 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Small-molecule axon-polarization studies enabled by a shear-free microfluidic gradient generator†
Hui
Xu
,
Meghaan M.
Ferreira
and
Sarah C.
Heilshorn
*
Department of Materials Science and Engineering, Stanford Cardiovascular Institute, Stanford University, 476 Lomita Mall, McCullough Building, Stanford, CA 94305-4045, USA. E-mail: heilshorn@stanford.edu
First published on 14th April 2014
Abstract
A deep understanding of the mechanisms behind neurite polarization and axon path-finding is important for interpreting how the human body guides neurite growth during development and response to injury. Further, it is of great clinical importance to identify diffusible chemical cues that promote neurite regeneration for nervous tissue repair. Despite the fast development of various types of concentration gradient generators, it has been challenging to fabricate neuron-friendly (i.e. shear-free and biocompatible for neuron growth and maturation) devices to create stable gradients, particularly for fast diffusing small molecules, which typically require high flow and shear rates. Here we present a finite element analysis for a polydimethylsiloxane/polyethylene glycol diacrylate (PDMS/PEG-DA) based gradient generator, describe the microfabrication process, and validate its use for neuronal axon polarization studies. This device provides a totally shear-free, biocompatible microenvironment with a linear and stable concentration gradient of small molecules such as forskolin. The gradient profile in this device can be customized by changing the composition or width of the PEG-DA barriers during direct UV photo-patterning within a permanently bonded PDMS device. Primary rat cortical neurons (embryonic E18) exposed to soluble forskolin gradients for 72 h exhibited statistically significant polarization and guidance of their axons. This device provides a useful platform for both chemotaxis and directional guidance studies, particularly for shear sensitive and non-adhesive cell cultures, while allowing fast new device design prototyping at a low cost.
Introduction
Regeneration of tissue in the central nervous system remains a critical unmet clinical challenge. Studies in the past 10 years have discovered a variety of mechanical and chemical cues that influence neuronal cell migration, neurite growth, and axon path-finding. In addition to traditional large molecules such as proteins, there has been persistent interest in small molecules for their potential clinical use to induce neuronal chemotaxis, neurite growth and axon guidance. Small molecules typically have better oral bioavailability, faster pharmacokinetics, and usually are less challenging for large-scale production at low cost.Forskolin is an example of a small molecule known to enhance neurite growth1 and neuronal survival.2,3 Forskolin promotes axon formation, growth cone turning and axon path finding through the activation of adenylyl cyclase, which increases intracellular levels of cyclic adenosine monophosphate (cAMP) and leads to activation of cAMP-sensitive pathways, such as protein kinase A (PKA) and exchange protein activated by cAMP (Epac).4–6 Previous in vivo studies have used local injections3 and genetic mutations in animals, while in vitro studies have used micropipette assays6,7 and substrates patterned with biochemical stripes5 to elucidate the effects of forskolin on neurons. However, multi-day in vitro exposure of mammalian neurons to stable, soluble forskolin gradients has not previously been possible.
A variety of in vitro chemotaxis assays have been developed to investigate how various factors act individually or collectively to regulate cell movement by creating a spatial concentration gradient of compounds of interest. Traditional in vitro chemotaxis assays (e.g., micropipette assays,7,8 diffusion chambers,9–11 Boyden chambers,12–14 Zigmond chambers,15 and Dunn chambers16) create gradient profiles that are often transient and unstable over multi-day time-scales. Mammalian neurons grow slowly in comparison to other cell cultures, thus precluding their use in studies that utilize these assays. In addition, many of these techniques are incompatible with direct cell-imaging, which prevents quantitative single-cell analysis, making it impossible to distinguish between chemokinesis, chemotaxis, and chemoproliferation.17 The advent of soft lithography18 for producing microfluidic devices led to the development of microfluidic gradient generators for studying chemotaxis. The first generation of these devices exposed the cells to shear flow, which can bias cell movement and is detrimental to shear-sensitive cells, such as neurons.19–25 The high diffusivity of small molecules, such as forskolin, exacerbates this issue by requiring higher flow rates to compensate for the rapid decay of the concentration gradient. Recently developed microfluidic platforms include a family of devices that utilize various strategies to restrict convective flow while still enabling Fickian diffusion to generate either soluble or surface-bound26,27 chemical gradients. Micro-jets28,29 take advantage of the incompressibility of fluid, which directs the micro-jet flow upward, providing a shear-free gradient-generating region everywhere except the boundary near the micro-jets. An alternative approach modified the original serpentine device designed by Whitesides et al.19,21 by layering it on top of a series of microwells, which creates a controllable gradient without the high shear stress. Other strategies use increased fluidic resistance to reduce shear, by designing a gradient-generating region with a significantly smaller height than the main channels30–34 or by separating the gradient-generating region from the main channels using microcapillaries.35–40 Membranes10,41–43 and hydrogels44–52 have also been incorporated into gradient-generating devices to increase fluidic resistance.
Hydrogels are an ideal candidate for restricting convective flow while still enabling diffusion of small molecules. Microfluidic device designs have used an assortment of both natural and synthetic hydrogels for generating chemical gradients, including agarose,45–47 Matrigel,44 collagen,48 and polyethylene glycol (PEG).49,50 Although prepolymers can be crosslinked via irradiation, chemical cross-linking agents, ion solutions, temperature control, or protein–protein interactions,53,54 light patterning allows precise temporal and spatial control of polymerization without direct contact between the UV light source and the precursor solution, as demonstrated by the work of Doyle et al. on stop-flow lithography.55 UV curable polymers include polyethylene glycol diacrylate (PEG-DA), dextran acrylate, Pluronic™-DA and many others.56–58 PEG is an FDA-approved material for medical use and is a well-known hydrophilic, biocompatible material widely used for surface modification of biomedical devices. PEG and/or PEG-DA crosslinked microstructures have been used in cell culture,59–61 drug delivery,62 pH sensing,63 mechanical gradient formation in materials,64,65 chemokine gradient formation in 2D,49 and 3D crosslinked integrin-ligand gradients.66
Despite the wide use of PEG and PEG-DA hydrogels in biotechnology applications, PEG structures do not bind strongly to PDMS, making it challenging to generate microfluidic pathways using these materials. Oxygen inhibits the crosslinking reaction at the PDMS boundary, creating a thin layer of uncrosslinked prepolymer between the PEG structures and the PDMS walls.55,67 For this reason, most microfluidic systems that use PEG structures require a separate manifold to form a good seal.49,50,68 This fabrication process requires external housing systems, which are prone to liquid leakage and can be challenging to assemble. To overcome this challenge, we have developed a novel fabrication process that utilizes surface silanization and vacuum filling to aid direct photopatterning of PEG-DA hydrogel barriers, which results in strong PDMS/PEG-DA anchoring.
