditions outside the usual bounds of chemical exploration. However, while much of the technology already exists for applying such techniques, uptake has been slow. Even outside the field of nanocrystal synthesis, there are relatively few examples of self-optimising microreactors being used to improve on manual procedures (refer to McMullen and Jensen for a comprehensive review38). In part this is because the techniques remain hard to apply, requiring a broader range of skills than are typically found in a single person or small group of researchers. Requisite expertise spans the fields of synthetic chemistry, microfluidics, sensor design/integration, computational science, and applied mathematics. To change this situation, easy-to-use software tools are needed that place automation technologies in the hands of the practicing chemist. Until that happens, we are unlikely to realise the full potential of microfluidics for nanocrystal synthesis.
3.3. Scale out
To date there has been little interest in microreactors as a potential production technology for nanocrystals due to a widespread perception that microreactors cannot satisfy industrial demand. This perception is certainly reinforced by the literature, where virtually all reports of materials synthesis in microreactors – nanocrystalline or otherwise – have involved small sub-gram levels of production. Throughput may be raised substantially by upping reagent concentrations, increasing flow rates and scaling out, i.e. operating N identical reaction channels in parallel to achieve an N-fold increase in output relative to a single-channel reactor. Scaling out is an especially attractive approach since it requires no changes to the reactor geometry, and so allows materials throughput to be increased without any changes to the underlying process chemistry. Hence, in contrast to the scaling up of batch reactions, scaling out can in principle be implemented without any detriment to product quality or yield.Despite its obvious importance, scale out has been a rather neglected aspect of microreactor technology, with only a handful of reports describing its application to large-scale materials production. The most notable success in this area was reported by Chambers and co-workers, who developed a thirty-channel device for the direct fluorination of organic molecules using fluorine gas.39 They noted that twenty such reactors would have a combined throughput of ~3 kg per day, comparable to many fine-chemical processes. The extension of this approach to nanocrystals is appealing, but nanocrystal synthesis raises additional challenges due to the substantial risk of reactor fouling and the need to maintain identical conditions in all channels at all times (so as to ensure product consistency). In recent work Nightingale and co-workers reported a five-channel droplet-based microfluidic reactor for CdTe synthesis designed specifically to address these issues (Fig. 3).40 The precursor solution and carrier fluids were drawn from separate reservoirs, and split five ways using passive flow dividers. Individual streams of the precursor solution and carrier fluid were then merged on a pairwise basis in five separate droplet generators, creating parallel droplet streams in five intertwined capillaries. The intertwined capillaries were passed into a heated oil bath to initiate nucleation and growth, and the entire droplet stream from each one was then collected in a single beaker. The carrier fluid sank to the bottom of the beaker, leaving the product as a discrete layer on top. (The carrier fluid was recycled in a closed-loop manner to minimize usage). During a nine hour production run, 54.4 g of material was produced corresponding to a production rate of 145 g per day. Absorption and fluorescence spectroscopy showed no change in product properties between channels and over time, confirming the stability of the multichannel droplet reactor.
