 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dynamics of counterion-induced attraction between vimentin filaments followed in microfluidic drops†
Christian
Dammann
and
Sarah
Köster
*
Institute for X-Ray Physics, Georg-August-Universität Göttingen, Germany and Center for Nanoscale Microscopy and Molecular Physiology of the Brain (CNMPB), Friedrich-Hund-Platz 1, 37077 Göttingen, Germany. E-mail: sarah.koester@phys.uni-goettingen.de; Fax: +49 (0)551399430; Tel: +49 (0)551399429
First published on 8th May 2014
Abstract
Intermediate filaments (IFs) are fiber-forming proteins and part of the cytoskeleton of eukaryotes. In vitro the network formation of purified IF systems is mediated, for example, by the interaction with multivalent ions. The understanding of these interaction mechanisms increases the knowledge of the cytoskeleton on a fundamental level. Here, we employ time-lapse fluorescence microscopy to directly image the evolution of network formation of vimentin IFs upon addition of divalent ions. We are thus able to follow the process starting a few seconds after the first encounter of free filaments and ions up to several minutes when the networks are in equilibrium. The local protein density in the compacted networks can reach a factor of 45 higher than the original solution concentration. The competition between mono- and divalent ion condensation onto the protein explains our observations and reveals the polyelectrolyte nature of vimentin as a reason for the protein attraction in the presence of small cations. The method for time-lapse studies in microfluidic drops presented here can be generalized to other dynamic systems.
Introduction
The cytoskeleton of eukaryotes provides mechanical stability to the cell and at the same time serves other purposes like cell division, motility and intracellular signal transduction.1 Three types of fibrous proteins – actin filaments (MF), microtubules (MT) and intermediate filaments (IFs) – form cross-linked, intracellular higher-order structures. Among these three classes of proteins, IFs are the least well studied from the biophysical point of view and also the most diverse in terms of specific amino acid sequence. Vimentin is an IF protein found in cells of mesenchymal origin. Like other IFs, vimentin is a polyelectrolyte which forms filaments with an overall negative line charge density of about −14 e nm−1. In vivo the regulation of IF networks is highly complex. Associated linker proteins connecting IFs, MFs and MTs to each other,2–5 protein phosphorylation,6–8 and the biochemical environment1,9 all play a role. This large number of influencing parameters makes it difficult to fully capture the system in vivo, and in vitro studies of isolated, purified components of the cytoskeleton provide a very fruitful bottom-up approach to understand the mechanisms behind the construction of the cytoskeleton.The influence of multivalent ions has been studied for MFs,10–13 MTs10,14 and IFs15–20 using fluorescence microscopy,13 light scattering,10,12,21 rheology,16,19 X-ray scattering11 and electron microscopy.22 All these studies focus primarily on an end state, into which the system has either equilibrated or into which it has been driven by external forces. In order to investigate the pathway by which the system reaches the end state, techniques are needed that allow for manipulation and concurrent observation. Microfluidic methods are extremely well-suited for this approach as they can be combined in a straightforward way with, for example, fluorescence microscopy. Microfluidic drops are widely used to confine biological systems like cells,23–27 cell extracts28 or proteins18,29,30 in three dimensions without having to attach them to a substrate. For time-resolved studies, the drops have to be kept in place and several methods for drop trapping have been presented, including constrictions,31 rails and anchors32 or ‘U’-shaped traps.33
Here, we combine microfluidic modules for composing drops,34 mixing the contents35 and trapping the individual drops33immediately thereafter. We study the influence of divalent cations and monitor the transition from freely diffusing vimentin IFs to densely aggregated networks. This investigation reveals a typical network formation time on the order of a few minutes. Here, we relate the attraction process of the filaments to competitive condensation of mono- and divalent cations onto the protein. We show that within this context the polyelectrolyte nature of vimentin36 drives the network formation.
