 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Liquid polystyrene: a room-temperature photocurable soft lithography compatible pour-and-cure-type polystyrene
Tobias M.
Nargang
a,
Lara
Brockmann
a,
Pavel Mitkov
Nikolov
b,
Dieter
Schild
c,
Dorothea
Helmer
a,
Nico
Keller
a,
Kai
Sachsenheimer
a,
Elisabeth
Wilhelm
a,
Leonardo
Pires
a,
Marian
Dirschka
a,
Alexander
Kolew
a,
Marc
Schneider
a,
Matthias
Worgull
a,
Stefan
Giselbrecht
b,
Christiane
Neumann
a and
Bastian E.
Rapp†
*a
aInstitute of Microstructure Technology (IMT), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: bastian.rapp@kit.edu
bInstitute for Biological Interfaces 1 (IBG-1), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
cInstitute for Nuclear Waste Disposal (INE), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 7th May 2014
Abstract
Materials matter in microfluidics. Since the introduction of soft lithography as a prototyping technique and polydimethylsiloxane (PDMS) as material of choice the microfluidics community has settled with using this material almost exclusively. However, for many applications PDMS is not an ideal material given its limited solvent resistance and hydrophobicity which makes it especially disadvantageous for certain cell-based assays. For these applications polystyrene (PS) would be a better choice. PS has been used in biology research and analytics for decades and numerous protocols have been developed and optimized for it. However, PS has not found widespread use in microfluidics mainly because, being a thermoplastic material, it is typically structured using industrial polymer replication techniques. This makes PS unsuitable for prototyping. In this paper, we introduce a new structuring method for PS which is compatible with soft lithography prototyping. We develop a liquid PS prepolymer which we term as “Liquid Polystyrene” (liqPS). liqPS is a viscous free-flowing liquid which can be cured by visible light exposure using soft replication templates, e.g., made from PDMS. Using liqPS prototyping microfluidic systems in PS is as easy as prototyping microfluidic systems in PDMS. We demonstrate that cured liqPS is (chemically and physically) identical to commercial PS. Comparative studies on mouse fibroblasts L929 showed that liqPS cannot be distinguished from commercial PS in such experiments. Researchers can develop and optimize microfluidic structures using liqPS and soft lithography. Once the device is to be commercialized it can be manufactured using scalable industrial polymer replication techniques in PS – the material is the same in both cases. Therefore, liqPS effectively closes the gap between “microfluidic prototyping” and “industrial microfluidics” by providing a common material.
Introduction
The choice of materials is of paramount importance in microfluidics. Given the high surface-to-volume ratios in microfluidics, the chemical and/or physical properties of the material from which the microfluidic system is made of significantly influence the behaviour of the system. Therefore, one would suppose that materials research is an essential component of the microfluidics community and that a wide choice of potential materials is being developed and characterized. However, the choice of materials is rarely motivated by the requirements of the application but rather by the ease of manufacturing. The incredible success story that siloxane-based polymers, most notably polydimethylsiloxane (PDMS), have seen since the introduction of this class of materials in 19981 bears witness of this fact. Without a doubt, PDMS has a number of advantageous properties such as its optical clarity and the ability to be bonded after corona or plasma activation.2 Furthermore, the elasticity of the material allows the creation of monolithic mechanical valves, pioneered by Kazuo and Ryutaro3 and most notably by the Quake group.4 However, the most prominently cited advantage of the material is its ease of manufacturing. PDMS can be cured by room-temperature casting from structured layers of photoresists such as SU-8. Duffy et al.1 referred to this process as rapid prototyping of microfluidic systems in PDMS and numerous microfluidic systems demonstrated in the literature have been created using this technique. Alternative methods based on direct structuring of PDMS using UV lithography have also been described.5 Interestingly most of these applications use PDMS primarily because it is so much easier to create microfluidic components from PDMS than from any other materials. However, PDMS suffers from numerous drawbacks, most notably its high solubility in most polar organic solvents which cause intense swelling of the material.6 This property is especially disadvantageous in analytical assays where PDMS may act as a selective absorber material resulting in sample cross-contamination.7 Furthermore, solvents (including water) readily evaporate through PDMS which is disadvantageous if liquid is to be kept inside of channels for prolonged experiments. This may be especially problematic for analytical assays where the change of the sample volume results in a change of the analyte concentration over time.Numerous materials have been described for microfluidic devices with the aim of replacing PDMS by solving (at least) one of its inherent disadvantages. In 1997 Priola et al.8 described a class of elastomers based on perfluorinated polyethers (PFPE) as photocurable solvent-resistant replacements for PDMS.9 Another type of PFPE-based polymers is SIFEL, a two-component curing siloxane/PFPE hybrid material.10 Even though these materials outperform PDMS in chemical resistance, they have not been widely adopted mostly due to the fact that they are significantly more expensive than PDMS and, due to their high chemical inertness, difficult to bond. Thiol-ene based polymers have been suggested by Kim et al.11 The most commonly used materials of this class are the commercially available optical adhesives by Norland (termed Norland Optical Adhesives, NOA) which can be photocured and bonded after plasma activation. Alvankarian et al. introduced polyurethane methacrylate (PUMA) as an alternative elastomer material for microfluidics.12 However, none of the alternative materials have reached the popularity and widespread application of PDMS. There are multiple reasons for this. Firstly, none of the described materials offers advantages over PDMS in prototyping. Secondly, they are mostly more expensive than PDMS. Thirdly, using a new material unknown to the community raises numerous questions concerning long-term (chemical and biochemical) stability under various experimental conditions, ease of manufacturing and biocompatibility. Researches more focussed on application development tend to avoid having to answer these questions by sticking with well-known (and characterized) materials.
However, we like to draw attention to another problem which PDMS microfluidics faces and which may be termed as the “three community problem”. The material was originally described for “rapid prototyping of microfluidic systems”1 and in fact it has found widespread application in the, so to speak, “microfluidic prototyping” community. Materials suitable for prototyping may (and often are) not be ideal once a device developed is scaled up to an industrial application. Due to its crosslinking chemistry which is based on platinum-catalysed hydrosilylation, PDMS cannot be manufactured at acceptable cycle times industrially as the curing process is too slow. The “industrial microfluidics” community rather favours materials which can be structured by scalable industrial polymer replication processes that allow creation of components in cycle times as short as several seconds. This requirement is best fulfilled by thermoplastic polymers. The third and potentially most important community, especially for microfluidic applications in the life sciences, can be termed as the “applied microfluidics” community which is mostly composed of researches with non-technical academic backgrounds. Cell biologist, biochemists as well as researchers from fundamental biology are well acquainted to one specific material: polystyrene (PS). In the “applied microfluidics” community, PS is without a doubt the most widely used and studied material. Protocols based on PS can built on decades of experimental results from well-established laboratory platforms, e.g., petri dishes, culture flasks and microwell plates, which have been used extensively especially for cell culture. PS is highly biocompatible, of high optical clarity, mechanically robust and among the cheapest polymers available.13 Furthermore, PS is a thermoplastic polymer which is compatible with the demands of the “industrial microfluidics” community. Suitable manufacturing methods have been reviewed by Becker and Locascio.14 These methods include injection moulding or hot embossing, potentially even from structurally less resilient templates such as crosslinked epoxy moulds.15 Laser structuring using, e.g., a CO2 laser system is another alternative.16 On a laboratory scale PS may be structured by melting the polymer against PDMS templates. This process involves prolonged heating of PS on a hot plate (185 °C for 9 h) and handling the hot melt.17 Reduced process times (in the range of a few minutes) can be achieved by clamping a thin sheet of PS and a PDMS mould between two glass slides and putting the stack into an oven.18,19 However, this soft embossing method is limited to low aspect ratios because of the elasticity of the mould material. Another method reported is “Shrinky-Dink microfluidics” where a (biaxially) stretched PS foil is first structured and bonded and then exposed to thermal treatment whereupon the material shrinks.20 The shrinking is not entirely homogenous and thus the final dimensions of the structures are difficult to adjust. Given that none of these processes is compatible with the requirements of the “microfluidic prototyping” community, PS has not been accepted as suitable prototyping material. Even most recent reviews only list PS as a polymer for industrial replication.21 Attempts have been made to use PS for prototyping, most notable by Wang et al. who described a solvent-based micromoulding technique for PS.22 In this process, PS is first dissolved in a solvent and cast onto a replication mould. After solvent extraction under reduced pressure, PS components are obtained. However, this process has not found wide adoption most likely due to the necessity for handling solvents and working under reduced pressure.