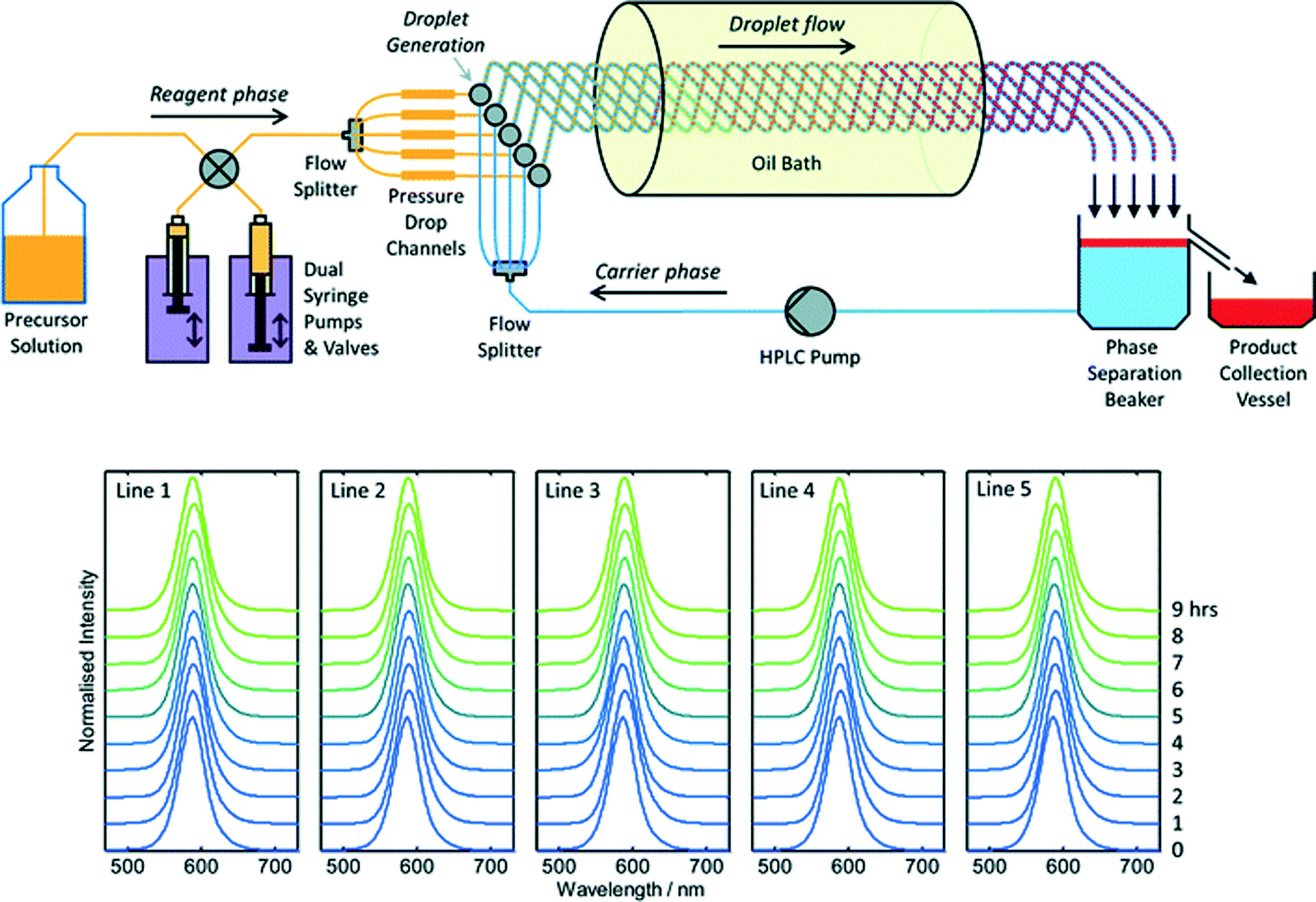 | ||
| Fig. 3 Top: schematic of a scaled-out five-channel droplet flow reactor developed by Nightingale et al. for the synthesis of CdTe quantum dots at a rate of 145 g per day. Bottom: normalised solution-phase emission spectra of CdTe collected at hourly intervals from each of the five reaction lines over a period of nine hours. Adapted from ref. 40 with permission from The Royal Society of Chemistry. | ||
The 145 g per day level of throughput – whilst in itself a significant production rate for nanomaterials – was obtained using a modest five-way level of parallelisation. With only minimal changes to the reactor architecture, it should be feasible to expand the architecture to forty channels or more, providing ready access to production rates in excess of 1 kg per day. A key challenge now is to apply scale out procedures to multistep chemistries, rather than the simple single-step synthesis used by Nightingale et al., in order to access a broader range of materials systems. This is likely to require the development of robust fault-tolerant architectures that can withstand the failure of individual channels so that localized problems in a single channel do not cause catastrophic failure of the entire system. Little work has previously been carried out to address these issues, and the feasibility of applying scale out methods on a massively parallel multistep basis remains an open question. The answer to this question may ultimately determine whether microfluidics has a role to play in future industrial production.
