Engineering safer-by-design silica-coated ZnO nanorods with reduced DNA damage potential
Georgios A.
Sotiriou
a,
Christa
Watson
a,
Kimberly M.
Murdaugh
ab,
Thomas H.
Darrah
c,
Georgios
Pyrgiotakis
a,
Alison
Elder
d,
Joseph D.
Brain
a and
Philip
Demokritou
*a
aDepartment of Environmental Health, Center for Nanotechnology and Nanotoxicology, School of Public Health, Harvard University, 665 Huntington Avenue, Boston, MA 02115, USA. E-mail: pdemokri@hsph.harvard.edu
bSchool of Engineering and Applied Sciences, Harvard University, 29 Oxford Street, Cambridge, MA 02138, USA
cSchool of Earth Sciences, 231 Mendenhall Laboratory, The Ohio State University, Columbus, OH 43210, USA
dDepartment of Environmental Medicine, University of Rochester, 601 Elmwood Avenue, Rochester, NY 14642, USA
First published on 3rd February 2014
Abstract
Zinc oxide (ZnO) nanoparticles absorb UV light efficiently while remaining transparent in the visible light spectrum rendering them attractive in cosmetics and polymer films. Their broad use, however, raises concerns regarding potential environmental health risks and it has been shown that ZnO nanoparticles can induce significant DNA damage and cytotoxicity. Even though research on ZnO nanoparticle synthesis has made great progress, efforts on developing safer ZnO nanoparticles that can maintain their inherent optoelectronic properties while exhibiting minimal toxicity are limited. Here, a safer-by-design concept was pursued by hermetically encapsulating ZnO nanorods in a biologically inert, nanothin amorphous SiO2 coating during their gas-phase synthesis. It is demonstrated that the SiO2 nanothin layer hermetically encapsulates the core ZnO nanorods without altering their optoelectronic properties. Furthermore, the effect of SiO2 on the toxicological profile of the core ZnO nanorods was assessed using the Nano-Cometchip assay by monitoring DNA damage at a cellular level using human lymphoblastoid cells (TK6). Results indicate significantly lower DNA damage (>3 times) for the SiO2-coated ZnO nanorods compared to uncoated ones. Such an industry-relevant, scalable, safer-by-design formulation of nanostructured materials can liberate their employment in nano-enabled products and minimize risks to the environment and human health.
Nano impactZnO nanoparticles are sought out for UV-filter applications due to their inherent optoelectronic properties and are, therefore, broadly used today in cosmetics and polymers. Preliminary toxicological data, however, point out that they can induce significant DNA damage and genotoxicity due to their Zn2+ ion leaching. It has become important for the nanotechnology industry to devise scalable, safer-by-design approaches to minimize the ZnO nanoparticle dissolution and toxicity without altering their desired optoelectronic properties. In this original work, we demonstrate such a safer-by-design approach for ZnO nanorods using a scalable flame aerosol process. This technology allows for controlled synthesis of high-purity ZnO nanorods with a highly crystalline core and a nanothin amorphous silica shell that improves their biocompatibility. The as-prepared nanorods exhibit high transparency in the visible range but strong absorption in the UV, rendering them suitable for use in sunscreens and polymers. Furthermore, it is demonstrated that the hermetic silica coating does not alter the desired optoelectronic properties of the core ZnO nanorods while their DNA damage potential has been decreased by 3-fold. |
Introduction
Zinc oxide (ZnO) is a wide bandgap semiconductor with optical, piezoelectric, and dielectric properties that render it useful for a variety of applications, ranging from transistors,1 light emitting diodes,2 sensors,3 photocatalysis,4 and ultraviolet (UV)-filters,5 to name a few. When in the nanoscale, ZnO exhibits several advantages. For example, the high electron mobility and diversity of ZnO nanostructures make it a suitable material for dye-sensitized solar cells,6 while its large surface-to-volume ratio may improve its bioavailability when employed in food-fortification applications.7 Furthermore, ZnO nanoparticles exhibit luminescence and therefore may be used as novel labels in vitro.8ZnO absorbs UV wavelengths ranging from 280 to 400 nm5,9 making it an ideal component in cosmetic creams and sunscreens that aim to block both UVA (320–400 nm) and UVB (280–320 nm) radiation.10 Traditional sunscreens use micron-sized ZnO particles that make them opaque with bright white color that is esthetically undesirable for consumers.11,12 In contrast, ZnO nanoparticles are transparent to visible light yielding a less opaque sunscreen. As a result, ZnO nanoparticle-based sunscreens are favored and their production has increased dramatically in recent years.11,12 Both wet- and gas-phase synthetic strategies have been employed to synthesize ZnO nanostructures, including chemical vapor deposition,13 hydrolysis,14 and flame synthesis15,16 with various morphologies explored.
Because of the wide range of ZnO nanoparticle applications, exposure to humans is inevitable.11 However, the potential adverse health effects of ZnO nanoparticle exposures have not been fully determined. In vivo and in vitro toxicological studies have shown that these particles are acutely toxic to alveolar epithelial cells and macrophages.17–19 Additionally, ZnO nanoparticles have been reported to induce significant cytotoxicity and genotoxicity in human neuronal cells.20 ZnO nanoparticles can also generate reactive oxygen species (ROS)21 within human skin melanoma cells in acute exposures which led to DNA damage, cell viability reduction, and apoptosis.22 Exposure via inhalation is of particular concern as reduced pulmonary function in humans was found 24 hours after exposure to ZnO nanoparticles.23 Furthermore, recent work in our group has shown that ZnO nanoparticles can cause high genotoxicity when compared to various industrially relevant metal nanomaterials such as silver, iron oxide, and cerium oxide.24 The reported cytotoxicity and DNA damage associated with ZnO exposures are attributed to the released ions from its rapid dissolution within aqueous solutions and also from direct particle interactions with cells through the induction of oxidative stress.19,25
Therefore, minimizing the ZnO nanoparticle dissolution and direct contact with the cells can greatly inhibit toxicity.26 Such a safer-by-design formulation concept for the gas-phase synthesis of metal oxide nanoparticles using a modified flame spray pyrolysis (FSP) reactor27 has been recently developed by the authors and involves the encapsulation of the core nanoparticles with a nanothin amorphous silica (SiO2) layer.17 The SiO2 nanothin layer is formed by the swirl injection of Si-precursor vapor within the reactor after the core nanoparticle growth has ceased, allowing for a precise control over the coating thickness.27 Such a nanothin amorphous SiO2 coating preserves the functional inherent properties of the core material.27
Amorphous fumed nanostructured SiO2 is considered biologically inert28 and commonly used in cosmetic and personal care products and as a negative control in nanoparticle toxicity screening assays.29 At high doses (>100 μg mL−1), amorphous SiO2 may exhibit some in vitro toxicity30,31 which is associated with the presence of strained three-membered rings and the subsequent free radical formation.31 However, in vivo studies using a rat model have verified that such a biological response is transient,32 and at lower, physiologically relevant doses, a flame-made SiO2 coating has minimal lung injury and inflammation.33 Furthermore, it was also demonstrated that the SiO2 coating improves nanoparticle biocompatibility in vitro on a variety of nanomaterials including Ag,34 Y2O3,35 and ZnO nanoparticles and mammalian cell lines.17
Here, we explore the gas-phase synthesis of free-standing uncoated and hermetically SiO2-coated (23 wt% SiO2) ZnO nanorods by the flame spray pyrolysis (FSP) based Harvard Versatile Engineered Nanomaterial Generation System (VENGES).36 The morphology and physicochemical characteristics of the as-prepared nanoparticles are investigated using X-ray diffraction, N2 adsorption, thermogravimetric analysis and electron microscopy, while the hermetic nature of the SiO2 coating is evaluated by X-ray photoelectron spectroscopy, the inhibition of the photocatalytic methylene blue degradation and by measuring the ζ-potential across a broad pH range. The effect of the SiO2 coating on the optoelectronic properties (UV-vis transmission and bandgap) of the core ZnO nanorods is also investigated in detail providing valuable information on the functionality of the SiO2-coated nanoparticles and their application as efficient UV blockers. Most importantly, the effect of the SiO2 shell on the genotoxicity of both uncoated and SiO2-coated ZnO nanorods is evaluated using the Nano-Cometchip24,37 bioassay while the cellular viability is also monitored by the reduction of a tetrazolium dye.