David Beebe recently superbly summarized the discrepancy in material preference between the “microfluidic prototyping” community and the “applied microfluidics” community with the phrase “engineers are from PDMS-land, biologists are from polystyrenia”.13 This paper intends to “merge PDMS-land and polystyrenia” by establishing a new structuring technique for PS which will make the material appealing for use in rapid prototyping. For this, we introduce a liquid precursor to polystyrene which we term as “Liquid Polystyrene” (liqPS). liqPS is a photocurable “pour-and-cure”-type polymer which will, once cured, turn into pure PS. We show that this material behaves identically to commercial polystyrene using X-ray photoelectron spectroscopy (XPS) and Raman spectroscopy as well as cell culture experiments on mouse fibroblasts L929. Using liqPS, the “microfluidic prototyping,” the “industrial microfluidics” as well as the “applied microfluidics” communities will gain access to a mutual material which should not only solve the “three community problem” by facilitating interdisciplinary research between the communities but also ease the scale up of microfluidic prototypes to industrial manufacturing.
Experimental
Materials and methods
Toluene, acetone, isopropanol, cyclohexanone, ethyl-L-lactate, styrene, 2,2′-azobis(2-methylpropionitrile) (AIBN), phenylbis(2,4,6-trimethylbenzoyl)phosphine oxide (PPO) and FC-40 were purchased from Sigma-Aldrich (Germany) and used as received. SU-8 2075 was purchased from Microchemicals (Germany). Wacker Elastosil M 4601 and Elastosil RT 601 were purchased from Wacker (Germany). Cyclic olefin copolymer (COC) substrates were purchased from Kunststoffzentrum Leipzig (Germany). The handheld corona discharger used for substrate cleaning and oxidation was of type BD-20V, purchased from Electro-Technic Products Inc. (USA). Commercially available PS reference samples were purchased in the form of disposable PS dishes (article #1722) from Semadeni (Germany). The L929 expansion was carried out in PS flasks (75 cm2, SPL Life Sciences, South Korea). The corresponding liqPS samples were mounted on the bottoms of 6-well plates (Thermo Scientific, Germany). Contact angles were measured using a G-1 contact angle microscope purchased from Erma Inc. (Japan). The white light source used for liqPS curing was an OSRAM Ultra-Vitalux 300 W purchased from Conrad Electronic (Germany). Light intensities were measured with a bolometer of type PowerMax-USB PM10 purchased from Coherent Inc. (USA). Viscosities were measured using a BROOKFIELD DV-II+ Pro purchased from BROOKFIELD Inc. (USA). All viscosity values given were measured at a constant rotation of 0.5 rpm using a total liquid volume of about 1 ml.Scanning electron microscope
Scanning electron microscope images were recorded using a Zeiss SUPRA60 VP (variable pressure, Carl Zeiss NTS GmbH Germany) at the Karlsruhe Nano Micro Facility, a Helmholtz Research Infrastructure at Karlsruhe Institute of Technology (KIT). Images were recorded at a beam voltage of 3 kV and varying magnifications (see scale bars for reference).Differential scanning calorimetry (DSC)
Thermal characterization of the liqPS samples was carried out using a DSC 204 F1 Phoenix system purchased from NETZSCH (Germany). Cured liqPS samples of approximately 15 mg were used during analysis. Samples were measured against empty trays as reference.UV/VIS spectroscopy
A portable USB UV/VIS spectrometer of type CCS100 (purchased from Thorlabs, Germany) was used for characterizing optical transmission spectra. Light was coupled into the instrument via a flexible light guide from a portable halogen lamp. Reference spectra (air, i.e., no sample inserted) were recorded in transmission mode. The transmission spectra of cured liqPS and commercially available PS of 2 × 2 cm2 lateral dimensions and 1 mm thickness were recorded.X-ray photoelectron spectroscopy
Polymer samples were analysed with an X-ray photoelectron spectrometer (XPS, ULVAC-PHI Inc., model PHI 5000 VersaProbe I, Japan) equipped with a scanning microprobe X-ray source (monochromatic Al Kα (1486.6 eV) X-rays) in combination with an electron flood gun and a floating ion gun generating low energy electrons and low energy argon ions for charge compensation (dual beam technique), respectively. The spectrometer was equipped with a hemispherical capacitor analyser (mean diameter 279.4 mm) and a microchannel detector with 16 anodes. Calibration of the binding energy scale of the spectrometer was performed using well-established binding energies of elemental lines of pure metals (monochromatic Al Kα: Cu 2p3/2 at 932.62 eV, Au 4f7/2 at 83.96 eV).23 Standard deviations of binding energies of isolating samples were within ±0.2 eV. The spectra were collected at a take-off angle of 45° (the angle between the sample surface and the analyser). Cut surfaces of the polymer samples were prepared by means of a cleaned scalpel for bulk analyses by surface sensitive XPS. Survey scans were recorded with an X-ray source power of 50 W and an analyser pass energy of 187.85 eV to identify the elements and to determine their atomic concentrations at the sample surface. The X-ray beam diameter was adjusted to 200 μm and scanned over an area of 0.5 × 0.5 mm2. To retrieve information about the chemical state of the elements, narrow scan spectra of the elemental lines and the valence band were recorded at a pass energy of 23.5 eV and a step size of 0.1 eV. All spectra were charge referenced to the C 1s elemental line of the hydrocarbon (CXHy) at 284.8 eV. Data analysis was performed using the ULVAC-PHI MultiPak program version 9.4.Raman spectroscopy
A Bruker Senterra Raman microscope (purchased from Bruker, Germany) was used for analyses of the samples. Two laser wavelengths, 532 nm and 785 nm, were used subsequently to identify Raman active lines at the spectra. Calibration of the wavenumber scale was checked by measurement of a polystyrene reference sample supplied by Bruker. During measurement of the samples, wavenumber calibration was automatically performed by the Bruker's SureCal technique.Cell cultures
Mouse L929 fibroblasts (NCTC clone 929, ATCC, USA) were grown in Eagle's Minimal Essential Medium (EMEM, with L-glutamate, ATCC, USA), supplemented with 10% Fetal Bovine Serum (FBS, ATCC, Germany) and 1% penicillin–streptomycin (100× stock, PAA, Austria) at 37 °C and 5% CO2. The cells were cultivated in 75 cm2 flasks (SPL Life Sciences, South Korea) for 1 week until reaching approximately 100% confluence. The liqPS samples were fixed on the bottom of 6-well plates (SPL Life Sciences, South Korea), covered with 2 ml of the medium and let to precondition at standard culturing conditions for 2 h. The PS well plates were preconditioned in the same manner. Some 0.05 × 106 L929 cells were inoculated per liqPS or PS sample and left to grow in L929-adjusted EMEM for 168 h at standard conditions. In all experiments, the L929 cells were used until passage 20.Live/dead staining and XTT proliferation assays