3.4. Thinking in flow
Until now nanoparticle synthesis in microreactors has largely involved simple – albeit well controlled – adaptations of conventional macroscale synthesis procedures. However, microfluidics offers the chance to rethink the entire approach to nanoparticle synthesis by using techniques that are not available in macroscale chemistry. There have already been some efforts to exploit these advantages.41 For instance Erdem et al. reported a silicon-based segmented flow microreactor for the synthesis of TiO2 nanoparticles, incorporating separate hot and cool temperature zones to define a short period of nucleation followed by a longer period of slow, controlled growth – processes that are hard to separate in a batch system due to the difficulty of changing temperatures quickly.42 However, this kind of thinking-in-flow needs to go much further, with new chemistries being developed that are specifically tailored to exploit the advantages of a flow-based microscale environment. One very interesting option here is to use non-photochemical laser-induced nucleation processes to initiate particle formation.43 In contrast to a batch environment (where this approach has previously been applied), illuminating a specific point on the flow profile would sequentially activate the entire reaction volume. By defining a sufficiently tight illumination zone, it should be possible to confine the nucleation phase to a millisecond duration or less, far shorter than could be achieved by heating and cooling (Fig. 4).44 Improvements in size and shape control could be significant.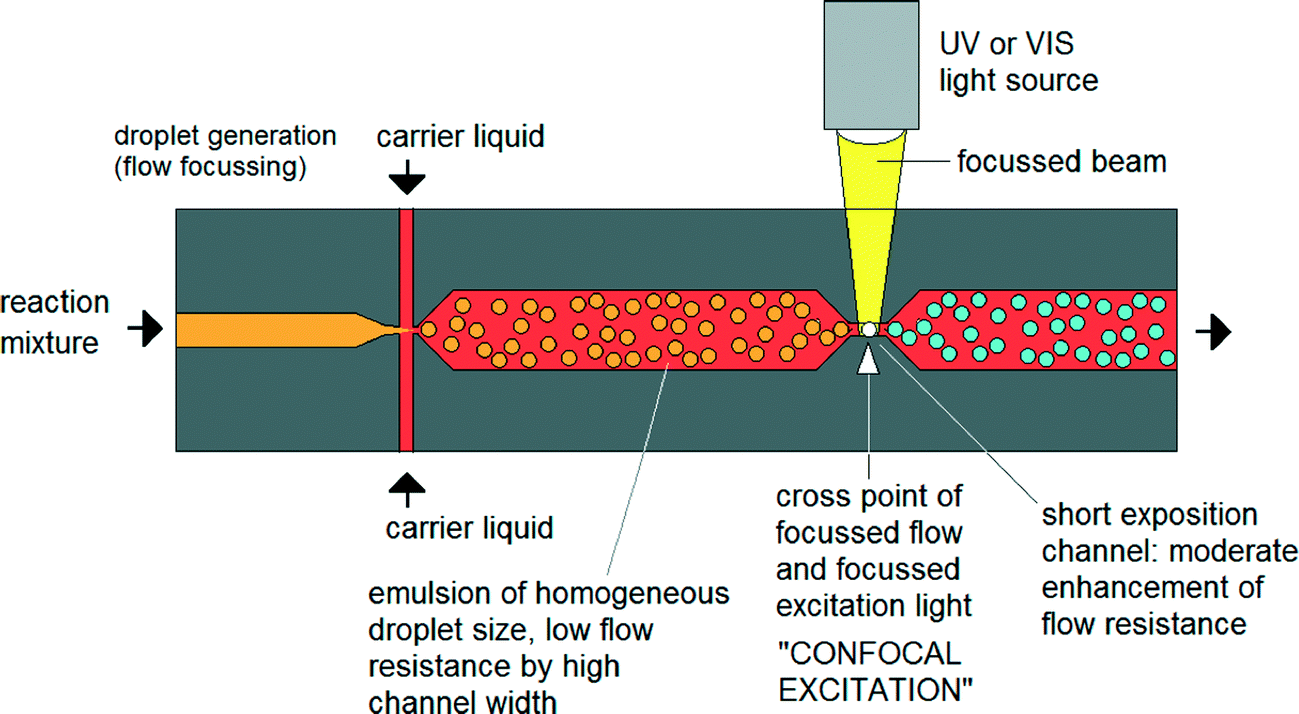 | ||
| Fig. 4 Concept of focused nucleation by confocal excitation as proposed by Köhler et al. Adapted from ref. 44 with permission from John Wiley and Sons. | ||
Many batch syntheses perform best under reflux conditions that cannot be used in flow. As an effective alternative, however, flow reactions can be readily performed under supercritical conditions through the use of a back pressure regulator that permits fluid flow only after a target pressure has been reached. In this way the boiling point of the solvent can be increased above its standard value, allowing higher reaction temperatures to be accessed. Early work by Marre, Park and co-workers confirmed the importance of supercritical reactions for nanocrystal synthesis. They developed a pressurized high-temperature microreactor to synthesise quantum dots under supercritical conditions (Fig. 5), showing that synthesis in supercritical hexane led to narrower emission line-widths and size distributions than the equivalent reaction in liquid (i.e. non-supercritical) squalane.45 There is a clear need to build on this work, with a view to exploring and exploiting the wider chemical parameter space that supercritical conditions unlock.
