
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Computed ligand effects on the oxidative addition of phenyl halides to phosphine supported palladium(0) catalysts†
Claire L.
McMullin‡
,
Natalie
Fey
* and
Jeremy N.
Harvey
*
School of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK. E-mail: Natalie.Fey@bristol.ac.uk; Jeremy.Harvey@bristol.ac.uk; Fax: +44 (0)117 9251295
First published on 5th August 2014
Abstract
The manifold of reaction pathways for the oxidative addition of phenyl bromide and phenyl chloride substrates to phosphine-modified palladium(0) complexes has been investigated with dispersion-corrected density functional theory (B3LYP-D2) for a range of synthetically relevant ligands, permitting the evaluation of ligand, substrate and method effects on calculated predictions. Bulky and electron-rich ligands PtBu3 and SPhos can access low-coordinate complexes more easily, facilitating formation of the catalytically active species throughout the cycle. While the bisphosphine oxidative addition step is reasonably facile for the smaller PCy3 and PPh3 ligands, the dissociation of these ligands to generate reactive palladium complexes becomes more important and the catalyst is more likely to become trapped in unreactive intermediates. This study demonstrates the feasibility of exploring the catalytic manifold for synthetically relevant ligands with computational chemistry, but also highlights the remaining challenges.
Introduction
Palladium-catalyzed cross-coupling reactions have been developed to accommodate a variety of substrates,1 permitting the formation of new C–C or C–Y bonds, where Y is a heteroatom. The importance of this group of reactions was internationally recognized in 2010, when the Nobel prize for Chemistry was jointly awarded to Heck,2 Negishi3 and Suzuki4 for their research on C–C bond formation. Other researchers who have also contributed to this field include Kumada,5 Stille,6 and Hiyama,7 as well as Buchwald8 and Hartwig,9 who have made significant contributions to a range of carbon–heteroatom bond forming reactions. Computational studies of C–C cross-coupling reactions have recently been reviewed,10 as have studies combining experimental and computational data to gain insight into palladium catalysis.11A key step of these reactions, in common with some palladium-catalyzed C–H activation reactions,12 is the oxidative addition of an aryl or alkyl halide to the palladium centre (Scheme 1). The activation of the Pd(0) catalyst, followed by insertion of the metal atom into the Cipso and halide (X) (or triflate, SO3CF3) bond, has been shown to be rate limiting in certain conditions.13
 | ||
| Scheme 1 Oxidative addition step for an aryl halide (ArX) with a palladium(0) catalyst. | ||
Broadly, catalyst design to support oxidative addition can be formulated as requiring an electron rich palladium centre, which supports the increase in oxidation state from Pd(0) to Pd(II).14 The catalyst also needs to tolerate an increase in coordination number during the oxidative addition by starting out as low-coordinate or through the facile loss of one/several ligands.15 Furthermore, the system must remain active through multiple cycles, either by avoiding resting states and inactive reservoir species,16 or by re-entering the catalytic cycle relatively easily.
Ligands are often key to the fine-tuning of the activity and selectivity of organometallic catalysts. In general, electron-donating spectator ligands are favoured for oxidative additions,17 and steric bulk can be used to support lower coordination numbers for the initial [PdLn] species; the ligands shown in Scheme 2 fulfill these criteria and patented systems for cross-coupling reactions have been reviewed.17
 | ||
| Scheme 2 Popular, experimentally used monodentate phosphine spectator ligands (L) in palladium catalyzed oxidative addition, including PtBu3,18 P(o-tol)319 and QPhos,20 as well as SPhos and XPhos ligands from the biaryl-alkylphosphine family developed by Buchwald and co-workers.21 | ||
With a view to exploring and enhancing the role computational chemistry can play in the process of ligand-driven catalyst discovery and design we have been pursuing a computational methodology for the analysis and prediction of ligand effects in homogenous organometallic catalysis, combining DFT-calculated ligand property parameters22 with the analysis and prediction of both experimental23 and calculated24 data capturing catalyst performance, such as yield, rates and barriers to reaction. Accurate computational studies of catalytic cycles, especially where a manifold of competing reaction pathways exists, are an important component of this methodology and we have evaluated the impact of computational method effects to establish and validate a theoretical approach suitable for the study of the key oxidative addition step for synthetically relevant organometallic catalysts.25 In this earlier work, we deliberately focused on the PtBu3 ligand because its large steric bulk restricts the number of accessible reaction pathways in the mechanistic manifold and limits the number of complexes and conformers which need to be considered.
For other, slightly smaller ligands such as PPh3 and PCy3, a number of additional steps/pathways may become energetically accessible and hence need to be taken into account (Scheme 3), making the study of these ligands computationally more demanding. In order to permit energetic comparison of competing pathways and comparison with experiment, the computational approach used must be sufficiently accurate, which requires it to take account of solvation,26 dispersion27 and vibrational corrections.28
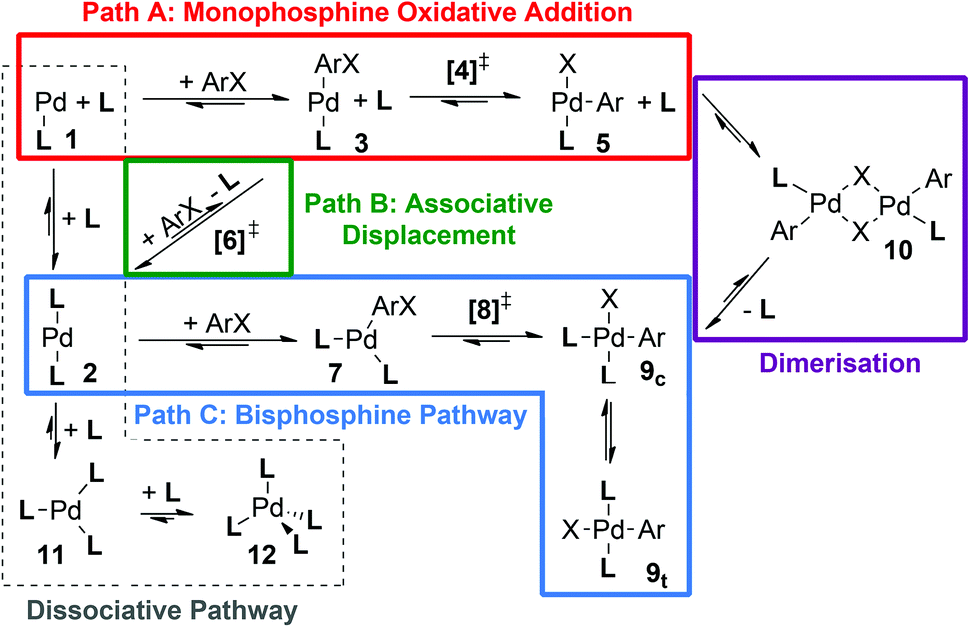 | ||
| Scheme 3 Reaction pathways and intermediates for the oxidative addition of ArX by a palladium catalyst [PdLn]. | ||
The exact energetic balance of such competing pathways will be influenced most profoundly by both the ligand and the aryl/alkyl halide used. Here we report calculated results for the oxidative addition of two aryl halides (PhBr and PhCl) to a palladium centre supported by a number of synthetically useful ligands (PCy3, PPh3, PtBu3 and the SPhos ligand as a representative of the family of biaryl ligands developed in Buchwald's group21). Results for the PCy3 ligand and a phenyl bromide substrate will be used to map out competing dissociative and associative pathways; this ligand has been used experimentally in a range of cross-coupling reactions with less activated aryl chloride and aryl triflate substrates29 and has been the subject of several experimental kinetic studies,13a,30 providing experimental data to validate calculated results. Ligand effects on the mechanism have also been considered for additional, synthetically relevant ligands (PPh3, SPhos, PtBu3) and the interplay between ligand and substrate effects has been evaluated by comparing data for PhBr and PhCl substrates in the presence of these ligands. These results have been used to illustrate what can be achieved with the methodology used, but also to discuss where computational improvements might be made in the future.
Mechanistic manifold
As indicated in Scheme 3, a number of different pathways and species can be envisaged as part of the mechanistic manifold for palladium-catalyzed oxidative addition. Depending on ligand size and donor strength, as well as the catalyst precursor used, the initial coordination number of the palladium(0) species [PdLn] can take values of n = 1–4 (1, 2, 11 or 12), although the monoligated complex 1 has not been detected experimentally for any ligand in solution;31 solvent coordination might occur in this case.13b,31a,32For large ligands it has often been assumed that the active species which undergoes oxidative addition is the monophosphine complex [PdL], 1, formed after ligand dissociation from the bisligated [PdL2] species (2),30a,33 potentially stabilized by solvent coordination.13b,31a,32 However, the trisligated and tetrakisligated [PdLn] species, 11 and 12 respectively, have been observed experimentally for ligands PCy3 and PPh3,30a,b,34 and this has recently been confirmed computationally for PPh3,35 with kinetic studies showing that in solution [PdL4] (12) easily loses one or two ligand molecules, to form a rapidly equilibrating mixture of [PdL3] (11) and [PdL2] (2).30a,36 [PdL2] (2) and [PdL] (1) have also recently been observed in the gas phase when L = [PPh2(m-C6H4SO3)]−.31b
One dissociative pathway and two associative pathways (Scheme 3) could lead to the oxidative addition of an aryl halide to the catalyst. In the dissociative pathway (Scheme 3, shown in pale grey outline), ligand loss would occur before coordination of the aryl halide substrate to form the [PdL(ArX)] adduct 3, and then undergo monoligated oxidative addition via [4]‡. The dissociative pathway links with monophosphine oxidative addition (Path A, red outline) via the [PdL] complex 1.