The viability and proliferation rates of the L929 cells grown on PS and liqPS samples were analysed at 3 time points: 24 h, 96 h and 168 h. The viability assay required washing of the attached cells in PBS+/+ buffer (3 times, 5 min), which were then stained with a master-mix solution, containing Calcein-AM (5 μM, Sigma-Aldrich, USA) and propidium iodide (PI, 500 nM, Sigma Life Sciences, Germany) in L929-adjusted EMEM. The cells were incubated for 20 min at 37 °C under a 5% CO2 atmosphere. The staining solution was removed and the samples were rinsed with 1× PBS+/+ buffer (3 times, 5 min each). All samples were subjected to live-cell imaging using an Axiovert 200M (Carl Zeiss, Germany) inverted fluorescence microscope, equipped with the AxioVision software version 4.7 (Carl Zeiss, Germany). The cell proliferation assays on liqPS and PS samples were conducted after 24 h, 96 h and 168 h, using Cell Proliferation Kit XTT (AppliChem, Germany). At the end of each incubation time, the cells were washed with PBS+/+ (3 times, 5 min each) and incubated with 1 ml of L929-adjusted EMEM containing XTT reaction solution. The cells were incubated for 5 h at standard conditions and the absorbance of each sample was measured according to the manufacturer's recommendations at a wavelength of 450 nm using a spectrophotometer (Molecular Devices, USA).Results and discussion
Synthesis of liquid polystyrene
liqPS was synthesized by a two-step process. First, styrene was polymerized in solution by free-radical polymerization. The resulting polymer was then dissolved in small quantities of the monomer resulting in a viscous solution. This solution was mixed with PPO, a very efficient phosphine-based visible-light photoinitiator which enables, once the solution is exposed to light, the final curing to solid bulk PS. We chose PPO over other commonly used radical photoinitiators (such as benzo- or acetophenones) because phosphine-based photoinitiators show virtually no residual fluorescence once initiated. We deemed this important in order to avoid creating materials with high autofluorescence. Furthermore, PPO is a visible-light photoinitiator that can be activated using light of wavelengths above UV which allows liqPS to be cured with any halogen light source and even sunlight. Under ambient laboratory lighting conditions, the curing process takes about 12 hours. We found it sufficient to process the liqPS within one hour after mixing with PPO. Longer storage times can be obtained by keeping the blended liqPS in the dark.For the first step, styrene (60 ml) and AIBN (2.13 g, thermal radical initiator) were dissolved in toluene (60 ml) in a two-necked flask. We have not found it necessary to strip the styrene from the stabilizer. If desired, typical phenolic inhibitors (most commonly hydroquinone or 4-tert-butylcatechol) can be removed by first washing styrene with 10% (w/v) aqueous sodium hydroxide solution twice and then with distilled water until the washings are neutral. The flask was then immersed in an oil bath and refluxed for 5 hours under a nitrogen flow. After cooling, the solution is filtered with a Büchner funnel (prefilter, mesh size ~10 μm) and stripped of the solvent under reduced pressure (10 mbar, 55 °C). During solvent removal the polystyrene expands to foam filling the entire flask. Once the solvent was completely removed, the polystyrene was fractured mechanically to small flakes and a total of about 49.3 g (reaction yield about 91% calculated from the amount of monomer used) of the polymer was obtained.
In the second step the polymer was dissolved in styrene (mass ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.75, polymer to styrene) and stirred until all solids had dissolved. In this form, liqPS can be stored (4 °C, in the dark) for several weeks. Prior to use, the viscous solution must be blended with 2 m% (referred to the amount of styrene added) of PPO. The resulting “Liquid Polystyrene” is a photocurable slightly yellowish solution (see Fig. 1) with a viscosity of about 560 ± 18 mPa s (mean value of 5 measurements). During the course of this work, several different mass ratios (polymer to styrene) were evaluated. Increasing the amount of polymer yielded solutions with higher viscosity which we found difficult to process. With reduced amounts of polymer, the solution became more fluid. However, the components made from these solutions suffered from significant shrinkage during polymerization due to the density difference between the monomer (styrene, density ~0.91 g ml−1) and the polymer (PS, density ~1.04 g ml−1) which results in brittle PS components with high internal stresses. We found the polymer-to-monomer mass ratio of 1:0.75 to be the most conveniently processable liqPS creating PS components with good mechanical properties. We measured the density of this liqPS mixture to be 0.977 g ml−1 and therefore very close to the density of PS. We found this material to show negligible shrinkage upon curing. This process of synthesizing liqPS is suitable for small scale (micro molar) but can conveniently be scaled up to industrial manufacturing where kilograms of the material can be produced.
:0.75, polymer to styrene) and stirred until all solids had dissolved. In this form, liqPS can be stored (4 °C, in the dark) for several weeks. Prior to use, the viscous solution must be blended with 2 m% (referred to the amount of styrene added) of PPO. The resulting “Liquid Polystyrene” is a photocurable slightly yellowish solution (see Fig. 1) with a viscosity of about 560 ± 18 mPa s (mean value of 5 measurements). During the course of this work, several different mass ratios (polymer to styrene) were evaluated. Increasing the amount of polymer yielded solutions with higher viscosity which we found difficult to process. With reduced amounts of polymer, the solution became more fluid. However, the components made from these solutions suffered from significant shrinkage during polymerization due to the density difference between the monomer (styrene, density ~0.91 g ml−1) and the polymer (PS, density ~1.04 g ml−1) which results in brittle PS components with high internal stresses. We found the polymer-to-monomer mass ratio of 1:0.75 to be the most conveniently processable liqPS creating PS components with good mechanical properties. We measured the density of this liqPS mixture to be 0.977 g ml−1 and therefore very close to the density of PS. We found this material to show negligible shrinkage upon curing. This process of synthesizing liqPS is suitable for small scale (micro molar) but can conveniently be scaled up to industrial manufacturing where kilograms of the material can be produced.
 | ||
| Fig. 1 Appearance of liqPS after synthesis. a) liqPS before being blended with the photoinitiator PPO. The material is a clear free-flowing viscous liquid. b) After blending with PPO the solution turns yellowish due to the absorbance of the photoinitiator. The white objects visible in both vials are Teflon®-coated magnetic stirrers used for blending the photoinitiator. c) View of liqPS with the photoinitiator being poured onto a PDMS template for replication. d) View of a cured liqPS block with a microfluidic structure used in this work. Three of such structures are arranged on the replication template shown in c). The replicated liqPS structures can be separated by cutting or mechanically breaking the substrates. Following breaking, the edges can be ground off if required. The resulting PS is again a clear material. As shown via UV/VIS spectroscopy and Raman spectroscopy the material does not show any residual fluorescence due to the presence of non-initiated PPO. Colour in the online version. | ||
SU-8 template creation
The negative resist SU-8 was used for the creation of replication templates. COC substrates (6 × 6 cm2) were cleaned thoroughly by rinsing with isopropanol and blown dry with compressed air. The substrates were subsequently activated by corona discharging for 20 s at a distance of about 2 cm. SU-8 (about 4 ml) was then spin-coated for 35 s at 500 rpm (ramp 100 rpm s−1), then 1 min at 0 rpm (rest) and finally 30 s at 3000 rpm (ramp 400 rpm s−1). The resist was then prebaked using the following protocol: heating from room temperature to 75 °C (ramp 110 °C h−1), then 1 h at 75 °C, then heating from 75 °C to 95 °C (ramp 40 °C h−1), then 4 h at 95 °C and finally cooling to room temperature (approximately 4 h). The resist was subsequently structured using a custom-built maskless projection lithography system based on a digital mirror device which we have described previously.24 A total exposure time of 5.5 s per frame was used. Exposure was performed using a band-pass wavelength filter (320–400 nm) and an i-line filter (365 nm). After exposure, the following postbake protocol was applied: heating from room temperature to 65 °C (ramp 250 °C h−1), then 2 min at 65 °C, then heating from 65 °C to 95 °C (ramp 250 °C h−1), then 10 min at 95 °C and finally cooling to room temperature (approximately 4 h). The resist was developed using ethyl-L-lactate as the solvent in an ultrasonic bath (6 min). Finally the substrate was blown dry using compressed air. The creation of SU-8 replication masters has been reported multiple times in the literature with the protocols varying depending on the type of SU-8 used and the lithography system employed. We have found the reported protocol to be very robust. If multiple replica are to be created from one SU-8 master, decreasing the free surface energy by creation of a non-stick coating may be required which can be carried out, e.g., via silanization protocols using fluorinated silanes. However, we have not found this necessary during the course of this work.Creation of PDMS replica
Standard protocols were applied for creating the PDMS replica from the SU-8 template. In short, the respective PDMS (Elastosil M 4601 or RT 601) was mixed in a 9:1 (m/m) ratio, stirred extensively and degassed under a vacuum to remove trapped air bubbles. The prepolymer was then poured onto the SU-8 template and cured at 60 °C in the oven for 2 hours. The ready-to-use PDMS replica was carefully peeled off the SU-8 master and cleaned with isopropanol.
liqPS replication
liqPS was structured by casting against PDMS moulds. For this the cleaned moulds were covered with liqPS layers of various thicknesses. liqPS was then cured by exposure to visible light (halogen white lamp or arc-lamp). We found a total dosage of 2 J cm−2 sufficient to cure a liqPS layer of approximately 30 μm thickness within 10 s with an OSRAM Ultra-Vitalux 300 W white light source for which we measured a light intensity of about 0.2 W cm−2. Upon curing, the yellow colour in the material disappears as a consequence of the decomposition of PPO. If very fine structures are to be replicated (or the PDMS is very soft), we found it useful to close the PDMS mould with a thin sheet of PDMS and a quartz glass plate. By applying light pressure to the quartz plate, the closed mould could be set under pressure thus ensuring replication of delicate features. Depending on the light transparency of the top layer and the thickness of the liqPS layer longer curing times may be required. We have cured bulk liqPS components of about 5 mm thickness using the described PDMS top layer within 45 minutes. The PDMS layer absorbs about 50% of the light intensity. We measured a remaining light intensity of only 0.1 W cm−2 below the PDMS layer. If shorter curing times are required, stronger light sources and/or materials with higher optical transmission than PDMS should be used.Once cured, the solid liqPS components can be peeled off the PDMS moulds. The moulds can then be cleaned again (using the protocol described) and reused immediately. We found that a typical PDMS replica is suitable for the creation of at least five liqPS replica. Cured liqPS samples were first immersed in isopropanol then rinsed with ethanol and water and dried using pressurized air. DSC measurements on cured liqPS samples found glass transition temperatures of approximately 72 °C. The glass transition temperature of PS varies with the molecular weight of the polymer. From the literature, an average molecular weight of approximately 3600 g mol−1 may be derived.25 Thus cured liqPS is in the range of commercially available average molecular weight PS.
Cured liqPS samples can be used directly after cleaning. During the course of this work we also created replicas directly from SU-8 masters. However, given that both cured SU-8 and cured liqPS are stiff materials damage to the SU-8 mould was observed on fine microstructures. In such cases, non-stick coatings should be applied to the SU-8 moulds prior to usage. Furthermore the chemical resistance of a replication mould (i.e., the mould material) master against liqPS should be assessed prior to replication. Fig. 2 shows a replication example on a microfluidic post array. As can be observed, the replication process is very accurate and even finest artifacts are transferred with high fidelity. We have found the aspect ratio limit which can be replicated to be between 5 and 10. As can be seen, thinner posts of the post array (with aspect ratios of ~10) are not entirely transferred to liqPS from the PDMS replica whereas posts with aspect ratios of 5 are transferred accurately.
 | ||
| Fig. 2 SEM images of structures created during replication of a microfluidic post array. a) SU-8 master created using a maskless projection lithography system, b) PDMS replica created by soft lithography replication from the SU-8 master, c) replica in liqPS. The height of the structures is 50 μm, the diameter of the posts 13 μm (excluding the last column which has only 5 μm) with 40 μm pitch. a-i) As can be seen, several of the posts have been broken off the mould during development of the SU-8. b-i) These posts are also missing on the PDMS. c-i) The liqPS replica transfers the defects with high fidelity. Even the remainder of the broken posts are replicated (bottom line). The rightmost row of the post array (with posts of 5 μm diameter) is not replicated entirely. These posts have aspect ratios of 10 which is the limit of what can be replicated in this two-step replication method from the PDMS replica. a-ii) Detail view of the post array. b-ii) Detail view of the holes in the PDMS replica. c-ii) Detail view of the posts in cured liqPS. As can be seen, the replication fidelity is high. a-iii) Detail view of a defect at the wall of a microfluidic channel by incomplete removal of SU-8 resists during development. c-iii) The same defect is replicated in cured liqPS. Even these miniature artefacts are transferred accurately. The cracks in c-ii) are artefacts of the sputtering process which caused (due to the low glass transition temperature of cured liqPS and therefore light material expansion) tension at the liqPS/gold interface. | ||
Surface contact angle measurement
The surface properties of freshly cured liqPS samples were characterised by measurement of the static contact angle of water. Comparative measurements on commercial PS were carried out and the values obtained compared to the literature. The respective surfaces were first thoroughly cleaned using isopropanol. Droplets of water (5 μl) were applied to the free surface and the static contact angle measured. Values of 87° ± 2° (5 measurements) were found for cured liqPS surfaces. Contact angles of commercial PS were found to be 93° ± 2°. These values are in accordance with values from the literature which states static contact angles of around 90° (e.g., 88° cited by Kwok et al.26 or 91° cited by Ellison and Zisman27). In order to increase cell adhesion and binding of suitable adhesion promoting proteins, PS is often surface treated in order to reduce the water contact angle rendering the material more hydrophilic.28 This is also possible with liqPS. We treated a cured liqPS surface for 1 min using the corona discharger and measured static water contact angles of 25° ± 3° (5 measurements). These results are in accordance with the values obtained for standard PS.29Bonding of cured liqPS structures
After replication, cured liqPS structures must be bonded in order to close open channel structures. As cured liqPS is effectively PS all bonding methods reported in the literature which can be used for bonding structures in PS can also be used to bond cured liqPS. In addition, given the fact that liqPS is cured by radical polymerization, techniques based on partial curing may also be applicable. In the course of this work, we successfully bonded cured liqPS microfluidic chips using three techniques: solvent-based bonding, bonding by radical polymerization curing and thermal bonding.For solvent-based bonding, a thin layer of liqPS (~30 μm) was created by spin-coating (about 2 ml of liqPS, 1300 rpm, 20 s) on top of an objective slide and cured by light exposure (~10 s). Then about 1 ml of a 1:1 (v/v) mixture of acetone and cyclohexanone was deposited onto the cured layer and spun off (6000 rpm, 10 s). The cured liqPS microfluidic chip was then carefully set onto the tacky layer and gently pressed for 60 s.
For bonding by radical polymerization curing liqPS was spin-coated onto an objective slide (about 2 ml of liqPS, 6000 rpm, 10 s). Following spin-coating the cured liqPS microfluidic chip is put onto the tacky layer and pressed onto it gently. After about 60 s the stack is subjected to light exposure (~60 s) which cures the bond.
The third bonding method is based on thermal bonding and we have found this method most convenient to use. For this a cured layer of liqPS (about 1 mm thick) is used as the sealing layer. This layer is preheated on a hot plate to a temperature around the glass transition temperature. In our experiments, we found the temperature of 68 °C to be most suitable. In order to reduce adhesion to the hot plate, a thin layer of PDMS can be placed under the liqPS layer. Upon heating, liqPS turns soft and pliable. The hot plate can then be switched off and the cured liqPS microfluidic chip gently pressed onto the sealing layer. After cooling, the stack can be removed from the hot plate.
Chemical compatibility and solvent resistant testing
Solvent compatibility was assessed on cured cylindrical liqPS samples (6.7 mm diameter, 7.4 mm length). The lateral dimensions of the samples prior to solvent exposure were recorded and the samples fully immersed in the respective solvent for 24 hours. After this time, half of the samples were removed from the solvents and the increase in length (due to solvent swelling) recorded. The remaining samples were immersed for a total of 7 days before being measured. Table 1 shows the results of the measurements.| Solvent | Length increase after 1 day (%) | Length increase after 7 days (%) | Comment |
|---|---|---|---|
| Acetone | — | — | Dissolved |
| Isopropanol | 0 | 0 | |
| Tetrahydrofuran | — | — | Dissolved |
| Ethanol | <0.2 | <0.2 | |
| Water | 0 | 0 | |
| Dichloromethane | — | — | Dissolved |
| Dimethylformamide | — | — | Dissolved |
| Chloroform | — | — | Dissolved |
| Cyclohexane | — | — | Dissolved |
Cured liqPS shows identical chemical resistance as reported for PS.30,31 It is resistant to alcohols and water whereas non-resistant to aromatic hydrocarbons, chlorinated solvents and dimethylformamide. As stated in the literature, PS is resistant to acids and bases (such as, e.g., hydrochloric acid and aqueous sodium hydroxide). The same compatibility is to be expected from liqPS as well.
UV/VIS spectroscopy
We selected a number of bulk and surface sensitive chemical and/or physical characterization techniques in order to compare cured liqPS to the reference samples of commercially available PS in order to demonstrate that cured liqPS cannot be distinguished from pure PS. By using liqPS researchers can effectively use a material whose physical and chemical properties have already been widely studied and understood.The first method chosen was UV/VIS spectroscopy which allows assessing optical properties of the material. The result is shown in Fig. 3. As can be seen, there is no distinguishable difference in the optical absorbance and transmission of cured liqPS in direct comparison to commercial PS. In particular, no effect from traces of the photoinitiator is seen. It was likely to assume that trace amounts of unreacted photoinitiator would have resulted in an increase of autofluorescence which we have found not to be the case. Further evidence of this was found using Raman spectroscopy.
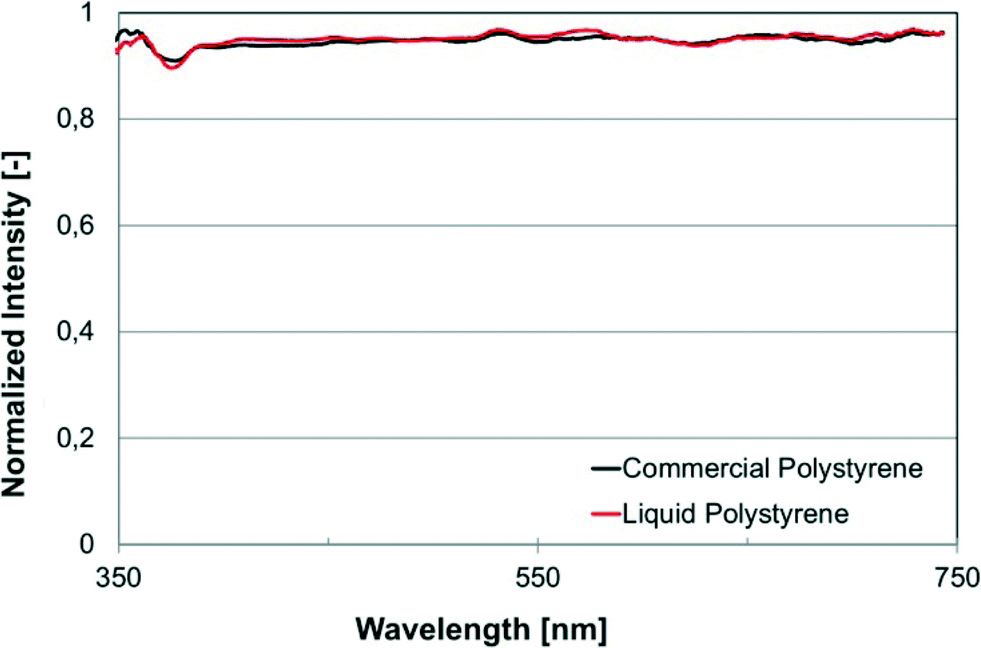 | ||
| Fig. 3 Results of UV/VIS transmission measurements on cured liqPS (“Liquid Polystyrene”) and a commercially available PS (“Commercial Polystyrene”). As can be seen, transmission spectra of both samples are almost indistinguishable. Colour in the online version. | ||
XPS and Raman spectroscopy
XPS and Raman spectroscopy are two of the most sensitive analytical techniques for assessing the chemical composition of surfaces. In this work, XPS and Raman spectroscopy were carried out on the freshly cut surfaces of cured liqPS as well as on commercially available PS (“Commercial Polystyrene”). We compared the data found to reference spectra from the literature (“Beamson et al.”).32 For Raman spectroscopy, a PS reference provided by Bruker was used for recording the reference spectra (“Bruker reference polystyrene”). These PS samples are used by Bruker for recording reference spectra and are of analytical purity.The results of the measurements are shown in Fig. 4. As can be seen, XPS survey scans as well as the narrow scans of C 1s elemental line and valence band of liqPS samples are almost identical to the commercial reference. The spectra also correlate well to literature reference values recorded for standard PS.32 Raman spectra were recorded for cured liqPS samples, the commercial reference PS and an analytical grade reference PS provided by Bruker as the reference for PS. As can be seen from Fig. 4d, the spectra match very well. Cured liqPS shows additional Raman lines at 1412 cm−1 und 774 cm−1 with about 1% of main line intensity. The commercial PS reference exhibits an intense overall autofluorescence which is (partially) removed by baseline correction in Fig. 4d for comparison reason. At lower wavelengths, the correction algorithm fails displaying the autofluorescence strongly. In contrast, cured liqPS (although being photochemically cured) does not show autofluorescence (above the autofluorescence of pure PS) which is proof of the effectiveness of the initiation of PPO. It is important to note that the spectra recorded for liqPS and the Bruker analytical PS reference sample correlate very well. As noted, the latter sample is of analytical quality which demonstrates that liqPS (judging from bulk and surface chemistry) cannot be distinguished from pure PS.
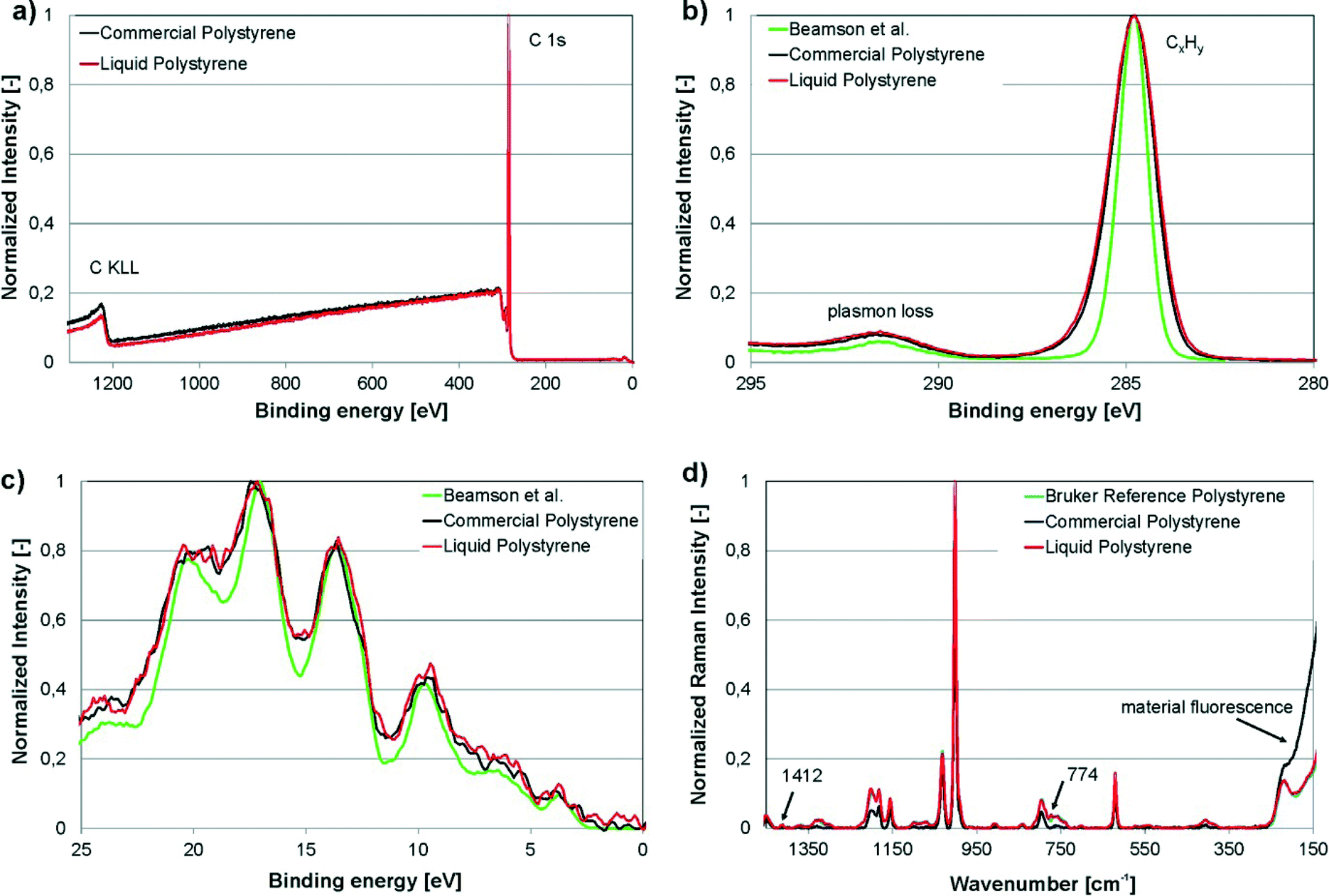 | ||
| Fig. 4 Experimental results of XPS and Raman spectroscopy on liqPS samples (“Liquid Polystyrene”), commercially available PS as reference (“Commercial Polystyrene”) as well as analytical reference samples for Raman spectroscopy provided by Bruker (“Bruker reference polystyrene”). a) XPS-survey plot, normalized. The plot shows the comparison of liqPS samples and commercial PS. b) Narrow scan of C 1s elemental line, normalized. The plot shows the comparison of liqPS samples, commercial PS as well as a literature reference spectrum (“Beamson et al.”32). c) Narrow scan of valence band spectra, Savitzky–Golay smoothed. The plot shows the comparison of liqPS samples, commercial PS as well as the literature reference spectrum. d) Raman spectra (785 nm laser, depolarized), baseline corrected, normalized. The plot shows the comparison of liqPS, commercial PS as well as an analytical reference PS provided by Bruker. The additional lines at 1412 and 774 cm−1 are highlighted. The commercial PS sample shows material fluorescence whereas liqPS samples do not show this effect. The spectra of liqPS and the analytical reference match very well throughout the whole spectrum. | ||
Cell culture studies
To evaluate whether there was a cellular cytotoxic response to liqPS, we cultured L929 cells on PS and liqPS surfaces (both having an area of 9.6 cm2) and compared the proliferation rates by XTT assay. The tetrazolium dye XTT can be efficiently used as a marker for proliferation and cytotoxicity as it is reduced to a soluble brightly coloured derivative by several cellular enzymatic systems.33 The XTT assays at 24 h, 96 h and 168 h showed that the L929 fibroblasts had comparable proliferation rates on both PS and liqPS surfaces under standard cell culture conditions (5% CO2, 37 °C)(Fig. 5).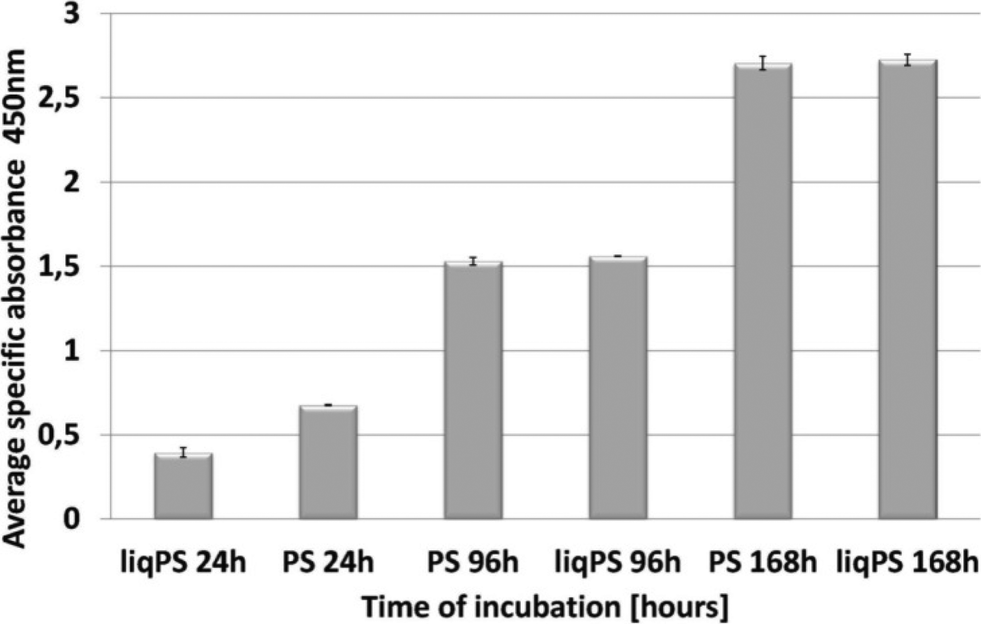 | ||
| Fig. 5 Comparison of cellular proliferation rates on PS and liqPS surfaces. Some 0.05 × 106 L929 cells were seeded on liqPS and PS surfaces and left to grow for 24 h, 96 h and 168 h under standard conditions. The XTT assay measured at 450 nm demonstrated that the proliferation rates of L929, grown on PS surfaces, were comparable with those of L929, grown on liqPS surface for 168 h under standard conditions. | ||
To confirm the potential biocompatibility of cured liqPS, we performed live/dead staining assay at 24 h, 96 h and 168 h. The Calcein-AM/Propidium Iodide (PI) combined protocol is used to distinguish the viable cells (green cytosol) from the dead cells (red nuclei) by fluorescence microscopy.34 Our results showed that over 168 h of incubation, the majority of L929 cells were green, indicating that the liqPS surface provided a cytocompatible environment for L929 growth and proliferation (Fig. 6b i–iii), comparable with the PS surfaces used in conventional cell cultures (Fig. 6a, i–iii). Notably, the fluorescent images also indicated a uniform cell distribution on both liqPS and PS surfaces, suggesting that the L929 cells could spread, adhere and grow on these surfaces.
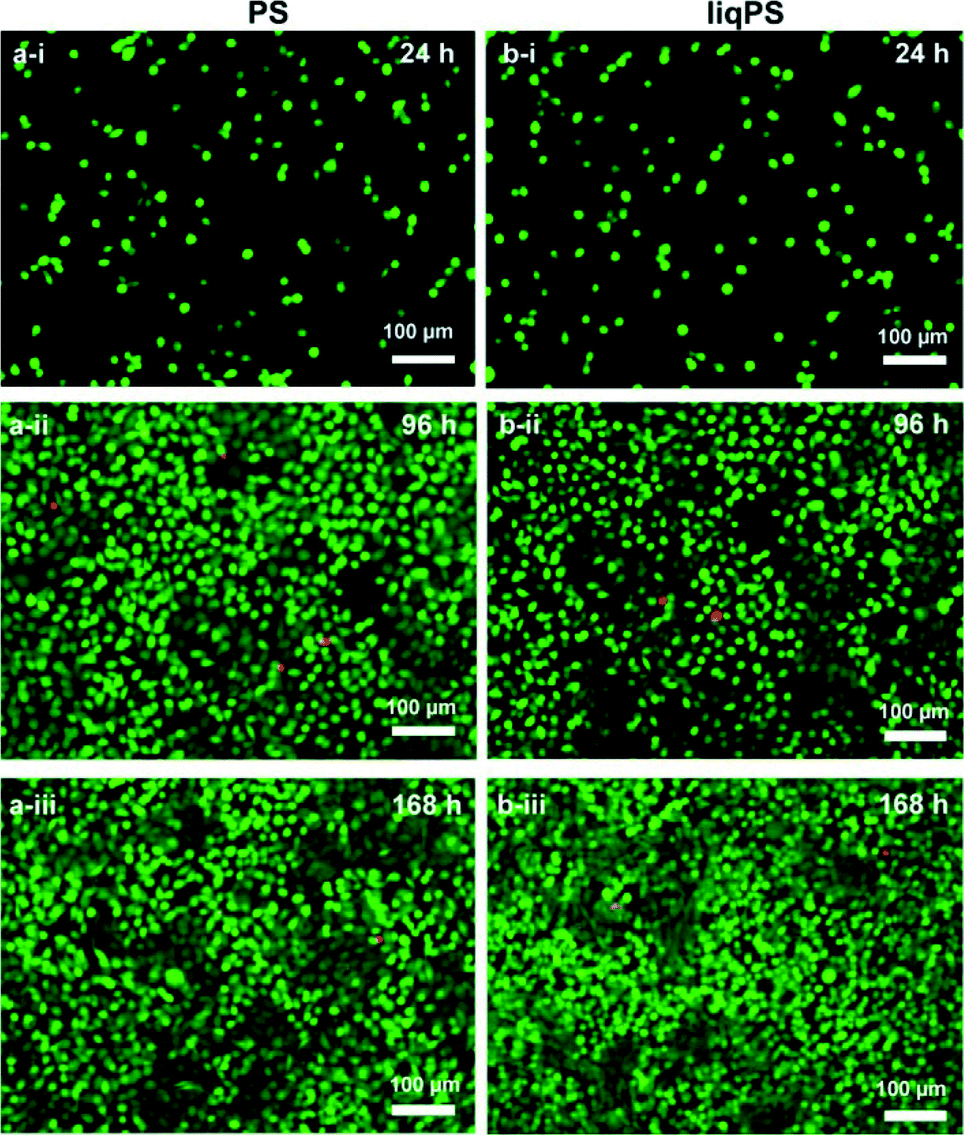 | ||
| Fig. 6 Viability of L929 cells on PS and liqPS surfaces. Some 0.05 × 106 L929 cells were cultured on PS and liqPS surfaces for 24 h, 96 h and 168 h. Calcein-AM (green) represents live cells, whereas the propidium iodide stains the nuclei of dead cells (red). a) The majority of cells on PS surfaces remained viable for 168 h (i–iii). b) No changes in cell viability of the L929 cells on liqPS surfaces were observed for 168 h. Moreover, the viability of cells on liqPS surfaces was comparable to that on PS surfaces for 168 h of incubation (i–iii). Colour in the online version. | ||
Microfluidic application example
Several microfluidic structures were created and used in exemplary applications in order to demonstrate the ease of microfluidic prototyping using liqPS (see Fig. 7). A SU-8 replication master with a simple microfluidic T-junction channel structure (channel width 800 μm, channel height ~60 μm) was created and replicated into liqPS as described. Holes were drilled into the block in order to access the microfluidic channel network. The channels were then sealed using thermal bonding as described. The microfluidic channel network was then used in a simple two-phase microfluidic experiment using fluorinated oil (FC-40) and water coloured with a blue dye. Several other microfluidic structures were also replicated in order to assess the replication quality when using finer structures. Fig. 7g shows one of these examples: a microfluidic Tesla mixer structure from a cascade mixer which we previously described when characterising the maskless projection lithography system used in this work.24 As can be seen, even fine structures are replicated with high fidelity. | ||
| Fig. 7 Exemplary microfluidic channel structure created in liqPS. a) T-junction channel in liqPS replicated from a PDMS mould created from a structured SU-8 layer. b) liqPS microfluidic chip sealed against a thin layer of cured liqPS mounted on an objective slide. The bonding method chosen was thermal bonding. c) Dyed water penetrating into the microfluidic channel network. d/e) Microscopic detail views of the filled channels. As can be seen the channels are replicated correctly with sharp edges and bonded successfully. f) Exemplary two-phase application example in the T-junction using dyed water and FC-40. A confluent laminar flow can be observed in the output channel. g) Tesla mixer structure replicated from a PDMS mould which we have previously described and used.24 Colour in the online version. | ||
Summary and conclusion
In this work we have demonstrated a new method for prototyping microfluidic structures in PS. For this, we synthesized “Liquid Polystyrene” (liqPS), a liquid PS prepolymer which can be poured onto templates to be replicated and cured using visible light in any laboratory. Using liqPS, researchers can create microfluidic chips using soft lithography by replication from structurally weak templates such as PDMS moulds created from SU-8 templates. Using surface as well as bulk analytical techniques, we showed that cured liqPS cannot be distinguished from commercial PS and can be considered (both chemically and physically) identical to PS. We also demonstrated this in cell culture experiments showing that L929 grown on liqPS and PS surfaces for 168 h remained viable on both samples and also exhibited comparable rates of proliferation and viability, as shown by the XTT and live/dead assays. In fact, no difference between L929 cultures maintained on cured liqPS samples and commercially available PS could be observed in these experiments.We believe that liqPS is a significant step towards the establishment of PS as prototyping material in microfluidics and an effective method for solving the “three community problem” by giving the “microfluidic prototyping”, the “industrial microfluidics” as well as the “applied microfluidics” communities access to a mutual material.