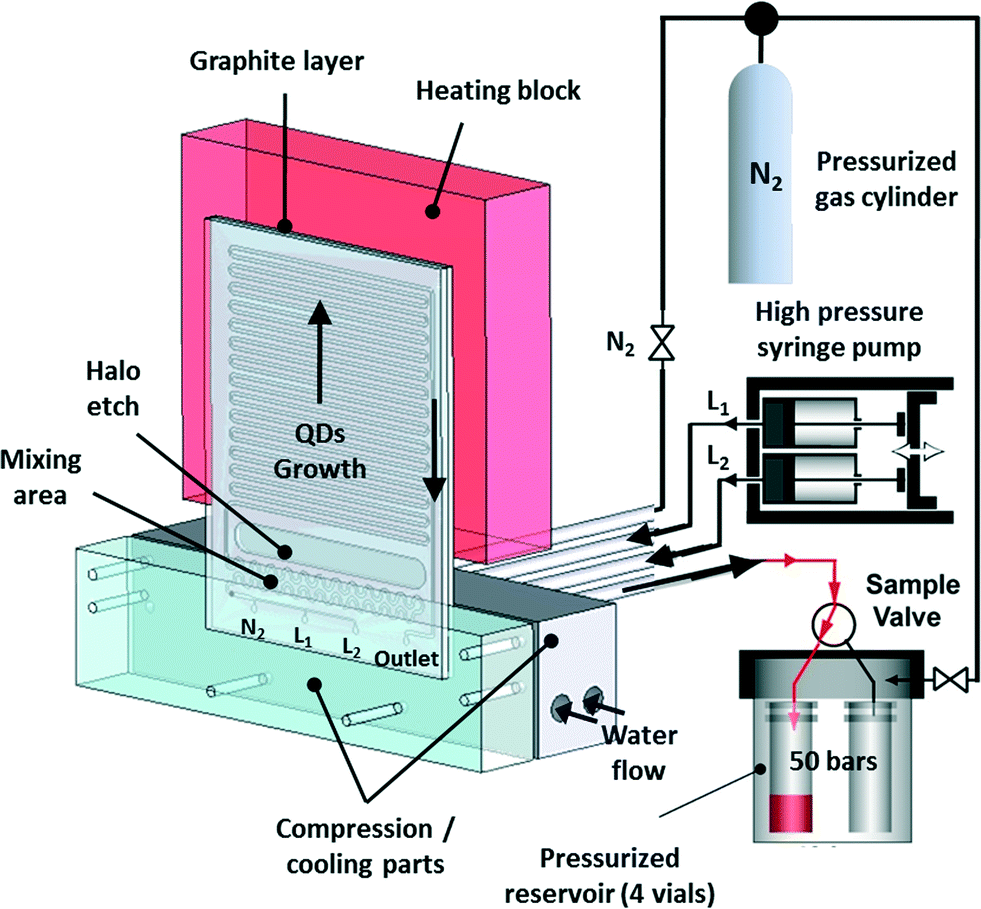 | ||
| Fig. 5 High-pressure, high-temperature microreactor developed by Marre, Park et al., comprising a compression-cooling aluminium part, a high-pressure syringe pump, a five-way high-pressure valve, and a high-pressure reservoir containing four vials. Reproduced from ref. 45 with permission from John Wiley and Sons. | ||
4. Conclusion
In summary the past twelve years have seen some significant advances in the application of microreactors to nanocrystal synthesis. Key innovations include the transition from single-phase to two-phase flow reactors for improved reactor stability, the use of supercritical reaction conditions, the integration of sensor technology for real-time product evaluation, the use of control algorithms to optimize nanocrystal properties, and the demonstration of scale out as a viable means of increasing production volumes. While each of these developments has individually been shown to offer important advantages in terms of improved reaction control and product quality, there is now a need to turn limited proof-of-principle demonstrations into mature technologies that can be routinely applied to real-world multistep chemical syntheses.It is evident that the microfluidic reactor is still a long way from displacing the round-bottomed flask as the principal reaction environment for nanocrystal synthesis. Much will need to change before there is a realistic prospect of it doing so. The current strategy of adapting simple one- and two-step batch procedures to flow has yielded promising early results. However, its limitations with regards to the palette of accessible materials are now becoming clear. The full benefits of microscale synthesis will only be realized when nanocrystal synthesis starts to be addressed from a flow-first perspective, using reaction schemes that have been designed from the ground-up for multistep synthesis in flow. Exactly what impact this will have on nanoscale science and technology remains to be seen, but we look forward to reflecting on the question again in another twelve years' time.