Materials and methods
Protein preparation
We investigate a human vimentin mutant that has an additional cysteine at its C-terminus and the sole cysteine in the rod domain is replaced by alanine. These point mutations do not influence the equilibrium filaments formed, and allow us to label the protein via maleimide chemistry without having the fluorophore interfering with filament assembly.37 Vimentin is expressed in Escherichia coli bacteria and then purified from inclusion bodies.38 Vimentin is stored at −80 °C in 8 M urea, 5 mM Tris–HCl (pH 7.5), 1 mM EDTA, 0.1 mM EGTA, 1 mM DTT and 10 mM methyl ammonium chloride (MAC). The protein is fluorescently tagged with Alexa Fluor 488 C5 Maleimide (Invitrogen GmbH).39 To achieve optimized assembly kinetics and imaging properties, labeled vimentin is mixed at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 with unlabeled vimentin prior to reconstitution of vimentin tetramers during the dialysis step. The dialysis is done stepwise against first 6 M urea, 0.8 mM phosphate buffer (PB) (pH 7.5) for 1 h and then two times against 2 mM PB for 3 h in total. Vimentin assembly is initiated by mixing vimentin (440 μg mL−1) solution 1:1 with 200 mM KCl solution in 2 mM PB. For assembly, the solution is kept at 37 °C for 6 h. This procedure results in filaments with an average contour length of ≈5 μm.
:4 with unlabeled vimentin prior to reconstitution of vimentin tetramers during the dialysis step. The dialysis is done stepwise against first 6 M urea, 0.8 mM phosphate buffer (PB) (pH 7.5) for 1 h and then two times against 2 mM PB for 3 h in total. Vimentin assembly is initiated by mixing vimentin (440 μg mL−1) solution 1:1 with 200 mM KCl solution in 2 mM PB. For assembly, the solution is kept at 37 °C for 6 h. This procedure results in filaments with an average contour length of ≈5 μm.
Microfluidics
Microfluidic devices are fabricated by standard PDMS (polydimethylsiloxane)-based soft-lithography,40,41 with a two-layer photoresist design. We use SU-8 3025 negative photoresist (MicroChem) for both layers and follow the instructions by the manufacturer with some modifications: for the first layer SU-8 3025 resist is spin coated onto a silicon wafer to obtain a layer thickness of 18 μm and the layer is exposed to UV-light through a photomask. After baking this layer on a hot plate at 95 °C for 5 min, we spin coat a second layer with a thickness of 15 μm onto the first layer. The mask for the second layer is aligned to the structures on the first layer using a mask aligner (MJB4, Süss MicroTec AG). After exposure and baking, the resist is developed.The ‘trap’ parts of two silicon wafers are shown in Fig. 1. The structure heights (which transfer to channel heights in the PDMS replicas) are determined with a profilometer (Dektak 6, Veeco Instruments Inc.). PDMS replicas of this ‘master’ are bonded to cover slips and flushed with Ombrello (Autoserv) to ensure hydrophobic channel walls. The coating with Ombrello has the additional advantage that the protein which is injected into the channels is unlikely to absorb to the channel walls during the travel to the drop production site.
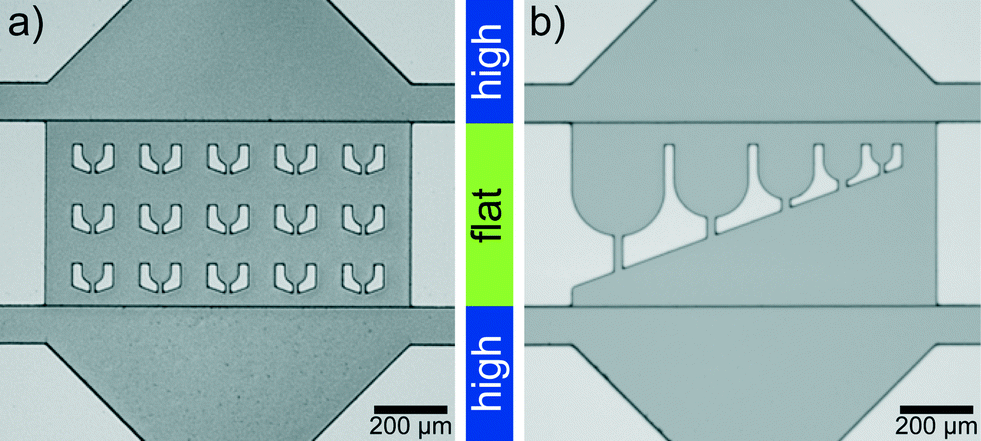 | ||
| Fig. 1 Two-layer silicon wafer imaged with an upright microscope. (a) Device with trap diameter (corresponds to drop diameter) 55 μm. (b) Device for drops of different diameter from 250 μm to 55 μm. The ‘trap’ region in the corresponding PDMS devices is flat compared to the rest of the device. | ||
The devices are connected to glass syringes (Hamilton) via polyethylene tubing (Intramedic™ PE20, BD). Fluorocarbon oil (Fluorinert™ FC-40, 3M) is used as the oil phase of the emulsion produced in the channels. The emulsion is stabilized by addition of a biocompatible surfactant to the oil phase (perfluoropolyether–polyethyleneglycol block-copolymer, 1.8% w/w, Raindance Technologies).42 The syringes are controlled by precise syringe pumps (neMESYS, cetoni GmbH).