Here we present the first multi-day study of forskolin gradients on guided axon growth of mammalian cortical neurons, which was enabled by the development of a shear-free PDMS/PEG-DA hybrid microfluidic device. The PDMS/PEG-DA device generates reproducible, linear concentration gradients for fast-diffusing small molecules. Exposure of neurons isolated from E18 rats to forskolin gradients resulted in guided axon growth that favoured the direction of higher forskolin concentrations. This microfluidic platform is universal and may be readily applied to chemotaxis studies of other small molecules with a variety of cell types, particularly shear-sensitive neurons and low-attaching suspension cell cultures.
Materials and methods
Device design and fabrication
The microfluidic pattern of the gradient generator was designed in AutoCAD (Autodesk, Inc., San Rafael, CA), and an SU8 master of the device was fabricated using standard soft lithography protocols. Briefly, a thin layer of SU8 (80 μm-thick, MicroChem Corp., Newton, MA) was spin-coated onto a silicon wafer, and the CAD patterns were transferred to this SU8 layer through a transparency photo mask (Infinite Graphics, MN). Upon curing, the SU8 master surface was then treated with 3-aminopropyl trimethoxysilane (Sigma, St. Louis, MO) to form a release layer. To make PDMS devices, a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 w/w mixture of Sylgard 184 monomer and hardener (Dow Corning, Corning, NY) was poured over the SU8 master in a dish, degassed under vacuum for 20 min to remove air bubbles, and then baked at 65 °C for 1 h to cure. Inlet and outlet fluidic ports were punched out using tissue biopsy punches (SYNEO Corp., Angleton, TX). Permanent bonding between PDMS chips and the cover glass of Lab-Tek™ chamber slides (Nalge Nunc International, Rochester, NY) was achieved by a 40 s oxygen plasma treatment at 80 watts (Branson IPC oxygen plasma Asher, Hayward, CA). A 10 min post-bake at 65 °C on a hot plate was used to strengthen and accelerate the covalent bonding.
:1 w/w mixture of Sylgard 184 monomer and hardener (Dow Corning, Corning, NY) was poured over the SU8 master in a dish, degassed under vacuum for 20 min to remove air bubbles, and then baked at 65 °C for 1 h to cure. Inlet and outlet fluidic ports were punched out using tissue biopsy punches (SYNEO Corp., Angleton, TX). Permanent bonding between PDMS chips and the cover glass of Lab-Tek™ chamber slides (Nalge Nunc International, Rochester, NY) was achieved by a 40 s oxygen plasma treatment at 80 watts (Branson IPC oxygen plasma Asher, Hayward, CA). A 10 min post-bake at 65 °C on a hot plate was used to strengthen and accelerate the covalent bonding.
Photopatterning of PEG-DA barriers
Immediately after PDMS/glass bonding, a surface silanization solution was injected into the microfluidic channels and allowed to sit for 3 h (Fig. 1A, step 1). Silanization of device inner surfaces was carried out using two types of surface silanization solutions, 3-(trichlorosilyl)propyl methacrylate (TPM, Sigma) or 3-(trimethoxysilyl)propyl methacrylate (TMSPMA, Sigma). TMSPMA was diluted in isopropyl alcohol at 1:200 (v/v, 1× dilution) or at 1:20 (v/v, 10× dilution), and 3% acetic acid was added to the solution immediately before surface treatment. TPM was diluted in perfluorooctane (20 μg to 1 g). After incubation, microfluidic channels were rinsed either with perfluorooctane for TPM coating or isopropyl alcohol for TMSPMA coating and then rinsed with water. Microfluidic channels were then emptied by vacuum and allowed to dry overnight at room temperature.
 | ||
| Fig. 1 Fabrication of gradient-generating devices with PEG barriers. A. Device fabrication includes standard soft lithography to generate and bond the PDMS layer to coverglass followed by silanization to modify the surfaces with methacrylate moieties (Fig. A1), vacuum filling of PEG-DA solution throughout the entire device (Fig. A2), UV patterning of PEG-DA barriers (Fig. A3) and water rinsing to remove uncrosslinked PEG-DA solution (Fig. A4). B. Without vacuum filling, significant gaps (arrows) remain between the PDMS and PEG-DA structures. C. Vacuum filling eliminates gaps between the PDMS and PEG-DA hydrogel barriers. D. A finished device had three fluidic chambers and two PEG-DA barrier structures (barrier on the left is outlined with a dashed line). The “finger” structures were designed to increase the contact surface area between PDMS and PEG-DA for a stronger bond. | ||
Before UV patterning of the PEG-DA barriers, the devices with silanized channel surfaces were first placed into a glass vessel on a Labconco lyophilizer (Kansas City, MO) and vacuum was applied at 0.133 mBar for 1 h (Fig. 1A, step 2). PEG-DA solution was prepared by mixing 20% PEG-DA (MW 700, Sigma), 30% water and 50% ethanol; 0.1% photoinitiator (Irgacure 2959, Ciba Specialty Chemicals, Switzerland) was added into the solution right before injection into the microfluidic devices. Ethanol was used to promote PEG-DA dissolution and to decrease solution viscosity.49,70 The PEG-DA solution was immediately added onto the device to cover all the inlet/outlet openings upon removal of the device from the vacuum chamber, and allowed to sit for 3 min, during which time the vacuum within the channels drew the PEG-DA solution uniformly into all open channels. The microfluidic device was then transferred onto an inverted fluorescence microscope (Zeiss 200M). The PEG-DA structures were generated by photocrosslinking the PEG-DA by focusing UV light from a mercury lamp (X-CITE, Lumen Dynamics, Canada) through an excitation filter G365 peak (excitation peak at 365 nm, Zeiss #49) using a 40× oil immersion objective (Fig. 1A, step 3). An adjustable diaphragm slide (Carl Zeiss LLC, Thornwood, NY) was inserted into the optical pathway to define the UV exposed region (70 to 600 μm wide, 600 μm long) and thus the dimensions of the crosslinked hydrogel barriers. Exposure time (500 ms to 1 s) was controlled by Axiovision software. Ten to 20 UV exposures were applied sequentially to form the elongated barrier structures (Fig. 1D) either with manual stage movement or with automatic position definition in the MosaiX module within the Axiovision software (Carl Zeiss LLC, Thornwood, NY). Two PEG-DA barrier structures were generated to form three adjacent channels in each device (Fig. 1D). Non-crosslinked PEG-DA solution was then rinsed out of the channels with sterile water (Fig. 1A, step 4).
The center cell culture chamber of the microfluidic device was coated with Matrigel (BD Bioscience; San Jose, CA) prior to neuron culture. Immediately after PEG-DA barrier writing, the device bonded to the cell culture chamber slide was sterilized with 70% ethanol and then rinsed with phosphate buffered saline (PBS). Matrigel was diluted to 0.5 mg ml−1 in complete neural culture medium, injected into the center cell culture chamber, and incubated first at room temperature for 10 min then at 37 °C in a 5% CO2 tissue culture incubator for a minimum of 3 days. Water droplets were added in the chamber slide to maintain humidity and to avoid evaporation from within the device. Neuron cells (cultured as described below) were injected into the center cell culture chamber and allowed to attach for one hour. Cell loading ports were then closed with solid PEEK plugs (Idex Health and Science), inlet/outlet tubing was connected to a syringe pump, and medium perfusion was initiated at 10 μl min−1. For cell experiments, the whole microfluidic setup was kept in a humidified tissue culture incubator for three days. Unnecessary door-opening was avoided to maintain optimal culture conditions including temperature, pH and humidity.