Acknowledgements
TOC graphic kindly supplied by Dr Adrian Nightingale.References
- D. V. Talapin, J.-S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389–458 CrossRef CAS PubMed.
- L. L. Chng, N. Erathodiyil and J. Y. Ying, Acc. Chem. Res., 2013, 46, 1825–1837 CrossRef CAS PubMed.
- J. A. Webb and R. Bardhan, Nanoscale, 2014, 6, 2502 RSC.
- J. B. Edel, R. Fortt, J. C. de Mello and A. J. de Mello, Chem. Commun., 2002, 1136–1137 RSC.
- A. Nightingale and J. C. de Mello, Adv. Mater., 2013, 25, 1813–1821 CrossRef CAS PubMed.
- S. Marre and K. F. Jensen, Chem. Soc. Rev., 2010, 39, 1183–1202 RSC.
- L. Zhang and Y. Xia, Adv. Mater., 2014, 26, 2600–2606 CrossRef CAS PubMed.
- A. Nightingale and J. C. de Mello, J. Mater. Chem., 2010, 20, 8454–8463 RSC.
- Y. Song, J. Hormes and C. S. S. R. Kumar, Small, 2008, 4, 698–711 CrossRef CAS PubMed.
- K. S. Krishna, Y. Li, S. Li and C. S. S. R. Kumar, Adv. Drug Delivery Rev., 2013, 65, 1470–1495 CrossRef CAS PubMed.
- C.-H. Chang, B. K. Paul, V. T. Remcho, S. Atre and J. E. Hutchison, J. Nanopart. Res., 2008, 10, 965–980 CrossRef CAS.
- V. Sebastian Cabeza, S. Kuhn, A. A. Kulkarni and K. F. Jensen, Langmuir, 2012, 28, 7007–7013 CrossRef CAS PubMed.
- S. Duraiswamy and S. A. Khan, Small, 2009, 5, 2828–2834 CrossRef CAS PubMed.
- Y. H. Kim, L. Zhang, T. Yu, M. Jin, D. Qin and Y. Xia, Small, 2013, 9, 3462–3467 CrossRef CAS PubMed.
- Z. Wan, W. Luan and S.-T. Tu, J. Phys. Chem. C, 2011, 115, 1569–1575 CAS.
- H. Yang, W. Luan, Z. Wan, S.-T. Tu, W.-K. Yuan and Z. M. Wang, Cryst. Growth Des., 2009, 9, 4807–4813 CAS.
- S. Krishnadasan, J. Tovilla, R. Vilar, A. J. de Mello and J. C. de Mello, J. Mater. Chem., 2004, 14, 2655–2660 RSC.
- B. K. H. Yen, A. Günter, M. A. Schmidt, K. F. Jensen and M. G. Bawendi, Angew. Chem., Int. Ed., 2005, 44, 5447–5451 CrossRef CAS PubMed.
- I. Shestopalov, J. D. Tice and R. F. Ismagilov, Lab Chip, 2004, 4, 316–321 RSC.
- E. M. Chan, A. P. Alivisatos and R. A. Mathies, J. Am. Chem. Soc., 2005, 127, 13854–13861 CrossRef CAS PubMed.
- A. Nightingale, S. H. Krishnadasan, D. Berhanu, X. Niu, C. Drury, R. Mcintyre, E. Valsami-Jones and J. C. de Mello, Lab Chip, 2011, 11, 1221–1227 RSC.