For the startup of the device we use a ≈70 cm long tubing and fill it with 45 μL protein solution, a 3 μL air cushion and finally 5 μL assembly buffer. The tubing is connected to the inlet of the protein channel. The air barrier prevents Taylor-dispersion-induced premature mixing of protein solution and assembly buffer.43 Injecting assembly buffer into the channel first, ensures a stable drop formation without protein potentially clogging the channels. Once the drop production is stable, the protein is encapsulated and the risk of clogging is notably reduced.
During the experiment, the aqueous phase (see Results sections and Fig. 3b) is composed of varying ratios of: (aq 1) assembly buffer with additional multivalent salt (100 mM KCl, 2 mM PB and either 31 mM MgCl2 or 7 mM (or 18 mM) hexamminecobalt-III-chloride Co(NH3)6Cl3), (aq 2) assembly buffer (100 mM KCl, 2 mM PB), and (aq 3) assembled vimentin in buffer (100 mM KCl, 2 mM PB). While the average flow velocity of aq 3 is kept at 1.2 mm s−1 (5 μL h−1) and the total flow velocity of aq 1, 2 and 3 combined is constant at 3.6 mm s−1 (15 μL h−1), the ratio of aq 1 and aq 2 is adjusted to modify the concentration of the multivalent salt. We thus keep the protein concentration, pH, monovalent salt and buffer conditions equal for different drops, but change the multivalent salt concentrations. To control the process of trapping and release of the drops (see Results section), the oil flow rates (top and bottom, see Fig. 3a) are adjusted. During drop trapping and imaging of the drop, the top oil flow velocity is 2.4 mm s−1 (20 μL h−1) and the oil flow velocity from the bottom 24 mm s−1 (100 μL h−1). The bottom oil velocity is shortly increased to ≈48 mm s−1 (200 μL h−1) to release the drops. These are typical values for drops of 100 μm in diameter. For drops of different diameter, the flow velocities are adjusted accordingly, e.g. a higher top oil flow rate decreases the drop diameter.
The vimentin IFs in the drops are imaged using a 60× oil-immersion objective and an inverted confocal microscope (IX81 with FV-1000 confocal unit, Olympus). For confocal fluorescence imaging, a photodiode is used, for bright field images of the wafer a Leica DLM 4000 microscope with included camera is used and bright field images of the microfluidics devices are recorded using a Hamamatsu Orca R2 camera.