In the first associative bisphosphine pathway (Path B), associative displacement of one ligand by the aryl halide substrate via [6]‡ can generate the [PdL(ArX)] adduct 3. At this stage the pathway merges with Path A and oxidative addition occurs to the monoligated metal center via TS [4]‡ (associative displacement pathway (Path B, connection to Path A shown in green in Scheme 3)). Alternatively, the metal remains bisligated and the oxidative addition occurs directly to [PdL2] (2) via [8]‡, possibly via a (transient) adduct of the form [PdL2(ArX)] (7); this will be denoted as the bisphosphine pathway (Path C, Scheme 3, shown in blue). A further alternative, where solvent is weakly coordinated by [PdL] 1,13b,31a,32 has also been considered for L = PtBu3 (section ESI2†).
Depending on the pathway followed, a range of different complexes can result from the oxidative addition. Monoligation during the oxidative addition transition state, accessed by pathways A and B, will initially produce three possible isomers of a T-shaped complex, [Pd(L)(Ar)(X)] 5. The isomer with the phosphine ligand trans to the halide has been observed crystallographically for PtBu3, whose steric bulk and ability to form γ-agostic interactions with the metal can protect the adjacent vacant coordination site on the metal.15,37 For smaller ligands, the monoligated product 5 can undergo ligand addition to form square-planar cis or trans isomers of [PdL2(Ar)(X)], c-9 or t-9; both isomers have been observed crystallographically (see, for example, ref. 38 (PPh3)). The cis isomer c-9 can also be reached directly by oxidative addition to a bisligated palladium center (Path C), which may be followed by isomerisation to give t-9.
Alternatively, 5 can coordinate a second T-shaped complex to form a halide bridged dinuclear complex [(μ-X)2Pd2(L)2(Ph)2] 10, which again adopts a square-planar geometry around each metal center for either an anti or a syn dimer (a-10 and s-10 respectively), observed crystallographically for PtBu3 and P(1-Ad)tBu2,13a as well as P(o-tol)319a,b and CataCXiumA.39 Based on a computational study by Lledós, Espinet and co-workers, albeit without consideration of dispersion corrections, the observed higher activity of bulky ligands may be related to avoiding the formation of side products and reservoir species such as 9 and 10.40
Method effects on prediction
Before describing our results on the different mechanisms and ligands, we first consider how the results depend on the computational protocol, for the case of speciation in solution between the different [Pd(PCy3)n] complexes (n = 1–3). As in our previous work,25b the geometry of all species has been optimized using the B3LYP functional in vacuum, with a medium basis set (denoted as BS1), followed by single-point energy calculations of the dispersion correction§ and with larger basis sets (BS2) (see computational details below and the ESI†). The effect of geometry optimisation with dispersion-corrected functionals was found to be generally modest, but might make a more important contribution for crowded complexes involved in Path C, see discussion below and in section ESI3.† We have then computed vibrational frequencies and used ideal gas statistical mechanics to compute relative free energies in vacuum. These have been corrected by solvation free energies computed using continuum methods.41We note that some authors have argued that this approach exaggerates entropic effects, which are smaller in solution than in the gas phase.42 However, such effects should in principle be treated adequately by the continuum model. In our experience, discrepancies with experiment observed when using this approach, instead of being due to such entropic factors, can be the result of inaccurate electronic structure theory. For relative energies, this can arise, for example, from the neglect of dispersion interactions in many density functional theory methods, underestimating the stabilization of higher coordination numbers. We note also that for complex systems such as those considered here, it is essential to identify correctly the lowest-energy isomer and conformer of each of the minima and transition states involved.43 Finally, while anionic metal complexes have been observed to play a role in Heck and cross-coupling reactions performed in polar solvents and with some palladium precursors (Pd(OAc)2, Pd(dba)2),44 here we focused on reactions in toluene, where these species are less likely to play a dominant role.30b The same applies to mechanisms for oxidative addition of alkyl halides, RX, in which the metal centre attacks the carbon atom to displace X−, which only subsequently adds to the metal. While there is evidence for such mechanisms with RX and in more polar conditions, they should play little role here.
Table 1a and Fig. 1 show calculated relative free energies of different coordination numbers for the three [Pd(PCy3)n] species (n = 1–3), at different levels of theory.¶ Experimentally, solutions are found to contain a mixture of the n = 2 and n = 3 species. Measurement of the equilibrium constant at temperatures between −68 and −85 °C yields an experimental enthalpy for binding of L to PdL2 in solution of about −5 kcal mol−1.30a This is not readily comparable to computation as it includes solvent enthalpic effects, which are not easily computed using continuum solvent methods. The standard Gibbs energy for binding is negative at lower temperatures, but increases at higher temperatures. Extrapolation of these experimental data suggests a ΔG° of binding of −0.7 kcal mol−1 at 25 °C, and of +0.3 kcal mol−1 at 100 °C.
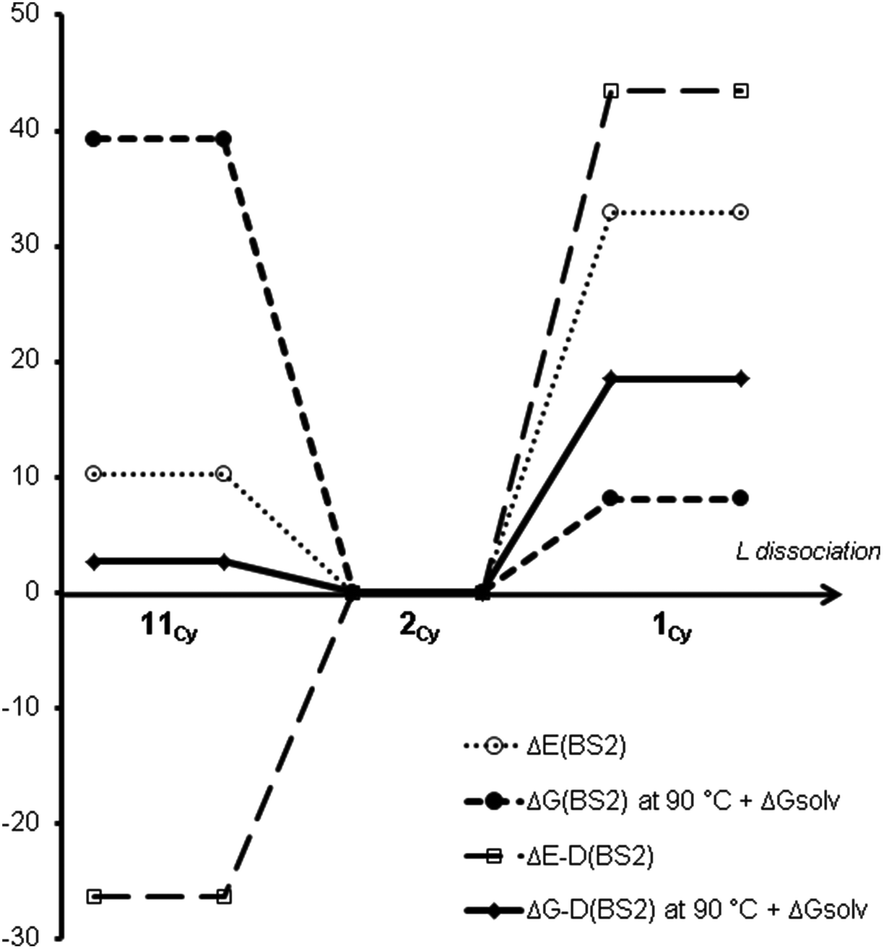 | ||
| Fig. 1 Method effects on the coordination number of [PdLn] for L = PCy3 at 90 °C (all energies are given in kcal mol−1 and are calculated relative to [PdL2], see also Table 1a). | ||
| (a) Method effects at 90 °C | |||||||
|---|---|---|---|---|---|---|---|
| n | ΔE (BS2) | ΔG° (BS2) | ΔG° (BS2) + ΔGsolv | ΔE-D (BS2) | ΔG°-D (BS2) | ΔG°-D (BS2) + ΔGsolv | |
| 1Cy | 1 | 32.9 | 20.1 | 8.1 | 43.4 | 30.6 | 18.5 |
| 2Cy | 2 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| 11Cy | 3 | 10.3 | 32.3 | 39.3 | −26.3 | −4.2 | 2.7 |
| (b) Temperature effects on favoured coordination number (note that the temperature dependence of the solvent contribution has been neglected here). See ESI for a more detailed breakdown of energy contributions‖ | ||||
|---|---|---|---|---|
| n | ΔG°-D (90 °C) + ΔGsolv | ΔG°-D (10 °C) + ΔGsolv | ΔG°-D (−60 °C) + ΔGsolv | |
| 1Cy | 1 | 18.5 | 20.9 | 23.5 |
| 2Cy | 2 | 0.0 | 0.0 | 0.0 |
| 11Cy | 3 | 2.7 | −1.5 | −5.8 |
It can be noted that at the B3LYP level of theory, PdL3 lies higher in energy than PdL2 + L, and much higher in Gibbs energy as ligand dissociation is entropically favourable. This clearly disagrees with the experimental values. This has also been observed for L = PPh3 by Ahlquist and Norrby.35
On the other hand, upon including an approximate treatment of dispersion (ΔE-D(BS2)),§ the [PdL3] species is found to lie much lower in potential energy compared to [PdL2] and L in vacuum. The computed Gibbs energy of binding is now close to zero, and, in excellent agreement with experiment, is calculated to be small and negative near room temperature. The agreement with experiment is less good at higher and lower temperatures, perhaps because the temperature dependence of the solvation Gibbs energy has been neglected here. The continuum model is parameterized to give accurate results at room temperature and may be less reliable at higher and lower temperatures. Nevertheless, the present tests suggest that our chosen “best” computational protocol yields results within a few kcal mol−1 of experiment. In the rest of the paper, only these “best” computed standard free energies will be discussed, with all results corrected for a reaction temperature of 90 °C. A breakdown of energy contributions for each ligand may be found Tables S2–5.†
Ligand effects
Table 2 shows results for ligands discussed here; PCy3, PPh3, and SPhos, with previously published25b and additional data for PtBu3 also included for comparison. Both PCy3 and SPhos ligands can adopt a number of different conformers and these have been explored by mining of the Cambridge Structural Database (CSD)45 and DFT calculations in different coordination environments, see section ESI3† for details. All energies shown are given relative to [PdL2] + PhX and corrected for a reaction temperature of 90 °C, chosen to allow direct comparison with previous work.25b| ΔG° (90 °C) + ΔGsolv | PCy3 | PPh3 | SPhos | PtBu3 | |
|---|---|---|---|---|---|
| a Optimisation unsuccessful. b Not attempted for this ligand. | |||||
| 1 | [PdL] + PhBr | 18.5 | 18.4 | 2.4 | 25.7 |
| 2 | [PdL2] + PhBr | 0.0 | 0.0 | 0.0 | 0.0 |
| 3 | [PdL(PhBr)] | 14.0 | 14.4 | 11.0 | 18.6 |
| [4]‡ | [PdL(Ph⋯Br)]‡ | 22.7 | 24.7 | 19.6 | 26.9 |
| 5 | [PdL(Ph)(Br)] | −9.3 | −6.4 | −19.5 | −1.2 |
| [6]‡ | [PdL2(PhBr)]‡ | 28.6 | 24.9 | 29.1 | 29.2 |
| 7 | [PdL2(PhBr)] | ||||
| [8]‡ | [PdL2(Ph⋯Br)]‡ | 20.6 | 21.8 | ||
| c-9 | [PdL2(Ph)(Br)] | −13.7 | |||
| t-9 | [PdL2(Ph)(Br)] | −27.3 | −19.8 | 10.0 | |
| a-10 | ½[(μ-Br)2Pd2L2(Ph)2] | −14.1 | −13.2 | 1.3 | |
| s-10 | ½[(μ-Br)2Pd2L2(Ph)2] | −19.9 | 1.0 | ||
| 11 | [PdL3] + PhBr | 2.7 | −1.9 | ||
| 12 | [PdL4] + PhBr | 23.5 | |||
Dissociative pathway (A). These results suggest that ligand dissociation to form the low-coordinate [PdL] complex 1Cy is energetically accessible (Fig. 2, labeled in grey, Tables 2 and S2†). This might exist as a transient species or as a solvated intermediate with a solvent molecule coordinated to the palladium centre.13b,30c,31a Note that the inclusion of continuum-based solvation free energies in our “best” computed energies should implicitly describe such solvent binding, at least roughly. We have argued previously25b that the continuum solvent method may slightly overestimate the solvent stabilization of this highly unsaturated metal centre. In contrast, several computational studies of other ligands have assumed that the active catalyst is the monophosphine complex 1, formed after ligand dissociation from bisligated [PdL2], 2.10a,33a,b,d,46
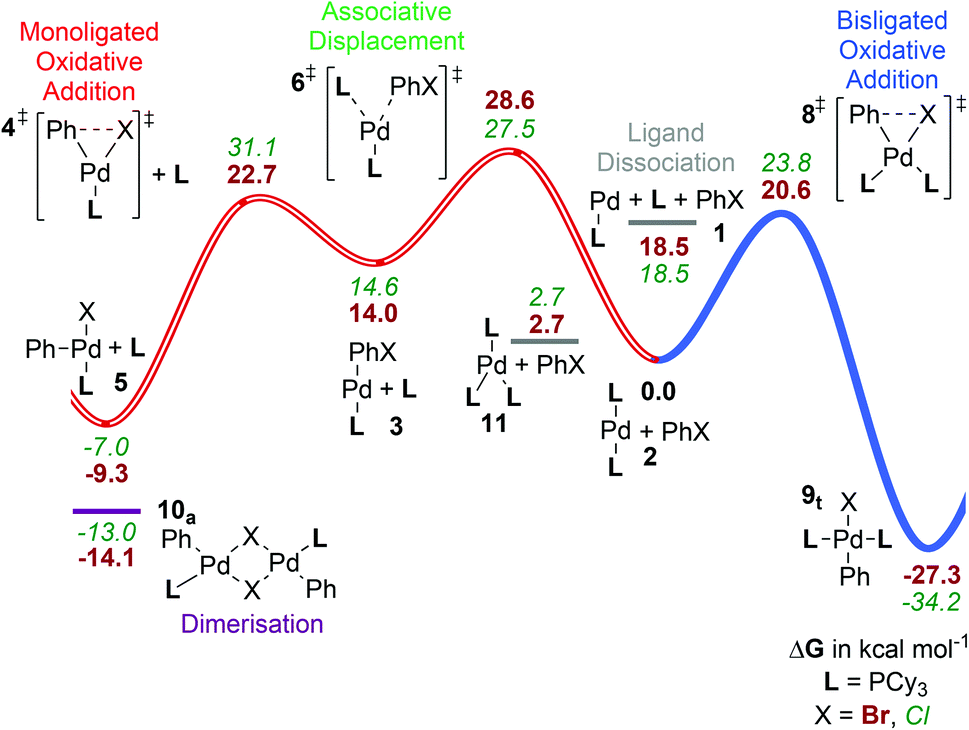 | ||
| Fig. 2 Gibbs energy surface (ΔG° (90 °C) + ΔGsolv) for the oxidative addition of PhBr to a [Pd(PCy3)n] catalyst. All energies are in units of kcal mol−1 and relative to [PdL2]. The blue pathway shown corresponds to the bisligated oxidative addition (C, see Scheme 3), while the red pathway (hollow line) shows the associative displacement route (B), which merges with the dissociative pathway (A) at complex 3 (Scheme 3). | ||
In addition to the dissociated complex 1Cy shown, there will be a point of maximum Gibbs energy along the reaction path for addition of a ligand, or of the aryl halide, to [PdL]. There is no potential energy barrier to such additions, with the Gibbs energy barrier being due to loss of entropy upon approach. As in our previous work,25b we estimate the Gibbs energy barrier to addition to be ca. 4.5 kcal mol−1 at 90 °C, based on the rate constants for reactions known to be diffusion-controlled. With this estimate, the transition state (TS) for oxidative addition to the monoligated metal centre [4Cy]‡ is predicted to be very slightly lower in Gibbs energy (at 22.7 kcal mol−1) than the TSs for ligand loss from PdL2 or for aryl halide addition to PdL (both at ca. 23.0 kcal mol−1). However, these values are very close, preventing calculations from distinguishing reliably between these options.
Associative displacement pathway (B). The highest Gibbs energy transition state on this pathway (B, Fig. 2) is for associative displacement of one PCy3 ligand by the PhBr substrate ([6Cy]‡, 28.6 kcal mol−1), which has the highest barrier of all the pathways considered here, presumably due to considerable ligand steric hindrance. This is one of the two mechanisms that are consistent with the experimentally observed order of reaction in Hartwig's kinetic study.13a It is also the mechanism predicted to be favoured with L = PtBu3.25b However, for PCy3 it can be discounted based on the present calculations, since the barrier is so much higher than for Path C; this is also consistent with experiment as discussed below.
Bisphosphine pathway (C). The bisligated oxidative addition, [8Cy]‡ (shown as the blue pathway in Fig. 2) has the lowest calculated barrier (20.6 kcal mol−1) of the competing transition states, [4Cy]‡, [6Cy]‡ and [8Cy]‡ studied here. This is in agreement with Hartwig's interpretation of the experimental kinetics,13a whereby the irreversible step during the oxidative addition involves a bisligated complex. It is to be noted, though, that this observation is not enough to exclude the associative displacement mechanism discussed above.
Mitchell and Baird carried out careful kinetics studies on the reaction of Pd(PCy3)2 with PhBr, both in the absence and presence of additional PCy3, at room temperature.30b Added PCy3 inhibits oxidative addition, decreasing the relative amounts of [Pd(PCy3)] (1Cy) and [Pd(PCy3)2] (2Cy) due to formation of [Pd(PCy3)3] (11Cy).30b In the absence of additional ligand, the Pd(0) is mostly present in the bisligated form. This is observed to disappear, with a pseudo-first order rate law, with an apparent rate constant given by k[PhBr] + k′. The first term here can be accounted for by the bisphosphine pathway (C). The value of k in their experiments is 1.3 × 10−3 M−1 s−1, corresponding to a Gibbs energy of activation of 21.3 kcal mol−1. This compares very well to our calculated Gibbs energy of activation (22.8 kcal mol−1 at 10 °C, see Table S2b†). The second term can be accounted for by the dissociative pathway (A), with phosphine loss assumed to be rate-limiting, given the low concentration of free L and high concentration of PhBr in these experiments. The value of k′ measured, 6.1 × 10−4 s−1, would correspond to a Gibbs energy of activation of 21.7 kcal mol−1. Again, this matches our calculated barriers reasonably well (ca. 24.5 kcal mol−1 at 10 °C from energy for 1Cy + Gibbs energy barrier to ligand addition estimated as detailed above). In experiments carried out in the presence of excess PCy3, the results suggest that the contribution from the monoligated Path A is suppressed, but the analysis is complicated due to the fact that some of the Pd(0) is now present as PdL3 which is unlikely to participate in the reaction.
Hartwig et al. have also studied the kinetics for this reaction, at the slightly lower temperature of 10 °C, and with an excess of ligand present. They observe a slight decrease in reactivity as more L is added, due to slight changes in the position of the equilibrium with unreactive PdL3. Reactivity is first-order with respect to [PhBr], and, neglecting the complications arising from equilibrium with PdL3, their observed reactivity corresponds to a rate constant of 3 × 10−4 M−1 s−1, or to a ΔG‡ of 21 kcal mol−1.|| This is consistent with the other experiments, and with our computational results predicting the bisphosphine pathway (C) to be most favourable. The considerable sensitivity to method effects discussed earlier makes it difficult to distinguish confidently between paths A and C based on computation alone, and both pathways could indeed in principle be operating to some extent, though the bisligated path C is more consistent with experiment.
Computational work by Schoenebeck and Houk29b has shown that, while calculations suggest the monoligated pathway (A) to be favoured in terms of Gibbs energies, experimentally observed selectivities can only be explained by considering a bisligated oxidative addition pathway (Table S6† gives an overview of the computational approaches used in computational studies referred to). An earlier study involving the sterically and electronically similar47 PiPr3 ligand that included solvation corrections, but not dispersion effects, had also ruled out the bisligated oxidative addition (C), as well as associative displacement (path B), as too high in terms of Gibbs energy.33a The discussion of method effects above helps to explain these discrepancies, with the neglect of dispersion effects artificially favouring the dissociative pathway (A).
Products of oxidative addition. The free energies of a range of monoligated and bisligated oxidative addition products are given in Table 2 (complexes 5, 9, 10). Isomer interconversion has been shown by Maseras to be facile for three- and five-coordinate PH3 complexes, which can be accessed by square-planar complexes if a ligand is dissociated in the former case or ligand/solvent is coordinated in the latter.42a Therefore, we have assumed here that interconversion can take place easily to reach the isomer of lowest Gibbs energy. In line with experimental results, where the trans complex of 9Cy has been observed as the only product of the oxidative addition,15,30b this complex is the only one which could be optimised for the PCy3 ligand.
 | ||
| Fig. 3 Gibbs energy surface (ΔG° (90 °C) + ΔGsolv) for the oxidative addition of PhBr to a [Pd(PPh3)n] catalyst. See caption of Fig. 2 for labelling conventions used. | ||
Complex 12Ph, the tetrakis triphenylphosphine palladium(0) complex, has been crystallographically characterized34b and is a commercially available precursor for PPh3-ligated catalysts. In solution, ligand dissociation to the [PdL3] species 11Ph is known to be near quantitative except at very low temperatures and in the presence of excess phosphine.36,50 Hence, the equilibrium with 12Ph, while it can be observed by 31P-NMR at low temperatures,50 is not relevant here. Dissociation of 11Ph to form bisligated 2Ph is found experimentally to be unfavourable, with a dissociation equilibrium constant of the order of 10−4 M.34a Our calculations underestimate the stability of 12Ph significantly: this species is predicted to be much higher in Gibbs energy than 11Ph + L. This is probably an artifact associated with carrying out geometry optimization at a level of theory that does not account for dispersion. Indeed, test optimisations with B3LYP-D2 (section ESI4†) show a significant change in geometry. This is also in agreement with previous work,27,35 showing reasonable stability for 12Ph when using methods that account for dispersion in optimization as well as energy calculations; for a more general overview, see e.g. a recent study by Jensen et al.51
It appears that this shortcoming in our calculations mainly affects the very crowded complexes, such as 12Ph; see the ESI† for a more complete discussion. The calculated ligand binding Gibbs energy required to transform 11Ph into 2Ph is 2.9 kcal mol−1 at 25 °C, corresponding to a dissociation equilibrium constant of 8 × 10−3. The experimental estimate for this dissociation constant places it as being significantly smaller than 10−4 M.34a
Considering the three possible oxidative pathways starting from the [PdL2], 2Ph, complex, as with PCy3 our calculations predict that the bisphosphine pathway C is favoured, proceeding via [8Ph]‡, with a barrier of 21.8 kcal mol−1vs. 2Ph, and a barrier of 23.7 kcal mol−1vs. 11Ph (90 °C, blue pathway in Fig. 3) to be favoured. The competing associative displacement route (B, red in Fig. 3) has a highest barrier ([6Ph]‡) of 24.9 kcal mol−1 above 2Ph, and the highest point of the monoligated oxidative addition pathway ([4Ph]‡) lies 24.7 kcal mol−1 above reactants. The trans-bisligated product complex (t-9) is predicted to be most stable for this ligand and has indeed been observed crystallographically.38b
The bisphosphine pathway (C) was also found to be favoured by Kozuch and Martin.16 An earlier study by Fu, Liu et al. compared the three different pathways, but, in the absence of dispersion corrections, found both paths B and C to involve much higher Gibbs energy barriers than path A, in line with the analysis of method effects detailed above.33a
Experimentally, kinetic data for oxidative addition of various aryl halides to solutions of 12Ph is available for comparison.30c,34a,52 For the case of PhBr, an apparent rate constant kapp = 9 × 10−4 M−1 s−1 was measured at 25 °C for reaction with 12Ph.30c In fact, under the conditions used, 12Ph will have fully dissociated to 11Ph, which will itself be in equilibrium with a small amount of 2Ph. Assuming that the latter reacts with PhBr with a rate constant k, then 11Ph will decay with kapp = kK/[PPh3]. Given that [PPh3] was equal to 0.002 M in the experiments, this means that kK equals 1.8 × 10−6 s−1, equivalent to an activation Gibbs energy of 25.2 kcal mol−1, in reasonable agreement with the value of 24.0 kcal mol−1 at 25 °C calculated for pathway C here.
Computational modelling of this ligand is challenging, as both the cyclohexyl and biaryl substituents can adopt a range of conformations and their motion is likely to be correlated. We have found that the nature of the preferred conformer changes for the different species involved in oxidative addition, as shown in Table 3. As discussed in ESI3,† both biaryl and cyclohexyl group orientations respond to the coordination environment and these have been sampled extensively. While the effect of biaryl rotation has been explored previously,56 consideration of cyclohexyl conformational preferences was less complete and our present work has used a more extensive approach, combining database mining of the CSD45 and DFT calculations to identify the lowest energy conformer for each complex (section ESI3†). Table 2 and Fig. 4 show results for the conformer compatible with all steps, assuming that barriers to conformational change are lower in Gibbs energy than ‘reactive’ barriers lying along the oxidative addition mechanistic route.
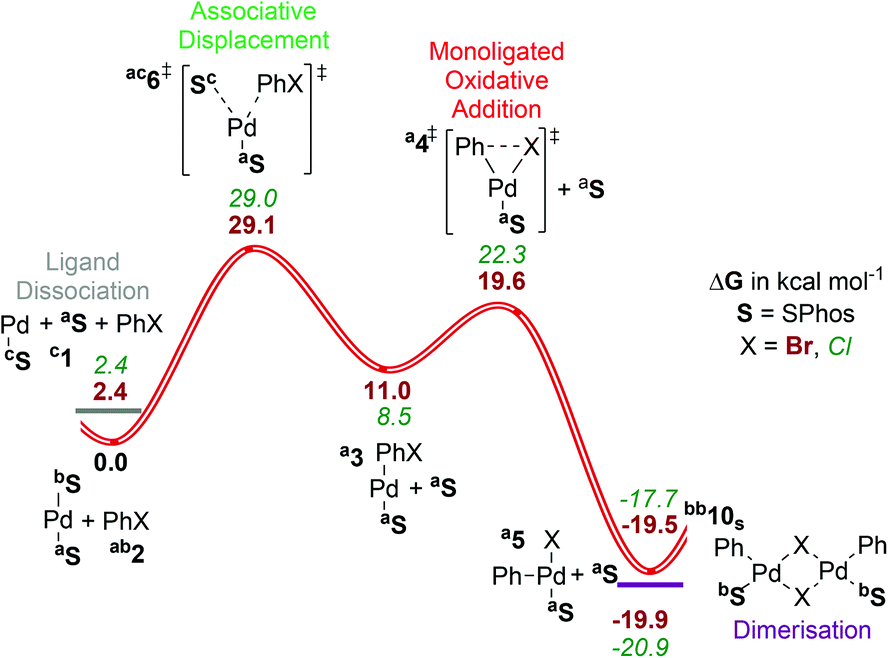 | ||
| Fig. 4 Gibbs energy surface (ΔG (90 °C) + ΔGsolv) for the oxidative addition of PhBr to a [Pd(SPhos)n] catalyst. See caption of Fig. 2 for labelling conventions used. | ||
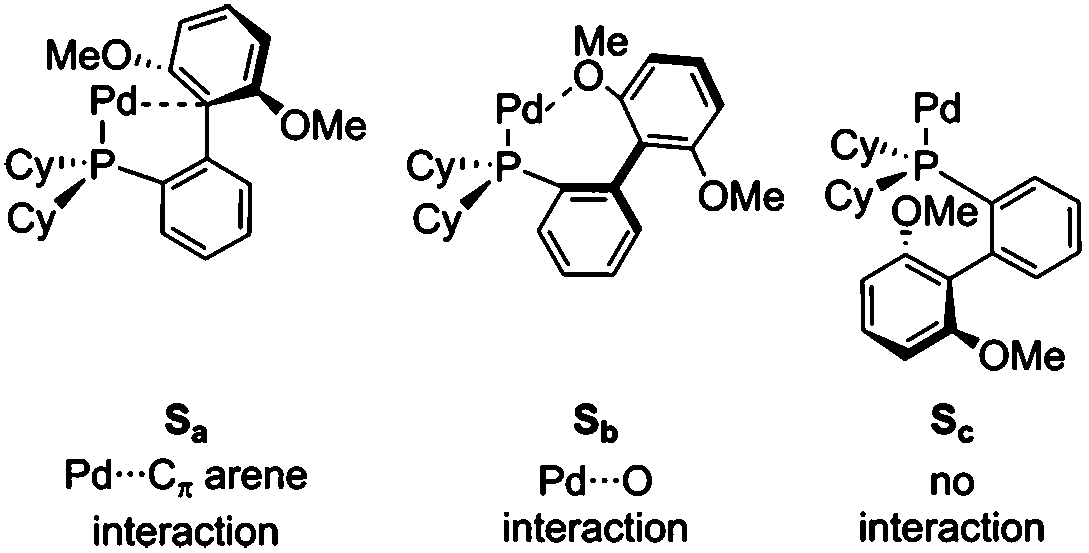
Low coordination numbers for this ligand are stabilized by the biaryl group coming into close proximity to the palladium centre,56 either providing sites for secondary interactions with the π-system (Sa) or the oxygens of the methoxy groups (Sb), as well as hampering the coordination of solvent/ligand/substrate through steric bulk. This is discussed in section ESI3.†
For SPhos the dissociative pathway (A, [4SP]‡, barrier = 19.6 kcal mol−1) presents the lowest barrier, while the associative displacement pathway (B, [6SP]‡) is significantly higher in Gibbs energy. We have not considered the bisligated pathway (path C) due to the steric hindrance exerted by two SPhos ligands in cis coordination sites. The overall calculated Gibbs energy of activation with this ligand is lower than with the other ligands covered in this study, accounting in part for the success of this ligand in catalysis. To the best of our knowledge, no detailed kinetic studies have been reported for this ligand, preventing further validation against experimental data.
The bromide bridged dimer is quite similar in energy to the monoligated T-shaped product, and indeed a chloride-bridged dimer of SPhos has been isolated and characterized crystallographically.57 This presumably also contributes to the synthetic utility of the ligand, by minimising the amount of metal complexes present as unreactive reservoir species under catalytic conditions.
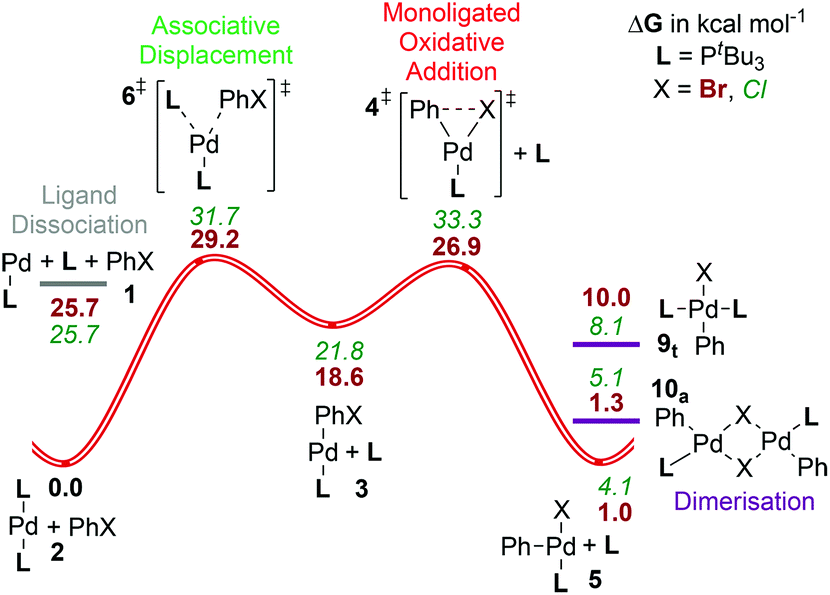 | ||
| Fig. 5 Gibbs energy surface (ΔG° (90 °C) + ΔGsolv) for the oxidative addition of PhBr to a [Pd(PtBu3)n] catalyst. See caption of Fig. 2 for labelling conventions used. | ||
For this ligand, the complex 2tBu [Pd(PtBu3)2] has been characterized crystallographically60 and no higher coordination numbers have been observed by NMR.30a In addition, both the T-shaped oxidative addition product 5tBu15 and a chloride bridged-analogue of 10 with an ortho-substituted aryl group13a have been isolated and characterized crystallographically. The three-coordinate product 5tBu is stabilized by agostic interactions between the ligand and the palladium center,15,61 which reduce the likelihood of dimerisation to form 10tBu,21c although the results here suggest that this may be finely balanced energetically, with 5tBu and both isomers of 10tBu lying within less than 2.5 kcal mol−1 of each other. Slight modifications of the ligand or the substrate might thus lead to dimerisation and could trap some of the palladium in an unproductive pathway. The trans square-planar complex t-9tBu lies higher in energy (10.0 kcal mol−1) than the other possible product species.
As we have reported previously,25b mechanistically, this ligand presents fewer pathways for evaluation, as the bisphosphine pathway (C) is not accessible due to the considerable steric hindrance of two PtBu3 ligands, which would hamper their cis coordination. The preferred mechanism with PhBr involves passing over the associative displacement TS (path B) followed by oxidative addition. The key TS lies higher in Gibbs energy ([6tBu]‡, barrier = 29.2 kcal mol−1) than the monoligated oxidative addition (path A, [4tBu]‡, barrier = 26.9 kcal mol−1). Our best estimate places formation of PdL at a Gibbs energy of around 30 kcal mol−1,25b which, with the variational TSs for addition of L or PhBr, means that path B, i.e. the associative displacement route, should be favoured over a dissociative pathway (A) for this ligand. The calculated barrier to associative displacement is in reasonable agreement with available experimental data (see ref. 25b for a more detailed discussion).
Before considering substrate effects in greater detail, we can take stock of ligand effects on the likely pathway and energetics of oxidative addition. The calculations reported here, validated where possible by comparison with available experimental data, suggest that very bulky ligands (PtBu3 and SPhos) favour low-coordinate pathways (A and B). Also, with these ligands, the T-shaped three-coordinate oxidative addition products are reasonably stable towards formation of dimer or bisligated complexes. This is presumably favourable for the reactions following on from oxidative addition in typical catalytic cycles. Smaller ligands, which are less “privileged” in experimental usage, can support higher coordination numbers around the metal centre. This means that prior to oxidative addition, ligand dissociation must occur. Nevertheless, contrary to the more bulky ligands where the C–X bond cleavage event occurs with only a single phosphine coordinated to the palladium centre, for PCy3 and PPh3, the bisphosphine pathway (C) is predicted to dominate. Though the barriers to oxidative addition are similar to or indeed lower than those calculated for bulkier ligands, unproductive product complexes are energetically much more accessible after this step, suggesting that the rate-limiting step for cross-coupling reactions might occur later in the catalytic cycle. Our calculated barriers are generally in good agreement with experimental data where this is available, although we note that detailed kinetic studies of oxidative addition are rare.
Halide effects
The cost of reagents for a chemical reaction can be an important factor, especially when considering the adoption of a new catalytic route. For oxidative addition, this can impact the choice of halide substrate used. Ideally, ligand design can enable the use of cheaper and structurally more versatile aryl chlorides as substrates.62 The geometries optimized with phenyl bromide provided a convenient starting point for expanding this study of ligand effects to also consider phenyl chloride as the substrate. “Best method” Gibbs energies for 90 °C to allow comparison with the bromide results are shown in Table 4, with a more detailed breakdown of method effects on energies for all ligands included in Tables S7–S10.†| ΔG° (90 °C) + ΔGsolv | PCy3 | PPh3 | SPhos | PtBu3 | |
|---|---|---|---|---|---|
| a Optimisation unsuccessful. b Not attempted for this ligand. c Looser convergence criteria used for BS2 single point calculation. | |||||
| 1 | [PdL] + PhCl | 18.5 | 18.4 | 2.4 | 25.7 |
| 2 | [PdL2] + PhCl | 0.0 | 0.0 | 0.0 | 0.0 |
| 3 | [PdL(PhCl)] | 14.6 | 14.4 | 8.5 | 21.8 |
| [4]‡ | [PdL(Ph⋯Cl)]‡ | 31.1 | 26.9 | 22.3 | 33.3 |
| 5 | [PdL(Ph)(Cl)] | −7.0 | −6.4 | −17.7 | 4.1 |
| [6]‡ | [PdL2(PhCl)]‡ | 27.5 | 24.6 | 29.0 | 31.7 |
| 7 | [PdL2(PhCl)] | ||||
| [8]‡ | [PdL2(Ph⋯Cl)]‡ | 23.8 | 24.7 | ||
| c-9 | [PdL2(Ph)(Cl)] | −13.8 | |||
| t-9 | [PdL2(Ph)(Cl)] | −34.2 | −18.5 | 8.1 | |
| a-10 | ½[(μ-Cl)2Pd2L2(Ph)2] | −13.0 | −12.1 | 5.1 | |
| s-10 | ½[(μ-Cl)2Pd2L2(Ph)2] | −20.9c | |||
| 11 | [PdL3] + PhCl | 2.7 | −1.9 | ||
| 12 | [PdL4] + PhCl | 21.1 | |||
Experimental and computational studies suggest that the rate limiting step for phenyl chloride oxidative addition is the monophosphine transition state [4]‡ (accessed by paths A and B), with limited ligand effects on the likely mechanism for the systems considered.10a,13a In Hartwig's experimental kinetic study exploring both ligand and substrate effects,13a different rate limiting steps were assigned for the different phenyl halides, with each rate law first order with respect to the concentration of [PdL2] species (2). Chloride substrates showed a dependence on the concentration of the ligand as well as the aryl chloride, thereby indicating that dissociation of a ligand must occur prior to rate-limiting ArCl oxidative addition ([4]‡); this transition state contributes to both the dissociative pathway (A) and the associative displacement pathway (B) considered here. As discussed above, the trisligated complex 11 is unlikely to contribute to experimental observations at higher temperature.
Comparison of the chloro species (Table 4) with the equivalent bromo complexes (Table 2) show that the former have higher relative free energies for transition states, and to some extent also for intermediates, relative to the bisligated reference complex, 2. The most pronounced substrate effects can be seen for the monophosphine and bisphosphine oxidative addition transition states, [4]‡ and [8]‡ respectively, as for these barriers different C–halide bonds are broken. For the monoligated oxidative addition, the increase in the barrier ranges from 2.2–8.4 kcal mol−1, with the largest difference observed for [4Cy]‡, whilst for the bisligated pathway (C) both PPh3 and PCy3 see barrier increases of 2.9 and 3.0 kcal mol−1 respectively. The lower reactivity of aryl chlorides has been attributed to the strength of the C–Cl bond,29a compared to weaker C–Br and C–I bonds.
Our previous work25b showed that for a PtBu3 catalyst the change in rate limiting step is observed for the associative displacement step (path B), with the bromo system limited by the associative displacement of PhBr and a ligand ([Br6tBu]‡), whilst the chloro system was restricted by the monophosphine oxidative addition of PhCl ([Cl4tBu]‡), part of paths A and B. For this ligand, dimerisation of the product becomes slightly less likely with the smaller chloride.
With additional results in hand, we can now consider the interplay between ligands and substrates more fully: In the case of PPh3 no change in favoured mechanism or rate limiting step is predicted by the calculations, although higher barriers suggest a slower rate of reaction, as expected for the chlorides. Similarly, for SPhos, there is no change for the different halide substrates, with the dissociative pathway (A) most likely.
Experimental data is available for the PCy3 and PCytBu2 cases, from two separate studies. For PCytBu2, the experiments (in toluene at 100 °C) yield rates that vary linearly with substrate concentration, and inversely with concentration of excess ligand. This suggests that path A or B is followed, with insertion into the C–Cl bond rate-limiting. The measured rate constant suggests an activation Gibbs energy of 29.3 kcal mol−1. We have not considered PCytBu2 computationally, but, as in our previous work,25b suggest that the calculated properties for PtBu3 should be rather similar. As seen in Table 4, our calculations agree well with experiment, showing [4]‡ as rate-limiting, and with a relative Gibbs energy of 30.7 kcal mol−1.
For PCy3, there is the complication that the PdL2/PdL3 equilibrium affects observed kinetics (as discussed in the section on method effects above). However, the available experimental data were obtained at fairly low concentration of free L, so that mostly PdL2 should be present, and we will not consider this equilibrium here. In the study by Hartwig et al.,13a the measured rates again vary in proportion to substrate concentration, and inversely to concentration of free ligand. This again supports mechanism A or B, with rate limiting oxidative addition through [4]‡; the measured rate constant at 70 °C, k = 1.07 × 10−6 s−1, yields a ΔG‡ = 29.4 kcal mol−1. In another experimental study,30c the data appear to have been analysed using a rate law that assumes no dependence on concentration of free ligand, and without added ligand. The proposed rate constant of 0.015 M−1 s−1 at room temperature is at first sight much larger than that reported by Hartwig et al.,13a but considering the likely very low concentration of free ligand in this study, there is probably no inconsistency. We thus focus our comparison with computation on the results of Hartwig et al.13a As for PtBu3, our calculated transition state ([Cl4Cy]‡) is found to lie at a relative Gibbs energy (31.1 kcal mol−1) that is consistent with experiment. However, our calculations show a significantly lower relative Gibbs energy for the bisligated transition state ([Cl8Cy]‡), suggesting that path C should instead be favoured. This mechanism is not compatible with the observed kinetics, so it appears that the computational protocol used is not sufficiently accurate for this particular ligand/substrate combination, for reasons that are not yet entirely clear. There are of course many possible sources of inaccuracy – basis set, functional, treatment of dispersion, statistical mechanics for entropy correction, and solvent treatment; some of these effects have been explored in greater detail (see ESI4b–d†). As stated earlier, these effects seem to combine to yield errors of a few kcal mol−1 in most cases, but the present case is more sensitive.
Conclusions
The manifold of reaction pathways for the oxidative addition of phenyl halide substrates to phosphine-modified palladium(II) complexes has been investigated with dispersion-corrected density functional theory for a range of synthetically relevant ligands. Three mechanistic possibilities were considered (Scheme 3): (A) a dissociative pathway accessing a low-coordinate [PdL] complex 1, which undergoes oxidative addition; (B) concerted associative displacement to [PdL2] 2, followed by monoligated oxidative addition; (C) a bisligated pathway, where oxidative addition occurs directly to the [PdL2] complex 2. Depending on ligand concentration, aryl halide substrate, solvent and temperature, different pathways can be accessed and in some cases, equilibria and competing pathways may need to be considered. Detailed experimental kinetic analysis can supply mechanistic insights and rate constants/barriers in these cases, and here we have been able to use such data to test and validate our computational methodology.On the whole, the calculated barriers and favoured pathways agree well with the available experimental data, allowing the computational prediction of likely reaction pathway (and hence a rate law), as well as the quantitative analysis of intermediates and transition states.†† In line with ligand design criteria derived from experimental studies, the bulky and electron-rich ligands PtBu3 and SPhos can access low-coordinate complexes, the most catalytically active species, easily throughout the cycle. While the bisphosphine oxidative addition step is reasonably facile for the smaller PCy3 and PPh3 ligands, ligand dissociation to access reactive palladium complexes becomes more important and the catalyst is more likely to become trapped in unreactive intermediates.
This work has demonstrated that a detailed evaluation of ligand effects is feasible and can support the interpretation of experimental data, allowing some pathways to be ruled out with certainty. In addition, the energetic balance of competing reaction pathways has been shown to be quite subtle, illustrating and illuminating experimentally observed sensitivity to both ligand and substrate effects. Finally, this work has highlighted that multiple competing mechanisms may need to be considered for a full evaluation of ligand effects, and that both Gibbs energy and dispersion corrections are necessary to achieve reasonable agreement with available experimental data.
Computational details
Structures were fully optimized in Gaussian (G03, see ESI for full citation†) with the standard B3LYP density functional63 and a flexible double-ξ (triple-ξ with ECP on Pd and Br) polarized basis set, denoted as BS1 (full details are given in the ESI†). B3LYP/BS1 harmonic frequencies were used to identify stationary points and compute zero-point energy, enthalpic and entropic corrections. A polarizable continuum model was used to obtain single point solvation free energies with toluene solvent. Single point energy calculations were also carried out with a larger (augmented triple-ξ) basis set, again with ECP on Pd and Br (denoted as BS2), and with the B3LYP-D2 functional64 as implemented in ORCA.65 Full details of the computational methodology used, as well as further details about transition state scans and conformational searches, are given in the ESI.†Acknowledgements
We thank Prof. Guy Orpen for helpful discussion of this work. NF thanks the EPSRC for the award of an Advanced Research Fellowships (EP/E059376/1), JNH holds a Royal Society Wolfson Research Merit Award and CLM thanks the CCDC, EPSRC and the School of Chemistry for funding, and CASCaM for the use of some computing facilities at the University of North Texas (CRIF, CHE-0741936).Notes and references
- (a) J. F. Hartwig, Nature, 2008, 455, 314–322 CrossRef CAS PubMed; (b) S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1439 CrossRef CAS PubMed; (c) C. A. Fleckenstein and H. Plenio, Chem. Soc. Rev., 2010, 39, 694–711 RSC; (d) C. Torborg and M. Beller, Adv. Synth. Catal., 2009, 351, 3027–3043 CrossRef CAS PubMed; (e) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- R. F. Heck, Nobel Lect., http://www.nobelprize.org/nobel_prizes/chemistry/laureates/2010/heck-lecture.html, 2010 Search PubMed.
- E.-i. Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738–6764 CrossRef CAS PubMed.
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed.
- M. Kumada, Pure Appl. Chem., 1980, 52, 669 CrossRef CAS.
- J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508 CrossRef PubMed.
- Y. Hatanaka and T. Hiyama, J. Org. Chem., 1988, 53, 918 CrossRef CAS.
- A. R. Muci and S. L. Buchwald, Top. Curr. Chem., 2002, 219, 131 CrossRef CAS.
- J. F. Hartwig, Acc. Chem. Res., 1998, 31, 852 CrossRef CAS.
- (a) K. C. Lam, T. B. Marder and Z. Y. Lin, Organometallics, 2007, 26, 758–760 CrossRef CAS; (b) M. García-Melchor, A. A. C. Braga, A. Lledós, G. Ujaque and F. Maseras, Acc. Chem. Res., 2013, 46, 2626–2634 CrossRef PubMed.
- K. J. Bonney and F. Schoenebeck, Chem. Soc. Rev., 2014 10.1039/C4CS00061G.
- (a) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed; (b) D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749–823 CrossRef CAS PubMed; (c) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed; (d) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed.
- (a) F. Barrios-Landeros, B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 8141–8154 CrossRef CAS PubMed; (b) J. C. Green, B. J. Herbert and R. Lonsdale, J. Organomet. Chem., 2005, 690, 6054–6067 CrossRef CAS PubMed; (c) S. Shekhar and J. F. Hartwig, Organometallics, 2007, 26, 340–351 CrossRef CAS.
- J. F. Hartwig, Pure Appl. Chem., 1999, 71, 1417–1423 CrossRef CAS.
- J. P. Stambuli, C. D. Incarvito, M. Bühl and J. F. Hartwig, J. Am. Chem. Soc., 2004, 126, 1184–1194 CrossRef CAS PubMed.
- S. Kozuch and J. M. L. Martin, ACS Catal., 2011, 1, 246–253 CrossRef CAS.
- J.-P. Corbet and G. Mignani, Chem. Rev., 2006, 106, 2651–2710 CrossRef CAS PubMed.
- A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 1998, 37, 3387 CrossRef CAS.
- (a) F. Paul, J. Patt and J. F. Hartwig, J. Am. Chem. Soc., 1994, 116, 5969–5970 CrossRef CAS; (b) F. Paul, J. Patt and J. F. Hartwig, Organometallics, 1995, 14, 3030–3039 CrossRef CAS; (c) J. Louie and J. F. Hartwig, Tetrahedron Lett., 1995, 36, 3609–3612 CrossRef CAS.
- J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534–1544 CrossRef CAS PubMed.
- (a) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed; (b) E. R. Strieter, D. G. Blackmond and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 13978–13980 CrossRef CAS PubMed; (c) D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338–6361 CrossRef CAS PubMed.
- (a) N. Fey, M. F. Haddow, J. N. Harvey, C. L. McMullin and A. G. Orpen, Dalton Trans., 2009, 8183–8196 RSC; (b) J. Jover, N. Fey, J. N. Harvey, G. C. Lloyd-Jones, A. G. Orpen, G. J. J. Owen-Smith, P. Murray, D. R. J. Hose, R. Osborne and M. Purdie, Organometallics, 2010, 29, 6245–6258 CrossRef CAS; (c) J. Jover, N. Fey, J. N. Harvey, G. C. Lloyd-Jones, A. G. Orpen, G. J. J. Owen-Smith, P. Murray, D. R. J. Hose, R. Osborne and M. Purdie, Organometallics, 2012, 31, 5302–5306 CrossRef CAS PubMed.
- G. J. J. Owen-Smith, J. Jover, N. Fey, J. N. Harvey, D. R. J. Hose, G. C. Lloyd-Jones, P. Murray, A. G. Orpen, R. Osborne and M. Purdie, in preparation for publication.
- (a) J. Jover, N. Fey, M. Purdie, G. C. Lloyd-Jones and J. N. Harvey, J. Mol. Catal. A: Chem., 2010, 324, 39–47 CrossRef CAS PubMed; (b) N. Fey, A. G. Orpen and J. N. Harvey, Coord. Chem. Rev., 2009, 253, 704–722 CrossRef CAS PubMed.
- (a) C. L. McMullin, B. Rühle, M. Besora, A. G. Orpen, J. N. Harvey and N. Fey, J. Mol. Catal. A: Chem., 2010, 324, 39–47 CrossRef PubMed; (b) C. L. McMullin, J. Jover, J. N. Harvey and N. Fey, Dalton Trans., 2010, 39, 10833–10836 RSC.
- (a) C. J. Cramer and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 760–768 CrossRef CAS PubMed; (b) J. N. Harvey, Faraday Discuss., 2010, 145, 487–505 RSC.
- N. Fey, B. M. Ridgway, J. Jover, C. L. McMullin and J. N. Harvey, Dalton Trans., 2011, 40, 11184–11191 RSC.
- R. F. Ribeiro, A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2011, 115, 14556–14562 CrossRef CAS PubMed.
- (a) G. C. Fu, Acc. Chem. Res., 2008, 41, 1555–1564 CrossRef CAS PubMed; (b) F. Schoenebeck and K. N. Houk, J. Am. Chem. Soc., 2010, 132, 2496–2497 CrossRef CAS PubMed.
- (a) E. A. Mitchell and M. C. Baird, Organometallics, 2007, 26, 5230–5238 CrossRef CAS; (b) E. A. Mitchell, P. G. Jessop and M. C. Baird, Organometallics, 2009, 28, 6732–6738 CrossRef CAS; (c) A. Kurbangalieva, D. Carmichael, K. K. M. Hii, A. Jutand and J. M. Brown, Chem. – Eur. J., 2014, 20, 1116–1125 CrossRef CAS PubMed.
- (a) P. Vidossich, G. Ujaque and A. Lledos, Chem. Commun., 2014, 50, 661–663 RSC; (b) K. Vikse, T. Naka, J. S. McIndoe, M. Besora and F. Maseras, ChemCatChem, 2013, 5, 3604–3609 CrossRef CAS PubMed.
- (a) E. Lyngvi and F. Schoenebeck, Tetrahedron, 2013, 69, 5715–5718 CrossRef CAS PubMed; (b) F. Proutiere and F. Schoenebeck, Angew. Chem., Int. Ed., 2011, 50, 8192–8195 CrossRef CAS PubMed.
- (a) Z. Li, Y. Fu, Q.-X. Guo and L. Liu, Organometallics, 2008, 27, 4043–4049 CrossRef CAS; (b) H. M. Senn and T. Ziegler, Organometallics, 2004, 23, 2980–2988 CrossRef CAS; (c) L. M. Alcazar-Roman, J. F. Hartwig, A. L. Rheingold, L. M. Liable-Sands and I. A. Guzei, J. Am. Chem. Soc., 2000, 122, 4618–4630 CrossRef CAS; (d) M. Ahlquist, P. Fristrup, D. Tanner and P.-O. Norrby, Organometallics, 2006, 25, 2066–2073 CrossRef CAS.
- (a) C. Amatore and F. Pfluger, Organometallics, 1990, 9, 2276–2282 CrossRef CAS; (b) V. G. Andrianov, I. S. Akhrem, N. M. Chistovalova and Y. T. Struchkov, Zh. Strukt. Khim., 1976, 17, 135 CAS.
- M. S. G. Ahlquist and P.-O. Norrby, Angew. Chem., Int. Ed., 2011, 50, 11794–11797 CrossRef CAS PubMed.
- J.-F. Fauvarque, F. Pflüger and M. Troupel, J. Organomet. Chem., 1981, 208, 419–427 CrossRef CAS.
- J. P. Stambuli, M. Bühl and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 9346 CrossRef CAS PubMed.
- (a) T. Aoki, Y. Ishii, Y. Mizobe and M. Hidai, Chem. Lett., 1991, 20, 615–618 CrossRef; (b) J. P. Flemming, M. C. Pilon, O. Y. Borbulevitch, M. Y. Antipin and V. V. Grushin, Inorg. Chim. Acta, 1998, 280, 87–98 CrossRef CAS.
- A. G. Sergeev, A. Zapf, A. Spannenberg and M. Beller, Organometallics, 2008, 27, 297–300 CrossRef CAS.
- S. Moncho, G. Ujaque, A. Lledos and P. Espinet, Chem. – Eur. J., 2008, 14, 8986–8994 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- (a) A. A. C. Braga, G. Ujaque and F. Maseras, Organometallics, 2006, 25, 3647–3658 CrossRef CAS; (b) H. Jacobsen and L. Cavallo, ChemPhysChem, 2012, 13, 562–569 CrossRef CAS PubMed.
- (a) M. Besora, A. A. C. Braga, G. Ujaque, F. Maseras and A. Lledos, Theor. Chem. Acc., 2010, 639–646 Search PubMed; (b) F. Ragone, A. Poater and L. Cavallo, J. Am. Chem. Soc., 2010, 132, 4249–4258 CrossRef CAS PubMed.
- (a) C. Amatore and A. Jutand, Acc. Chem. Res., 2000, 33, 314–321 CrossRef CAS PubMed; (b) C. Amatore, G. Le Duc and A. Jutand, Chem. – Eur. J., 2013, 19, 10082–10093 CrossRef CAS PubMed; (c) L. J. Goossen, D. Koley, H. L. Hermann and W. Thiel, Organometallics, 2006, 25, 54–67 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef PubMed.
- M. Ahlquist and P.-O. Norrby, Organometallics, 2007, 26, 550–553 CrossRef CAS.
- N. Fey, A. C. Tsipis, S. E. Harris, J. N. Harvey, A. G. Orpen and R. A. Mansson, Chem. – Eur. J., 2006, 12, 291–302 CrossRef CAS PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- A. Ariafard and B. F. Yates, J. Am. Chem. Soc., 2009, 131, 13981–13991 CrossRef CAS PubMed.
- B. E. Mann and A. Musco, J. Chem. Soc., Dalton Trans., 1975, 1673–1677 RSC.
- Y. Minenkov, A. Singstad, G. Occhipinti and V. R. Jensen, Dalton Trans., 2012, 41, 5526–5541 RSC.
- A. Jutand and A. Mosleh, Organometallics, 1995, 14, 1810–1817 CrossRef CAS.
- (a) D. W. Old, J. P. Wolfe and S. L. Buchwald, J. Am. Chem. Soc., 1998, 120, 9722 CrossRef CAS; (b) J. P. Wolfe, S. Wagaw, J.-F. Marcoux and S. L. Buchwald, Acc. Chem. Res., 1998, 31, 805–818 CrossRef CAS; (c) J. P. Wolfe, H. Tomori, J. P. Sadighi, J. Yin and S. L. Buchwald, J. Org. Chem., 2000, 65, 1158 CrossRef CAS PubMed.
- T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685–4696 CrossRef CAS PubMed.
- S. D. Walker, T. E. Barder, J. R. Martinelli and S. L. Buchwald, Angew. Chem., Int. Ed., 2004, 43, 1871–1876 CrossRef CAS PubMed.
- (a) T. E. Barder, M. R. Biscoe and S. L. Buchwald, Organometallics, 2007, 26, 2183–2192 CrossRef CAS; (b) T. E. Barder and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 12003–12010 CrossRef CAS PubMed; (c) S. Kozuch and J. M. L. Martin, Chem. Commun., 2011, 47, 4935–4937 RSC.
- M. R. Biscoe, T. E. Barder and S. L. Buchwald, Angew. Chem., Int. Ed., 2007, 46, 7232 CrossRef CAS PubMed.
- F. Barrios-Landeros, B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 5842–5843 CrossRef CAS PubMed.
- W.-J. Sun, W. Chu, L.-J. Yu and C.-F. Jiang, Chin. J. Chem. Phys., 2010, 23, 175–179 CrossRef CAS.
- M. Tanaka, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1992, 48, 739–740 CrossRef.
- (a) J. Vignolle, H. Gornitzka, B. Donnadieu, D. Bourissou and G. Bertrand, Angew. Chem., Int. Ed., 2008, 47 Search PubMed; (b) D. V. Partyka, T. J. Robilotto, M. Zeller, A. D. Hunter and T. G. Gray, Organometallics, 2008, 27, 28–32 CrossRef CAS.
- B. Schlummer and U. Scholz, Adv. Synth. Catal., 2004, 346, 1599–1626 CrossRef CAS PubMed.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed; (c) C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS; (d) S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS PubMed; (e) B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS; (f) J. C. Slater, Quantum Theory of Molecules and Solids, Vol. 4: The Self-Consistent Field for Molecules and Solids, McGraw-Hill, New York, 1974 Search PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- ORCA, An Ab Initio, DFT, and Semiempirical Electronic Structure Package Version 2.6.35, F. Neese, Universität Bonn, Germany, 2007 Search PubMed.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full computational details, further calculation results and detailed breakdown of results for all ligands at all levels of theory, conformer classifications and xyz coordinates of all optimised structures. See DOI: 10.1039/c4dt01758g |
| ‡ Present address: Institute of Chemical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS. E-mail: E-mail: c.l.mcmullin@hw.ac.uk. |
| § We have used Grimme's “D2” correction here to facilitate comparison with our PtBu3 results published previously (ref. 25b). We note that the “D3” dispersion correction has since been described (in S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104), which Grimme recommends as superior. Our own test calculations (some of which are summarised in Table ESI4f†) suggest that the version of the dispersion correction does not substantially alter the trends and conclusions described here. |
| ¶ In figures and tables, free ligand L needed to achieve stoichiometrically correct notation will frequently be left out, e.g. where the Gibbs energy of “[PdL3]” is compared to that of “[PdL2]”, the calculated number will be based on comparing the calculated Gibbs energy of [PdL3] to the sum of those for [PdL2] + L. |
| || They also studied the reaction with PhI, in the presence of excess L and at the low temperature of −80 °C. Under these conditions, the Pd will be present predominantly as PdL3, and accordingly inverse first-order kinetics with respect to L are observed. The free energy of dissociation of L at this temperature is 2.3 kcal mol−1.30a The observed rate-constant obtained by Hartwig et al.,13a is 8.5 × 10−4 M−1 s−1, corresponding to a ΔG‡ of 13.8 kcal mol−1. This would correspond to a ΔG‡ with respect to PdL2 of 11.5 kcal mol−1, which is lower than calculated here for PhBr – as expected for the more reactive iodide. |
| ** Different steric measures can be considered, e.g. PPh3 has a Tolman cone angle66 of 145° and a He8_steric parameter47 of 8.0 kcal mol−1, whereas the corresponding data for PCy3 are 170°, and 15.5 kcal mol−1 respectively. Both sets of data indicate that PCy3 is larger. |
| †† With the exception of the PhCl/PCy3 system, where the calculated barrier for monoligated oxidative addition (path A) matches experimental data well, but calculations suggest that the bisligated oxidative addition of path C provides an alternative route with lower barriers. |
| This journal is © The Royal Society of Chemistry 2014 |