- K. Kumar, A. Nightingale, S. H. Krishnadasan, N. Kamaly, M. Wylenzinska-Arridge, K. Zeissler, W. R. Branford, E. Ware, A. J. deMello and J. C. de Mello, J. Mater. Chem., 2012, 22, 4704–4708 RSC.
- I. Lignos, L. Protesescu, S. Stavrakis, L. Piveteau, M. J. Speirs, M. A. Loi, M. V. Kovalenko and A. J. deMello, Chem. Mater., 2014, 26, 2975–2982 CrossRef CAS.
- M. Takagi, T. Maki, M. Miyahara and K. Mae, Chem. Eng. J., 2004, 101, 269–276 CrossRef CAS PubMed.
- S. Li, J. Xu, Y. Wang and G. Luo, Langmuir, 2008, 24, 4194–4199 CrossRef CAS PubMed.
- S.-Y. Teh, R. Lin, L.-H. Hung and A. P. Lee, Lab Chip, 2008, 8, 198 RSC.
- L.-H. Hung, K. M. Choi, W.-Y. Tseng, Y.-C. Tan, K. J. Shea and A. P. Lee, Lab Chip, 2006, 6, 174–178 RSC.
- L. Frenz, A. El Harrak, M. Pauly, S. Bégin-Colin, A. D. Griffiths and J.-C. Baret, Angew. Chem., Int. Ed., 2008, 47, 6817–6820 CrossRef CAS PubMed.
- Y. Wang, E. Tumarkin, D. Velasco, M. Abolhasani, W. Lau and E. Kumacheva, Lab Chip, 2013, 13, 2547–2553 RSC.
- A. Nightingale, T. W. Phillips, J. H. Bannock and J. C. de Mello, Nat. Commun., 2014, 5, 3777 Search PubMed.
- J. Yue, J. C. Schouten and T. A. Nijhuis, Ind. Eng. Chem. Res., 2012, 51, 14583–14609 CrossRef CAS.
- G.-T. Wei, F.-K. Liu and C. R. C. Wang, Anal. Chem., 1999, 71, 2085–2091 CrossRef CAS PubMed.
- F.-K. Liu, Chromatographia, 2010, 72, 473–480 CAS.
- M. Hanauer, S. Pierrat, I. Zins, A. Lotz and C. Sönnichsen, Nano Lett., 2007, 7, 2881–2885 CrossRef CAS PubMed.
- Z. Hens and J. C. Martins, Chem. Mater., 2013, 25, 1211–1221 CrossRef CAS.
- S. Krishnadasan, R. J. C. Brown, A. J. de Mello and J. C. de Mello, Lab Chip, 2007, 7, 1434–1441 RSC.
- A. Toyota, H. Nakamura, H. Ozono, K. Yamashita, M. Uehara and H. Maeda, J. Phys. Chem. C, 2010, 114, 7527–7534 CAS.
- J. P. McMullen and K. F. Jensen, Annu. Rev. Anal. Chem., 2010, 3, 19–42 CrossRef CAS PubMed.
- R. D. Chambers, M. A. Fox, D. Holling, T. Nakano, T. Okazoe and G. Sandford, Lab Chip, 2005, 5, 191–198 RSC.
- A. Nightingale, J. H. Bannock, S. H. Krishnadasan, F. T. F. O'Mahony, S. A. Haque, J. Sloan, C. Drury, R. McIntyre and J. C. de Mello, J. Mater. Chem. A, 2013, 1, 4067 CAS.
- H. Yang, W. Luan, S.-T. Tu and Z. M. Wang, Lab Chip, 2008, 8, 451–455 RSC.
- E. Y. Erdem, J. C. Cheng, F. M. Doyle and A. P. Pisano, Small, 2013, 10, 1076–1080 CrossRef PubMed.
- B. Garetz, J. Aber, N. Goddard, R. Young and A. Myerson, Phys. Rev. Lett., 1996, 77, 3475–3476 CrossRef CAS.
- J. M. Köhler, S. Li and A. Knauer, Chem. Eng. Technol., 2013, 36, 887–899 CrossRef.
- S. Marre, J. Park, J. Rempel, J. Guan, M. G. Bawendi and K. F. Jensen, Adv. Mater., 2008, 20, 4830–4834 CrossRef CAS.
Footnote |
| † These authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2014 |