Data analysis
Confocal time series of the aggregating vimentin IFs in the drops are recorded with a typical frame-to-frame time of 1.6 s, pixel size of 207 nm and a dwell time per pixel of 4 μs in the 512 × 512 px2 sized image. Image processing is performed using MATLAB® (MathWorks®). For each image sequence 10 reference regions in different frames are determined as background (containing only filament precursors that cannot be resolved in confocal microscopy; for an example see Fig. 2a, selection indicated by blue line). From these reference regions the average background intensity distribution is determined. The raw data (Fig. 2b) are analyzed by distinguishing pixels that belong to assembled vimentin (‘signal’) from background (Fig. 2c). From the binary images, the convex hull of the aggregated protein is calculated. Here we include the smallest number of binary islands such that at least 90% of the filament intensity in the drop is covered (Fig. 2d, red and black region) to make the convex hull robust against individual filaments (Fig. 2c, in blue circle) that have not been included into the larger aggregate(s) yet (Fig. 2d, blue regions). | ||
| Fig. 2 Data analysis. (a) In the inverted gray scale fluorescence images of the protein in the drops ‘background’ regions are selected (blue line) to obtain the intensity signature. Using this signature of the background region, the raw images (b) are analyzed and those pixels, which belong to filaments and filament networks are identified (c). (d) The convex hull is taken over the protein aggregates that mainly contribute to the intensity of the networks (black and red regions). The total drop area is shown in purple. | ||
Results and discussion
Drop trapping for rapid imaging start of time-lapse series
Rapid drop content imaging is achieved by the device shown in Fig. 3. Aqueous drops are produced in a flow focusing geometry (Fig. 3b). Prior to the drop production, the aqueous phase is composed of three different aqueous liquids.34 Due to the late contact of the aqueous phases, the first salt–protein contact is retarded to the very moment before drop production. Appropriate mixing in the drop is easily achieved for fast diffusing small cations. However, the component that diffuses the slowest is the filamentous protein with an estimated diffusion constant of 6 × 10−13 m2 s−1 for a 5 μm filament.44 To accelerate the mixing process, the drops pass a 2.5 mm long straight channel where they are plug-like. This plug-like motion induces internal drop vortices by which mixing becomes much faster35 and is completed within two seconds after drop production. The drops flow into a broadened region where they face a flat region with the channel height reduced from 33 μm to 18 μm (Fig. 3d; Materials and methods). In front of this barrier most of the drops leave to one of the lateral outlets. However, some cross the barrier and enter the flat region with ‘U’-shaped traps33 (Fig. 3a and d). In this hydrodynamic trap the content of a drop can be imaged over time as the drop dwell time is on average to 350 s. This dwell time has large deviations allowing to occasionally image for more than 10 min. The deviation is due to the possibility of drop squeezing through the small slit of the traps. To free the traps for a next set of drops in a controlled way, a higher oil counter flow can be applied to hydrodynamically release the drops (Fig. 3c). This allows for serial drop content imaging (movie S1†). | ||
| Fig. 3 Microfluidic device. (a) Aqueous drops are produced in a PDMS microfluidic device using a flow focusing design. (b) The aqueous dispersed phase is composed of three different aqueous components ‘aq 1’, ‘aq 2’ and ‘aq 3’. (c) Shortly after their production a fraction of the drops enters a flat region where some of them are trapped. They are stably localized and their content can be imaged for several minutes. The micrograph shows a detail of the larger field of view shown in Fig. 1a. (d) By applying a higher oil counter flow from downstream, the drops can be released from the traps again, freeing space for the next set of drops. | ||
The lag time between first contact of multivalent salt and protein to the first image in the trap is between 1 s and 5 s for the drops in the first row of traps, which are the fastest reached and therefore the most interesting traps (Fig. 3d). The flat region is needed since the squeezed drops are more stably trapped and the number of additionally incoming drops is reduced for undisturbed drop content imaging. The trap design is not restricted to a specific size and the principle also works for traps of various sizes (Fig. 1b). This way the intended size of the drops can be modified during the running experiment. The drop size does not influence the local interaction mechanisms between individual filaments, mediated by the ions, since they take place on the nanometer scale and thus several orders of magnitude below typical drop diameters. However, transport mechanisms like diffusion, which bring the filaments into proximity are in principle dependent on the total drop volume. We expect that for the droplet sizes used here the effect is not too large.
Time scales for network formation
The microfluidic device proves to be very suitable to image vimentin aggregation dynamics on relevant time scales. Image series of vimentin protein in trapped drops are taken at different multivalent salt concentrations. We show two example series with [MgCl2] = 4.1 mM in Fig. 4a–c and [MgCl2] = 14.5 mM in Fig. 4d–f. For both concentrations, the filaments are distributed over the entire drop at the beginning of the image series (t = 0 s). However, the dynamics of filament interaction differs dramatically. Whereas the filaments just diffuse freely for all times at the low [MgCl2], at higher [MgCl2] filaments and salt undergo strong interactions as they form a network that is highly compacted over time. We perform these experiments for a set of [MgCl2] concentrations in the range of 0.8–14.5 mM. For comparison of the aggregation behavior we calculate the area of convex hulls for the networks (see Materials and methods). These areas are normalized to the area of each drop, leading to the relative hull area A (Fig. 5a/b). For several experiments with [MgCl2] = 0.8–4.1 mM we observe that A ≈ 0.9 is roughly constant. This corresponds to the freely fluctuating filaments independent of time (Fig. 4a and 5a). In contrast, we observe a qualitatively different behavior of the protein at [MgCl2] = 14.5 mM (Fig. 5b). In all these cases the filaments form networks. This direct observation is quantitatively supported by the decrease of A from initially ≈0.9 to notably smaller values over time. The resulting networks can be classified into two morphologies: point-like networks that show very high compaction and scaffold-like networks which are more widely spread in the drop. At the same time the temporal evolution of A provides the speed of network formation and compaction, which starts from right at the beginning of observation and leads to strong aggregation within the first 10 min (Fig. 5b). In the cases where the network morphologies are point-like, the aggregation is faster compared to the scaffold-like networks. In these cases the compaction is almost finished after 200 s. | ||
| Fig. 4 Time-lapse series of vimentin at different salt conditions. a–c) At comparatively low [MgCl2] = 4.1 mM filaments diffuse freely for all times. d–f) At [MgCl2] = 14.5 mM the filaments aggregate strongly over time from being distributed all over the drop to a point-like aggregate (movie S2†). (Scale bars 20 μm.) | ||
 | ||
| Fig. 5 Time-lapse study of vimentin aggregation. a) The relative convex hull area A is constant over time for low [MgCl2]. b) At [MgCl2] = 14.5 mM strong aggregation is observed within ≈200 s. The morphologies can be classified into very dense point-like and scaffold-like aggregates. c) The mass density of the network in the drop increases during aggregation (inset). For the most pronounced compaction we observe an increase in mass density of 45 times of its initial density. | ||
From microscopic images to nanoscopic network structure
The impact of small divalent cations on vimentin filaments becomes obvious in the light of maximum compaction. The initial filament concentration at the beginning can be used to approximate the mass density considering the spreading of the network in the drop over time (Fig. 5c). We observe a 45-fold mass density compared to the initial value in case of maximum compaction (Fig. 5c). Giving a diameter of 7–11 μm of the final point-like aggregate in this situation, the persistence length of vimentin (2 μm (ref. 45)) becomes important as a barrier for further compaction. Further bending is energetically unfavorable.Due to the stabilization of the networks by aggregation, not all protein aggregates become point-like but we find another class which seems to be ‘trapped’ in a scaffold-like shape. Whereas the point-like aggregates show only little potential for further compaction, the scaffold-like ones are more widely spread. In both cases we observe that a) only one main aggregate is formed and b) over time almost all filaments are incorporated into the main aggregate very firmly as they show no longer their pronounced thermal fluctuations as expected for free filaments (Fig. 4a–c). We observe a clear filament–filament attraction mechanism that outperforms thermal fluctuations in all cases where a certain Mg2+ threshold is exceeded.
The polyelectrolyte nature of vimentin
In our effort to explain the observed aggregation process, we consider vimentin as a polyelectrolyte46 and discuss the condensation of counterions on the filaments. Monovalent K+ ions at the same concentration for all experiments are always present in solution since they are needed for filament stabilization. Additionally, divalent Mg2+ ions at varying concentrations are added. We therefore discuss the competitive condensation of the two ion types onto the filaments. Using the model of Rouzina and Bloomfield,47 we consider vimentin as a cylinder of filament radius R = 5 nm (ref. 48) and linear charge density λ = −14 e nm−1. The model predicts that the surface concentration of K+ and Mg2+ are equal on the filament at a bulk ion concentration of [Mg2+] = 14 mM. For a bulk ion concentration higher than 14 mM, the Mg2+ ion concentration is higher on the filament, and below, K+ ions outnumber Mg2+ ions. For the range of [Mg2+] = 0–4.1 mM, where we observe no attraction between the filaments, the relative amount of Mg2+ ions is always below 30%. Ion correlations are only effective for multivalent ions and these correlations are known as the reason for attraction between polyelectrolytes.49,50 Thus we hypothesize that in the case of vimentin when around half of the condensed ions are multivalent, filament attraction is mediated. Our hypothesis is further supported by additional experiments in which Mg2+ is replaced by trivalent Co(NH3)63+ (Fig. S3†). In these experiments we determine a threshold for aggregation at [Co(NH3)63+] ≈ 0.5–2.4 mM. This corresponds well to the model of Rouzina and Bloomfield as for the trivalent ions we expect an equal surface concentration of K+ and Co(NH3)63+ at a bulk concentration of [Co(NH3)63+] = 1.9 mM. Therefore, we can explain the concentration dependence of the onset of aggregation and we emphasize the importance of the polyelectrolyte nature of vimentin. Here, vimentin is included into the group of biological filaments like F-actin12 or DNA,51 whose behavior in the presence of cations is determined by fundamental electrostatics. The importance of polyelectrolyte properties of filamentous biopolymers for physiology has recently been reviewed by Janmey et al.52 As the secondary and tertiary structure of IFs is highly conserved,53 the behavior of vimentin we observe here may well be representative for other IFs, which also form negatively charged filaments.Conclusions
Our drop-based device meets the challenges of rapid imaging of vimentin aggregation in the presence of multivalent salt. Importantly, by contrast to previous studies where the static end point of the networks was imaged,18 the device allows us to image the drop content directly after mixing and at high temporal and spatial resolution, only limited by the microscopy technique used. We can change the drop contents multiple times during the experiments, thereby allowing for testing a large parameter range (e.g. ion concentrations). Such a device can be applied for studying dynamic phenomena using droplets in general. We show that within 10 min a distinct compaction of the filament networks takes place, given a critical Mg2+ or Co(NH3)63+ concentration. Considering vimentin as a polyelectrolyte, the onset of filament–filament attraction is explained. We find that when around 50% of the counterions on the filament are multivalent, attraction is mediated. Consequently, our approach emphasizes the role of competitive counterion binding in the discussion of the polyelectrolyte nature of vimentin. Our study opens the door for both more studies on time-resolved aggregation processes and a better understanding of the polyelectrolyte nature of IFs.Acknowledgements
We thank Harald Herrmann, Bernd Nöding, Britta Weinhausen, Rabea Sandmann, Jens-Friedrich Nolting, Jean-Christophe Baret, and Philipp Gruner for fruitful discussions. We thank Viktor Schroeder for proofreading the manuscript and Susanne Bauch for technical support. This work was supported by the German Research Foundation (DFG) in the framework of SFB 755 ‘Nanoscale Photonic Imaging’, project KO 3752/5-1, the Cluster of Excellence and DFG Research Center Nanoscale Microscopy and Molecular Physiology of the Brain (CNMPB), and the excellence initiative.References
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, Garland Science, 5th edn, 2008 Search PubMed.
- F. A. Steinböck and G. Wiche, Biol. Chem., 1999, 380, 151–158 CrossRef PubMed.
- G. Wiche, J. Cell Sci., 1998, 111, 2477–2486 CAS.
- J. W. Mack, A. C. Steven and P. M. Steinert, J. Mol. Biol., 1993, 232, 50–66 CrossRef CAS PubMed.
- C. L. Leung, K. J. Green and R. K. Liem, Trends Cell Biol., 2002, 12, 37–45 CrossRef CAS.
- J. E. Eriksson, T. He, A. V. Trejo-Skalli, A.-S. Hrml-Braskn, J. Hellman, Y.-H. Chou and R. D. Goldman, J. Cell Sci., 2004, 117, 919–932 CrossRef CAS PubMed.
- M. B. Omary, N.-O. Ku, G.-Z. Tao, D. M. Toivola and J. Liao, Trends Biochem. Sci., 2006, 31, 383–394 CrossRef CAS PubMed.
- M. Inagaki, Y. Matsuoka, K. Tsujimura, S. Ando, T. Tokui, T. Takahashi and N. Inagaki, BioEssays, 1996, 18, 481–487 CrossRef CAS.
- F. Lang, G. Busch, M. Ritter, H. Völkl, S. Waldegger, E. Gulbins and D. Häussinger, Physiol. Rev., 1998, 78, 247–306 CAS.
- J. X. Tang, S. Wong, P. T. Tran and P. A. Janmey, Berichte der Bunsengesellschaft, 1996, 100, 796–806 CAS.
- G. C. L. Wong, A. Lin, J. X. Tang, Y. Li, P. A. Janmey and C. R. Safinya, Phys. Rev. Lett., 2003, 91, 018103 CrossRef.
- J. X. Tang and P. A. Janmey, J. Biol. Chem., 1996, 271, 8556–8563 CrossRef CAS PubMed.
- F. Huber, D. Strehle and J. Käs, Soft Matter, 2012, 8, 931–936 RSC.
- D. J. Needleman, M. A. Ojeda-Lopez, U. Raviv, H. P. Miller, L. Wilson and C. R. Safinya, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16099–16103 CrossRef CAS PubMed.
- J.-F. Leterrier and J. Eyer, Biochem. J., 1987, 245, 93–101 CAS.
- Y.-C. Lin, C. P. Broedersz, A. C. Rowat, T. Wedig, H. Herrmann, F. C. MacKintosh and D. A. Weitz, J. Mol. Biol., 2010, 399, 637–644 CrossRef CAS PubMed.
- R. Beck, J. Deek, J. B. Jones and C. R. Safinya, Nat. Mater., 2009, 9, 40–46 CrossRef PubMed.
- C. Dammann, B. Nöding and S. Köster, Biomicrofluidics, 2012, 6, 022009 CrossRef PubMed.
- Y.-C. Lin, N. Y. Yao, C. P. Broedersz, H. Herrmann, F. C. MacKintosh and D. A. Weitz, Phys. Rev. Lett., 2010, 104, 058101 CrossRef.
- K. E. Kasza, G. H. Koenderink, Y. C. Lin, C. P. Broedersz, W. Messner, F. Nakamura, T. P. Stossel, F. C. MacKintosh and D. A. Weitz, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2009, 79, 041928 CrossRef CAS.
- J. X. Tang, T. Ito, T. Tao, P. Traub and P. A. Janmey, Biochemistry, 1997, 36, 12600–12607 CrossRef CAS PubMed.
- J. Kayser, H. Grabmayr, M. Harasim, H. Herrmann and A. R. Bausch, Soft Matter, 2012, 8, 8873–8879 RSC.
- S. Köster, F. E. Angile, H. Duan, J. J. Agresti, A. Wintner, C. Schmitz, A. C. Rowat, C. A. Merten, D. Pisignano, A. D. Griffiths and D. A. Weitz, Lab Chip, 2008, 8, 1110–1115 RSC.
- E. Brouzes, M. Medkova, N. Savenelli, D. Marran, M. Twardowski, J. B. Hutchison, J. M. Rothberg, D. R. Link, N. Perrimon and M. L. Samuels, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14195–14200 CrossRef CAS PubMed.
- B. El Debs, R. Utharala, I. V. Balyasnikova, A. D. Griffiths, A. Merten and Christophe, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11570–11575 CrossRef CAS PubMed.
- M. Chabert and J.-L. Viovy, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3191–3196 CrossRef CAS PubMed.
- B. L. Wang, A. Ghaderi, H. Zhou, J. Agresti, D. A. Weitz, G. R. Fink and G. S. Stephanopoulos, Nat. Biotechnol., 2014, 32, 473–478 CrossRef CAS PubMed.
- A. M. Jimenez, M. Roche, M. Pinot, P. Panizza, L. Courbin and Z. Gueroui, Lab Chip, 2011, 11, 429–434 RSC.
- H. M. Evans, E. Surenjav, C. Priest, S. Herminghaus, R. Seemann and T. Pfohl, Lab Chip, 2009, 9, 1933–1941 RSC.
- M. M. A. E. Claessens, M. Bathe, E. Frey and A. R. Bausch, Nat. Mater., 2006, 5, 748–753 CrossRef CAS PubMed.
- C. H. J. Schmitz, A. C. Rowat, S. Köster and D. A. Weitz, Lab Chip, 2009, 9, 44–49 RSC.
- P. Abbyad, R. Dangla, A. Alexandrou and C. N. Baroud, Lab Chip, 2011, 11, 813–821 RSC.
- A. Huebner, D. Bratton, G. Whyte, M. Yang, A. J. deMello, C. Abell and F. Hollfelder, Lab Chip, 2009, 9, 692–698 RSC.
- H. Song and R. F. Ismagilov, J. Am. Chem. Soc., 2003, 125, 14613–14619 CrossRef CAS PubMed.
- M. R. Bringer, C. J. Gerdts, H. Song, J. D. Tice and R. F. Ismagilov, Philos. Trans. R. Soc., A, 2004, 362, 1087–1104 CrossRef CAS PubMed.
- O. I. Wagner, S. Rammensee, N. Korde, Q. Wen, J.-F. Leterrier and P. A. Janmey, Exp. Cell Res., 2007, 313, 2228–2235 CrossRef CAS PubMed.
- H. Herrmann, DKFZ Heidelberg, Germany, personal communication.
- H. Herrmann, L. Kreplak and U. Aebi, in Intermediate Filament Cytoskeleton, ed. M. B. Omary and P. A. Coulombe, Academic Press, 2004, vol. 78, pp. 3–24 Search PubMed.
- S. Winheim, A. R. Hieb, M. Silbermann, E.-M. Surmann, T. Wedig, H. Herrmann, J. Langowski and N. Mücke, PLoS One, 2011, 6, e19202 CAS.
- S. R. Quake and A. Scherer, Science, 2000, 290, 1536–1540 CrossRef CAS.
- D. Duffy, J. McDonald, O. Schueller and G. Whitesides, Anal. Chem., 1998, 70, 4974–4984 CrossRef CAS PubMed.
- C. Holtze, A. C. Rowat, J. J. Agresti, J. B. Hutchison, F. E. Angilè, C. H. J. Schmitz, S. Köster, H. Duan, K. J. Humphry, R. A. Scanga, J. S. Johnson, D. Pisignano and D. A. Weitz, Lab Chip, 2008, 8, 1632–1639 RSC.
- G. Taylor, Proc. R. Soc. London, Ser. A, 1953, 219, 186–203 CrossRef CAS.
- G. Li and J. X. Tang, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2004, 69, 061921 CrossRef.
- B. Nöding and S. Köster, Phys. Rev. Lett., 2012, 108, 088101 CrossRef.
- Q. Wen and P. A. Janmey, Curr. Opin. Solid State Mater. Sci., 2011, 15, 177–182 CrossRef CAS PubMed.
- I. Rouzina and V. A. Bloomfield, Biophys. Chem., 1997, 64, 139–155 CrossRef CAS.
- S. Portet, N. Mücke, R. Kirmse, J. Langowski, M. Beil and H. Herrmann, Langmuir, 2009, 25, 8817–8823 CrossRef CAS.
- A. Y. Grosberg, T. T. Nguyen and B. I. Shklovskii, Rev. Mod. Phys., 2002, 74, 329–345 CrossRef CAS.
- A. Naji, S. Jungblut, A. G. Moreira and R. R. Netz, Phys. A, 2005, 352, 131–170 CrossRef PubMed.
- V. A. Bloomfield, Biopolymers, 1997, 44, 269–282 CrossRef CAS.
- P. A. Janmey, D. R. Slochower, Y.-H. Wang, Q. Wen and A. Cēbers, Soft Matter, 2014, 10, 1439–1449 RSC.
- S. Kim and P. A. Coulombe, Nat. Rev. Mol. Cell Biol., 2010, 11, 75–81 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3lc51418h |
| This journal is © The Royal Society of Chemistry 2014 |