 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Density based visualization for molecular simulation
Dmitri
Rozmanov
*,
Svetlana
Baoukina
and
D. Peter
Tieleman
Department of Biological Sciences and Centre for Molecular Simulation, University of Calgary, 2500 University Drive NW, Calgary, Alberta T2N 1N4, Canada. E-mail: rozmanov@gmail.com; tieleman@ucalgary.ca
First published on 12th June 2014
Abstract
Molecular visualization of structural information obtained from computer simulations is an important part of research work flow. A good visualization technique should be capable of eliminating redundant information and highlight important effects clarifying the key phenomena in the system. Current methods of presenting structural data are mostly limited to variants of the traditional ball-and-stick representation. This approach becomes less attractive when very large biological systems are simulated at microsecond timescales, and is less effective when coarse-grained models are used. Real time rendering of such large systems becomes a difficult task; the amount of information in one single frame of a simulation trajectory is enormous given the large number of particles; at the same time, each structure contains information about one configurational point of the system and no information about statistical weight of this specific configuration. In this paper we report a novel visualization technique based on spatial particle densities. The atomic densities are sampled on a high resolution 3-dimensional grid along a relatively short molecular dynamics trajectory using hundreds of configurations. The density information is then analyzed and visualized using the open-source ParaView software. The performance and capability of the method are demonstrated on two large systems simulated with the MARTINI coarse-grained force field: a lipid nanoparticle for delivering siRNA molecules and monolayers with a complex composition under conditions that induce monolayer collapse.
1 Introduction
Molecular visualization of structural information obtained from computer simulations is an important part of research work flow. A good visualization technique should be capable of eliminating unnecessary redundant information and highlight important effects clarifying the key phenomena for the researcher as well as for the audience.Current methods of presenting molecular structural information are generally limited to variants of the traditional “ball-and-stick” representation or various kinds of molecular surfaces. This approach, while good for simple chemical systems, becomes less attractive when very large biological systems, such as lipid nano-particles (LNP) for drug delivery,1 are simulated at microsecond timescales. In such simulations the structural aspects of the entire system are of interest and the visualization cannot be reduced to a detailed representation of a relatively small active site as is the case in some ribosome simulations,2 for example. Real time rendering of such large systems becomes a difficult task even on high end workstations; the amount of information in one single frame of a molecular dynamics trajectory is enormous at full atomic details. In addition, in coarse-grained simulations the features of interest typically are not at the level of individual atoms, and visualization methods that are based on the detailed chemical structure are by default not necessarily the best choice.
There have been previous attempts to improve the visualization quality and perception for structural data. For biologically relevant molecules, for example, there are several molecular representations which greatly help with interpretation of protein structures, such as various cartoon-based representations implemented in the VMD3 molecular visualization software. Such representations reduce the amount of redundant chemical information and highlight the features of the protein secondary structure relevant to its biological function. A limitation of this and similar methods is that they are only applicable to proteins.
A more general approach is to use average configurations for the visualization of ordered systems with low mobility.4,5 The method helps to remove structural variation of the visualized structures due to thermal vibrational motion and highlights the underlying ordered structure. At the same time, the method is poorly suited for disordered systems with significant molecular mobility at the time scale of the averaging and may result in non-physically looking structures. Mobility information is not a part of the average configuration but can be partially communicated by appropriate particle coloring.5
In this paper we report a novel visualization technique based on spatial atomic densities. The method can be considered a natural extension and combination of the average configuration4,5 and the spatial distribution function6 approaches for molecular visualization. The power of this method is demonstrated on two large-scale test systems simulated using the MARTINI coarse-grained force field7,8 on a length-scale of ca. 50 nm. Software tools and utilities necessary for application of this technique, as well as the workflow are also reported in this paper.
The rest of the paper is organized as follows. The following section 2 details the method and the workflow required to apply the proposed technique. An application of the method to a case study of the structural features and dynamics of a drug delivery lipid nano-particle will be given in section 3.1. The section 3.2 will discuss the visualization and analysis of the collapse of the lipid monolayers at the air–water interface. Finally, our conclusions will be summarized in section 4.
2 The method
2.1 Property grids
The method is based on visualization of spatial information sampled from molecular simulations and mapped on a high-resolution 3-dimensional property grid for some specific time frame of interest. Such a grid represents a 3-dimensional field and can contain average information about some property of the system in each volume element for the duration of the sampling time frame [see Fig. 1 (A)]. Therefore, the averaging is performed for a spatial location rather than for a specific particle. Generally, such grids can contain various types of data and represent scalar, vector, or tensor property fields such as, for example, density, potential energy, mobility, order parameter, linear momenta, force, or stress tensor. Each property of interest is sampled on a separate grid using a large number of snapshots of the system. This paper mainly focuses on the density as the sampled property of the system for illustration purposes but much of the power of the approach is based on the ability to combine density maps with arbitrary other grids, such as the densities of different entities or an average energy grid, for example, and do calculations on them.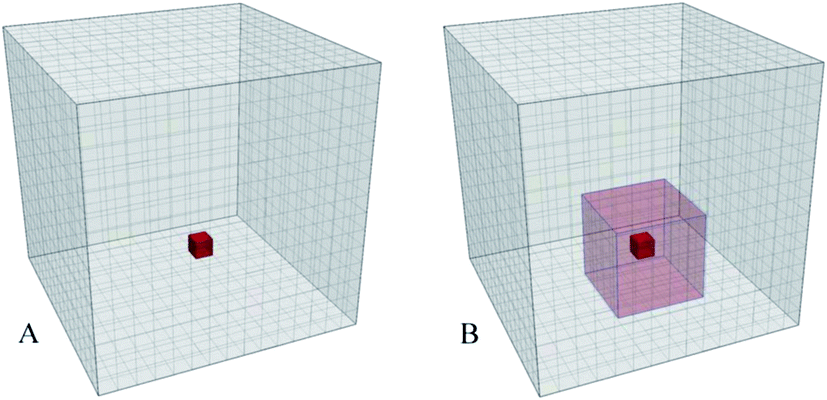 | ||
| Fig. 1 (A) A simulation box is mapped to a 3D grid; Properties of the particles found in a volume element (red) are sampled and averaged to produce the grid value for that volume element using a large number of the simulation trajectory snapshots for a specific time interval. (B) 3D grid of a locally averaged property. Values of the cells are obtained by applying an 3-dimensional moving average to the original grid. The values inside the local 3D domain (red transparent box) are averaged and assigned to the central cell (opaque red cell). The local domains are subjected to the periodic boundary conditions. | ||
In this method the resolution of the 3D grid should be fine enough to resolve the smallest structural feature of interest; in atomistic simulations the grid spacing can be 0.5 Å so that individual atoms can be resolved. For coarse grained (CG) simulations using the MARTINI7 force field the grid spacing can be in the range 1.5–2.0 Å to resolve the larger particles (beads) representing several atoms. The time frame for sampling should be short enough to prevent averaging of the temporal features of interest, such as structural elements or dynamical or configurational heterogeneities.9–11 At the same time the sampling period must be sufficiently long to allow for representative sampling of the accessible phase space of the system. The 3D grids obtained using this approach are essentially 3-dimensional histograms with the number of bins significantly larger (by 1–2 orders of magnitude) than the number of samples (particles) in a single configurational snapshot of the system. Hence, in order to generate smooth and representative density grids the number of sampled configurations should be large, typically, several hundreds or even thousands, depending on the grid resolution, number of particles in the system, and the desired quality of the density grid.