Acknowledgements
This work was partly carried out with support from the Karlsruhe Nano Micro Facility (KNMF, http://www.kit.edu/knmf), a Helmholtz Research Infrastructure at Karlsruhe Institute of Technology (KIT, http://www.kit.edu). We gratefully acknowledge funding by the Deutsche Forschungsgemeinschaft DFG, the German Federal Ministry of Education and Research (BMBF) and the “Heidelberg Karlsruhe Research Partnership (HEiKA)”. This research work is part of the project “Molecular Interaction Engineering: From Nature's Toolbox to Hybrid Technical Systems,” which is funded by BMBF, funding code 031A095B. P.M.N. has been supported by a DAAD (German Academic Exchange Service) doctoral grant.Notes and references
- D. C. Duffy, J. C. McDonald, O. J. A. Schueller and G. M. Whitesides, Anal. Chem., 1998, 70, 4974–4984 CrossRef CAS PubMed.
- K. Haubert, T. Drier and D. Beebe, Lab Chip, 2006, 6, 1548–1549 RSC.
- H. Kazuo and M. Ryutaro, J. Micromech. Microeng., 2000, 10, 415 CrossRef.
- M. A. Unger, H. P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Science, 2000, 288, 113–116 CrossRef CAS.
- T. Scharnweber, R. Truckenmuller, A. M. Schneider, A. Welle, M. Reinhardt and S. Giselbrecht, Lab Chip, 2011, 11, 1368–1371 RSC.
- J. N. Lee, C. Park and G. M. Whitesides, Anal. Chem., 2003, 75, 6544–6554 CrossRef CAS PubMed.
- D. G. Bessarabov, R. D. Sanderson, E. P. Jacobs and I. N. Beckman, Ind. Eng. Chem. Res., 1995, 34, 1769–1778 CrossRef CAS.
- A. Priola, R. Bongiovanni, G. Malucelli, A. Pollicino, C. Tonelli and G. Simeone, Macromol. Chem. Phys., 1997, 198, 1893–1907 CrossRef CAS.
- J. P. Rolland, R. M. Van Dam, D. A. Schorzman, S. R. Quake and J. M. DeSimone, J. Am. Chem. Soc., 2004, 126, 8349–8349 CrossRef CAS.
- G. Maltezos, E. Garcia, G. Hanrahan, F. A. Gomez, S. Vyawhare, R. M. van Dam, Y. Chen and A. Scherer, Lab Chip, 2007, 7, 1209–1211 RSC.
- S. H. Kim, Y. Yang, M. Kim, S. W. Nam, K. M. Lee, N. Y. Lee, Y. S. Kim and S. Park, Adv. Funct. Mater., 2007, 17, 3493–3498 CrossRef CAS.
- J. Alvankarian and B. Y. Majlis, J. Micromech. Microeng., 2012, 22, 035006 CrossRef.
- E. Berthier, E. W. K. Young and D. Beebe, Lab Chip, 2012, 12, 1224–1237 RSC.
- H. Becker and L. E. Locascio, Talanta, 2002, 56, 267–287 CrossRef CAS.
- E. W. K. Young, E. Berthier, D. J. Guckenberger, E. Sackmann, C. Lamers, I. Meyvantsson, A. Huttenlocher and D. J. Beebe, Anal. Chem., 2011, 83, 1408–1417 CrossRef CAS PubMed.
- H. Li, Y. Fan, R. Kodzius and I. Foulds, Microsyst. Technol., 2012, 18, 373–379 CrossRef CAS.
- A. S. Johnson, K. B. Anderson, S. T. Halpin, D. C. Kirkpatrick, D. M. Spence and R. S. Martin, Analyst, 2013, 138, 129–136 RSC.
- A. P. Russo, D. Apoga, N. Dowell, W. Shain, A. P. Turner, H. Craighead, H. Hoch and J. Turner, Biomed. Microdevices, 2002, 4, 277–283 CrossRef CAS.
- V. N. Goral, Y.-C. Hsieh, O. N. Petzold, R. A. Faris and P. K. Yuen, J. Micromech. Microeng., 2011, 21, 017002 CrossRef.
- C.-S. Chen, D. N. Breslauer, J. I. Luna, A. Grimes, W.-C. Chin, L. P. Lee and M. Khine, Lab Chip, 2008, 8, 622–624 RSC.
- K. Ren, J. Zhou and H. Wu, Acc. Chem. Res., 2013, 46, 2396–2406 CrossRef CAS PubMed.
- Y. Wang, J. Balowski, C. Phillips, R. Phillips, C. E. Sims and N. L. Allbritton, Lab Chip, 2011, 11, 3089–3097 RSC.
- M. P. Seah, I. S. Gilmore and G. Beamson, Surf. Interface Anal., 1998, 26, 642–649 CrossRef CAS.
- A. Waldbaur, B. Carneiro, P. Hettich, E. Wilhelm and B. Rapp, Microfluid. Nanofluid., 2013, 15, 625–635 CrossRef.
- T. G. Fox and P. J. Flory, J. Polym. Sci., 1954, 14, 315–319 CrossRef CAS.
- D. Y. Kwok, C. N. C. Lam, A. Li, K. Zhu, R. Wu and A. W. Neumann, Polym. Eng. Sci., 1998, 38, 1675–1684 CAS.
- A. H. Ellison and W. A. Zisman, J. Phys. Chem., 1954, 58, 503–506 CrossRef CAS.
- A. Curtis, J. Forrester, C. McInnes and F. Lawrie, J. Cell Biol., 1983, 97, 1500–1506 CrossRef CAS.
- T. G. van Kooten, H. T. Spijker and H. J. Busscher, Biomaterials, 2004, 25, 1735–1747 CrossRef CAS PubMed.
- A. Waldbaur, H. Rapp, K. Länge and B. E. Rapp, Anal. Methods, 2011, 3, 2681–2716 RSC.
- D. R. Lide, CRC handbook of physics and chemistry, The Chemical Rubber Company, Cleveland, USA, 2003 Search PubMed.
- The XPS of polymers database, ed. G. Beamson and D. Briggs, SurfaceSpectra Ltd., Manchester, UK, 2000 Search PubMed.
- M. V. Berridge, P. M. Herst and A. S. Tan, in Biotechnology Annual Review, ed. M. R. El-Gewely, Elsevier, 2005, vol. 11, pp. 127–152 Search PubMed.
- A. Lévesque, A. Paquet and M. Pagé, Cytometry, 1995, 20, 181–184 CrossRef PubMed.
Footnote |
| † For requests of liqPS please contact the corresponding author B.E.R. |
| This journal is © The Royal Society of Chemistry 2014 |