Materials and methods
Synthesis and characterization
Uncoated and SiO2-coated ZnO particles were synthesized by flame spray pyrolysis (FSP) of zinc naphthenate (Sigma-Aldrich) dissolved in ethanol (Sigma-Aldrich) at a precursor molarity of 0.5 M. The precursor solution was fed through a stainless steel capillary at 5 mL min−1, dispersed by 5 L min−1 O2 (air gas, purity >99%, pressure drop at nozzle tip: pdrop = 2 bar) and combusted. A premixed methane–oxygen (1.5 L min−1, 3.2 L min−1) supporting flame was used to ignite the spray. O2 (air gas, purity >99%) sheath gas was used at 40 L min−1. Core particles were coated in-flight by the swirl-injection of HMDSO vapor (Sigma Aldrich) through a torus ring with 16 jets at an injection height of 200 mm above the FSP burner. The torus ring jet injection angles were 20° in the downstream direction, in order to avoid stagnation flow, and 10° away from the centerline, in order to induce mixing. The reactor was enclosed by two quartz tubes: below and above the torus ring. A total gas flow of 16 L min−1, consisting of N2 carrying HMDSO vapor and pure N2, was injected through the torus ring jets. HMDSO vapor was obtained by bubbling N2 gas through liquid HMDSO (500 mL), maintained at a controlled temperature using a water bath. A theoretical coating thickness of ~5 nm was targeted assuming saturation conditions. After synthesis, particles were collected on a water-cooled glass fiber filter (Whatman) located 800 mm above the reactor with the aid of a vacuum pump.For the electron microscopy analysis, uncoated and SiO2-coated ZnO nanoparticles were dispersed in ethanol at a concentration of 1 mg mL−1 in 50 mL polyethylene conical tubes and sonicated for 10 minutes with a sonication power of 1.75 watt (Branson Sonifier S-450A). The samples were deposited onto lacey carbon TEM grids. All grids were imaged using a JEOL 2100.
X-ray diffraction (XRD) patterns for uncoated ZnO and SiO2-coated ZnO nanoparticles were obtained using a Bruker D8 diffractometer (Cu Kα, λ = 0.154 nm, 40 kV, 40 mA, step size = 0.02°). 100 mg of each sample was placed onto the diffractometer stage and analyzed in a range of 2θ = 20–70°. Major diffraction peaks were identified using the Inorganic Crystal Structure Database (ICSD) for the wurtzite (ZnO) crystal. The crystal size was determined by applying the Rietveld analysis of the major diffraction peaks. The specific surface area was obtained according to Brunauer–Emmet–Teller (BET) by five-point N2 adsorption at 77 K (Micrometrics Tristar 3000). Prior to that, samples were degassed in N2 for at least 1 h at 150 °C.
For the X-ray photoelectron spectroscopy analysis, 5 mg of powder was deposited on a metal sample holder coated with electrical tape and placed on the XPS sample stage (ESCA SSX-100). Soft X-ray from an Al anode is used to bombard the sample and eject electrons from the surface (monochromatic Al Kα, 10 kV, 10 mA). The KE of the ejected electrons was determined using a detector hemispherical electron energy spectrometer (spot size: 600 μm). The samples were scanned (BE range: 0–1100 eV, pass energy: 100 eV, step size: 0.65 eV), and the resulting spectra were analyzed using the CASA XPS software and calibrated using the C 1s hydrocarbon contamination peak (BE: 284.6 eV). XPS was also used to determine the ratio of the elemental concentration of the core particle to the total elemental concentration of the entire nanoparticle.17 This ratio, Xel, is defined as follows (eqn (1)):
 | (1) |
For the ζ-potential measurements, uncoated ZnO and SiO2-coated ZnO nanoparticles were dispersed in deionized water (5 mg mL−1) and placed in a 3 in cup horn and sonicated using a Branson Sonifier S-450A (Branson Ultrasonics, Danbury, CT) for 10 minutes as described above. The liquid conical tube was submersed in the DI H2O so that the level of the tube contents aligned with the surrounding DI H2O. Hydrochloric acid (HCl) and potassium hydroxide (KOH) (both 0.1 M) were added to the suspensions to vary the pH between 2 and 13 pH, as measured using a SympHony pH meter (VWR International, Radnor, PA). 1 mL aliquots of each sample were placed in a cuvette and were analyzed for their zeta potential using a Malvern Zetasizer Nano ZS (Malvern Instruments, Worcestershire, UK). Zeta potential measurements were repeated in triplicate.
Methylene blue (Aldrich, MB) was employed as a model dye to evaluate the photocatalytic activity of the as-prepared nanoparticles. For each condition, 18 mg of particles was dispersed in 20 mL of 10 ppm MB aqueous solution. The pH during the photocatalytic activity experiments was ~6.8 ± 0.5. The beaker containing the particles and solution was placed on a magnetic stirrer plate and a stirrer bar placed in the solution ensured full suspension of the particles throughout the experiment. Prior to this, the suspensions were sonicated in a water bath (Branson Sonifier S-450A) for 30 s. Before UV-irradiation, the suspensions were left for 30 min in the dark to equilibrate any adsorption–desorption effects. The photocatalytic reaction was conducted at room temperature under UV light from a single 9 W UV tube at 254 nm (Philips) positioned horizontally above (2 cm) the liquid surface. Each experiment was conducted for 45 min with 1 mL sample aliquots extracted after 10, 20, 30 and 45 min and were immediately centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g. The decomposition of MB was monitored by measuring the absorbance of the supernatant using a UV–vis spectrophotometer (Agilent Cary 50, at 661 nm) in liquid cuvette configuration with deionized water as the reference.
000g. The decomposition of MB was monitored by measuring the absorbance of the supernatant using a UV–vis spectrophotometer (Agilent Cary 50, at 661 nm) in liquid cuvette configuration with deionized water as the reference.
For the UV-vis transmission measurements, nanoparticles were dispersed in DI H2O (0.5 mg mL−1) in 50 mL polyethylene conical tubes. The samples were placed in a 3 in cup horn and sonicated for 10 minutes as previously described. The samples were dispersed using a cup sonicator immediately prior to spectral analysis. 2 mL of the each dispersion was placed in a quartz cuvette and placed in the UV–vis spectrophotometer (Agilent Cary 50). UV-vis transmission measurements were performed over a spectrum of 190 to 900 nm with a reference background for DI H2O. Dynamic light scattering (DLS) of such suspensions was performed using a Zetasizer Nano-ZS (Malvern Instruments, Worcestershire, UK). Diffuse reflectance measurements of the dry powders were performed using a Varian Cary 500 spectrophotometer. Samples were placed in a dry powder sample holder (Praying Mantis).
For the dissolution investigation of both uncoated and SiO2-coated ZnO nanorods, nanoparticles were dispersed in culture media (RPMI-1640 with L-glutamine supplemented with 10% horse serum and 100 units mL−1 penicillin and streptomycin, pH = 7.4, total concentration of inorganic salts [ionic strength] = 139.1 mM) by sonication38 at a concentration of 100 mg L−1 of ZnO. Samples (1 mL) were taken over time for 24 h and immediately centrifuged at 10000g for 1 h. Then the supernatant was taken and stored for further analysis. The experiment was conducted in triplicate.
All sample preparation for ICP-MS analysis was conducted in a class 100 trace metal-free clean laboratory. For the dissolution fractions, approximately 1.0 mL of each sample was transferred to the corresponding pre-labeled analytical vials and verified gravimetrically to ±0.002 mg using a Perkin Elmer AD6 microbalance. Next, the samples were digested in ultra-pure nitric acid (15.9 mol L−1) and concentrated hydrochloric acid (12.4 mol L−1) (Thermo Fisher Scientific, Waltham, MA, USA) and baked at 80 °C for 6 hours. After sample digestion was verified visually, the samples were decanted by baking at 60 °C until dry (~12 hours). All samples (and standards) were diluted and prepared for analysis using water purified to 18.2 MΩ cm resistance using a Milli-Q water purification system (Millipore, Bedford, MA, USA), acidified using trace metal-free concentrated (15.9 mol L−1) ultra-pure nitric acid (Thermo Fisher Scientific, Waltham, MA, USA), and spiked with internal standards consisting of known quantities of indium (In) and bismuth (Bi), to monitor the instrumental behavior and the sample response similar to methods reported previously.39
Silicon and zinc concentrations were measured using a Perkin Elmer DRC II ICP-MS at Ohio State University consistent with previous methods.39,40 External calibration standards used to determine Si, Zn, and other metal concentrations in analytical samples were spiked with known quantities of each analyte (e.g., Si, Zn, etc.) in a linear range (e.g. from 0.05 to 20.0 μg g−1). All standards were prepared from 1000 mg L−1 single element standards obtained from SCP Science, USA. Isobaric corrections were performed on-line using the ICP-MS software. Five duplicate analyses (n = 5) were performed for each analyte for all sample solutions. Average reproducibility based on replicate analysis of known–unknown external calibration standards was 3.06%. The LOD and LOQ for Si were 0.108 ng g−1 and 0.213 ng g−1, respectively. The LOD and LOQ for Zn were 0.016 ng g−1 and 0.034 ng g−1, respectively. Method detection limits were calculated according to the two-step approach using the t99SLLMV method at 99% CI (t = 3.71). The calculated MDL for Si and Zn were 0.890 ng g−1 and 0.080 ng g−1, respectively. The external precision for all analytes was less than 5% for all reported analysis.
DNA damage and cytotoxicity evaluation
For the cell culture and treatment, human lymphoblastoid cells, TK6, were a kind gift of Bevin Engelward from MIT. Human lymphoblastoid cell line, TK6, was chosen for these experiments as they are routinely used in the comet assay method and genotoxicity assessments.41 Additionally, TK6 cells are p53 proficient and genetically stable, which are essential attributes in the detection of genotoxic agents.42 Moreover, the decision to use this particular cell line was not based on physiological relevance but in the effort to detect minute differences in the genotoxic response to these similar materials. Cells were maintained in RPMI-1640 with L-glutamine supplemented with 10% horse serum and 100 units mL−1 penicillin and streptomycin (pH = 7.4, total concentration of inorganic salts [ionic strength] = 139.1 mM) at 37 °C in an atmosphere of 5% CO2/95% air. Cells were seeded for treatment in a 96-well plate at a density of 2 × 106 cells per well in 100 μL. Nanoparticle suspensions were prepared and sonicated as mentioned below by Cohen et al.38 in 3 mg mL−1 concentrated stocks in sterile distilled water. The stocks were then diluted in RPMI media containing 10% horse serum to the appropriate concentration (5, 10, 20 μg mL−1). Each suspension was vortexed prior to adding to the cells at a volume of 100 μL for 4 hours.DNA damage evaluation was performed as described by Watson et al.,24 in which the customization of the Cometchip platform37,43 was successfully performed for investigating the genotoxicity of various metal and metal oxide nanoparticles. For the Nano-Cometchip preparation and cell loading, negative silicon molds for polydimethylsiloxane (PDMS) casting of patterned microposts were made. The resulting PDMS mold was allowed to set in molten 1% normal melting point agarose applied to a gel bond film for 20 minutes. After agarose polymerization, the PDMS stamp was removed revealing a thick gel consisting of arrayed microwells. The gel was then clamped between a bottomless 96-well plate and a glass plate. Pre-exposed cells were then added to the microwell array and allowed to load for 30 minutes. After gravitational settling of cells had occurred, excess cells were aspirated and the gel was rinsed three times with warm phosphate buffered saline. Molten low melting agarose was used to cover microwell array/cells and allowed to set for 5 minutes at room temperature and 5 minutes at 4 °C. The gel was then submerged into lysis solution (containing 2.5 M NaCl, 100 mM Na2EDTA, 10 mM Tris, pH 9.5 with 0.5% Triton X-100) overnight at 4 °C. After lysis, gels were washed two times in PBS to remove the surfactant. Gels were then adhered to the electrophoresis inner tank well with double sided tape (gel side up). Electrophoresis was then performed using alkaline buffer (90 mM Tris, 90 mM boric acid, 2 mM Na2EDTA, pH 8.5) at 4 °C for 30 min at 25 V and 300 mA. Imaging and data collection was performed using an automated fluorescent microscope (Axiovert Zeiss) and the proprietary MatLab software was used to analyze data sets. The median of %DNA in tail was utilized to obtain the amount of DNA damage within the cell population at each dosage. The reported values are the average of at least six experiments along with the standard error. P-values less than 0.05 were considered to be significant.
For the cellular viability evaluation, the MTT assay (Roche) was used to assess the mitochondrial dehydrogenase activity of intact and injured TK6 cells exposed to nanoparticles. TK6 cells, a suspension lymphoblastoid cell line, were seeded into 96-well plates at a density of 1 × 104 cells per well in RPMI media in 100 μL containing 10% horse serum. Nanoparticle suspensions were added at 100 μL at twice the concentration needed to obtain 5, 10, and 20 μg mL−1 for 4 h at 37 °C in 5% CO2. After exposure, cells were spun down at 250g and spent media were aspirated. Fresh media were then added and the MTT reagent (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) (10 μL) was added to each well for a period of 4 hours at 37 °C. A solubilization reagent (150 μL) was added to dissolve the formazan crystals produced from the reduction of the tetrazolium salt or the MTT reagent in viable cells. Absorbance was measured at 550 nm using a Molecular Devices microplate reader. Acellular experiments were performed to ensure reagent integrity and to rule out NP interference. The data obtained revealed no interaction between test nanoparticles and MTT reagents during the 4 hours exposure time point. Percent cell viability (relative viability compared to untreated cells) was calculated as mean value ± standard error of the mean (SEM) as a result of three independent experiments performed in triplicate.
Results and discussion
Nanoparticle morphology
The morphology of the as-prepared nanoparticles was evaluated qualitatively by electron microscopy. Fig. 1 shows transmission electron microscopy (TEM) images of the uncoated (a, b) and SiO2-coated (c, d) ZnO nanoparticles. From the TEM images, it appears that the ZnO nanoparticles are not spherical but rather rod-like. Flame-made nanoparticles typically exhibit a log-normal particle size distribution (σg = 1.45),44 while the ZnO nanorod aspect ratio ranges from 2 to 8 with an average of ~3 and this is in agreement with similar studies reported in the literature.45 ZnO nanorods have been made before by flame spray pyrolysis in the presence of dopants (indium and tin up to 10 at%).46 Such doping, however, influences their optoelectronic properties,46 thus making these nanorods unsuitable for a number of applications where purity is important. In contrast, here, pure ZnO nanorods are made and their formation is attributed to particle annealing and rearrangement within the flame,46 and the long residence time at high temperatures typical for enclosed FSP reactors.27 Even though the nanorod morphology is not required specifically for their employment as UV-filters, such nanostructures may find applications in other fields, such as nanoelectronics.45 Furthermore, for the SiO2-coated sample (c, d) there is a ~4.5 nm thick homogeneous amorphous layer encapsulating the core crystalline ZnO nanoparticles.17 The SiO2 coating thickness distribution on the ZnO nanorods is shown in the inset of Fig. 1d, along with the average value ± the standard deviation, as well as the total number N of coatings counted. Furthermore, there are few, if any, free SiO2 nanoparticles further verifying the coating efficiency of the reactor.47 | ||
| Fig. 1 Transmission electron microscopy images of the uncoated (a, b) and SiO2-coated ZnO nanoparticles (c, d). In both cases, the ZnO forms nanorods with aspect ratio ~3:1. Furthermore, there is a nanothin amorphous SiO2 shell encapsulating the core particles for the SiO2-coated ZnO nanorods. The inset of (d) shows the SiO2 coating thickness distribution along with the average coating thickness ± standard deviation and the total number N of coatings counted. | ||
X-ray diffraction (XRD) was utilized to assess the crystal structure and size of the as-prepared pristine nanoparticles. Fig. 2 shows the diffraction patterns of both uncoated (black) and SiO2-coated (red) ZnO nanorods. Both patterns have the characteristic peaks corresponding to the wurtzite crystal structure.16 Furthermore, the diffraction peaks of both coated and uncoated samples coincide, verifying that the underlying ZnO core particles have identical crystallinity. The average crystal size for both samples is ~30 nm. This further indicates that the SiO2 coating formation does not influence the ZnO core nanoparticle growth that had already stopped before the injection of the SiO2 precursor vapor within the coating reactor.17,27
 | ||
| Fig. 2 X-ray diffraction patterns of the uncoated (black line) and SiO2-coated ZnO nanorods (red line). Their average crystal size (dXRD) and specific surface area (SSA) values are also displayed. | ||
It is also worth mentioning that there are no diffraction peaks corresponding to Zn–silicates. Under specific conditions during the nanoparticle synthesis, Zn–silicate formation can occur, especially when high specific combustion enthalpy solvents are used that can increase the reactor temperature profile.17,48 However, a low specific combustion enthalpy solvent was deliberately chosen (please see materials and methods section) since the Zn–silicate formation was undesired. There are also no signs of any highly toxic32 crystalline SiO2 diffraction peaks, further verifying that SiO2 is amorphous.27 This is expected for flame-made SiO2 nanoparticles.49 Furthermore, the specific surface area (SSA) values of the uncoated and SiO2-coated ZnO nanorods are similar (Fig. 2, inset) indicating that there are few or no separate SiO2 nanoparticles that would lead to larger (>100 m2 g−1) SSA values.47,50 This is also in qualitative agreement with the absence of such free SiO2 particles from the TEM analysis (Fig. 1c).
SiO2 coating encapsulation efficiency
The hermetic nature of the SiO2 coating was evaluated using multiple techniques. First, X-ray photoelectron spectroscopy (XPS) was employed, a surface sensitive technique that allows for the coating efficiency quantification on nanostructured materials.17 The XPS spectra of both uncoated (black) and SiO2-coated (red) ZnO nanorods are shown in Fig. 3a. The characteristic peaks corresponding to the Zn (diamonds) and Si (circles) metal electron transitions are also shown. When both spectra are compared, the Zn peaks are significantly reduced for the SiO2-coated sample, even though the SiO2 mass fraction is 23 wt%. It should be noted that the Zn peaks do not completely disappear. This could be attributed to two factors: (i) the inelastic mean free path of lower energy Zn electrons that can penetrate through the nanothin SiO2 layer51 and (ii) a small fraction of the core particles escape the coating process and remain partially uncoated.52 This is also evidenced by the coating efficiency which was found to be 95% here (Fig. 3a, inset), as calculated by the XPS spectra using the elemental concentrations of Zn and Si atoms (please see materials and methods section, eqn (1)).17 | ||
| Fig. 3 (a) X-ray photoelectron spectra of uncoated (black line) and SiO2-coated ZnO nanorods (red line). The Zn (diamonds) and Si (circles) binding energy peaks are also shown. The SiO2 coating efficiency is also displayed. (b) The ζ-potential of both uncoated (black line) and SiO2-coated ZnO nanorods. (c) The methylene blue (MB) degradation over time of the uncoated (black line) and SiO2-coated ZnO nanorods (red line) upon UV irradiation. The MB degradation of TiO2 (green line, P25) is also shown as a positive control. (d) Thermogravimetric analysis result of the SiO2-coated ZnO nanorods. The mass loss is less than 1% indicating no organic species on the SiO2 surface. | ||
Due to the aforementioned limitation of XPS, the hermetic nature of the SiO2 coating was further evaluated by monitoring the ζ-potentials of the uncoated and SiO2-coated nanoparticles over a range of pH values. Fig. 3b shows the ζ-potentials of uncoated (triangles) and SiO2-coated (circles) nanoparticles for pH = 2.5–8. Pure ZnO nanorods have an isoelectric point around pH = 8,53 while the SiO2-coated nanorods retain a negative ζ-potential for the same pH range, which is in agreement with the ζ-potential of pure SiO2.47 This indicates that there is no significant ZnO surface exposed, and most importantly, such SiO2-coated ZnO nanorods behave like SiO2 nanoparticles in aqueous solutions and in biological media.17,54