The property grids then can be visualized directly using appropriate visualization tools or manipulated to produce more complex fields, such as cross-correlation densities or local average property grids.12
2.2 Locally averaged property grids
Property grids generated using the approach outlined above produce continuous property fields which contain information about the density distribution of a specific set of particles in the system for a specified duration of time. At short sampling times the diffusive mass transfer is normally insignificant and the accessible configurational space is mostly determined by the molecular rattling motion determined by the nearest molecular neighbours. Essentially, under these conditions, the system's density distribution is an ensemble of weakly overlapping single particle density distributions.When coexisting phases in the system are of interest such short time density grids may not show a sufficiently clear difference in the density (or any other property) between these phases. Following a definition of a phase as a uniform continuous domain of matter with similar properties, a phase property is a collective quantity determined by many molecules comprising the phase. An obvious solution then is to provide additional averaging of the density grid at the characteristic length scale for the phases of interest. In practice, application of a 3-dimensional moving average over such a characteristic local domain around each grid cell is a convenient method to achieve such local averaging as shown in Fig. 1(B). The resulting property grid has the same dimensions and resolution as the original grid but its cell values are averaged over spatial domains local to each of the cells. The size of the domain necessary to provide the desired averaging can be estimated from the particle density and a typical number of molecules required to distinguish the phases.
2.3 Visualization workflow
To generate density grids suitable for the visualization approach described in this paper a number of steps are required:■ The starting point is a single initial configuration of the system to be visualized. The configuration has to contain all required information so that a new simulation can be initiated from it to generate enough additional configurations for the averaging procedure, if the original trajectory was not saved at a high enough frequency.
■ The atomic densities are sampled on a high resolution grid in this method. The grid resolution should be appropriately selected. Typically each atom or particle is expected to occupy the volume of at least 3 × 3 × 3 grid cells. Many configurational snapshots are necessary for a sufficient density sampling on such a high resolution grid. Normally, hundreds or thousands of configurations are needed to generate smooth density maps. A good rule for a uniform system is to have the number of particles times the number of configurations at least ten times the number of grid cells.
■ The simulation length is chosen so that it is short enough to avoid smearing of the structural features of interest due to diffusion, but sufficiently long to provide adequate sampling of the system's accessible volume. It can be 10 ps for an atomistic simulation of liquid water and 10 ns for a coarse grained simulation of a lipid membrane. By increasing the sampling time one can shift the focus of attention from short time behaviour in the system to processes occurring at longer time scales.
■ Once the number of required configurations and the length of the simulation for grid sampling is known, the simulation is performed and the configurations are saved to a trajectory data file. The GROMACS simulation package was used in this work, and the configurations were saved as an XTC trajectory file.13
■ The saved trajectory information is then used to sample the particle locations for density grid generation. For each density of interest a separate density grid is generated, so they can be analyzed and visualized independently. In this work for each grid to be sampled a particle index was generated and added to a GROMACS index file. The trajectory and the index files were used by software developed in our group to generate and save the density grids.14 Such high resolution grids carry a large amount of information, of the order of hundreds of megabytes per grid, so the data is compressed before it is written to disk to reduce the amount of storage space required.
■ Once the grids are generated they can be used for numerical analyses or manipulated to produce new grids, such as cross density products.
■ For visualization, normally, the grids have to be converted to an appropriate format first. In this work, ParaView,15 an open source multi-platform data analysis and visualization application, was used to generate most of the figures. The grids, therefore, were converted to the XML base VTI format, native to the VTK visualization library16 ParaView is based on, using software developed in our group.14
If an animation is to be generated using this density base approach, then all these steps have to be repeated for each frame of the animation.
3 Results and discussion
3.1 Structural features of a lipid nano-particle
To demonstrate an application of the proposed density based technique, a simulation of a lipid nano-particle (LNP) in an aqueous environment will be discussed in this section. The LNP studied in this work is designed for therapeutic siRNA drug delivery to a target cell to induce specific gene silencing following the RNA interference pathway.17 In addition to water and the siRNA drug, the particle is mainly composed of cholesterol and a specially designed ionizable cationic lipid, as well as a small amount of a structural phospholipid. A protective shell of a polyethylene glycol (PEG) grafted lipid is required for the particles to prevent aggregation and to protect the particles from being degraded by the immune system during systemic delivery.1,17 A more detailed understanding of the structure and the roles of all the components of the particle is required for designing more efficient and precisely targeted drug delivery systems. We have previously built a model of this particular particle and described initial simulations, as well as characterized its structure using traditional visualization and analysis techniques,1 but the simulations presented here are new and start from a different procedure to make the initial model.The simulations were performed in a 57.63 nm3 cubic simulation box under periodic boundary conditions using the GROMACS simulation package13 and the coarse grained MARTINI force field.7,8 The system was composed of 15984 molecules of 2,2-dilinoleyl-4-(2-dimethylaminoethyl)-[1,3]-dioxolane (DLin-KC2-DMA),1 an ionizable cationic lipid, 15984 cholesterol (CHOL) molecules,7 3996 molecules of 1,2-distearoyl-sn-glycerophosphocholine (DSPC),7 a phospholipid, 3996 PEG-lipid molecules (PEG-C2),1 and 1179533 water particles. The siRNA in this simulation was represented by 216 12 base pair double stranded RNA MARTINI molecules, in the current model indistinguishable from DNA,18 and 11232 chloride anion particles were added to the system to neutralize the system. In the MARTINI force field the coarse graining is implemented by representing 4 heavy atoms, typically, by a single effective particle.7 In total the simulation system contained 1714349 MARTINI particles, and represented a system of approximately 20 million atoms.
The particle was self-assembled from a mixture of the DLin-KC2-DMA, DSPC, cholesterol, and RNA molecules randomly distributed inside a sphere 25.5 nm in radius. The initial configuration for the LNP simulations was built using the bio.b-gen simulation box generator developed in our group.19 The PEG-lipid molecules were placed randomly around the sphere in a spherical shell between radii 26.0 and 26.5 nm. 130950 water particles were placed inside a sphere of radius 27.0 nm and 1059815 water particles were added to the rest of the system to achieve the desired level of lipid hydration and to maintain a uniform density throughout the system. After that, 11232 randomly selected water particles were replaced by Cl− ions to neutralize the system. The system was propagated for 200 ns at a temperature of 320 K using a 10 fs time step, during which the nano-particle self-assembled and equilibrated. The spacing for the density grids in this study was slightly less than 2 Å with grid dimensions of 300 × 300 × 300 cells. The grid resolution was a compromise between the captured structural details and the computational effort of visualization: a typical MARTINI bead has an effective diameter of 4.7 Å and can be resolved at this grid spacing; at the same time a grid of 27,000,000 cells still can be manipulated on current hardware. The grids were sampled using 1000 configurational snapshots of the system during a 100 ps simulation.
 | ||
| Fig. 2 An overview of the lipid nano-particle studied in this work. (A) The external view of the particle. (B) A cross-sectional view represented by a 3.5 nm slab cut from the middle of the particles. The molecular densities of the components are shown by a set of semi-transparent iso-surfaces: magenta, the protective PEG-lipid (PEG-C2), red, the therapeutic siRNA (RNA), green, cholesterol (CHOL), yellow, the phospholipid (DSPC), and blue, the ionizable cationic lipid (DLin-KC2-DMA). All iso-surfaces were generated using 5 nm−3 density values. | ||
The density of water (solvent) shown in Fig. 3(A) provides a representation of the external interface of the particle with the surrounding water. At the 100 ps time scale the diffusive contribution to molecular motion is small, so that the high resolution density grid contains most of the molecular level details cluttering the representation. By averaging the density grid locally the amount of detail is reduced, so that the general shape of the particle can be seen more clearly, see Fig. 3(B). This representation of the particle external surfaces reveals these details much more clearly when compared with a more conventional particle view shown in Fig. 2(A). Fig. 3(C) shows the same water density iso-surface as Fig. 3(B) but with a semi-transparent surface. Such a representation shows the water cavities inside the particle, their distribution, and sizes. Apparently, water entrapped inside the particle during the particle construction process aggregated into compact water pockets. Significant variation of the water pocket sizes can also be noticed. A search for continuous domains in the averaged grid revealed 69 water cavities inside the particle. The plot of the estimated cavity diameters in Fig. 4 shows a two-tier cavity organization, with the larger cavities being in the 6–10 nm diameter range and the smaller cavities in the 0.5–2 nm range. The total volume of the internal cavities is 8172 nm3 and their total estimated area is 6638 nm2. The domain of the solvent density lower than the threshold value corresponds to the rest of the particle and had a volume of 50475 nm3. The sum of these two volumes gives the total particle volume which can be used to estimate the particle radius of 24.1 nm. Thus, the external area of the LNP assuming its spherical shape can be approximated as 7300 nm2, and the total area of the interface with the solvent of approximately 14000 nm2. This analysis shows that the areas of the external and internal interfaces of the particle are similar, with 52 and 48% respective contributions to the total area.
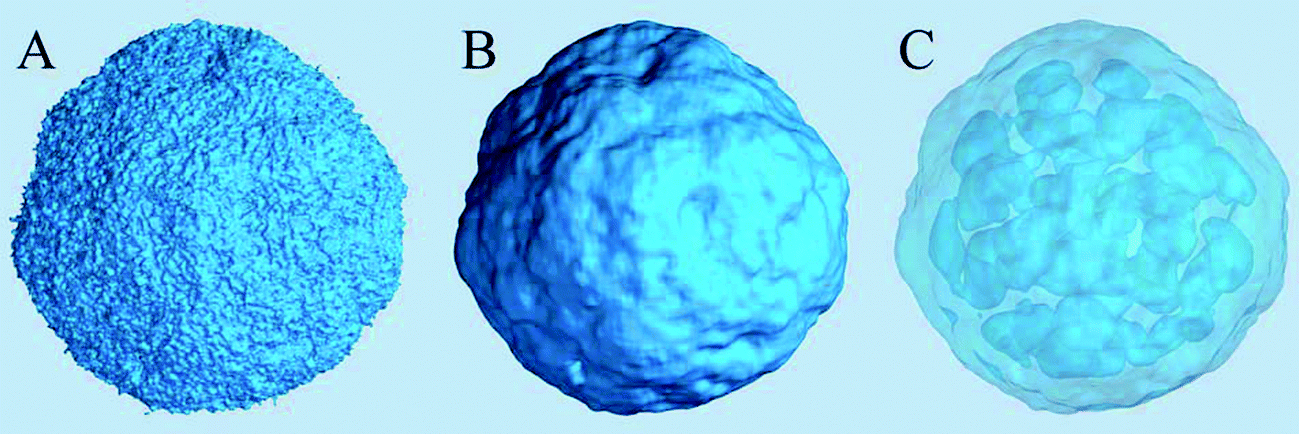 | ||
| Fig. 3 Iso-surfaces of the solvent density in the lipid nano-particle system at 5 nm−3 density values. (A) The original density shows too many molecular details, obscuring the overall topology of the particle surface. (B) A locally averaged water density using 93 cell averaging (≈ 1.73 nm3) shows the particle surface topology more clearly. (C) A semi-transparent representation of the same local-average density iso-surface showing water cavities inside the particle. | ||
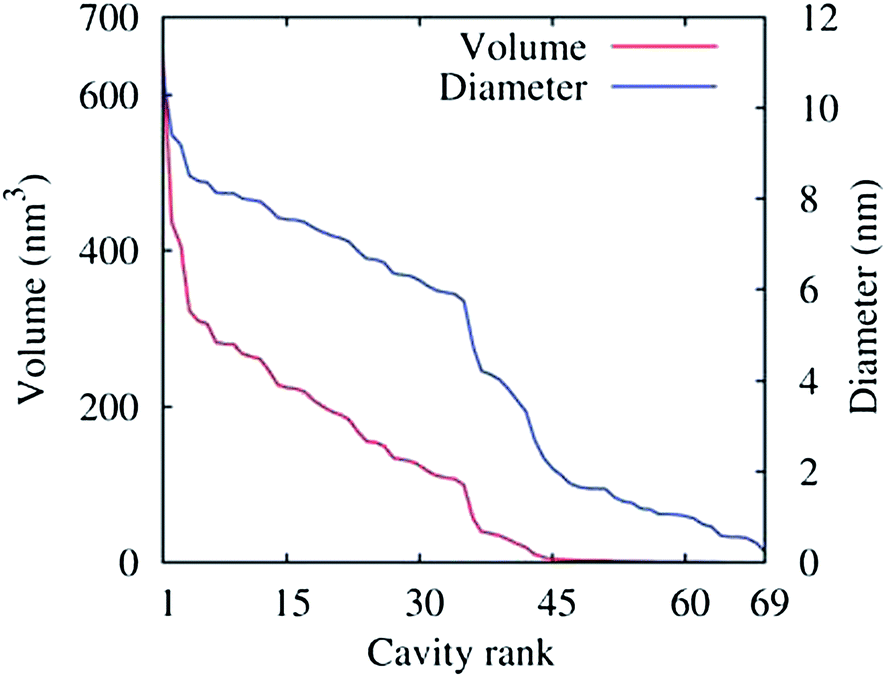 | ||
| Fig. 4 Volumes (red curve) of the 69 water cavities identified inside the lipid nano-particle shown in Fig. 3(C) sorted by size. The corresponding cavity diameters are also shown (blue curve), assuming a spherical shape for the cavities. The threshold density value for the density domain analysis was 5 nm−3. | ||
The distribution and structural ordering of the main lipid components of the nano-particle can be evaluated from their particle densities shown in Fig. 5. The short rigid cholesterol molecules demonstrate relatively low mobility inside the particle and produce well formed distinctive density “sticks” at this time scale, Fig. 5(A). One can see that cholesterol is strongly ordered in the lipid membranes separating the water cavities in the particle. Although the DSPC molecules are longer and more flexible than the cholesterol molecules, the amount of DSPC is four times smaller than that of cholesterol in the system, so that the densities of individual molecules can still be distinguished in Fig. 5(B). Some water cavities can be distinguished in the DSPC density plot in Fig. 5(B) but, generally, the DSPC ordering is less apparent than in the case of cholesterol.
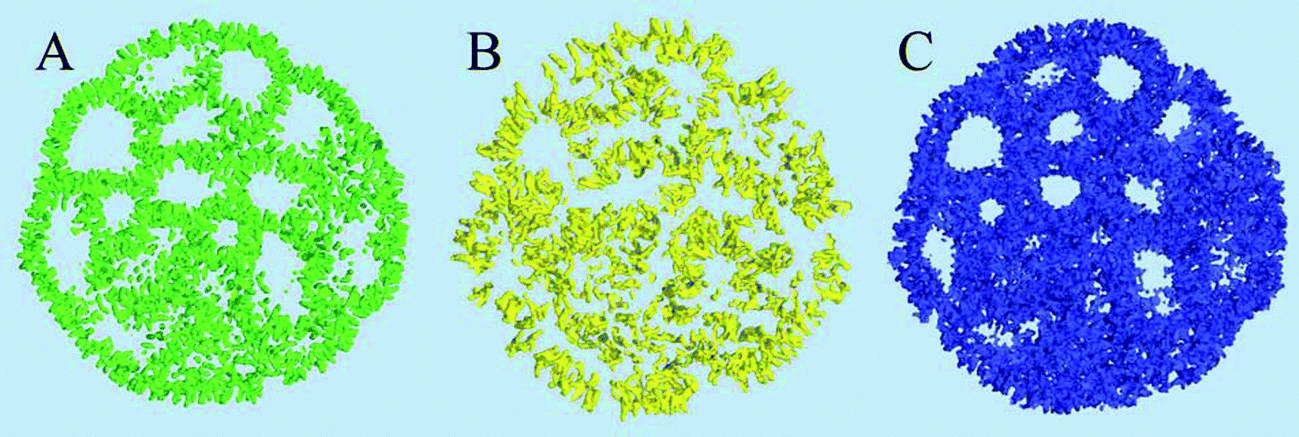 | ||
| Fig. 5 Molecular densities of the components represented by density iso-surfaces: (A) Cholesterol, (B) the DSPC lipid, (C) the DLin-KC2-DMA cationic lipid. Shown in a 3.5 nm cross-sectional slab using 5 nm−3 density iso-values. | ||
Fig. 5(C) shows the density representation of the main component of the particle, the DLin-KC2-DMA cationic lipid. Its molar fraction in the particle is the same as that of cholesterol, but the DLin-KC2-DMA molecules are longer and more flexible, so that its density in Fig. 5(C) is fused together and the ordering around the cavities is significantly more difficult to see. To address the question about DLin-KC2-DMA orientational ordering in the particle the densities of its hydrophilic head, linker, and hydrophobic tail groups were sampled separately and plotted in Fig. 6. This representation clearly demonstrates strong ordering of the cationic lipid head groups around the water pockets.
 | ||
| Fig. 6 The density of the DLin-KC2-DMA cationic lipid in the nano-particle. The densities of the three parts of the lipid molecules were sampled on separate grids and plotted as separate iso-surfaces through 3 nm−3 densities: the hydrophilic head groups (red), linker groups (blue), and the hydrophobic tail groups (yellow). | ||
A more careful examination of the component densities in Fig. 5 shows that the water cavity openings in the cholesterol density seem larger than in either the DSPC and DLin-KC2-DMA density plots. This observation suggests that the cholesterol molecules must reside deeper in the lipid membranes separating the water pockets and are not present at the lipid–water interface. To observe the effect more clearly the iso-surface of DLin-KC2-DMA density shown in Fig. 6(C) was colored by the cholesterol density and plotted in Fig. 7. In this representation it becomes apparent that cholesterol molecules tend to be absent from the interfaces with water, as can be seen by the dark blue color of the cavity surfaces; all the cholesterol molecules are deeper inside the lipid content of the particle.
 | ||
| Fig. 7 A fragment of the DLin-KC2-DMA density iso-surface from Fig. 5(C) colored by the cholesterol density. The volumes with the DLin-KC2-DMA density above the surface iso-value are shown by a haze of the same coloring. The DLin-KC2-DMA iso-density value is 3 nm−3, the units for the color scale of the cholesterol density are nm−3. | ||
 | ||
| Fig. 8 A detailed view of a water cavity. (A) The interface surface. The component densities are shown as 5 nm−3 iso-surfaces: red, RNA, green, CHOL, yellow, DSPC, and blue, DLin-KC2-DMA. The cutting plane is shown by a semi-transparent grey color. (B) The same cavity represented by the iso-surfaces of water (semi-transparent blue, 5 nm−3), chloride ion (green, 1 nm−3), and RNA (red, 5 nm−3) densities. Shown from a slightly different angle. | ||
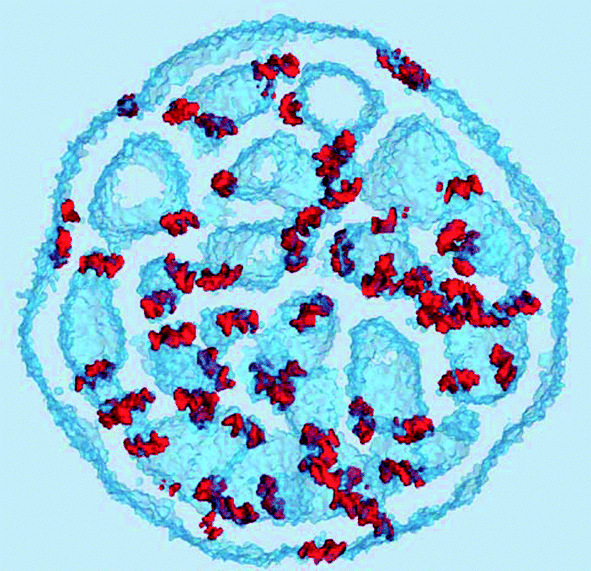 | ||
| Fig. 9 RNA molecules adsorbed on water interfaces. The RNA molecules are shown as an iso-surface of the RNA density (red), the water interfaces are represented by the iso-surface of water density (semi-transparent blue). Shown in a 3.5 nm cross-sectional slab using 5 nm−3 iso-densities. | ||
An iso-surface of water and the chloride ion densities, as well as the RNA density provide a different representation of the same water cavity in Fig. 8(B). Apparently, the water pockets carry a negative charge spread at the interface due to the chloride ions entrapped inside the cavities. The ions are adsorbed at the interface from the water side. The lipid side of the interface is mainly formed by the cationic, DLin-KC2-DMA, and zwitterionic, DSPC, lipids and is expected to be positively charged, explaining this attraction. Interestingly, the RNA molecules carry a negative charge of −22 and despite this fact they still prefer to adsorb onto the negatively charged water cavities.
It has been previously noted that the vicinity of the RNA molecules in the nano-particle contains approximately fourteen times more DLin-KC2-DMA lipid molecules than DSPC molecules.1 This ratio was estimated from RNA–lipid radial distribution functions and was significantly higher than the 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of these lipids in the particle composition. This observation indicated a higher affinity of the DLin-KC2-DMA lipid for RNA molecules in comparison with the DSPC lipid. In this work the number of RNA–lipid contacts were visualized directly using spatial density correlations between the RNA molecules and the DSPC and DLin-KC2-DMA lipids (Fig. 10). The number of RNA-DLin-KC2-DMA (blue) contacts is higher than RNA-DSPC contacts (yellow); their ratio is visually also higher than the 4:1 ratio expected from the composition, in qualitative agreement with previous work.1 Domain analysis indicates that the ratio of the number of RNA-DLin-KC2-DMA contacts to the number of RNA-DSPC contacts is approximately 40:1, or 10 times higher than the molar ratio of these lipids. Apparently, the RNA molecules are mostly surrounded by the cationic DLin-KC2-DMA lipid and DSPC is forced from these regions; under such circumstances DSPC is mostly expected to be present at water interfaces rather than in the vicinity of the RNA molecules. This observation is illustrated by Fig. 11 where an RNA molecule is shown bound to the surface of a water cavity. The RNA molecule lays flat on the water surface and there is practically no DSPC density in its immediate vicinity.
:1 molar ratio of these lipids in the particle composition. This observation indicated a higher affinity of the DLin-KC2-DMA lipid for RNA molecules in comparison with the DSPC lipid. In this work the number of RNA–lipid contacts were visualized directly using spatial density correlations between the RNA molecules and the DSPC and DLin-KC2-DMA lipids (Fig. 10). The number of RNA-DLin-KC2-DMA (blue) contacts is higher than RNA-DSPC contacts (yellow); their ratio is visually also higher than the 4:1 ratio expected from the composition, in qualitative agreement with previous work.1 Domain analysis indicates that the ratio of the number of RNA-DLin-KC2-DMA contacts to the number of RNA-DSPC contacts is approximately 40:1, or 10 times higher than the molar ratio of these lipids. Apparently, the RNA molecules are mostly surrounded by the cationic DLin-KC2-DMA lipid and DSPC is forced from these regions; under such circumstances DSPC is mostly expected to be present at water interfaces rather than in the vicinity of the RNA molecules. This observation is illustrated by Fig. 11 where an RNA molecule is shown bound to the surface of a water cavity. The RNA molecule lays flat on the water surface and there is practically no DSPC density in its immediate vicinity.
 | ||
| Fig. 10 The spatial correlations or contacts of the RNA molecules with the DLin-KC2-DMA and DSPC lipids inside the nano-particle shown as the iso-surfaces of the product of the RNA and the lipid density fields. The RNA density is represented by the red iso-surface at 5 nm−3; the DLin-KC2-DMA-RNA density product field is shown in blue, and the DSPC-RNA product field is shown in yellow. Both the product field surfaces are plotted using 0.001 nm−6 values. | ||
 | ||
| Fig. 11 A cut-away representation showing an RNA (red) molecule on the surface of a water pocket inside the lipid particle. The water interface is shown as the iso-surface of water density (blue). The yellow density iso-surface represents the DSPC lipid molecules. The surfaces were plotted using 5 nm−3 iso-densities. | ||
The actual role of the DSPC lipid in formation of lipid nano-particles is not yet fully understood, but it is known that some amount of DSPC is required for nano-particle formulation to have in vivo drug activity.20 Based on the observations made in this work we can speculate that the DSPC lipid is responsible for formation of more stable interfaces between the lipid content and water.
3.2 Structure and dynamics of lipid monolayers with phase coexistence
The second case study demonstrating the application of the density based technique for visualization is a simulation study of lipid monolayers at the air–water interface. The simulations were performed using the GROMACS simulation package13 and the MARTINI coarse grained force field.7,8 The system used in this study is shown in Fig. 12. Each monolayer was composed of 3840 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) molecules, 2304 1,2-(cis,cis-9,12-octadecadienoyl)-sn-glycero-3-phosphocholine (DOPC) molecules, and 3072 cholesterol (CHOL) molecules. Monolayers with these specific compositions in simulations separate into liquid-ordered (Lo) and liquid-disordered (Ld) phases at surface tensions in the range from 0 to 30 mM m−1 at a temperature of 290 K.21 | ||
| Fig. 12 The system for simulations of lipid monolayers. (A) A general view of the simulation box and two monolayers. The lipid components of the monolayers are shown by the iso-surfaces of their atomic densities sampled over 1 ns and using the density value of 5 nm−3: DPPC (yellow), DOPC (blue), and cholesterol (red). The water slab is between the monolayers in the middle of the box, shown by a semi-transparent blue haze. Above and below the monolayers is the vapor phase. (B) A side view of the system demonstrating lipid semi-vesicles caused by the monolayers' collapse. Water is not shown. (C) A top view of the upper monolayer from the hydrophilic side. | ||
The system set-up comprised a water slab in a vacuum with two symmetric monolayers at the two vapor–water interfaces (see Fig. 12(A)). Partial collapse of the monolayer was induced by applying a near-zero (<5 mN m−1) surface tension using a surface-tension coupling scheme, after which the monolayer compression was stopped by switching to constant volume simulation. This led to monolayer folding22 into bilayer discs in water; the bilayer discs bent and formed semi-vesicles attached to the monolayers (and filled with water), shown in Fig. 12(B) and (C). This resulted in a complex 3D structure with the lipid components heterogeneously distributed between the monolayer at the interface and the bilayers in water. This structure is directly relevant for lung surfactant, a mixture of lipids with a small amount of proteins lining the gas exchange interface in the lungs. Lung surfactant monolayer at the air–water interface is connected to the bilayer reservoirs in water; this connectivity provides low surface tensions on interface compression and expansion, which is necessary for breathing.23–25 The phase distribution in the monolayers and their properties are the main focus of this paper. The results reported in this section are a part of a larger study of lipid monolayers (Baoukina et al., in preparation).
The simulation box was 61.7 × 61.7 × 60.0 nm3 (with periodic boundary conditions); the spacing for the density grids in this study was slightly less than 2 Å with dimensions of 315 × 315 × 305 grid cells. The grids were sampled using two different time frames, 1 ns and 10 ns, with 1000 configurations in each time frame.
The cholesterol ordering in the DPPC (yellow) lipid domain can be clearly seen; the red dots of cholesterol can also be seen in the blue disordered (DOPC) phase. While cholesterol ordering is very different in the two phases, the actual cholesterol mobility for the 1 ns sampling time appears similar as can be judged from the size of the accessible cholesterol volume. A cross-sectional view of the same cholesterol density in Fig. 13(A) confirms this similarity: the cholesterol density distributions in the Ld phase are comparable to those in the Lo phase on the 1 ns time scale. However, the cholesterol density on the 10 ns time scale, shown in Fig. 13(B), demonstrates a significant difference in mobility between the two phases: the more mobile cholesterol molecules in the Ld phase produce diffuse delocalized density clouds at this time scale, while the character of the density of less mobile cholesterol molecules in the Lo phase has not changed. A similar insight can be gained from the total lipid density plots shown in Fig. 14, where the Lo phase maintains the molecular details on a 10 ns scale while in the Ld phase these details are completely smeared out by the molecular motion. Thus, by employing different time scales for density grid generation it is possible to visualize the difference in molecular mobility in two lipid phases.
 | ||
| Fig. 13 The atomic density of cholesterol shown on a plane cutting the system along the middle of the top monolayer. (A) 1 ns sampling time. (B) 10 ns sampling time. The units for the color scale are nm−3. | ||
 | ||
| Fig. 14 A close-up of the total lipid density 3 nm−3 iso-surface sampled over a 10 ns trajectory. The hydrophobic (lipid tail) side of the top monolayer is shown. The circular formation in the top right part is the spot where the lipid semi-vesicle is attached to the other side of the monolayer. (A) A shaded representation demonstrating the difference between Lo and Ld phases. Crystal lattice-like formations are seen in the Lo phase while the surface of the Ld phase appears smooth due to the diffusive averaging. (B) The same fragment of the iso-surface colored by the cholesterol density, for comparative purpose. | ||
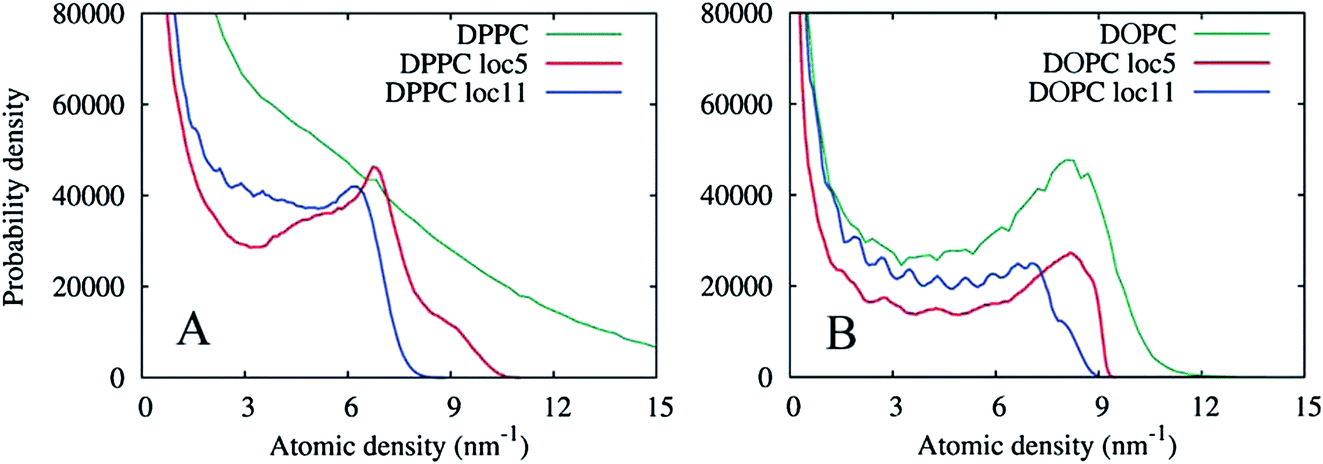 | ||
| Fig. 15 Probability distributions of the grid density values in the lipid monolayers from a 10 ns simulation. (A) For the DPPC lipid. The original density (DPPC) is shown as well as two locally averaged densities using 5 × 5 × 5 cell (loc5) and 11 × 11 × 11 cell (loc11) averaging volumes. (B) Corresponding probability distributions of the atomic density of the DOPC lipid. | ||
The graphs in Fig. 15(A) show negligible overlap in the original DPPC density grid, and the presence of a distinct phase in the grid averaged over 113 cell local volumes (≈ 2.23 nm3), with an average density of approximately 6.5 nm−3. The grid averaged over smaller local regions of 53 cells (≈ 1.03 nm3), shows a more complex shape with at least two regions of enhanced density probability, in the ranges of 4–8 and 8–11 nm−3. These distributions can be explained by the presence of small clusters of another phase within the Lo phase, with a characteristic size of 1.0–2.2 nm. Fig. 16 shows the isosurfaces for the DPPC density corresponding to these density values.
 | ||
| Fig. 16 Density iso-surfaces of the DPPC lipid demonstrating DPPC domains of different locally averaged densities: 1 nm−3 (semi-transparent yellow), 4 nm−3 (semi-transparent orange), 8 nm−3 (solid red). Top monolayer, 10 ns sampling time. | ||
The part of the probability distribution with density less than 4 nm−3 corresponds to interface regions (and the Ld phase); the main peak with a higher density corresponds to the bulk Lo phase; and the sub-peak of the highest density is due to the DPPC clusters inside the bulk phase. A better insight into the nature of the DPPC clusters can be obtained from Fig. 17 where the original DPPC density is shown together with the cluster surfaces. These clusters are formed by several DPPC molecules, with no other lipids. Interestingly, clusters of closely packed and ordered DPPC molecules with low mobility have the same density as a liquid condensed (LC) phase formed by pure DPPC. The density of the LC phase was calculated for lipid monolayers with coexisting LC and liquid-expanded (LE) phases (results not shown). Due to their small size, we believe the clusters represent compositional fluctuations in the vicinity to the transition to the LC phase, rather than a bulk LC phase. The monolayers at the conditions in the simulations are likely close to a three phase coexistence region, similar to the gel-Lo-Ld coexistence region in the ternary phase diagrams of DPPC:DOPC:cholesterol mixtures forming lipid bilayers.26,27
 | ||
| Fig. 17 The iso-surfaces of the original and a locally averaged (over 53 cells) DPPC density. The original density is shown by the yellow semi-transparent surface through 9 nm−3 density values. The locally averaged density is shown by the red semi-transparent surface with an iso-value of 8 nm−3. The semi-transparent blue surface represents the cholesterol density with an iso-value of 3 nm−3. | ||
To contrast the differences between the immobile Lo phase and the mobile Ld phase, Fig. 15(B) shows analogous density distributions for the DOPC lipid prevalent in the Ld phase. Apparently, at this timescale, the natural diffusion in the phase provides sufficient averaging, so that the presence of a bulk phase can be seen in the probability distribution of the original density. Local averaging of the density grids does not produce new evidence of any additional phases enriched in DOPC.
4 Conclusions
A density based visualization technique has been applied to the results of molecular dynamics simulation of two different systems in the framework of the MARTINI force field. It has been shown that very large systems can be visualized because the density based approach to visualization does not depend on the number of particles but on the density grid resolution. The grid resolution can be adjusted according to available resources.Density fields contain information about short term statistics of particle motion and accessible volume but still retain the main structural features of the system when an appropriate time scale is used. This aspect of generated visual representations is similar to the statistical nature of most experimental methods of structural analysis. Many experimental methods produce signals proportional to the volumes of phases with specific properties and averaged over time, such as X-ray diffraction for example. The volumes of the domains with specific densities can be related to the intensities of such experimental signals. The lipid density surface shown in Fig. 14(A) shows a striking resemblance with images produced in atomic force or scanning tunneling microscopy experiments. This similarity of the proposed visualization approach should simplify making connections between the results from simulations with the results obtained in experimental studies, the most crucial step of any simulation study. In addition, moving the data from the discrete space of molecular or atomic properties to the domain of continuous property fields offers new opportunities for more advanced structural analysis.
The density based approach to visualization and analysis of the simulation results reported in this paper have assisted us in revealing structural features of a drug delivery nano-particle as well as gaining new insights into the structure and dynamics of lipid monolayers at the air–water interface. The continuous nature of the density fields allowed for the structural correlational analysis of the RNA–lipid distribution, quantitative analysis of the water cavities and the lipid–water interfacial areas, and the clear visualization of the cholesterol distribution relative to the cationic lipid inside a lipid nano-particle in Section 3.1. The DPPC clusters in the lipid monolayers in Section 3.2 were identified after the probability distributions of the locally averaged grid density values were plotted and then used for targeted density visualization. The approach for data analysis and visualization proposed in this work undoubtedly offers a number of unique options which can potentially benefit any modern molecular dynamics simulation study.
Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council (Canada). Simulations were carried out on Compute Canada facilities. DPT is an Alberta Innovates Health Solutions Scientist and Alberta Innovates Technology Futures Strategic Chair in (Bio)Molecular Simulation.References
- A. K. K. Leung, I. M. Hafez, S. Baoukina, N. M. Belliveau, I. V. Zhigaltsev, E. Afshinmanesh, D. P. Tieleman, C. L. Hansen, M. J. Hope and P. R. Cullis, Lipid Nanoparticles Containing siRNA Synthesized by Microfluidic Mixing Exhibit an Electron-Dense Nanostructured Core, J. Phys. Chem. C, 2012, 116, 18440–18450 CAS.
- L. V. Bock, C. Blau, G. F. Schroeder, I. I. Davydov, N. Fisher, H. Stark, M. V. Rodina, A. C. Vaiana and H. Grubmueller, Energy barriers and driving forces in tRNA translocation through the ribosome, Nat. Struct. Mol. Biol., 2013, 20(12), 1390–1397 CAS.
- W. Humphrey, A. Dalke and K. Schulten, VMD – Visual Molecular Dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- M. S. G. Razul, J. G. Hendry and P. G. Kusalik, Mechanisms of heterogeneous crystal growth in atomic systems: Insights from computer simulations, J. Chem. Phys., 2005, 123, 204722 CrossRef PubMed.
- J. Vatamanu and P. G. Kusalik, Molecular insights into the heterogeneous crystal growth of sI methane hydrate, J. Phys. Chem. B, 2006, 110, 15896–15907 CrossRef CAS PubMed.
- I. M. Svishchev and P. G. Kusalik, Crystallization of Liquid Water in a Molecular Dynamics Simulation, Phys. Rev. Lett., 1994, 73(7), 975–978 CrossRef CAS.
- S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. de Vries, The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations, J. Phys. Chem. B, 2007, 111, 7812–7824 CrossRef CAS PubMed.
- S. J. Marrink and D. P. Tieleman, Perspective on the Martini model, Chem. Soc. Rev., 2013, 42, 6801–6822 RSC.
- A. Widmer-Cooper, P. Harrowell and H. Fynewever, How reproducible are dynamic heterogeneities in a supercooled liquid?, Phys. Rev. Lett., 2004, 93(13), 135701 CrossRef.
- A. Widmer-Cooper and P. Harrowell, On the relationship between structure and dynamics in a supercooled liquid, J. Phys.: Condens. Matter, 2005, 17, S4025–S4034 CrossRef CAS.
- G. S. Matharoo, M. S. G. Razul and P. H. Poole, Structural and dynamical heterogeneity in a glass-forming liquids, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2006, 74, 050502 CrossRef.
- D. Rozmanov and P. G. Kusalik, Isocofigurational molecular dynamics study of the kinetics of ice crystal growth, Phys. Chem. Chem. Phys., 2012, 14, 13010–13018 RSC.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, GROMACS 4: Algorithms for Highly Efficient Load-Balances, and Scalable Molecular Simulations, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS.
- “Grid3D Toolbox for molecular visualization.” on-line: http://sourceforge.net/projects/grid3d, Jan 2014.
- A. Henderson, “ParaView Guide, A Parallel Visualization Application,” Kitware Inc., 2007 Search PubMed.
- W. Shroeder, K. Martin, and B. Lorensen, The Visualization Toolkit (4th edn), Kitware Inc., 2006 Search PubMed.
- L. Huang and Y. Liu, In vivo delivery of RNAi with lipid-based nanoparticles, Annu. Rev. Biomed. Eng., 2011, 13, 507–530 CrossRef CAS PubMed.
- S. Khalid, P. J. Bond, J. Holyoake, R. W. Hawtin and M. S. P. Sansom, DNA and lipid bilayers: self-assembly and insertion, J. R. Soc. Interface, 2008, 5, 241–250 CrossRef PubMed.
- “Bio.B-Gen, a simulation box generator for biological simulations.” on-line: http://sourceforge.net/projects/biobgen, Jan 2014.
- K. T. Love, K. P. Mahon, C. G. Levins, K. A. Whitehead, W. Querbes, J. R. Dorkin, J. Qin, W. Cantley, L. L. Qin, T. Racie, M. Frank-Kamenetsky, K. N. Yip, R. Alvarez, D. W. Y. Sah, A. de Fougerolles, K. Fitzgerald, V. Koteliansky, A. Akinc, R. Langer and D. G. Anderson, Lipid-like materials for low-dose in vivo gene silencing, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(5), 1864–1869 CrossRef CAS PubMed.
- S. Baoukina, E. Mendez-Villuendas and D. P. Tieleman, Molecular View of Phase Coexistence in Lipid Monolayers, J. Am. Chem. Soc., 2012, 134, 17543–17553 CrossRef CAS PubMed.
- S. Baoukina, L. Monticelli, H. J. Risselada, S. J. Marrink and D. P. Tieleman, The molecular mechanism of lipid monolayer collapse, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10803–10808 CrossRef CAS PubMed.
- Y. Y. Zuo, R. A. W. Veldhuizen, A. W. Newmann, N. O. Petersen and F. Possmayer, Current perspectives in pulmonary surfactant – Inhibition, enhancement and evaluation, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 1947–1977 CrossRef CAS PubMed.
- B. Piknova, V. Schram and S. B. Hall, Pulmonary surfactant: phase behaviour and function, Curr. Opin. Struct. Biol., 2002, 12, 487–494 CrossRef CAS.
- J. Perez-Gil and T. E. Weaver, Pulmonary Surfactant Pathophysiology: Current Models and Open Questions, Physiology, 2010, 25, 132–141 CrossRef CAS PubMed.
- J. H. Davis, J. J. Clair and J. Juhasz, Phase Equilibria in DOPC/DPPC-d(62)/Cholesterol Mixtures, Biophys. J., 2009, 96, 521–539 CrossRef CAS PubMed.
- P. Uppamoochikkal, S. Tristram-Nagle and J. F. Nagle, Orientation of Tie-Lines in the Phase Diagram of DOPC/DPPC/Cholesterol Model Biomembranes, Langmuir, 2010, 26, 17363–17368 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |