 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluoropolymer surface coatings to control droplets in microfluidic devices†
Carson T.
Riche
,
Chuchu
Zhang
,
Malancha
Gupta
* and
Noah
Malmstadt
*
Mork Family Department of Chemical Engineering and Materials Science, University of Southern California, Los Angeles, CA 90089, USA. E-mail: malanchg@usc.edu; malmstad@usc.edu
First published on 28th March 2014
Abstract
We have demonstrated the application of low surface energy fluoropolymer coatings onto poly(dimethylsiloxane) (PDMS) microfluidic devices for droplet formation and extraction-induced merger of droplets. Initiated chemical vapor deposition (iCVD) was used to pattern fluoropolymer coatings within microchannels based on geometrical constraints. In a two-phase flow system, the range of accessible flow rates for droplet formation was greatly enhanced in the coated devices. The ability to controllably apply the coating only at the inlet facilitated a method for merging droplets. An organic spacer droplet was extracted from between a pair of aqueous droplets. The size of the organic droplet and the flow rate controlled the time to merge the aqueous droplets; the process of merging was independent of the droplet sizes. Extraction-induced droplet merging is a robust method for manipulating droplets that could be applied in translating multi-step reactions to microfluidic platforms.
Introduction
Fluid flow in microfluidic devices is highly sensitive to the surface chemistry at the walls of the channels due to characteristically high surface area-to-volume ratios. Poly(dimethylsiloxane) (PDMS) has been a broadly applied material for microfluidic device fabrication due to its ease of mold replication and facile bonding.1,2 However, there are several disadvantages to working with PDMS, including its tendency to swell in many organic solvents, its capacity to absorb low molecular weight species from flowing streams, and its moderate hydrophobicity.3 Many schemes to alter the surface chemistry of PDMS microfluidic devices have been proposed, but they generally present limitations and challenges that hinder their widespread adoption. Modifying the PDMS prior to assembling the molded slabs precludes bonding by silanol condensation4,5 and requires subsequent processing to seal the device.6,7 Alternatively, coating pre-assembled devices can alter the cross-sectional geometry of the channels and restrict flow.8 Methods aimed at constraining modification to the walls of the channels require pretreatments to activate the surfaces9 or UV exposure for photo-initiated reactions.10,11 Vapor phase approaches to surface modification are inherently appealing because they are adsorption-limited processes that only modify the channel surfaces.12 Vapor phase precursors diffuse into the channels, adsorb to the walls of the device, and subsequently react. Pre-assembled channels have been coated with modified poly(p-xylenes)13 and we recently demonstrated that initiated chemical vapor deposition (iCVD) can be used to deposit a multi-functional, cross-linked fluoropolymer coating.14 This coating acted as a barrier to organic phase permeation into the PDMS14 and reduced the surface energy of the channel walls to facilitate droplet breakup of an ionic liquid phase.15In this paper, we have demonstrated that a vapor-deposited fluoropolymer coating facilitates aqueous droplet formation at a wide range of flow rates and can be patterned to control the extraction of an organic phase for extraction-induced droplet merger. Previously, patterned coatings within microfluidic channels had been achieved by light exposure through masks or using flow patterns within the device to control the location of surface modification.6,11,16 We exploited the mass transfer limitations of iCVD polymerization to create a discontinuous pattern within pre-assembled microchannels without the use of a mask. In our system, reactants enter the channels through holes at the inlets and their concentration in the channel decreases with distance from the inlets until the concentration falls below the threshold for polymerization.
We considered the formation of droplets in T-junction devices where the dispersed phase was injected perpendicular to the continuous phase.17 Previously, the two-phase flow of immiscible fluids has been characterized as occurring in three regimes: dripping, jetting, and co-flow.18–20 These categories describe droplets that form at the intersection of the two phases, droplets that form downstream of the intersection, and the absence of droplets, respectively. The most stable and predictable regime is dripping. These regimes have been characterized on the basis of channel geometry, capillary number, flow rates, and fluid properties.21–28 The surface energy of channels also affects droplet formation. Given the proper flow conditions, channels with an advancing water contact angle greater than 92° are capable of forming water-in-oil droplets due to preferential wetting of the oil phase on the walls of the channels.29 The native PDMS surface has a water contact angle of 112 ± 1°.5 We decreased the surface energy of the channels by coating them with the fluoropolymer and increased the water contact angle to 135 ± 3°. The decreased surface energy extended the range of accessible capillary numbers for droplet formation, allowing droplets to be formed in the dripping regime at much higher flow rates than were accessible in uncoated channels.
We also demonstrated that we could pattern the fluoropolymer coating to facilitate a new method for the controlled merger of droplets in microfluidic channels: extraction-induced droplet merger. Droplet microfluidic platforms have been exploited as miniaturized reactors for synthesis,15,30 mixing,31 and rapid fluid exchange.32 However, these reactors typically produce one-step reaction products. Multi-step reactions require merging droplets. Previously, droplet merger has been achieved via pillar-induced droplet hold-up,33 surface energy mediated droplet hold-up,16 and expansion channels to modulate droplet velocities.34–36 The merging within these devices was sensitive to flow rates, droplet sizes, fluid properties, and channel geometries. Merging times were hard-wired into the geometry of the devices and could not be easily adjusted by varying the operating parameters. Our method to merge droplets uses an organic droplet as a spacer between two aqueous droplets. By using diffusion-restricted patterning to apply the fluoropolymer only near the channel inlets, we can facilitate droplet formation there while allowing for extraction downstream. Many hydrocarbons readily swell the PDMS matrix and partition from flowing streams into the surrounding device.37,38 We have shown that coating the channels with a fluoropolymer creates a barrier to prevent an organic phase from partitioning into the PDMS. However, the organic spacer is extracted spontaneously in the uncoated downstream region of the channel, bringing the two aqueous droplets into contact. We can achieve a range of merging times using a single device geometry by modulating flow rates or changing the composition of the organic phase. The merging times are insensitive to the size of the merged droplets.
Experimental
Microfluidic devices were made in poly(dimethylsiloxane) (PDMS) using standard photolithography and polymer molding procedures. SU-8 50 negative photoresist (MicroChem) was spun onto a silicon wafer (University Wafer) and exposed through a patterned transparency mask (Output City). The unexposed regions were developed and rinsed. The resulting molds were exposed to vapors of trichloro(1H,1H,2H,2H-perfluorooctyl)silane (Aldrich) to prevent PDMS adhesion. Subsequently, PDMS was mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 (curing agent:base elastomer) ratio, degassed, poured over the treated mold, and cured for 4 h at 65 °C. A piece of PDMS was also cured on a blank silicon wafer. Afterwards, both pieces of PDMS were peeled off their respective wafers and a 2.5 mm diameter punch was used to create holes in the slab with the channel pattern to form the inlets and outlets. The surfaces to be bonded were cleaned with packing tape (3M) and treated with a hand held corona generator (BD20-AC, Electro-Technic Products, Inc.), and then placed into intimate contact and lightly pressed together. The pre-assembled devices were cured for 2 h at 65 °C.
:10 (curing agent:base elastomer) ratio, degassed, poured over the treated mold, and cured for 4 h at 65 °C. A piece of PDMS was also cured on a blank silicon wafer. Afterwards, both pieces of PDMS were peeled off their respective wafers and a 2.5 mm diameter punch was used to create holes in the slab with the channel pattern to form the inlets and outlets. The surfaces to be bonded were cleaned with packing tape (3M) and treated with a hand held corona generator (BD20-AC, Electro-Technic Products, Inc.), and then placed into intimate contact and lightly pressed together. The pre-assembled devices were cured for 2 h at 65 °C.
Pre-assembled devices were then coated with a poly(1H,1H,2H,2H-perfluorodecyl acrylate-co-ethylene glycol diacrylate) coating as previously described.14 We used an initiated chemical vapor deposition process where the pre-assembled devices were placed on a stainless steel stage that was maintained at 30 °C within a pancake-shaped vacuum chamber (GVD Corp., 250 mm diameter, 48 mm height). The vacuum chamber was maintained at 50 mTorr by a throttle valve (MKS Instruments). Initiator – di-tert butyl peroxide (Sigma) – and monomer – 1H,1H,2H,2H-perfluorodecyl acrylate (SynQuest) and ethylene glycol diacrylate (Monomer–Polymer) – vapors were flown into the chamber and passed over a nichrome (Omega) wire array heated to 220 °C where the thermally labile initiator molecules decomposed into free radicals and the monomer molecules remained stable. The precursors diffused into the pre-assembled channels via the holes punched at the inlets/outlets of the channels. Precursor molecules diffused down the channels, adsorbed to the PDMS surface, and polymerized via a free radical chain mechanism.12,39 The reaction was run for 60 minutes and then terminated by turning off the wire array, stopping the flow of precursors, and pumping out any excess monomer and initiator molecules until the chamber reached its original base pressure.
Microfluidic devices with a T-junction geometry were used to form aqueous droplets within a fluorocarbon oil, poly(chlorotrifluoroethylene) (PCTFE) (Halocarbon). The fluorinated oil is available in various viscosities; we blended Halocarbon 6.3 with Halocarbon 95 to create a fluorous phase with a viscosity of 100 mPa s. For droplet formation experiments, we used T-junction devices with a 1.5 cm long channel, a main channel width of 200 or 400 μm, inlet channel widths of either 50 or 100 μm, and channel heights of 400 μm. The PCTFE was injected via the continuous phase inlet and the aqueous stream was injected via the dispersed phase inlet (Fig. 1b). The other inlets were plugged and the entire device was primed with PCTFE before starting the aqueous flow. After both the dispersed and continuous phase flow rates were set, the flow was allowed to stabilize for a few minutes before capturing bright field images. Flow rates were modulated by syringe pumps (Harvard Apparatus) and interfaced with the device using PEEK tubing (McMaster). Images of droplets for size analysis were taken on a Nikon TI-E inverted microscope. Over 500 images (1–8 droplets per image) of discrete droplets were analyzed in Matlab to determine the droplet lengths at each condition (ESI† contains an explanation of image analysis). High speed images of droplets (Fig. 2b) were captured on a Phantom V711 camera (Vision Research) at 159250 frames per second with a 1.76 μs exposure time.
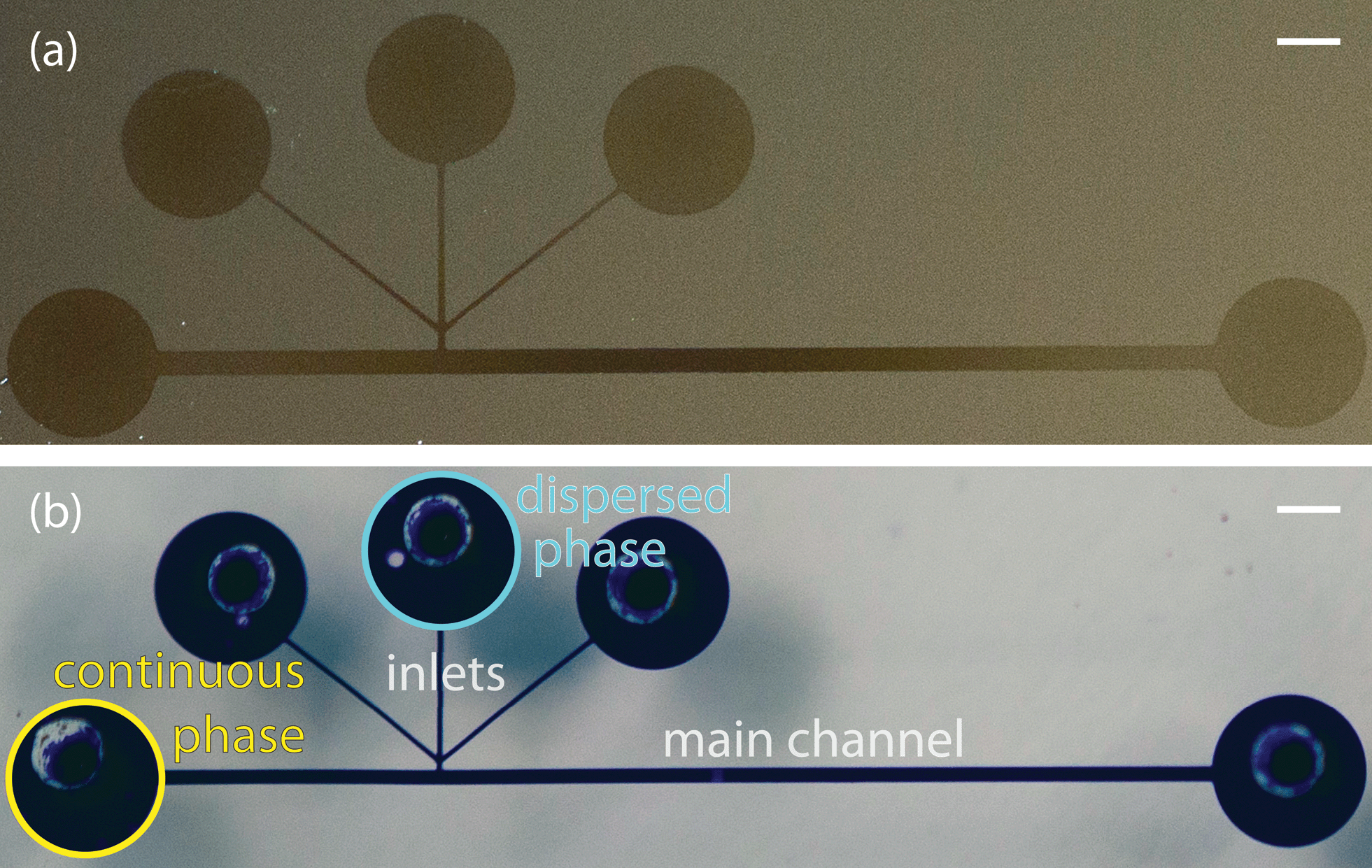 | ||
| Fig. 1 (a) Silicon wafers were masked with a PDMS slab imprinted with the channel pattern, coated with fluoropolymer using iCVD, and the PDMS was peeled off revealing the polymer coating on the silicon wafer. (b) Devices made from the same channel patterns were filled with a solution of crystal violet to elucidate the morphology of the channel. Scale bars indicate 1 mm. | ||
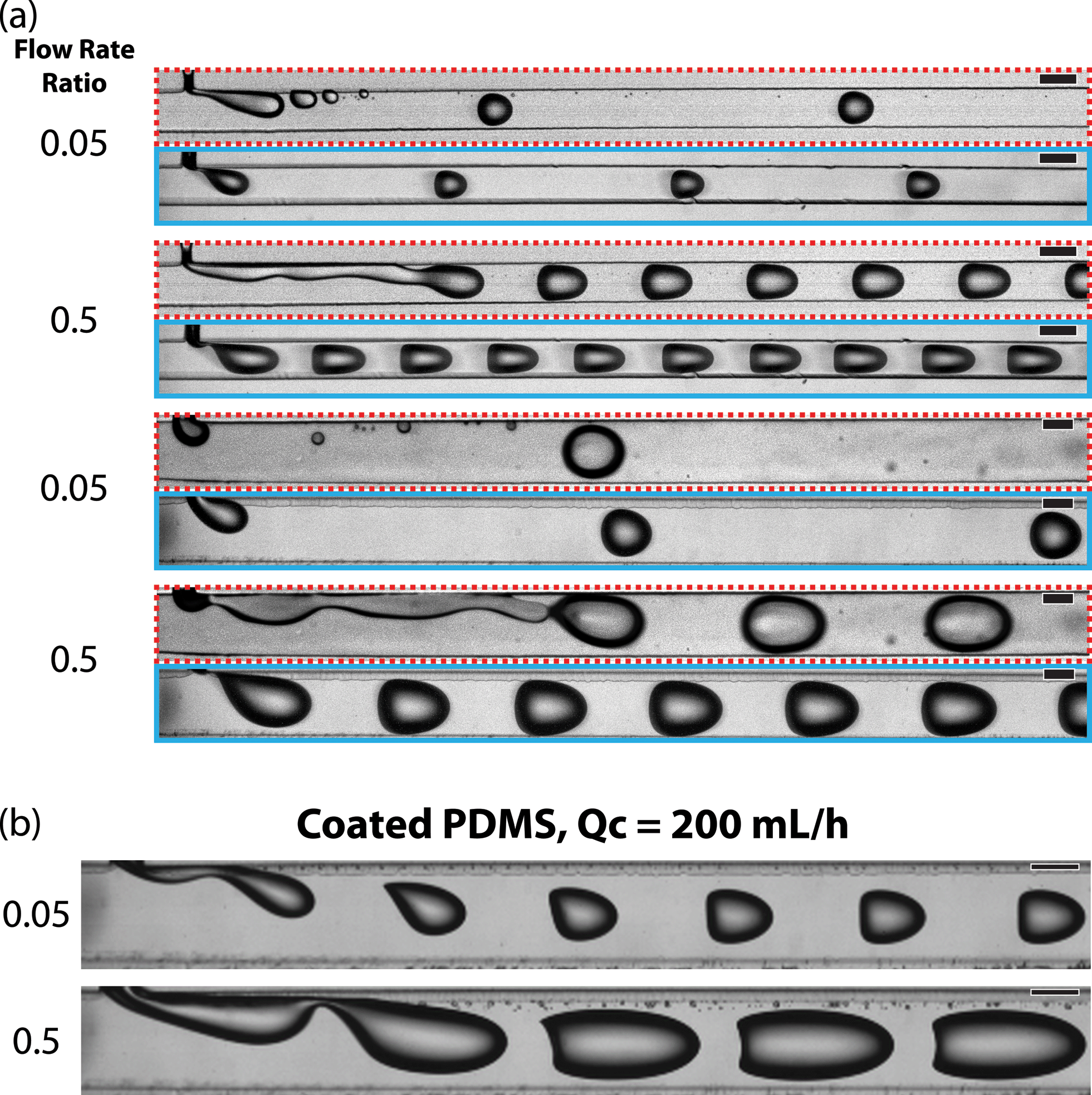 | ||
| Fig. 2 (a) Micrographs contrast the droplet formation process in uncoated (red/dashed outline) and fluoropolymer coated (cyan/solid outline) channels. The continuous phase flow rate was 10 mL h−1 and the channel widths were 200 or 400 μm. (b) Droplet formation was imaged at a continuous phase flow rate (Qc) of 200 mL h−1 in coated channels with a width of 400 μm. Scale bars indicate 200 μm | ||
Organic phase extraction was performed in serpentine channels with a height of 130 μm and inlet widths of 50 and 400 μm. FC-40 (3M) was injected via the fluorous phase inlet. Dyed aqueous streams were injected via the aqueous inlets and octanol, dodecane, and hexane were injected via the organic phase inlet (Fig. 5c).
Fluids were injected using a solenoid valve-actuated control system.40 Each liquid was contained within a reservoir that was connected, at the top, to a back pressure of nitrogen (10–15 psi) and, at the bottom, to an inline solenoid valve (Lee). Nitrogen displaced liquid from the reservoirs. This liquid flowed through LabView-controlled solenoid valves. Each droplet in a given set of droplets was successively injected such that the time each valve remained open determined the droplet size. The FC-40 flow was stopped while the other liquids were injected. New sets of droplets were not formed until the previous sets had exited the channel. The pressure applied to the FC-40 reservoir determined the flow rate of the droplets. The velocity was calculated by measuring the displacement of aqueous droplets for each pressure applied to the fluorous phase. Three separate channels were tested at each flow condition.
Results and discussion
Fluoropolymer coated droplet formation devices
Two sets of channel dimensions were examined, both with an inlet width to main channel width ratio of 1:4. These PDMS channels were pre-assembled and then coated with a fluorinated polymer, poly(1H,1H,2H,2H-perfluorodecyl acrylate-co-ethylene glycol diacrylate), using initiated chemical vapor deposition as previously described.14 The contact angle of a water droplet on the polymer film on a silicon wafer was 120°, matching our previously reported value, which corresponded to a cross-linked barrier film that was impermeable to organics.14 These T-junction devices were completely coated with the fluoropolymer. The combination of the short channel length and the large cross-sectional area of the main channel, along with the fact that both the inlets and outlets were left open during iCVD, allowed the precursors to diffuse down the entire length of the channels. This was visualized by sealing a slab of PDMS with the channel pattern to a silicon wafer, performing iCVD polymerization, and peeling off the PDMS. This revealed a continuous coating on the silicon wafer (Fig. 1a). The polymer outline matched that of a device filled with a solution of crystal violet. (Fig. 1b).
Droplets formed better in coated channels than in uncoated channels. Surface energy and wetting have been shown to play a key role in controlling the conditions under which droplets can be formed in two-phase flows in microchannels.29 We have previously demonstrated that fluoropolymer-coated channels can enable the formation of droplets of ionic liquid that would otherwise wet the uncoated PDMS walls. In that case, the static contact angle of the ionic liquid transitions from 75° on bare PDMS to 110° on the fluoropolymer-coated PDMS, crossing the empirical 92° minimum for droplet formation to occur.15,29 In the case of an aqueous dispersed phase, the static contact angle on PDMS was measured as 112 ± 1°. This increased to 135 ± 3° on the fluoropolymer on PDMS due to the formation of a rough coating. The operating regime in which stable droplets formed was greatly extended by coating the channels. We examined the formation of aqueous droplets (the dispersed phase) within an immiscible fluorocarbon oil, poly(chlorotrifluoroethylene) (PCTFE) (the continuous phase).
In the uncoated channels, at a flow rate ratio (water/oil) of 0.5, droplets formed by a jetting mechanism where breakup occurred downstream from the intersection of the dispersed and continuous phases (Fig. 2a, red/dashed outline). In uncoated channels at a flow rate ratio of 0.05, droplet holdup events occurred where small droplets, either satellite droplets or pieces of the main droplets, remained pinned to the surface of the channel. These would then recombine with passing droplets over time. This transfer of fluid between droplets is a mechanism for “droplet cross-talk” which has been described as mass transfer between discrete droplet volumes.32 The exchange of fluid creates issues when using the droplets as individual reaction vessels. Droplet formation was more stable in the fluoropolymer coated channels; droplets formed in the stable dripping regime and we did not observe any droplet holdup events (Fig. 2a, cyan/solid outline).
Coated devices formed droplets in the stable dripping regime18,20,41 over a wide range of continuous phase flow rates (0.25–200 mL h−1) and dispersed-to-continuous-phase flow rate ratios (0.05–2). This full range was achieved in the devices with a main channel width of 400 μm. The 200 μm devices could not be operated at the highest flow rates due to mechanical failure of PDMS bonding at high pressures. In the coated 400 μm channels, aqueous droplets were formed at higher capillary numbers (Ca ≈ 1) than previously reported for multi-phase flows in PDMS devices without surfactant stabilization (Fig. 2b, see ESI† for corresponding videos). Typically, the upper limit for the dripping regime is Ca ≈ 5 × 10−2 here we report stable droplet formation at Ca of 0.8.18,20
At flow rate ratios of both 0.05 and 0.5 and the highest continuous phase flow rate of 200 mL h−1, uniformly sized droplets were formed that assumed a shape conforming to the parabolic velocity profile of the carrier stream (Fig. 2b). The high flow rate overcame the interfacial tension and deformed the liquid–liquid droplet interface.42,43 The cross-section of these channels was square, allowing droplets to adopt a more rounded geometry as compared to channels with a low aspect ratio that forces droplets into a compressed, pancake shape. At the flow rate ratio of 0.05, the droplets did not contact the walls of the channel and exhibited a flat edge on their upstream side. Droplets formed at a flow rate ratio of 0.5 were larger due to the higher dispersed phase flow rate and their upstream sides were concave.
For Ca < 5 × 10−2, the size of droplets formed within the coated channels followed well studied trends as a function of the capillary number.18 Droplet size was approximated by the length of the droplets imaged through the top of the transparent PDMS. Droplet size could be tuned by changing the flow rate ratio. At a constant capillary number, the droplet size increased with increasing flow rate ratio. Increasing capillary number resulted in a decreased droplet size for flow rate ratios of 0.05, 0.25 and 0.5 (Fig. 3). At a constant flow rate ratio, the droplet size appeared to plateau above Ca ≈ 5 × 10−2. Above this threshold, droplet formation remained in the dripping regime in the coated channels. In the uncoated channels, droplet formation was observed to transition to the jetting regime above Ca ≈ 1 × 10−2. At lower capillary numbers in the uncoated channels, droplet formation shifted from dripping to jetting over time as the dispersed phase began to wet the channel walls. This transition was not observed in the coated channels.
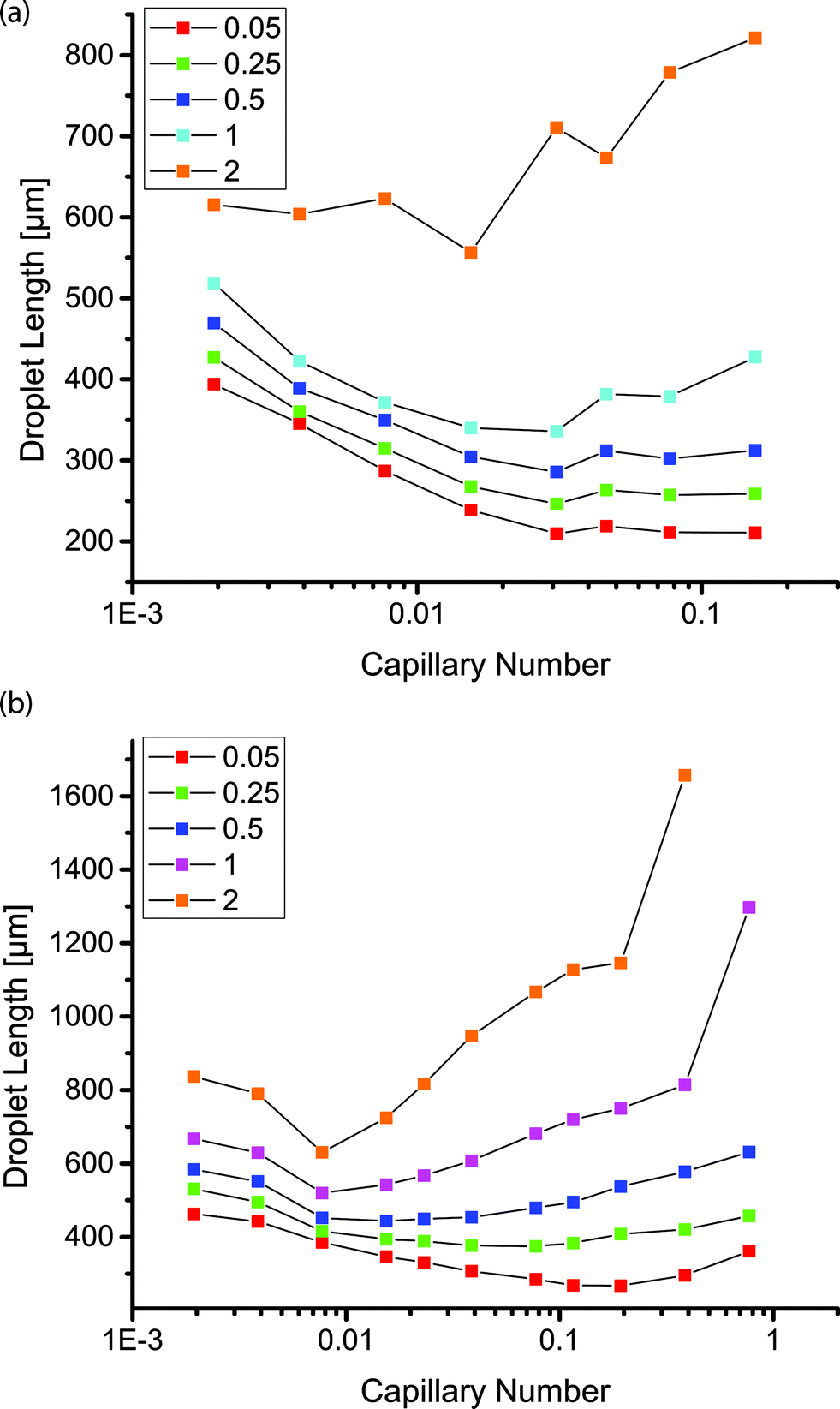 | ||
| Fig. 3 Plot of droplet length as a function of capillary number for different flow rate ratios as indicated in the legends in channels with inlet and main channel widths of (a) 50 and 200 μm and (b) 100 and 400 μm. | ||
In the coated channels, we observed that droplet formation was possible at flow rate ratios of 1 and 2, such that the flow rate of the aqueous phase equaled or surpassed that of the continuous phase. These droplets appeared stable within the channels despite the short separation between droplets and without the use of any stabilizing surfactants. At these higher flow rate ratios, the droplet size increased above Ca ≈ 5 × 10−2 or Ca ≈ 1 × 10−2 in the geometries with a main channel width of 200 or 400 μm, respectively. At the higher flow rate ratios and higher capillary numbers, the size of the continuous phase segments separating the dispersed phase droplets decreased, resulting in larger droplets. In the uncoated PDMS channel, droplets were not observed at flow rate ratios of 1 or 2 for any of the capillary numbers we tested; the two phases were observed as co-flowing streams with no jetting at any point.
As mentioned above, a key benefit of the low surface energy coating was more stable droplet formation over extended periods of operation. We examined the size distribution of droplets formed over the duration of an hour in coated and uncoated channels at two different flow rate ratios (0.05 and 0.25) at a continuous phase flow rate of 10 mL h−1. A comparison at a higher flow rate ratio of 0.5 and continuous phase flow rate of 10 mL h−1 was not possible because the uncoated devices did not produce droplets. The distribution of sizes was noticeably wider in the uncoated channels than in the fluoropolymer-coated devices. Comparing identical flow conditions, the statistical F-test revealed that the standard deviation of droplet lengths in the uncoated PDMS channels was larger than the standard deviation of droplet lengths in the coated channels (p < 0.01). This analysis examined the ratio of variances of droplet sizes in the coated and uncoated channels. We represented this graphically by plotting the cumulative deviation from the mean versus time 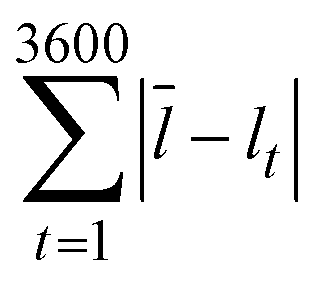 , where l is the droplet length (Fig. 4). The mean
, where l is the droplet length (Fig. 4). The mean ![[l with combining macron]](https://www.rsc.org/images/entities/i_char_006c_0304.gif) was a constant value calculated based on all droplets observed. We expect some deviation attributable to variation in the syringe pumps. However, the deviation was larger in the uncoated channels (red) than in the coated channels (cyan) under identical conditions.
was a constant value calculated based on all droplets observed. We expect some deviation attributable to variation in the syringe pumps. However, the deviation was larger in the uncoated channels (red) than in the coated channels (cyan) under identical conditions.
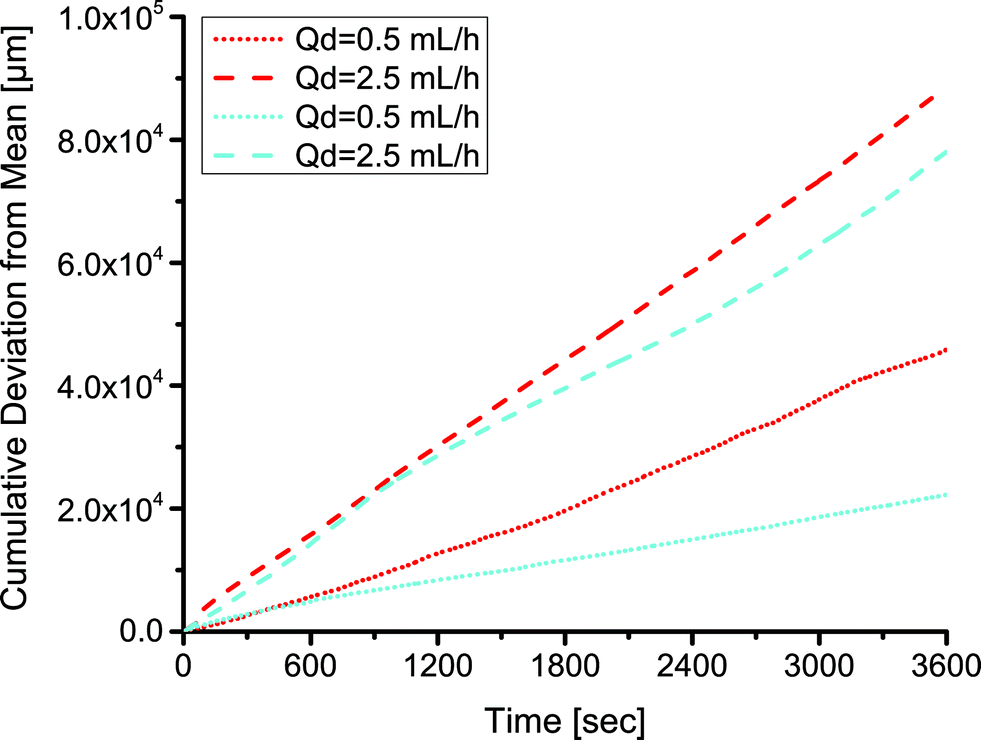 | ||
| Fig. 4 The deviation in the size of droplets in coated (cyan) and uncoated (red) channels during an hour of operation at two different flow rate ratios and a continuous flow rate of 10 mL h−1 and various dispersed phase flow rates (Qd) as indicated in the legend. The deviations are calculated based on a constant, total mean. | ||
Extraction-induced droplet merger
In addition to facilitating droplet formation, the fluoropolymer coating functioned as a barrier to control where an organic phase partitioned into the PDMS for extraction-induced merger of droplets. We utilized serpentine channels that were 40 cm long with a main channel width of 400 μm, inlet widths of 50 μm, and a channel height of 130 μm. We patterned the polymeric coating to restrict the fluoropolymer to the vicinity of the inlets of the channel. To limit polymerization to only this region, the outlet was sealed with tape. The inlets remained open for precursors to diffuse in and polymerize on the luminal surfaces. At our operating pressure, the calculated mean free path of the precursors was on the order of 100 μm. We achieved a patterned coating by exploiting our previous work which showed that a channel with a width of 200 μm and a height of 50 μm had an extent of coating of approximately 1.5 cm.14 The extent of coating is defined as the location where the polymer thickness is greater than 10 nm. When a silicon wafer was used to seal the channels, the extent of coating was observed to extend until the first turn in the channel (Fig. 5b). The combination of the winding channel and the distance from the inlets led to a concentration gradient of precursors that ultimately decreased to a point below which a thin film polymer coating could be observed. The full extent of the channel can be seen when it is filled with a solution of crystal violet (Fig. 5c). Leaving the outlet uncovered did not change the extent of polymer deposition near the inlets, indicating that mass transfer of reactants was via diffusion rather than convective flow through the channel. Near the uncovered outlet, polymer deposited within the channel. Increasing the deposition time resulted in a thicker polymer film but it did not significantly change the extent of coating down the channel (see ESI,† Fig. S2). This result was consistent with previous work on vapor deposition of modified poly(p-xylenes) where the polymer thickness within microchannels increased linearly with time.13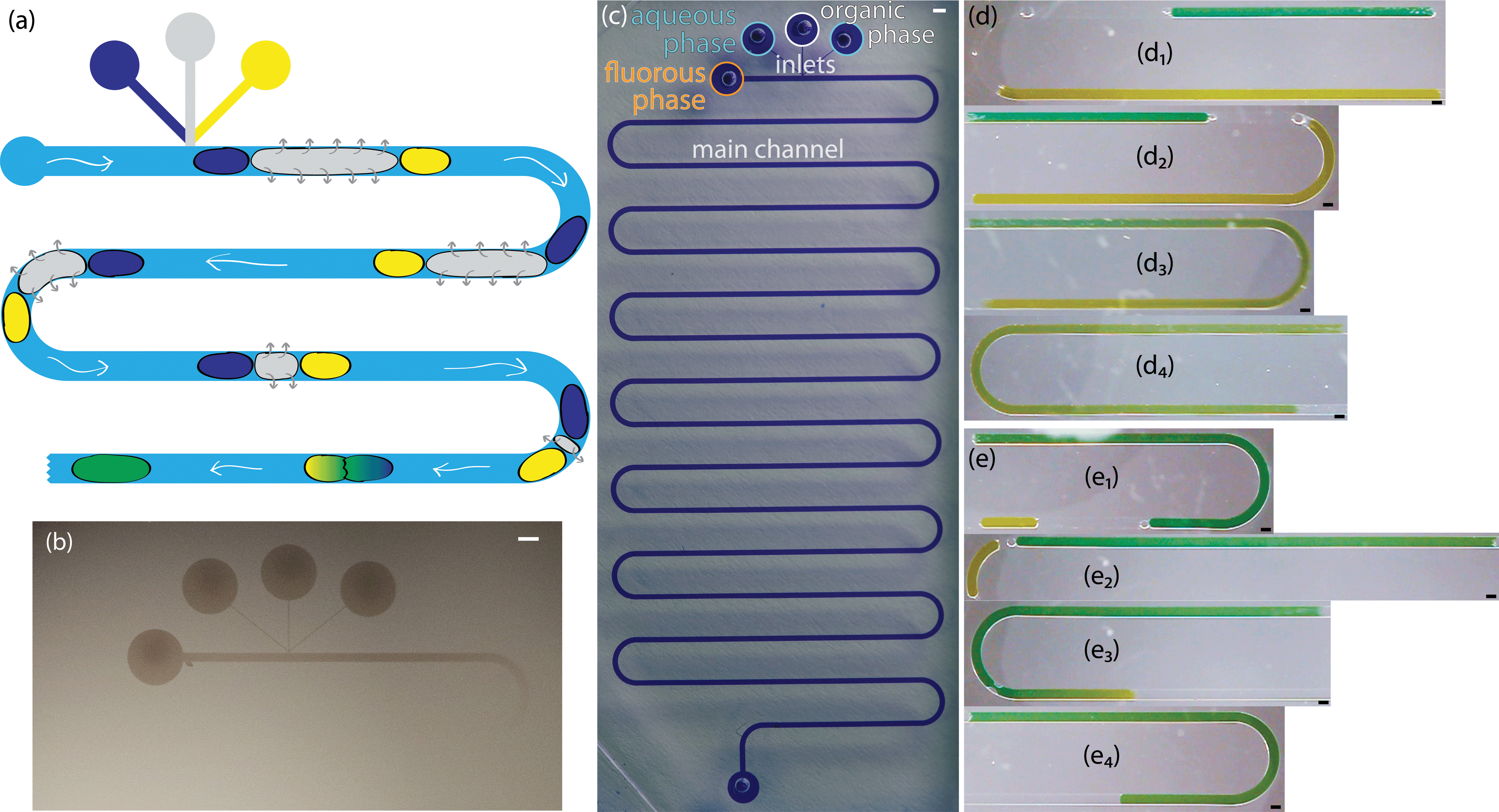 | ||
| Fig. 5 (a) Cartoon of the extraction-induced process to merge two aqueous droplets (navy and yellow) separated by an organic droplet (gray) within a fluorous phase (blue). (b) Silicon wafers were masked with a PDMS slab imprinted with the channel pattern, coated with fluoropolymer using iCVD, and the PDMS was peeled off revealing the polymer coating on the silicon wafer. (c) Devices made from the same channel patterns were filled with a solution of crystal violet to elucidate the morphology of the channel. (d/e) Micrographs of two different sets of droplets (two dyed aqueous droplets surrounding a transparent organic droplet). Each set is shown at different stages in the merger process (1–4). Scale bars represent 400 μm. | ||
In this channel geometry, active control over the input streams was used to control droplet size. We controlled the size and frequency of droplets independent of the continuous, fluorous phase flow rate. Thus, we could modulate the size of each droplet independent of the flow conditions. We used solenoid valve-actuated control on each stream to form discrete trains of droplets with two aqueous droplets surrounding an organic spacer droplet. This active control of droplet size is useful in designing reaction schemes because passive formation of droplets by shear breakup produces a limited range of droplet sizes and frequencies. We merged the two aqueous droplets by extracting and eliminating the organic droplet (Fig. 5a, d, e). We utilized an organic (i.e., dodecane, hexane, or octanol) spacer droplet to control the timing of the merger event. The extraction occurred in the uncoated region of the channel, downstream from the inlets. Sets of droplets flowed downstream under the pressure of the fluorous phase. The PCTFE oil used for droplet formation experiments proved inadequate for this work because it was miscible with a wide range of organics tested. Therefore, we used FC-40 (a tertiary amine with three perfluoroalkane substituents) as the fluorous phase. This fluorinated oil was immiscible with hexane, dodecane, and octanol. The pressure drop across the length of the channel exerted a force on the organic droplet that drove the organic phase into the walls of the channel. Typical of low Reynolds number flows in microfluidic channels, the pressure drop across the channel was proportional to the flow rate.44 At higher flow rates, the pressure drop across the device increased and the pressure on the organic droplet was greater. Therefore, there was a higher driving force for extraction at high flow rates. For an organic droplet of constant size, the time to merge the outer aqueous droplets decreased with increasing velocity (Fig. 6a). We tested four different flow rates resulting in droplet velocities of 1.0 to 2.0 mm s−1. The time to merge two droplets increased as the length of the organic phase droplet increased for a constant flow rate. This trend was consistently observed for all four flow rates. In these experiments, the size of the aqueous droplets remained approximately constant.
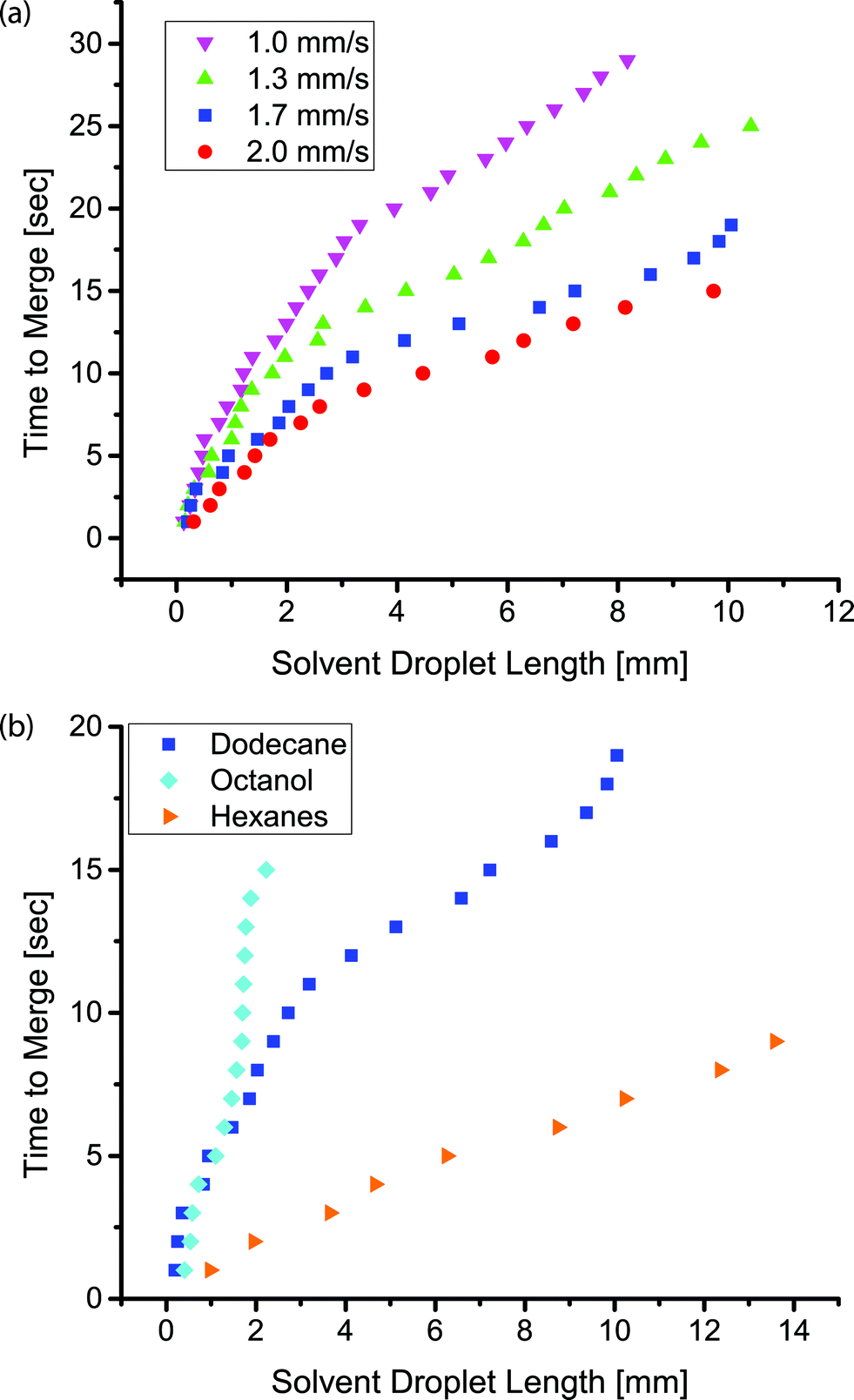 | ||
| Fig. 6 The relation between the time to merge two aqueous droplets and the length of the organic droplet (a) for different droplet velocities and an organic phase of dodecane and (b) for different organics at a constant droplet velocity of 1.7 mm s−1. | ||
The composition of the organic droplet also affected the time to merge droplets. Generally, liquids with a solubility parameter similar to that of PDMS (14.9 MPa0.5) will partition into the PDMS more quickly due to an ability to swell the PDMS to a higher degree.37 We examined three organics: octanol, dodecane, and hexane. These three liquids form a series with solubility parameters increasingly approaching that of PDMS.37 As expected, we observed that the merger time for a given droplet length decreased as the solubility parameter of the organic phase approached that of PDMS (Fig. 6b). Each droplet train was flowed down the channel at the same flow rate, thus the same pressure drop existed in all three cases. The droplet velocity was 1.7 mm s−1 and the dodecane data is the same as that shown in Fig. 6a. The PDMS was not saturated with the organic phase during the course of these experiments (see ESI†).
Finally, we showed the merger times in this system were insensitive to the size of the aqueous droplets. We injected an aqueous droplet of varying length, followed by an organic droplet of constant length, and finally an aqueous droplet of constant length. The time to merge the two aqueous droplets depended only on the initial size of the organic droplet (Fig. 7). We showed various sized droplets merging in the same channel geometry (Fig. 5d, e). Previous techniques for merging droplets have relied on decreasing the velocity of a lead droplet to allow a second to catch up and merge; however, they are sensitive to droplet size and fluid properties.16,33,35 In our extraction-induced merging, the merging was only dependent on the organic phase composition and the flow rate. Neither of these depends on the properties of the merged droplets.
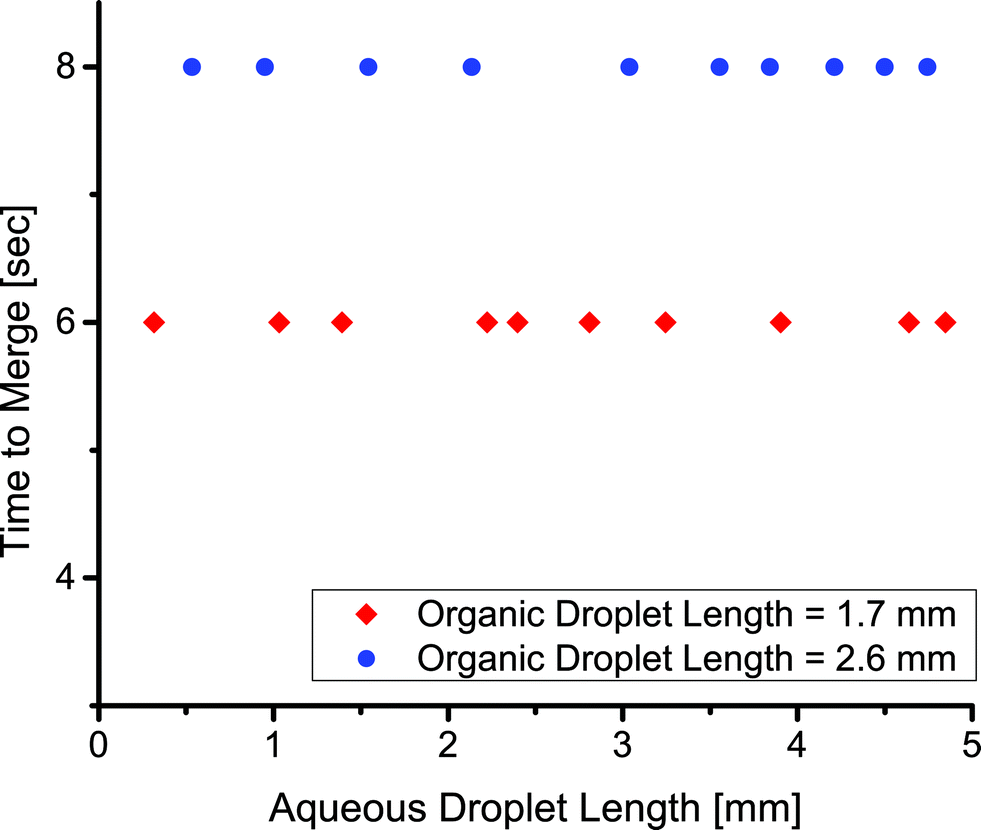 | ||
| Fig. 7 The relation between the time to merge two aqueous droplets and the length of one of the aqueous droplets for a droplet velocity of 2.0 mm s−1. | ||
Conclusions
We have demonstrated a robust extraction-induced droplet merging technique that works with a wide range of droplet sizes and flow rates. In contrast to previously demonstrated methods for droplet merger, our extraction-induced method is not tied to the device geometry. We patterned a cross-linked fluoropolymer coating using a solventless, maskless process to facilitate droplet formation. The polymer was deposited near the inlets of serpentine channels while leaving the downstream region unmodified. The time to merge droplets depended on the composition of the organic phase and the velocity of the droplets. The organic phase could be changed to a wide range of fluids because the fluorous phase, FC-40, is immiscible with various organics. Additionally, the aqueous droplets could be replaced with another liquid phase that is immiscible with both the organic droplet and the fluorous phase. Extraction-induced merger could be used in translating many multi-step reactions onto microfluidic platforms. Additionally, in fully coated channels, we formed droplets at unusually high continuous phase flow rates up to 200 mL h−1 and dispersed-to-continuous-phase flow rate ratios up to 1 and 2. This allows for high density and high frequency droplet generation and greatly increases droplet device throughput beyond what has previously been demonstrated.Acknowledgements
CTR is supported by a fellowship from the von Liebig center (CETCF). We thank Professor Veronica Eliasson for use of the Phantom camera. This work was based on a project initially funded by the National Science Foundation (award CMMI-0926969).Notes and references
- J. R. Anderson, D. T. Chiu, R. J. Jackman, O. Cherniavskaya, J. C. McDonald, H. Wu, S. H. Whitesides and G. M. Whitesides, Anal. Chem., 2000, 72, 3158–3164 CrossRef CAS.
- D. C. Duffy, J. C. McDonald, O. J. A. Schueller and G. M. Whitesides, Anal. Chem., 1998, 70, 4974–4984 CrossRef CAS PubMed.
- M. W. Toepke and D. J. Beebe, Lab Chip, 2006, 6, 1484–1486 RSC.
- K. Haubert, T. Drier and D. Beebe, Lab Chip, 2006, 6, 1548–1549 RSC.
- S. Bhattacharya, A. Datta, J. M. Berg and S. Gangopadhyay, J. Microelectromech. Syst., 2005, 14, 590–597 CrossRef CAS.
- H. Y. Chen and J. Lahann, Anal. Chem., 2005, 77, 6909–6914 CrossRef CAS PubMed.
- J. M. Jeong, M. S. Oh, B. J. Kim, C. H. Choi, B. Lee, C. S. Lee and S. G. Im, Langmuir, 2013, 29, 3474–3481 CrossRef CAS PubMed.
- A. R. Abate, D. Lee, T. Do, C. Holtze and D. A. Weitz, Lab Chip, 2008, 8, 516–518 RSC.
- S. Hu, X. Ren, M. Bachman, C. E. Sims, G. P. Li and N. L. Allbritton, Anal. Chem., 2004, 76, 1865–1870 CrossRef CAS PubMed.
- S. Hu, X. Ren, M. Bachman, C. E. Sims, G. P. Li and N. L. Allbritton, Langmuir, 2004, 20, 5569–5574 CrossRef CAS.
- A. R. Abate, A. T. Krummel, D. Lee, M. Marquez, C. Holtze and D. A. Weitz, Lab Chip, 2008, 8, 2157–2160 RSC.
- S. Seidel, C. Riche and M. Gupta, Encyclopedia of Polymer Science and Technology, 2011 Search PubMed.
- H. Y. Chen, Y. Elkasabi and J. Lahann, J. Am. Chem. Soc., 2006, 128, 374–380 CrossRef CAS PubMed.
- C. T. Riche, B. C. Marin, N. Malmstadt and M. Gupta, Lab Chip, 2011, 11, 3049–3052 RSC.
- L. L. Lazarus, C. T. Riche, B. C. Marin, M. Gupta, N. Malmstadt and R. L. Brutchey, ACS Appl. Mater. Interfaces, 2012, 4, 3077–3083 CAS.
- L. M. Fidalgo, C. Abell and W. T. Huck, Lab Chip, 2007, 7, 984–986 RSC.
- T. Schneider, D. R. Burnham, J. VanOrden and D. T. Chiu, Lab Chip, 2011, 11, 2055–2059 RSC.
- G. F. Christopher, N. N. Noharuddin, J. A. Taylor and S. L. Anna, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2008, 78, 036317 CrossRef.
- G. F. Christopher and S. L. Anna, J. Phys. D: Appl. Phys., 2007, 40, R319–R336 CrossRef CAS.
- M. De Menech, P. Garstecki, F. Jousse and H. A. Stone, J. Fluid Mech., 2008, 595, 141–161 Search PubMed.
- W. Lee, L. M. Walker and S. L. Anna, Phys. Fluids, 2009, 21, 032103 CrossRef PubMed.
- S. L. Anna, N. Bontoux and H. A. Stone, Appl. Phys. Lett., 2003, 82, 364 CrossRef CAS PubMed.
- J. D. Tice, A. D. Lyon and R. F. Ismagilov, Anal. Chim. Acta, 2004, 507, 73–77 CrossRef CAS PubMed.
- J. D. Tice, H. Song, A. D. Lyon and R. F. Ismagilov, Langmuir, 2003, 19, 9127–9133 CrossRef CAS.
- A. Rotem, A. R. Abate, A. S. Utada, V. Van Steijn and D. A. Weitz, Lab Chip, 2012, 12, 4263–4268 RSC.
- V. van Steijn, M. T. Kreutzer and C. R. Kleijn, Chem. Eng. Sci., 2007, 62, 7505–7514 CrossRef CAS PubMed.
- S. van der Graaf, T. Nisisako, C. G. Schroen, R. G. van der Sman and R. M. Boom, Langmuir, 2006, 22, 4144–4152 CrossRef CAS PubMed.
- P. Garstecki, M. J. Fuerstman, H. A. Stone and G. M. Whitesides, Lab Chip, 2006, 6, 437–446 RSC.
- W. Li, Z. Nie, H. Zhang, C. Paquet, M. Seo, P. Garstecki and E. Kumacheva, Langmuir, 2007, 23, 8010–8014 CrossRef CAS PubMed.
- R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef CAS PubMed.
- E. Um, M. E. Rogers and H. A. Stone, Lab Chip, 2013, 13, 4674–4680 RSC.
- X. Casadevall i Solvas and A. deMello, Chem. Commun., 2011, 47, 1936–1942 RSC.
- X. Niu, S. Gulati, J. B. Edel and A. J. deMello, Lab Chip, 2008, 8, 1837–1841 RSC.
- Y.-C. Tan, Y. L. Ho and A. P. Lee, Microfluid. Nanofluid., 2006, 3, 495–499 CrossRef.
- N. Bremond, A. R. Thiam and J. Bibette, Phys. Rev. Lett., 2008, 100, 024501 CrossRef.
- L. H. Hung, K. M. Choi, W. Y. Tseng, Y. C. Tan, K. J. Shea and A. P. Lee, Lab Chip, 2006, 6, 174–178 RSC.
- J. N. Lee, C. Park and G. M. Whitesides, Anal. Chem., 2003, 75, 6544–6554 CrossRef CAS PubMed.
- N. Malmstadt, M. A. Nash, R. F. Purnell and J. J. Schmidt, Nano Lett., 2006, 6, 1961–1965 CrossRef CAS PubMed.
- M. Gupta and K. K. Gleason, Langmuir, 2006, 22, 10047–10052 CrossRef CAS PubMed.
- K. W. Bong, S. C. Chapin, D. C. Pregibon, D. Baah, T. M. Floyd-Smith and P. S. Doyle, Lab Chip, 2011, 11, 743–747 RSC.
- J. H. Xu, S. W. Li, J. Tan and G. S. Luo, Microfluid. Nanofluid., 2008, 5, 711–717 CrossRef CAS.
- M. T. Kreutzer, F. Kapteijn, J. A. Moulijn, C. R. Kleijn and J. J. Heiszwolf, AIChE J., 2005, 51, 2428–2440 CrossRef CAS.
- C. P. Tostado, J. Xu and G. Luo, Chem. Eng. J., 2011, 171, 1340–1347 CrossRef CAS PubMed.
- S. A. Vanapalli, A. G. Banpurkar, D. van den Ende, M. H. Duits and F. Mugele, Lab Chip, 2009, 9, 982–990 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Videos of droplet formation at a continuous phase flow rate of 200 mL h−1 and description of image analysis. See DOI: 10.1039/c4lc00087k |
| This journal is © The Royal Society of Chemistry 2014 |