COMSOL modeling and gradient simulation
COMSOL Multiphysics (Burlington, MA), a commercial finite element modeling software, was used to simulate fluid dynamics and diffusion within the microfluidic device. The diffusion coefficient of fluorescein (MW 372), in water, Dfluorescein/water = 6.4 × 10−6 cm2 s−1,69 and in crosslinked PEG-DA, Dfluorescein/PEG = 1.5 × 10−6 cm2 s−1, was used in all simulations. Dfluorescein/PEG was estimated based on the reported diffusion coefficient of casamino acid (MW 250, 1.5 ± 0.5 × 10−6 cm2 s−1 (ref. 36)) and 2-NBDG (MW 342, 1.5 × 10−6 cm2 s−1 (ref. 49)) in 20% PEG-DA (MW 575).Gradient verification
Syringes (1 ml, Hamilton Co, 1001 TLL-XL) and tubing (Hamilton Co, catalog #90676) were rinsed with 70% ethanol followed by PBS, before filling with neural culture medium with or without 1 mM forskolin. To quantify the concentration profile, 1 mM fluorescein (MW 372), which has a similar molecular weight to forskolin (MW 410.5), was added into the medium in the source channel as a tracer molecule. Fluorescence images were taken and intensity across the cell culture chambers was quantified with NIH ImageJ software. Stable gradients formed in less than 15 min for all devices.Cortical neuron isolation, culture, and immunostaining
Rat cortex tissue was microsurgically dissected from E18 Sprague Dawley Rats, immersed in Hibernate E (Invitrogen and BrainBits) medium and kept on ice until use. The tissue was incubated with Tryple dissociation solution for 30 min at 37 °C and then disassociated by sequential triturating with a 1000 μl pipette tip and glass pasteur pipette (9 inch) with fire polished tip. Undispersed pieces were allowed to settle for 1 min. Supernatant with cell suspension was then transferred to a sterile 15 ml tube, and the cells were spun down at 1000 rpm for 1 min. The cell pellet was resuspended in neural basal medium (Invitrogen 21103) supplemented with 2% v/v B27 and 1% v/v GlutaMAX (Invitrogen) at either a high density of 4 million cells per ml or a low density of 0.4 million cells per ml. Cells were seeded in the microfluidic devices and cultured for 72 h within the forskolin gradient before immunostaining.Immunostaining of the neuron cells cultured in the microfluidic devices was performed with extra care to avoid introducing air bubbles into the cell culture chamber and the side channels, especially before the cells are fixed by paraformaldehyde (PFA). Air bubbles, as well as high-speed flow, may shear and dislodge cells and delicate neurite extensions from the culture surface. At the end of the microfluidic experiments, the inlet/outlet tubes and the center plug pieces were gently removed. Microfluidic channels were rinsed first with gravity flow of PBS buffer. Then the cells were fixed with 5% PFA for 10 min, followed by a PBS buffer rinse. Cells were permeabilized with a blocking solution (PBS with 0.1% Triton X-100 and 5% donkey serum) for 2 h. The cells were then incubated with primary antibodies for 1 h, followed by two PBS washes (5 min incubation each wash), and then two washes with the blocking solution (1 and 30 min, respectively). The secondary antibodies and/or DAPI staining were introduced and incubated for 1 h followed by three PBS washes. Cells were imaged immediately or mounted with ProLong® Gold Antifade Reagent (Life Technologies) for later assessment.
The following antibodies and reagents were used: anti-MAP2 (Covance, Berkley, CA); monoclonal-pan-axonal neurofilament marker SMI-312 (Covance). Normal donkey serum; Cy3-conjugated, donkey-anti-mouse IgG and FITC-conjugated, donkey-anti-rabbit IgG antibodies were purchased from Santa Cruz Biotechnology (Dallas, Texas). Primary antibodies were diluted 1:1000 in blocking solution (PBS with 0.1% Triton X-100 and 5% serum). Secondary antibodies were diluted 1:250 in blocking solution.
Contact angle and surface protein adsorption measurement
Cover glass was cleaned by sonication for 30 min in 70% ethanol. Cover glass and PDMS surfaces were silanized for 3 h before static contact angle and protein adsorption measurements. Static contact angle was measured by quantifying the slope of the tangent line to a water droplet (20 μl) at the liquid–solid–vapour interface. For Matrigel coating, Matrigel solution was added into each well of 24-well plates to immerse the coverglass and allowed to stand for one hour or 3 days at 37 °C in a 5% CO2 tissue culture incubator. Protein adsorbed to the coverslip surfaces was measured using the total protein BCA assay (Thermo Scientific Inc., Rockford, IL).Data and statistical analyses
An ImageJ plugin, NeuriteTracer, was used to track neurite growth. Axon tracks were analyzed using the Chemotaxis and Migration tool from Ibidi for the calculation of axon length and axon orientation. The starting coordinate of each axon track was defined as the origin (0,0) to create the plot of multiple axon tracks and angular histograms of axon endpoint positions. Axons shorter than 10 μm after the 48 h experiments were excluded from further analysis. The starting growth angle (θ1, Fig. 5B) and the final growth angle (θ2) was calculated for each axon, and the cumulative turning angle was defined as: Δθ = θ1 − θ2.Non-paired, non-parametric analysis of variance (ANOVA) was used to evaluate the statistical significance of protein adsorption and neuron polarization. Mean and standard deviation are reported in plots created with GraphPad Prism (GraphPad Inc, La Jolla, CA). Statistical significance was determined with the student t test (p < 0.05, n ≥ 3). Statistically significant asymmetry in the angular histogram of axon endpoint positions was determined using a Rayleigh test with p-value < 0.05.
Results and discussion
Microfluidic device fabrication
The microfluidic, gradient-generator device described here is fabricated from PDMS, cover glass, and PEG-DA. The pattern in the PDMS layer outlines the devices while leaving space for further UV patterning of the PEG-DA barriers (Fig. 1A). The “finger” structures (Fig. 1B–D) were designed to increase the contact surface area of PDMS/PEG-DA for increased bond strength. The PDMS layer was first permanently bonded to cover glass with oxygen plasma treatment. The hydroxylated inner surfaces of the devices were then reacted with TPM or TMSPMA to attach surface-tethered methacrylate groups for covalent bonding with PEG-DA upon free-radical-induced photocrosslinking. Both TPM and TMSPMA have been used for surface silanization of glass, PDMS, and silicone rubber surfaces to add functional groups or to modify surface hydrophobicity and biocompatibility.71We are the first to use vacuum filling to improve PEG structure formation in a microfluidic device. The vacuum-filling step was used to introduce the viscous PEG-DA solution into the channels. It is a critical step to ensure 1) complete filling of the channels, and 2) good anchoring between the PEG-DA hydrogel structures and the PDMS substrate. Vacuum filling uses a degassed PDMS mold to pull fluids through the microfluidic channels without external pumping.72,73 It is particularly useful to fill dead-end channels as well as PDMS structures with sharp corners, such as the finger structures in this device, which tend to trap air bubbles. Without the vacuum-filling step, small gaps were consistently observed between the PEG-DA structure and the PDMS layer after UV crosslinking (Fig. 1B), which caused liquid leakage and convective flow into the cell culture chamber. The presence of the thin, non-crosslinked PEG-DA layer is likely due to inhibition of photocrosslinking by oxygen released from the PDMS.67 When vacuum filling is used, the negative pressure present inside the PDMS layer functions in two ways: 1) it maintains a low local oxygen concentration that promotes complete polymerization of PEG-DA near the PDMS layer and 2) it enhances the surface contact between the PEG-DA solution and the PDMS surface, thereby overcoming the surface hydrophobicity and creating a seamless adhesion between PDMS and PEG (Fig. 1C).
PEG-DA was selected as the material for the diffusion barrier because of its low protein adsorption and its UV crosslinking capability. As a strongly hydrophilic polymer, PEG (and its functional variants such as PEG-DA) is very resistant to protein and chemical adsorption and has been used to minimize mammalian and bacterial cell adhesion.71 PEG-DA also has great biocompatibility and has been used in conjunction with other protein-modified polymers to generate microstructures for 3D cell culture.74 The unique capability for precise spatial control of photocrosslinking allows fast, low-cost prototyping to modify microfluidic structures. Although PEG hydrogels have been used previously in microfluidic devices, previous fabrication strategies generated hydrogel structures in non-permanently bonded PDMS devices using screws, clamps or extra housing pieces to maintain glass/PDMS/PEG contact.45,49 In contrast, our approach uses a unique vacuum filling/UV crosslinking process to generate PEG-DA barriers that are covalently bonded to both PDMS and glass surfaces, negating the need for external clamping or housing. This simple fabrication process allows both bubble-free filling and seamless PEG-PDMS bonding in one single step. The completed gradient-generator devices were found to be very stable and had a shelf life of at least one month with no compromise in performance if kept hydrated in water. No hydrogel detachment or delamination was observed during the course of all experiments (3 to 7 days).
Finite element simulation and gradient validation
A commercial finite element modelling software, COMSOL Multiphysics, was used to simulate the two dimensional concentration gradient profiles at equilibrium for various microfluidic designs (device dimensions, barrier positions and barrier widths), and to optimize operating conditions (e.g., flow rates) to produce a stable linear gradient for a given biochemical diffusivity.Initial simulations modeled devices with 100 μm wide hydrogel barriers fabricated from 20% (v/v) PEG-DA 575. Our mathematical models suggested that a critical flow rate of approximately 1 μl h−1 is required to maintain the concentration gradient (Fig. S1†). Below this critical flow rate, diffusion of forskolin across the hydrogel barriers dominates over the influx of the compound into the side channels. As a result, the chemical concentration decreases along the direction of flow, causing the concentration gradient (dC/dx) to vary along the y-distance at low flow rates (Fig. S1A†). As the flow rate increases, the change in dC/dx along the y-distance decreases and eventually disappears, resulting in a uniform concentration gradient in the microfluidic device (Fig. S1B†).
Diffusivity across the PEG-DA hydrogel barrier is another critical factor that shapes the equilibrium gradient profile. Diffusivity is determined by both the hydrodynamic radius of the solute molecules and the hydrogel mesh size, which is further controlled by the polymer molecular weight, polymer concentration, photoinitiator concentration, and extent of crosslinking reaction. In a device with 100 μm-wide hydrogel barriers, sharper equilibrium gradients across the cell culture chamber were predicted as the diffusivity was increased from 0.1 to 0.64 × 10−6 cm2 s−1 (Fig. S1D†). This observation suggested that gradient profiles could be tailored by adjusting the MW of PEG-DA, polymer concentration, and crosslinking density to control the hydrogel mesh size. Larger polymer molecular weight and lower polymer concentration will result in larger mesh sizes and faster diffusion. For example, the mesh size for PEG MW 575 was reported to be 15 Å, while that for PEG 4000 was 50 Å.60 At the same time, hydrogels with larger mesh sizes are typically mechanically weaker and withstand less pressure. All of these factors need to be carefully balanced for device design and materials selection.
The width of the PEG barriers, which can be controlled easily during photocrosslinking by adjusting the size of the diaphragm slide on the microscope, is another variable that influences the final gradient profile. As barrier width increases, diffusion across the barrier into the cell culture chamber is slower. This increased resistance results in a reduced dynamic concentration range, and hence decreased gradient steepness in the cell culture chamber. COMSOL simulation results confirmed this theory and showed that tapering the PEG-DA barriers around a center cell culture chamber of fixed width would generate predictable concentration gradients of different slopes within a single microfluidic device (Fig. 2A, B). Such a heterogeneous gradient field would allow high-throughput screening of cell responses to a range of concentration gradients simultaneously in a single test. The tapered barriers can be generated with either an optical mask or by direct pattern writing using multiple exposures. A variety of other barrier shapes could also be employed to achieve complex two-dimensional concentration profiles.
 | ||
| Fig. 2 Simulation and experimental verification of the gradient response to varied PEG-DA hydrogel barrier widths. A–B. Devices with tapered barriers generate concentration gradients of different slopes within a single microfluidic device. C–D. Simulations (blue) of fluorescein gradient profiles within devices with 70 μm and 400 μm PEG-DA barrier widths are confirmed by experimental measurements (red). Relative fluorescein concentration is calculated as a percentage of fluorescein in the source channel. | ||
To verify the computational simulation, we fabricated microfluidic devices with two barrier widths, 70 μm and 400 μm. The total width of the PEG barrier and the center cell culture chamber in these devices was fixed at 1600 μm. Fluorescein was used to visualize the gradient profile. Relative fluorescein concentration is calculated as a percentage of local fluorescence intensity relative to the fluorescence intensity in the source channel. The experimentally measured gradient profiles correlated well with the simulated concentration profiles, which confirmed our mathematical modeling setup (Fig. 2C, D).
Primary neuron culture in the microfluidic device
The microfluidic devices were first tested for their capability to support neuronal culture. Primary neuronal cultures are sensitive to a variety of factors including shear stress, surface protein coatings, humidity, and pH, as well as chemicals in their surroundings.75,76 The two PEG-DA hydrogel barriers physically separate the cell culture chamber from the flow channels, thus totally eliminating the presence of convective flow and shear stress in the cell culture chamber. We then characterized various surface modifications to enable sustained neuronal culture within the device. TPM and TMSPMA were used to functionalize the glass and PDMS surfaces with acrylate functional groups to enable direct covalent bonding with PEG-DA. However, this surface functionalization also alters the surface energy, hydrophobicity, and protein adsorption, all of which impact neuron adhesion.Both TPM and TMSPMA treatment altered the water contact angles on PDMS and glass surfaces (Fig. 3A). Untreated PDMS is highly hydrophobic with a contact angle close to 90 degrees. While TPM-treatment did not alter the PDMS contact angle, TMSPMA-treatment decreased the angle to about 40 degrees. On the other hand, clean glass has a relatively small contact angle of less than 30 degrees, while treatment with either TPM or TPSPMA increased the angle to close to 40 degrees. These results suggest that TMSPMA is more effective than TPM for surface PEGylation of PDMS, while both worked well for glass surfaces.
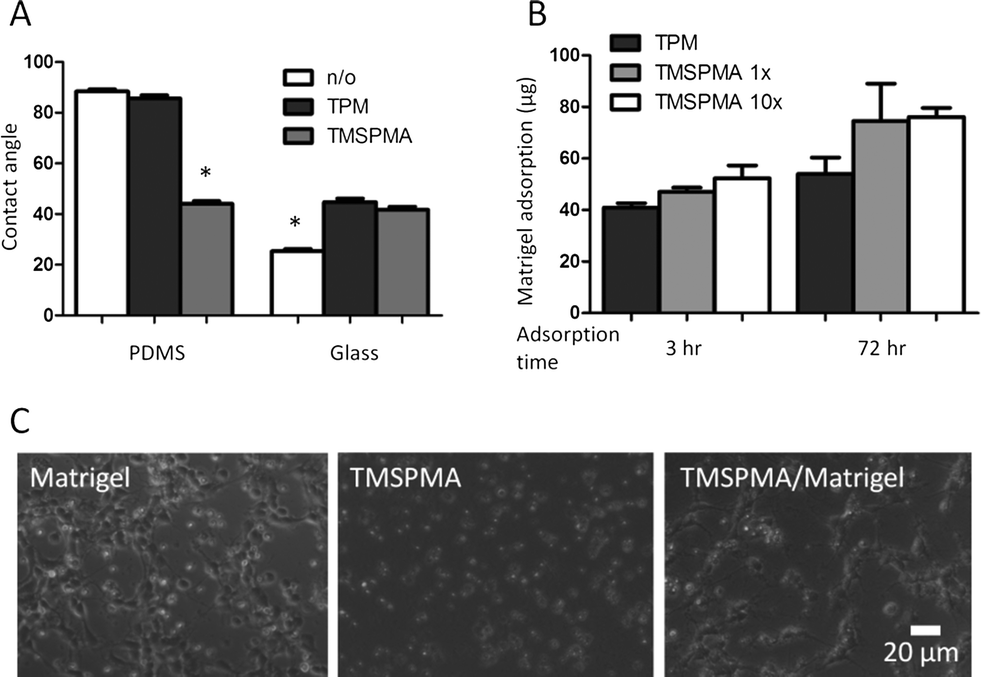 | ||
| Fig. 3 Surface modifications to enable device fabrication and primary neuron cell culture. A. Water contact angles were measured after surface silanization of PDMS and glass to present methacrylate moieties (*p < 0.05). B. Matrigel adsorption after exposures of 3 h or 3 days on various silanized glass surfaces. C. Primary cortical neuron adhesion and neurite extension were similar between control cultures (Matrigel-adsorbed coverglass) and microfluidic devices (Matrigel-adsorbed, TMSPMA-functionalized glass). In contrast, without Matrigel coating, neurons on TMSPMA-functionalized glass remained rounded and refractile under phase contrast microscopy and were unable to extend neurites. | ||
In vitro cultures of neurons on glass substrates typically require sufficient surface coating with poly(L-lysine) (PLL) or Matrigel to enhance cell viability, cell adhesion, and neurite outgrowth.75,76 PLL adsorption to glass surfaces is mainly mediated through electrostatic charge interactions between the positively charged polymer and the negatively charged glass. Because the negative surface charge on glass surfaces is lost after silanization, we predicted this would result in poor PLL adsorption. Experimental studies confirmed that both TPM- and TMSPMA-functionalized glass adsorbed with PLL did not support neuron culture (data not shown). Instead, we found that Matrigel was more effective as a surface-coating protein for neuron culture than PLL within this microfluidic device. In addition, longer coating times (3 days) resulted in 30 to 60% more protein adsorption than 3 h Matrigel coating on both the TPM- and TMSPMA-functionalized surfaces (Fig. 3B). Primary neurons seeded onto the Matrigel-modified microfluidic devices were found to adhere and extend neurites at 72 h that were morphologically similar to control cultures on Matrigel in standard tissue-culture Petri dishes. In contrast, without Matrigel modification, neurons on TMSPMA- functionalized glass remained rounded and refractile under phase contrast microscopy and were unable to extend neurites (Fig. 3C). Immunocytochemical labelling of fixed neuron cultures further confirmed the neuronal phenotype and neurite extension (Fig. 4A). A low neuron cell seeding density of 0.4 million cells ml−1 or ~30 cells mm−2, was chosen in this study to minimize direct cell–cell contact, and thereby enable identification and tracking of individual axons extended from each soma (i.e., cell body, Fig. 4B). In contrast, neurons grown at a higher density (4 million cells ml−1) formed a dense network of neurites (Fig. 4A), thus preventing the assignment of each axon to its originating soma. For neurons cultured inside the microfluidic devices, axon outgrowth became significant on day 2 and usually extended to a typical length of 10 cell bodies (or 100 μm) on day 3. After 72 h exposure to the forskolin gradient, neuron cells were immunostained for axons. We then quantified the axon growth and analyzed axon orientation by tracing the entire axon for each individual cell. A total of 58 neurons were analyzed. Each axonal track was overlaid onto a single plot with the soma centered at the origin (Fig. 4C). Our results revealed that 67.3% of axons oriented towards higher concentrations of forskolin, while 32.7% were observed to extend along the opposite direction (Fig. 4C), suggesting that two times more axons were attracted, rather than repelled, by the forskolin source. As an alternative analysis, the final location of each axonal growth cone (i.e. the tip of the extending axon) relative to the soma was plotted as an angular histogram (Fig. 4D). A Rayleigh test for statistical significance confirmed the asymmetric distribution of axonal growth cones (p = 0.011) towards higher forskolin concentrations.
 | ||
| Fig. 4 Forskolin-induced axon polarization within the gradient-generating device. Immunostaining was performed in the microfluidic device to visualize the neurons (neuronal MAP2 marker, green; nuclear DAPI stain, blue) and their axons (monoclonal-pan-axonal neurofilament marker SMI-312, red). A. A high seeding density (4 million cells ml−1) resulted in a dense neurite network that prevented the assignment of each neurite to its originating soma. B. A low seeding density (0.4 million cells ml−1) allowed easy identification of the individual axon extended from each cell body. C. Individual axonal tracks were plotted with all neuronal soma positioned at the origin (0,0). Axons that oriented towards higher concentrations of forskolin (39 of 58) are shown in red, while axons that extended towards lower concentrations (19 total) are shown in black. D. The final position of each axonal tip (i.e. growth cone) is shown in an angular histogram. A Rayleigh test for asymmetry confirmed that the distribution of growth cone positions was significantly skewed towards higher forskolin concentrations (p = 0.011 < 0.05). | ||
Interestingly, despite their preferred orientation, the axons did not display significantly different lengths when extended towards higher or lower forskolin concentrations (Fig. 5A, p = 0.28). This suggests that axonal orientation preference was a result of growth cone turning within the forskolin gradient rather than simply being caused by changes in axonal growth rate. To further evaluate this notion, we quantified the cumulative turning angle (Δθ = θ1−θ2) for each individual axon (Fig. 5B). An axon that displayed guidance towards higher forskolin concentrations would have a positive Δθ value, while an axon that turned away from the forskolin gradient would have a negative Δθ value. Axons with a starting growth angle (θ1) greater than 90° (i.e. those that initiated their growth against the gradient) had an average cumulative turning angle of 12.0° (Fig. 5C). This indicates that axons initiated in the “wrong” direction (i.e. against the gradient) had a propensity to “correct” their orientation during the 72 h experiment. In contrast, the average cumulative turning angle was nearly 0 for axons that had starting growth angles (θ1) less than 90° (i.e. those that initiated their growth towards the gradient). This indicates that axons initiated with the “correct” orientation (i.e. with the gradient) were likely to persist along their original direction. These two populations of neurons had statistically distinct (p = 0.016) cumulative turning angle distributions, suggesting two different types of axonal outgrowth that nonetheless result in axons of similar lengths.
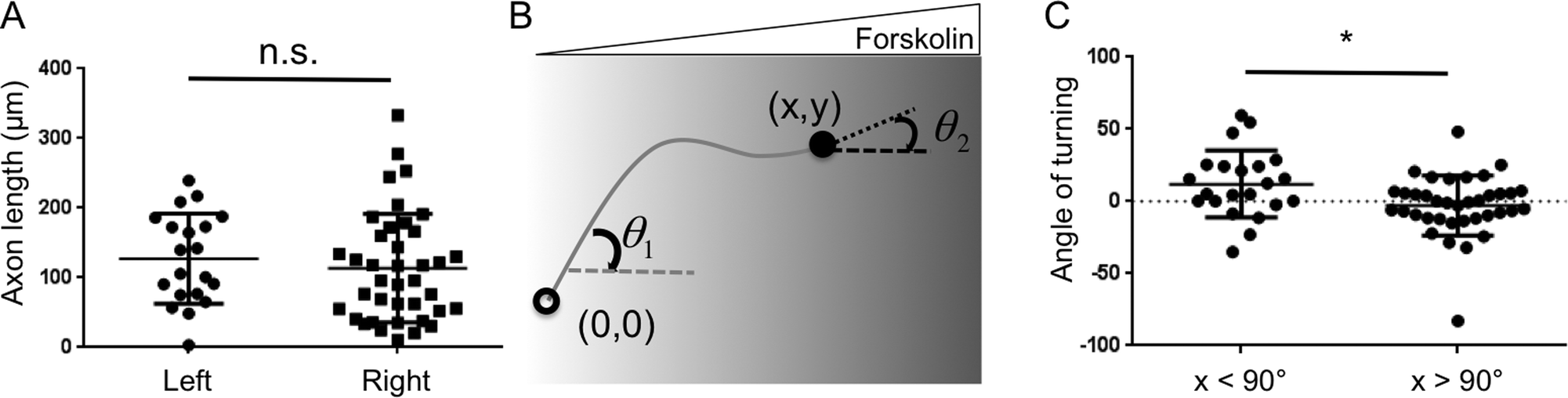 | ||
| Fig. 5 Quantification of axon lengths and cumulative turning angles. A. The length of each axon was quantified individually for all 58 neurons. Despite the preferred orientation of the axons, there was no statistically significant difference (p = 0.28) between the lengths of axons oriented towards higher or lower forskolin concentrations. B. The cumulative turning angle (Δθ = θ1 − θ2) for each individual axon was calculated as the difference between the starting growth angle (θ1) and the final growth angle (θ2). C. The distribution (individual points), average (horizontal bar) and standard deviation (error bars) of cumulative turning angles for axons with starting growth angles (θ1) greater than 90° (i.e. initiation towards higher forskolin concentrations) or less than 90° (i.e. initiation towards lower concentrations) (*p = 0.016). | ||
Our axon polarization results are consistent with previous observations that forskolin serves as an axonal chemoattractant. Forskolin is known to activate adenylyl cyclase, thereby increasing the levels of intracellular cyclic adenosine monophosphate (cAMP)4 and activating cAMP-sensitive pathways, such as protein kinase A (PKA) and exchange protein activated by cAMP (Epac), to promote axon formation, growth cone turning and axon path finding.4–6 Forskolin has also been found to enhance neurite growth1 and neuronal survival.2,3 Many of these studies are based on experimental observations upon local injection in animals, genetic mutations, in vitro formation of transient gradients with micropipettes6,7 or patterned in vitro substrates with stripes of biochemicals.5 It has been particularly difficult to form a stable gradient of small molecules such as forskolin due to their small molecular size, fast diffusion and thus fast decay of the slope of the gradient in both traditional micropipette assays and many microfluidic assays. The PEG-DA hydrogel-based device described here allowed us to generate a stable forskolin gradient without exposing the neurons to shear stress, which can cause cell damage and influence neurite outgrowth. We are the first to directly quantify cortical neuron response to a long-term (72 h) stable gradient of forskolin to demonstrate its axon guidance function. This device will enable future mechanistic studies of the temporal requirements for stimulation (e.g., duration of signal during exposure, persistence of signal after exposure) as well as the competing action of multiple, simultaneous chemical signals.
Concluding remarks
This study analyzed the long-term effects of a stable, soluble forskolin gradient on mammalian cortical neurons. We directly quantified the effect of a forskolin gradient on axon polarization, length, and turning. Our studies supported previous work suggesting that forskolin induces axonal polarization. They also revealed that while forskolin affects turning angle it does not alter neurite outgrowth in our experimental platform. These studies were enabled by the development of a PEG-DA-based microfluidic gradient-generator capable of generating long-term, stable, linear gradients of small molecules.Our optimized fabrication process for the PDMS/PEG-DA devices includes PDMS molding, surface silanization, vacuum-aided PEG-DA solution filling, UV photopatterning of PEG-DA hydrogel barriers, and surface modification with Matrigel. The vacuum-filling step aids in bubble-free filling despite the presence of dead-end patterns in the device and overcomes the oxygen inhibition of radical-induced polymerization, thereby allowing formation of a stable PEG-DA/PDMS interface within a permanently bonded PDMS device. This fabrication process is flexible, as it allows generation of microfluidic devices with tailored PEG-DA barriers of different properties (hydrogel mesh size and diffusivity, width, shape, and location) for each experiment without redesigning the PDMS mold. We identified a combination of TMSPMA silanization and Matrigel surface treatment that was effective at maintaining primary rat cortical neuron cell cultures.
The excellent biocompatibility of the device and lack of convective flow within the culture chamber enabled multi-day culture of primary rat cortical neurons. The slow growth and sensitivity of mammalian cortical neurons to shear flow, combined with the high diffusivity of forskolin, made the development of this tool a necessary component to successfully conduct these experiments.
Acknowledgements
We thank Sally Kim, Segio Leal-Ortiz and Prof. Craig Garner in the Department of Psychiatry and Behavioral Sciences at Stanford School of Medicine for providing us with primary neurons and culture protocols and for insightful discussions. This work was supported by NIH 1T32-HL098049-01A1 to the Stanford Cardiovascular Institute (H.X.), NIH R21-AR062359, DP2-OD006477-01, and R01-DK085720, and NSF DMR-0846363 (S.C.H.).References
- C. Richter-Landsberg and B. Jastorff, J. Cell Biol., 1986, 102, 821–829 CrossRef CAS.
- C. Galli, O. Meucci, A. Scorziello, T. M. Werge, P. Calissano and G. Schettini, J. Neurosci., 1995, 15, 1172–1179 CAS.
- M. Watanabe, Y. Tokita, M. Kato and Y. Fukuda, Neuroscience, 2003, 116, 733–742 CrossRef CAS.
- A. K. Jassen, H. Yang, G. M. Miller, E. Calder and B. K. Madras, Mol. Pharmacol., 2006, 70, 71–77 CAS.
- M. Shelly, B. K. Lim, L. Cancedda, S. C. Heilshorn, H. Gao and M.-M. Poo, Science, 2010, 327, 547–552 CrossRef CAS PubMed.
- H. J. Song, G. L. Ming and M. M. Poo, Nature, 1997, 388, 275–279 CrossRef CAS PubMed.
- G. L. Ming, H. J. Song, B. Berninger, C. E. Holt, M. Tessier-Lavigne and M. M. Poo, Neuron, 1997, 19, 1225–1235 CrossRef CAS.
- J. Adler, Science, 1966, 153, 708–716 CAS.
- T. Tani and Y. Naitoh, J. Exp. Biol., 1999, 202, 1–12 CAS.
- V. V. Abhyankar, M. A. Lokuta, A. Huttenlocher and D. J. Beebe, Lab Chip, 2006, 6, 389–393 RSC.
- X. Cao and M. S. Shoichet, Neuroscience, 2001, 103, 831–840 CrossRef CAS.
- T. N. Hartmann, V. Grabovsky, R. Pasvolsky, Z. Shulman, E. C. Buss, A. Spiegel, A. Nagler, T. Lapidot, M. Thelen and R. Alon, J. Leukocyte Biol., 2008, 84, 1130–1140 CrossRef CAS PubMed.
- J. Imitola, K. Raddassi, K. I. Park, F.-J. Mueller, M. Nieto, Y. D. Teng, D. Frenkel, J. Li, R. L. Sidman, C. A. Walsh, E. Y. Snyder and S. J. Khoury, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 18117–18122 CrossRef CAS PubMed.
- E. Kokovay, S. Goderie, Y. Wang, S. Lotz, G. Lin, Y. Sun, B. Roysam, Q. Shen and S. Temple, Cell Stem Cell, 2010, 7, 163–173 CrossRef CAS PubMed.
- S. H. Zigmond, Methods Enzymol., 1988, 162, 65–72 CAS.
- D. Zicha, G. Dunn and G. Jones, Methods Mol. Biol., 1997, 75, 449–457 CAS.
- C. Zhang, S. Jang, O. C. Amadi, K. Shimizu, R. T. Lee and R. N. Mitchell, BioMed Res. Int., 2013, 2013, 373569 Search PubMed.
- D. C. Duffy, J. C. McDonald, O. J. A. Schueller and G. M. Whitesides, Anal. Chem., 1998, 70, 4974–4984 CrossRef CAS PubMed.
- S. K. W. Dertinger, D. T. Chiu, N. L. Jeon and G. M. Whitesides, Anal. Chem., 2001, 73, 1240–1246 CrossRef CAS.
- B. G. Chung, L. A. Flanagan, S. W. Rhee, P. H. Schwartz, A. P. Lee, E. S. Monuki and N. L. Jeon, Lab Chip, 2005, 5, 401–406 RSC.
- N. L. Jeon, S. K. W. Dertinger, D. T. Chiu, I. S. Choi, A. D. Stroock and G. M. Whitesides, Langmuir, 2000, 16, 8311–8316 CrossRef CAS.
- C. Joanne Wang, X. Li, B. Lin, S. Shim, G.-L. Ming and A. Levchenko, Lab Chip, 2008, 8, 227 RSC.
- C. Urbich, Arterioscler., Thromb., Vasc. Biol., 2002, 22, 69–75 CrossRef CAS.
- G. M. Walker, J. Sai, A. Richmond, M. Stremler, C. Y. Chung and J. P. Wikswo, Lab Chip, 2005, 5, 611 RSC.
- B. Wojciak-Stothard, J. Cell Biol., 2003, 161, 429–439 CrossRef CAS PubMed.
- L. J. Millet, M. E. Stewart, R. G. Nuzzo and M. U. Gillette, Lab Chip, 2010, 10, 1525 RSC.
- H. Cho, T. Hashimoto, E. Wong, Y. Hori, L. B. Wood, L. Zhao, K. M. Haigis, B. T. Hyman and D. Irimia, Sci. Rep., 2013, 3, 1823 Search PubMed.
- T. M. Keenan, C.-H. Hsu and A. Folch, Appl. Phys. Lett., 2006, 89, 114103 CrossRef PubMed.
- N. Bhattacharjee, N. Li, T. M. Keenan and A. Folch, Integr. Biol., 2010, 2, 669–679 RSC.
- D. Irimia, G. Charras, N. Agrawal, T. Mitchison and M. Toner, Lab Chip, 2007, 7, 1783 RSC.
- S. Paliwal, P. A. Iglesias, K. Campbell, Z. Hilioti, A. Groisman and A. Levchenko, Nature, 2007, 446, 46–51 CrossRef CAS PubMed.
- W. Saadi, S. W. Rhee, F. Lin, B. Vahidi, B. G. Chung and N. L. Jeon, Biomed. Microdevices, 2007, 9, 627–635 CrossRef PubMed.
- B. Mosadegh, C. Huang, J. W. Park, H. S. Shin, B. G. Chung, S.-K. Hwang, K.-H. Lee, H. J. Kim, J. Brody and N. L. Jeon, Langmuir, 2007, 23, 10910–10912 CrossRef CAS PubMed.
- H. Cho, B. Hamza, E. A. Wong and D. Irimia, Lab Chip, 2014, 14, 972 RSC.
- M. T. Breckenridge, T. T. Egelhoff and H. Baskaran, Biomed. Microdevices, 2010, 12, 543–553 CrossRef PubMed.
- H. Xu and S. C. Heilshorn, Small, 2013, 9(4), 585–595 CrossRef CAS PubMed.
- A. Shamloo and S. C. Heilshorn, Lab Chip, 2010, 10, 3061–3068 RSC.
- A. Shamloo, H. Xu and S. Heilshorn, Tissue Eng., Part A, 2012, 18, 320–330 CrossRef CAS PubMed.
- F. Kuhnert, M. R. Mancuso, A. Shamloo, H.-T. Wang, V. Choksi, M. Florek, H. Su, M. Fruttiger, W. L. Young, S. C. Heilshorn and C. J. Kuo, Science, 2010, 330, 985–989 CrossRef CAS PubMed.
- A. Shamloo, M. Manchandia, M. Ferreira, M. Mani, C. Nguyen, T. Jahn, K. Weinberg and S. Heilshorn, Integr. Biol., 2013, 5, 1076–1085 RSC.
- J. Diao, L. Young, S. Kim, E. A. Fogarty, S. M. Heilman, P. Zhou, M. L. Shuler, M. Wu and M. P. DeLisa, Lab Chip, 2006, 6, 381 RSC.
- T. Kim, M. Pinelis and M. M. Maharbiz, Biomed. Microdevices, 2009, 11, 65–73 CrossRef CAS PubMed.
- J. Y. Park, S.-K. Kim, D.-H. Woo, E.-J. Lee, J.-H. Kim and S.-H. Lee, Stem Cells, 2009, 27, 2646–2654 CrossRef CAS PubMed.
- A. P. Wong, R. Perez-Castillejos, J. Christopher Love and G. M. Whitesides, Biomaterials, 2008, 29, 1853–1861 CrossRef CAS PubMed.
- S.-Y. Cheng, S. Heilman, M. Wasserman, S. Archer, M. L. Shuler and M. Wu, Lab Chip, 2007, 7, 763 RSC.
- H. Wu, B. Huang and R. N. Zare, J. Am. Chem. Soc., 2006, 128, 4194–4195 CrossRef CAS PubMed.
- U. Haessler, M. Pisano, M. Wu and M. A. Swartz, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5614–5619 CrossRef CAS PubMed.
- S. Chung, R. Sudo, P. J. Mack, C.-R. Wan, V. Vickerman and R. D. Kamm, Lab Chip, 2009, 9, 269–275 RSC.
- D. C.-W. Tan, L.-Y. L. Yung and P. Roy, Biomed. Microdevices, 2010, 12, 523–532 CrossRef CAS PubMed.
- M. A. Traore and B. Behkam, J. Micromech. Microeng., 2013, 23, 085014 CrossRef.
- M. Kim and T. Kim, Anal. Chem., 2010, 82, 9401–9409 CrossRef CAS PubMed.
- X. Zhu, G. Si, N. Deng, Q. Ouyang, T. Wu, Z. He, L. Jiang, C. Luo and Y. Tu, Phys. Rev. Lett., 2012, 108, 128101 CrossRef.
- J. L. Ifkovits and J. A. Burdick, Tissue Eng., 2007, 13, 2369–2385 CrossRef CAS PubMed.
- C. T. S. Wong Po Foo, J. S. Lee, W. Mulyasasmita, A. Parisi-Amon and S. C. Heilshorn, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22067–22072 CrossRef PubMed.
- D. Dendukuri, S. S. Gu, D. C. Pregibon, T. A. Hatton and P. S. Doyle, Lab Chip, 2007, 7, 818–828 RSC.
- G. M. Cruise, D. S. Scharp and J. A. Hubbell, Biomaterials, 1998, 19, 1287–1294 CrossRef CAS.
- K. T. Nguyen and J. L. West, Biomaterials, 2002, 23, 4307–4314 CrossRef CAS.
- G. Pitarresi, F. S. Palumbo, G. Giammona, M. A. Casadei and F. Micheletti Moracci, Biomaterials, 2003, 24, 4301–4313 CrossRef CAS.
- J. A. Burdick, A. Khademhosseini and R. Langer, Langmuir, 2004, 20, 5153–5156 CrossRef CAS.
- W.-G. Koh and M. V. Pishko, Anal. Bioanal. Chem., 2006, 385, 1389–1397 CrossRef CAS PubMed.
- D. Gao, J. Liu, H.-B. Wei, H.-F. Li, G.-S. Guo and J.-M. Lin, Anal. Chim. Acta, 2010, 665, 7–14 CrossRef CAS PubMed.
- D. Missirlis, R. Kawamura, N. Tirelli and J. A. Hubbell, Eur. J. Pharm. Sci., 2006, 29, 120–129 CrossRef CAS PubMed.
- W. Zhan, G. H. Seong and R. M. Crooks, Anal. Chem., 2002, 74, 4647–4652 CrossRef CAS.
- N. Zaari, P. Rajagopalan, S. K. Kim, A. J. Engler and J. Y. Wong, Adv. Mater., 2004, 16, 2133–2137 CrossRef CAS.
- S. Nemir, H. N. Hayenga and J. L. West, Biotechnol. Bioeng., 2010, 105, 636–644 CrossRef CAS PubMed.
- J. He, Y. Du, J. L. Villa-Uribe, C. Hwang, D. Li and A. Khademhosseini, Adv. Funct. Mater., 2010, 20, 131–137 CrossRef CAS PubMed.
- D. Dendukuri, P. Panda, R. Haghgooie, J. M. Kim, T. A. Hatton and P. S. Doyle, Macromolecules, 2008, 41, 8547–8556 CrossRef CAS.
- D. R. Albrecht, V. L. Tsang, R. L. Sah and S. N. Bhatia, Lab Chip, 2005, 5, 111–118 RSC.
- P. Galambos and F. Forster, in Micro Total Analysis Systems ’98, ed. D. J. Harrison and A. Berg, Springer Netherlands, 1998, ch. 46, pp. 189–192 Search PubMed.
- 2011 IEEE/RSJ International Conference on Intelligent Robots and Systems, September 25–30, 2011, San Francisco, CA, T. Yue, M. Nakajima, M. Ito, M. Kojima and T. Fukuda, 2011, pp. 433–438 Search PubMed.
- J. H. Silver, J. C. Lin, F. Lim, V. A. Tegoulia, M. K. Chaudhury and S. L. Cooper, Biomaterials, 1999, 20, 1533–1543 CrossRef CAS.
- J. T. Nevill, A. Mo, B. J. Cord, T. D. Palmer, M.-M. Poo, L. P. Lee and S. C. Heilshorn, Soft Matter, 2011, 7, 343–347 RSC.
- K. Hosokawa, K. Sato, N. Ichikawa and M. Maeda, Lab Chip, 2004, 4, 181–185 RSC.
- H. Xu, J. Wu, C.-C. Chu and M. L. Shuler, Biomed. Microdevices, 2012, 14, 409–418 CrossRef CAS PubMed.
- S. Kaech and G. Banker, Nat. Protoc., 2007, 1, 2406–2415 CrossRef PubMed.
- K. T. Tisay and B. Key, J. Neurosci., 1999, 19, 9890–9899 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4lc00162a |
| This journal is © The Royal Society of Chemistry 2014 |