The photocatalytic activity of the ZnO nanorods was also examined. Upon UV irradiation on ZnO nanorods, the electron–hole pairs on the particle surface participate in redox reactions with organic species, such as methylene blue (MB), that lead to their degradation.55 Therefore, monitoring the MB degradation over time in aqueous solutions upon UV irradiation gives a good estimate on the ZnO surface availability and reactivity. Fig. 3c shows the MB degradation as a function of time using the as-prepared uncoated (black line) and SiO2-coated ZnO nanorods (red line). Photocatalytic TiO2 nanoparticles (green line, P25) were used as a positive control. It can be seen that the uncoated ZnO nanorods are highly photocatalytic4 and they slightly outperform the “gold standard” P25 TiO2 nanoparticles by completely decomposing MB after 30 min. In contrast, the SiO2-coated ZnO nanorods show no photocatalytic activity because of the small uncoated ZnO surface fraction (<5%). This verifies that the SiO2 coating hermetically encapsulates the core ZnO nanorods.17
It is noteworthy that gas-phase synthesis results in high purity products56 and that there is no organic contamination on the SiO2 surface. This is further verified here by thermogravimetric analysis of the SiO2-coated ZnO nanorods. Fig. 3d shows the sample mass percentage as a function of time. The measured mass loss is <1% for up to 700 °C, typical for impurity-free flame-made nanoparticles.57 Furthermore, given that the surface chemistry of SiO2 is very well understood,58,59 the SiO2 shell on the ZnO core can facilitate further surface (bio)functionalization which can increase the nanorod functionality and versatility for a variety of bio-applications. It is noteworthy that typical wet-made SiO2 coatings on nanoparticles are porous,60 therefore allowing ion release. However, dry-coated27,34 nanoparticles typically yield a dense non-porous nanothin coating completely isolating the core material from its environment. Fig. 4 shows the dissolved Zn fraction (%) as a function of time for both uncoated and SiO2-coated ZnO nanorods (ZnO concentration of 100 mg L−1) in the cell culture medium (pH = 7.4, total concentration of inorganic salts [ionic strength] = 139.1 mM). The uncoated ZnO nanorods continuously dissolve up to 24 h without reaching equilibrium. In contrast, the SiO2-coated ZnO nanorods also exhibit an initial ZnO dissolution; however, it appears that after 6 h the system has reached equilibrium. In the case of the SiO2-coated ZnO nanorods the core dissolution most probably originates from the small uncoated ZnO surface fraction (<5%). This indicates that even though the surface coating has a high efficiency (>95%), as soon as there is some core surface exposed, a large fraction of the core ZnO is dissolved, as expected, but it is still lower than the case of uncoated ZnO nanorods. Furthermore, the SiO2 coating is rather stable in the cell medium, as over 24 h, it only dissolves <8% (data not shown). These results are in agreement with dissolution data of both SiO2 and ZnO in the culture media published in the literature.61
 | ||
| Fig. 4 The dissolution kinetics of uncoated (black circles) and SiO2-coated ZnO nanorods (red triangles) in RPMI-1640 cell culture medium (pH = 7.4, total concentration of inorganic salts [ionic strength] = 139.1 mM) for an initial ZnO concentration of 100 mg L−1. | ||
Effect of the SiO2 coating on optoelectronic properties
A fundamental prerequisite for the successful utilization of these safer-by-design ZnO nanorods is the preservation of their desired optoelectronic properties. Fig. 5a shows the transmission (%) spectra of aqueous suspensions of uncoated (black line) and SiO2-coated (red line) ZnO nanorods (0.5 mg mL−1 of ZnO). Both samples exhibit relatively high transmittance in the visible range 400–700 nm with the SiO2-coated ZnO nanorods outperforming (>80%) the uncoated ones. This increase in transmittance is attributed to the lower hydrodynamic size and better dispersibility in aqueous solution of SiO2-coated ZnO nanorods in comparison to uncoated ones: the coated ones have slightly lower agglomerate size in pure DI water than the uncoated ones (165 vs. 220 nm), while in cell media both particles have similar agglomeration states (210 vs. 220 nm).17 The ζ-potential of ZnO nanorods at pH = 6.5–8.5 is close to zero that allows the formation of larger agglomerates (Fig. 3b, black line). In contrast, the pH of SiO2-coated ZnO nanorods at this pH range is highly negative (Fig. 3b, red line), resulting in a highly-stable suspension with low hydrodynamic diameter. The lower hydrodynamic diameter of the SiO2-coated ZnO nanorods results in less light scattering and high transmittance. However, both samples show a strong decrease in transmittance for wavelengths below 400 nm indicating the strong UV absorption of ZnO nanorods.10 | ||
| Fig. 5 (a) Optical transmission spectra of aqueous suspensions containing the uncoated (black line) and SiO2-coated ZnO nanorods (red line) at a concentration of 0.5 mg mL−1. The SiO2-coated ZnO nanorods are more transparent in the visible spectrum because of their smaller agglomerate size. (b) Diffuse-reflectance measurements of dry powders of the uncoated (black line) and SiO2-coated ZnO nanorods (red line). Both curves are identical and exhibit the same bandgap energy. | ||
The optoelectronic properties of both uncoated and SiO2-coated ZnO nanorods are further evaluated by measuring their bandgap energy, Eg. Fig. 5b shows the diffuse-reflectance spectra (Kubelka–Munk function F(R)) of both uncoated (black line) and SiO2-coated ZnO nanorods (red line). The curves for both samples overlap and therefore these two samples have identical bandgaps (~3.3 eV), which are in agreement with the theoretical value.10 This is in line with the identical crystallinity of these samples (Fig. 2) and further verifies that the presence of the SiO2 coating does not influence the optoelectronic properties of the core ZnO nanorods, rendering them highly transparent (in the visible range) UV-filters that may be employed in sunscreens and polymer nanocomposites.
Effect of the SiO2 coating on DNA damage and cytotoxicity
The DNA damage in human lymphoblastoid cells (TK6) exposed to both uncoated and SiO2-coated ZnO nanorods was evaluated using the Nano-Cometchip cellular assay.24,37,43Fig. 6a shows the %DNA tail (damage) of cells incubated for 4 hours with uncoated and SiO2-coated ZnO nanorods at ZnO concentrations of 5 (blue bars), 10 (green bars) and 20 μg mL−1 (red bars). H2O2 (100 μM) was used as a positive control, and pure SiO2 nanoparticles (SSA = 185 m2 g−1) and the cell culture medium (white bar, no particles) were used as negative controls. For all concentrations, pure SiO2 nanoparticles do not exhibit strong DNA damage (≤20%), further verifying their relatively inert nature. The SiO2 DNA damage is slightly higher than that of the pure medium, which could be attributed to the high SiO2 surface area concentration in solution (1–4 m2 L−1). The ZnO nanorods are not fully dissolved for the incubation period here, thus it is expected that both the uncoated and SiO2-coated nanorods are internalized by the cells. Such SiO2-coated nanoparticle uptake can be qualitatively detected by TEM analysis. As indicated in a study recently published by the authors, SiO2-coated ZnO nanorods were indeed internalized by cells.62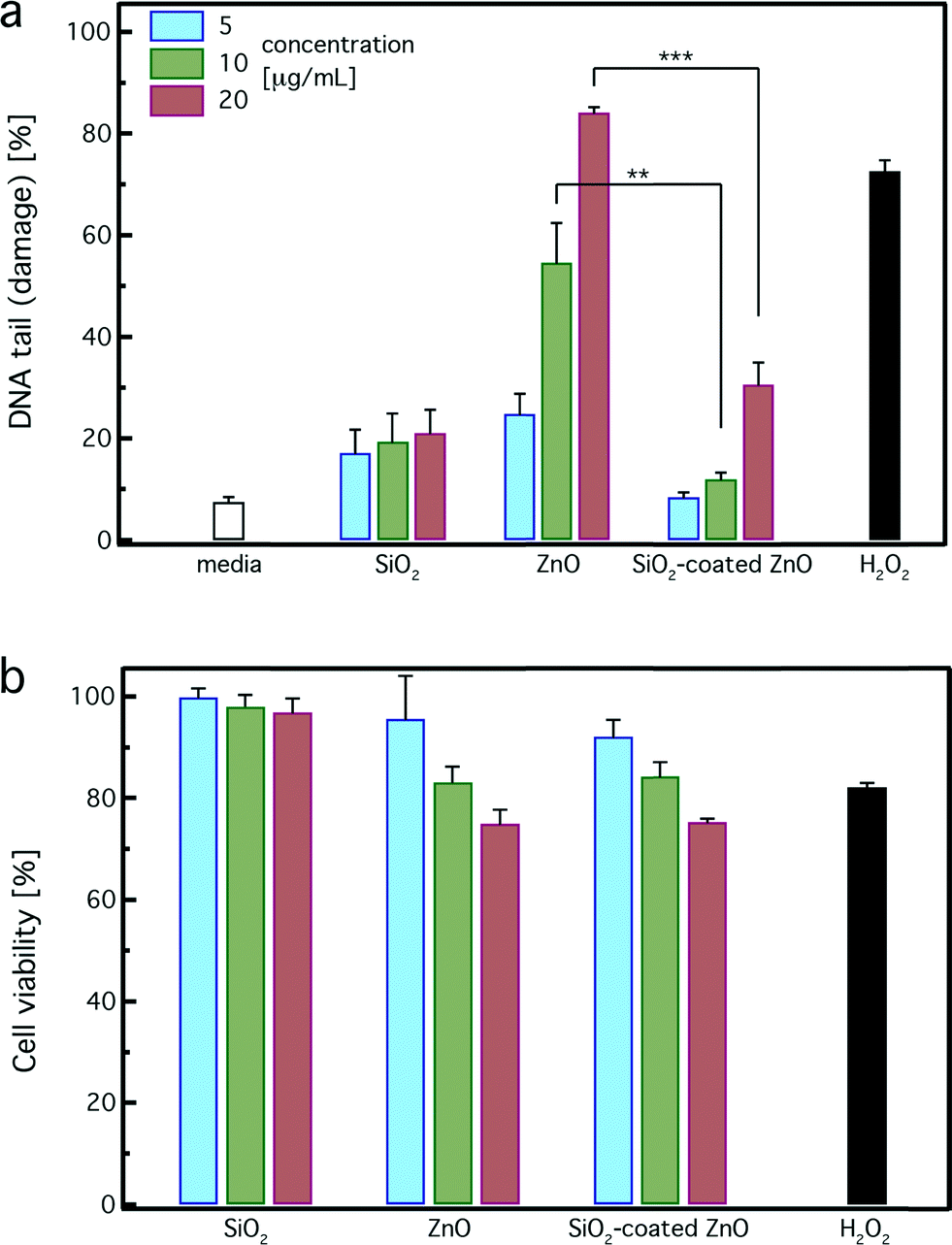 | ||
| Fig. 6 (a) Single stranded DNA damage in TK6 cells after zinc oxide exposure using the Nano-Cometchip technology. Cells were treated with 5, 10, and 20 μg mL−1 of SiO2-coated or uncoated ZnO nanorods for 4 hours. The positive control was 100 μM of hydrogen peroxide (H2O2) for 20 minutes. The results are expressed as mean + standard error of three independent experiments performed in triplicate. Thus, approximately 800 comets were analyzed for each treatment/dose. Values are significantly different in comparison to each treatment (SiO2-coated vs. uncoated); **p-value < 0.01, ***p-value < 0.001. (b) Cell viability was determined using the MTT assay after 4 hours of incubation. | ||
Pure uncoated ZnO nanorods induce a strong dose-dependent DNA damage and at the highest dose are comparable to the positive control (H2O2), which is in agreement with published studies.20,22,24 In contrast, SiO2-coated ZnO nanorods exhibit significantly less DNA damage in comparison to the uncoated ones. For concentrations up to 10 μg mL−1, the SiO2-coated ZnO nanorods do not exhibit any DNA damage at all, similar to the pure medium. At the highest concentration of 20 μg mL−1, the DNA damage of the SiO2-coated ZnO nanorods is slightly higher than that of the control and pure SiO2. However, the DNA damage induced by the uncoated ZnO nanorods at that concentration (20 μg mL−1) is much higher (~3 times) than that of the SiO2-coated ZnO nanorods. Therefore, the nanothin amorphous SiO2 coating on the ZnO surface clearly exhibits a protective effect and inhibits the strong DNA damage induced by the core ZnO nanorods.
Fig. 6b illustrates the cell viability of pure SiO2, uncoated, and SiO2-coated ZnO nanorods for 5 (blue bars), 10 (green bars) and 20 μg mL−1 (red bars) as determined by the MTT assay after 4 hours of incubation. None of the particles exhibited strong cytotoxicity. Reductions in viability were observed in TK6 cells exposed to both uncoated and SiO2-coated ZnO nanorods at the highest dose (20 μg mL−1). This decrease, however, was less than the half maximal inhibitory concentration or IC50, as only 25% reductions were found. This is expected here for all samples because of their limited exposure period (4 hours). For longer exposure period (24 h), a clear protective effect was detected for the SiO2-coated ZnO nanorods.17 This result highlights that even though there is no significant cytotoxicity observed, DNA damage (Fig. 6a) may be present at low, sublethal cellular doses. It is worth pointing out that in genotoxicity studies, it is important to evaluate cytotoxicity in correlation with DNA damage, as they are both interrelated. When assessing cytotoxicity using metabolic function as an endpoint, cellular viability of 50% and above are acceptable levels of cell death, which do not interfere with genotoxicity evaluations.63 Thus, the observed results using the MTT analysis suggest that moderate amounts of cytotoxicity were induced by exposure to the utilized concentrations for both coated and uncoated nanoparticles and no interference with genotoxicity assessment occurs here.
Conclusions
The above DNA damage results indicate that a hermetic SiO2 shell on ZnO nanorods minimizes their nanobiointeractions significantly reducing their genotoxicity, which is in agreement with the biologically inert nature of amorphous SiO2. This SiO2 shell protective effect might be attributed to the inhibition of the core ZnO dissolution, as well as the direct contact minimization of the cells with the ZnO surface. Amorphous SiO2 is quite stable in water and biological media,61 not allowing, therefore, the core ZnO nanorod dissolution. It should be noted that it is possible to reduce the ZnO nanoparticle dissolution and mitigate the toxicity of similar flame-made ZnO nanoparticles by iron-doping (1–10 at% Fe) during their synthesis.26,64 However, such doping changes the electronic configuration, bandgap and color of the ZnO nanoparticles, reducing their transparency in the visible spectrum. This effect makes the iron-doped ZnO nanoparticles unsuitable for their employment as transparent UV-filters in cosmetic products or polymer films. In contrast, by applying a nanothin amorphous SiO2 coating on ZnO nanoparticles, the DNA damage is minimized while all desired optoelectronic properties are maintained.In conclusion, we present here the rational design and engineering of safer functional ZnO nanorods that fulfill all performance requirements for their broad employment as UV-filters in cosmetics and personal care products as well as polymers. By hermetically encapsulating these ZnO nanorods during their synthesis with a nanothin, amorphous SiO2 shell, the core ZnO nanobiointeractions are minimized. Furthermore, the SiO2 coating facilitates the nanoparticle dispersion in solution, improving their transparency in the visible spectrum. The presence of SiO2 does not influence the optoelectronic properties of the core ZnO nanorods so they retain their desired high transparency in the visible spectrum and UV absorption rendering them suitable for UV blocking applications. Most importantly, the SiO2-coating on the ZnO nanorods significantly reduces the strong DNA damage that is otherwise observed for the pure uncoated ZnO nanorods. Such safer-by-design core–shell ZnO nanoparticles can be broadly employed in commodities without any performance loss and with a reduced hazard to the environment and human health.
Acknowledgements
The authors thank Samuel Gass for the help with the nanoparticle synthesis, Profs. Sotiris E. Pratsinis (ETH Zurich) and Evelyn Hu (Harvard University) for discussions, and B. Engelward (MIT) for providing the TK6 cells and the Cometchip assay. This research was supported by NSF grant (ID#1235806) and NIH grant (P30ES000002). This work was performed in part at the Harvard Center for Nanoscale Systems (CNS), a member of the National Nanotechnology Infrastructure Network (NNIN), which is supported by the National Science Foundation under NSF award number ECS-0335765. Georgios A. Sotiriou gratefully acknowledges the Swiss National Science Foundation for the Advanced Researcher fellowship (grant no. 145392). Christa Watson is funded by the NIH–NHLBI Ruth L. Kirschstein T32 training grant (NIH HL007118). Kimberly M. Murdaugh was supported with an NSF Graduate Research Fellowship grant (DGE-1144152).References
- B. Sun and H. Sirringhaus, Nano Lett., 2005, 5, 2408–2413 CrossRef CAS PubMed.
- R. Konenkamp, R. C. Word and M. Godinez, Nano Lett., 2005, 5, 2005–2008 CrossRef CAS PubMed.
- J. X. Wang, X. W. Sun, Y. Yang, H. Huang, Y. C. Lee, O. K. Tan and L. Vayssieres, Nanotechnology, 2006, 17, 4995–4998 CrossRef CAS.
- M. J. Height, S. E. Pratsinis, O. kasuwandumrong and P. Praserthdam, Appl. Catal., B, 2006, 63, 305–312 CrossRef CAS PubMed.
- A. Becheri, M. Durr, P. Lo Nostro and P. Baglioni, J. Nanopart. Res., 2008, 10, 679–689 CrossRef CAS.
- J. A. Anta, E. Guillen and R. Tena-Zaera, J. Phys. Chem. C, 2012, 116, 11413–11425 CAS.
- F. M. Hilty, M. Arnold, M. Hilbe, A. Teleki, J. T. N. Knijnenburg, F. Ehrensperger, R. F. Hurrell, S. E. Pratsinis, W. Langhans and M. B. Zimmermann, Nat. Nanotechnol., 2010, 5, 374–380 CrossRef CAS PubMed.
- X. Tang, E. S. G. Choo, L. Li, J. Ding and J. Xue, Langmuir, 2009, 25, 5271–5275 CrossRef CAS PubMed.
- D. M. King, X. Liang, C. S. Carney, L. F. Hakim, P. Li and A. W. Weimer, Adv. Funct. Mater., 2008, 18, 607–615 CrossRef CAS.
- A. Janotti and C. G. Van de Walle, Rep. Prog. Phys., 2009, 72, 126501 CrossRef.
- G. J. Nohynek, E. K. Dufour and M. S. Roberts, Skin Pharmacol. Physiol., 2008, 21, 136–149 CrossRef CAS PubMed.
- G. J. Nohynek, J. Lademann, C. Ribaud and M. S. Roberts, Crit. Rev. Toxicol., 2007, 37, 251–277 CrossRef CAS PubMed.
- E. Galoppini, J. Rochford, H. Chen, G. Saraf, Y. Lu, A. Hagfeldt and G. Boschloo, J. Phys. Chem. B, 2006, 110, 16159–16161 CrossRef CAS PubMed.
- Q. Zhang, T. R. Chou, B. Russo, S. A. Jenekhe and G. Cao, Angew. Chem., Int. Ed., 2008, 47, 2402–2406 CrossRef CAS PubMed.
- L. Madler, W. J. Stark and S. E. Pratsinis, J. Appl. Phys., 2002, 92, 6537–6540 CrossRef CAS PubMed.
- T. Tani, L. Madler and S. E. Pratsinis, J. Nanopart. Res., 2002, 4, 337–343 CrossRef CAS.
- S. Gass, J. M. Cohen, G. Pyrgiotakis, G. A. Sotiriou, S. E. Pratsinis and P. Demokritou, ACS Sustainable Chem. Eng., 2013, 1, 843–857 CAS.
- D. B. Warheit, C. M. Sayes and K. L. Reed, Environ. Sci. Technol., 2009, 43, 7939–7945 CrossRef CAS PubMed.
- T. Xia, M. Kovochich, M. Liong, L. Maedler, B. Gilbert, H. Shi, J. I. Yeh, J. I. Zink and A. E. Nel, ACS Nano, 2008, 2, 2121–2134 CrossRef CAS PubMed.
- V. Valdiglesias, C. Costa, G. Kilic, S. Costa, E. Pasaro, B. Laffon and J. P. Teixeira, Environ. Int., 2013, 55, 92–100 CrossRef CAS PubMed.
- H. Shu-Feng, D. Bello, D. F. Schmidt, A. K. Pal, A. Stella, J. A. Isaacs and E. J. Rogers, Small, 2013, 9, 1853–65 CrossRef PubMed.
- S. Alarifi, D. Ali, S. Alkahtani, A. Verma, M. Ahamed, M. Ahmed and H. A. Alhadlaq, Int. J. Nanomed., 2013, 8, 983–993 CrossRef PubMed.
- W. S. Beckett, D. F. Chalupa, A. Pauly-Brown, D. M. Speers, J. C. Stewart, M. W. Frampton, M. J. Utell, L. S. Huang, C. Cox, W. Zareba and G. Oberdorster, Am. J. Respir. Crit. Care Med., 2005, 171, 1129–1135 CrossRef PubMed.
- C. Watson, J. Ge, J. Cohen, G. Pyrgiotakis, B. Engelward and P. Demokritou, ACS Nano, 2014 DOI:10.1021/nn404871p, in press.
- V. Sharma, R. K. Shukla, N. Saxena, D. Parmar, M. Das and A. Dhawan, Toxicol. Lett., 2009, 185, 211–218 CrossRef CAS PubMed.
- T. Xia, Y. Zhao, T. Sager, S. George, S. Pokhrel, N. Li, D. Schoenfeld, H. Meng, S. Lin, X. Wang, M. Wang, Z. Ji, J. I. Zink, L. Madler, V. Castranova, S. Lin and A. E. Nel, ACS Nano, 2011, 5, 1223–1235 CrossRef CAS PubMed.
- A. Teleki, M. C. Heine, F. Krumeich, M. K. Akhtar and S. E. Pratsinis, Langmuir, 2008, 24, 12553–12558 CrossRef CAS PubMed.
- Cabot Corp., “CAB-O-SIL® Fumed silica in cosmetic and personal care products” Report; 2001 Search PubMed.
- T. J. Brunner, P. Wick, P. Manser, P. Spohn, R. N. Grass, L. K. Limbach, A. Bruinink and W. J. Stark, Environ. Sci. Technol., 2006, 40, 4374–4381 CrossRef CAS.
- D. Napierska, L. C. J. Thomassen, V. Rabolli, D. Lison, L. Gonzalez, M. Kirsch-Volders, J. A. Martens and P. H. Hoet, Small, 2009, 5, 846–853 CrossRef CAS PubMed.
- H. Zhang, D. R. Dunphy, X. Jiang, H. Meng, B. Sun, D. Tarn, M. Xue, X. Wang, S. Lin, Z. Ji, R. Li, F. L. Garcia, J. Yang, M. L. Kirk, T. Xia, J. I. Zink, A. Nel and C. J. Brinker, J. Am. Chem. Soc., 2012, 134, 15790–15804 CrossRef CAS PubMed.
- D. B. Warheit, T. A. McHugh and M. A. Hartsky, Scand. J. Work, Environ. Health, 1995, 21, 19–21 CAS.
- P. Demokritou, S. Gass, G. Pyrgiotakis, J. M. Cohen, W. Goldsmith, W. McKinney, D. Frazer, J. Ma, D. Schwegler-Berry, J. Brain and V. Castranova, Nanotoxicology, 2013, 7, 1338–1350 CrossRef CAS PubMed.
- G. A. Sotiriou, T. Sannomiya, A. Teleki, F. Krumeich, J. Voeroes and S. E. Pratsinis, Adv. Funct. Mater., 2010, 20, 4250–4257 CrossRef CAS PubMed.
- G. A. Sotiriou, D. Franco, D. Poulikakos and A. Ferrari, ACS Nano, 2012, 6, 3888–3897 CrossRef CAS PubMed.
- P. Demokritou, R. Buechel, R. M. Molina, G. M. Deloid, J. D. Brain and S. E. Pratsinis, Inhalation Toxicol., 2010, 22, 107–116 CrossRef CAS PubMed.
- D. M. Weingeist, J. Ge, D. K. Wood, J. T. Mutamba, Q. Huang, E. A. Rowland, M. B. Yaffe, S. Floyd and B. P. Engelward, Cell Cycle, 2013, 12, 907–915 CrossRef CAS PubMed.
- J. Cohen, G. DeLoid, G. Pyrgiotakis and P. Demokritou, Nanotoxicology, 2013, 7, 417–431 CrossRef CAS PubMed.
- T. H. Darrah, J. J. Prutsman-Pfeiffer, R. J. Poreda, M. E. Campbell, P. V. Hauschka and R. E. Hannigan, Metallomics, 2009, 1, 479–488 RSC.
- M. Sprauten, T. H. Darrah, D. R. Peterson, M. E. Campbell, R. E. Hannigan, M. Cvancarova, C. Beard, H. S. Haugnes, S. D. Fossa, J. Oldenburg and L. B. Travis, J. Clin. Oncol., 2012, 30, 300–307 CrossRef CAS PubMed.
- A. Kimura, A. Miyata and M. Honma, Mutagenesis, 2013, 28, 583–590 CrossRef CAS PubMed.
- R. Walmsley and M. Tate, The GADD45a-GFP GreenScreen HC Assay, in Genetic Toxicology, ed. J. M. Parry and E. M. Parry, Springer, New York, 2012, vol. 817, pp. 231–250 Search PubMed.
- D. K. Wood, D. M. Weingeist, S. N. Bhatia and B. P. Engelward, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10008–10013 CrossRef CAS PubMed.
- G. D. Ulrich, Chem. Eng. News, 1984, 62, 22–29 CAS.
- K. Hembram, D. Sivaprakasam, T. N. Rao and K. Wegner, J. Nanopart. Res., 2013, 15, 1461 CrossRef.
- M. J. Height, L. Madler, S. E. Pratsinis and F. Krumeich, Chem. Mater., 2006, 18, 572–578 CrossRef CAS.
- A. Teleki, M. K. Akhtar and S. E. Pratsinis, J. Mater. Chem., 2008, 18, 3547–3555 RSC.
- T. Tani, L. Madler and S. E. Pratsinis, Part. Part. Syst. Charact., 2002, 19, 354–358 CrossRef CAS.
- L. Madler, H. K. Kammler, R. Mueller and S. E. Pratsinis, J. Aerosol Sci., 2002, 33, 369–389 CrossRef CAS.
- G. A. Sotiriou, M. Schneider and S. E. Pratsinis, J. Phys. Chem. C, 2012, 116, 4493–4499 CAS.
- R. Jung, J. C. Lee, G. T. Orosz, A. Sulyok, G. Zsolt and M. Menyhard, Surf. Sci., 2003, 543, 153–161 CrossRef CAS PubMed.
- A. Teleki, B. Buesser, M. C. Heine, F. Krumeich, M. K. Akhtar and S. E. Pratsinis, Ind. Eng. Chem. Res., 2008, 48, 85–92 CrossRef.
- S. Liufu, H. Xiao and Y. P. Li, Powder Technol., 2004, 145, 20–24 CrossRef CAS PubMed.
- G. A. Sotiriou, A. M. Hirt, P.-Y. Lozach, A. Teleki, F. Krumeich and S. E. Pratsinis, Chem. Mater., 2011, 23, 1985–1992 CrossRef CAS PubMed.
- M. J. Height, S. E. Pratsinis, O. Mekasuwandumrong and P. Praserthdam, Appl. Catal., 2006, 63, 305–312 Search PubMed.
- S. E. Pratsinis, AIChE J., 2010, 56, 3028–3035 CrossRef CAS.
- R. Mueller, H. K. Kammler, K. Wegner and S. E. Pratsinis, Langmuir, 2003, 19, 160–165 CrossRef CAS.
- Y. Deligiannakis, G. A. Sotiriou and S. E. Pratsinis, ACS Appl. Mater. Interfaces, 2012, 4, 6609–6617 CAS.
- A. Teleki, M. Suter, P. R. Kidambi, O. Ergeneman, F. Krumeich, B. J. Nelson and S. E. Pratsinis, Chem. Mater., 2009, 21, 2094–2100 CrossRef CAS.
- Y. Han, J. Jiang, S. S. Lee and J. Y. Ying, Langmuir, 2008, 24, 5842–5848 CrossRef CAS PubMed.
- H. Zhang, Z. Ji, T. Xia, H. Meng, C. Low-Kam, R. Liu, S. Pokhrel, S. Lin, X. Wang, Y.-P. Liao, M. Wang, L. Li, R. Rallo, R. Damoiseaux, D. Telesca, L. Maedler, Y. Cohen, J. I. Zink and A. E. Nel, ACS Nano, 2012, 6, 4349–4368 CrossRef CAS PubMed.
- J. M. Cohen, R. Derk, L. Rojanasakul, J. Godleski, L. Kobzik, J. Brain and P. Demokritou, Nanotoxicology, 2014 DOI:10.3109/17435390.2013.879612 , in press.
- E. Kiskinis, W. Suter and A. Hartmann, Mutagenesis, 2002, 17, 37–43 CrossRef CAS PubMed.
- S. George, S. Pokhrel, T. Xia, B. Gilbert, Z. Ji, M. Schowalter, A. Rosenauer, R. Damoiseaux, K. A. Bradley, L. Maedler and A. E. Nel, ACS Nano, 2010, 4, 15–29 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |