 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New avenues in the directed deprotometallation of aromatics: recent advances in directed cupration
Philip J.
Harford
a,
Andrew J.
Peel
a,
Floris
Chevallier
b,
Ryo
Takita
cd,
Florence
Mongin
*b,
Masanobu
Uchiyama
*cd and
Andrew E. H.
Wheatley
*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: aehw2@cam.ac.uk; Fax: +44 (0)1223 336362; Tel: +44 (0)1223 763966
bUMR 6226, Institut des Sciences Chimiques de Rennes, Chimie et Photonique Moléculaires, Université de Rennes 1-CNRS, Campus de Beaulieu, Bâtiment 10A, Case 1003, 35042 - Rennes Cedex, France. E-mail: Florence.Mongin@univ-rennes1.fr
cAdvanced Elements Chemistry Research Team, RIKEN Center for Sustainable Resource Science (CSRS), and Elements Chemistry Laboratory, RIKEN, 2-1 Hirosawa, Wako-shi, Saitama 351-0198, Japan. E-mail: uchiyama@mol.f.u-tokyo.ac.jp
dGraduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan
First published on 4th June 2014
Abstract
Recent advances in the selective deprotometallation of aromatic reagents using alkali metal cuprates are reported. The ability of these synergic bases to effect deprotonation under the influence of a directing group is explored in the context of achieving new and more efficient organic transformations whilst encouraging greater ancillary group tolerance by the base. Developments in our understanding of the structural chemistry of alkali metal cuprates are reported, with both Gilman cuprates of the type R2CuLi and Lipshutz and related cuprates of the type R2Cu(X)Li2 (X = inorganic anion) elucidated and rationalised in terms of ligand sterics. The generation of new types of cuprate motif are introduced through the development of adducts between different classes of cuprate. The use of DFT methods to interrogate the mechanistic pathways towards deprotonative metallation is described. Theoretical modelling of in situ rearrangements undergone by the cuprate base are discussed, with a view to understanding the relationship between R2CuLi and R2Cu(X)Li2, their interconversion and the implications of this for cuprate reactivity. The advent of a new class of adduct between different cuprate types is developed and interpreted in terms of the options for expelling LiX from R2Cu(X)Li2. Applications in the field of medicinal chemistry and (hetero)arene derivatization are explored.
Philip Harford obtained an MSc degree in Natural Sciences in 2008 from the University of Cambridge and is currently studying for his PhD there under the supervision of Dr Andrew Wheatley. His research is focussing on the synthesis of heterobimetallic lithium cuprate bases with particular interest in their structures as derived from X-ray crystallographic studies. |
Andrew Peel completed his degree in Natural Sciences at the University of Cambridge in 2013. He is currently undertaking a PhD in Chemistry in the Wheatley group. His research presently focuses on lithium amidocuprate chemistry, with his principal interests being in chemical crystallography. His other interests include growing exotic chilli peppers and playing guitar. |
Floris Chevallier is an Associate Professor at the University of Rennes 1 (Rennes, France). He studied (organic) chemistry at the universities of Orléans and Nantes. He received his PhD in 2004 under the supervision of Prof. Jean-Paul Quintard, for whom he worked on organotin chemistry. In 2005, he joined the group of Prof. Bernhard Breit at the Albert-Ludwigs-Universität Freiburg (Freiburg, Germany) as a postdoctoral researcher, where he focused on supramolecular catalysis. His current research interests include organometallic methodologies and molecular self-assemblies. |
Ryo Takita obtained his PhD in 2006 from the University of Tokyo with Prof. Masakatsu Shibasaki. From 2006 to 2007, he worked as a postdoctoral Fellow at Massachusetts Institute of Technology with Professor Timothy M. Swager. He became an Assistant Professor at Kyoto University (2007–2010) and University of Tokyo (2010–2012), and was promoted to Lecturer (2012–2014). He is currently a Deputy Team Leader at RIKEN Center for Sustainable Resource Science (CSRS). His research interests include organic chemistry for unexplored reactivity and functions. |
 Florence Mongin | Florence Mongin obtained her PhD in Chemistry in 1994 from the University of Rouen (France) under the supervision of Prof. Guy Queguiner. After two years at the Institute of Organic Chemistry of Lausanne (Switzerland) as a postdoctoral Fellow with Prof. Manfred Schlosser, she returned to the University of Rouen as an Assistant Professor in 1997 (HDR in 2003). She took up her present position in 2005 as Professor at the University of Rennes (France) and was appointed Junior Member of the Institut Universitaire de France in 2009. Her interests include the functionalization of aromatic compounds including heterocycles with recourse to mixed lithium-metal bases. |
 Masanobu Uchiyama | Masanobu Uchiyama obtained his PhD in 1998 from the University of Tokyo with Prof. Koichi Shudo. He worked as an Assistant Professor at Tohoku University (1995–2001) and University of Tokyo (2001–2003), and was promoted to lecturer (2003–2006). He became Associate Chief Scientist at RIKEN in 2006. He is currently a Full Professor at the University of Tokyo (from 2010) and Chief Scientist/Team Leader at RIKEN and RIKEN CSRS (from 2013). His research interests are in the area of synthetic organic chemistry with an emphasis on organometallic, physical, and computational chemistry. |
 Andrew Wheatley | Andrew Wheatley obtained his BSc from the University of Kent at Canterbury in 1995. He then worked at Cambridge under the guidance of Dr Ron Snaith, receiving his PhD in 1999 and an MA (Cantab) in 2002. After his PhD studies he undertook a Research Fellowship at Gonville & Caius College before becoming a University Lecturer at Cambridge (2000) and a Fellow of Fitzwilliam College. He has received the Harrison Memorial Medal from the RSC and more recently he has been a Visiting Associate Professor at Tohoku University, Japan. He is now a University Senior Lecturer at Cambridge. |
1. General introduction
Deprotolithiation has proved to be an enormously powerful method by which to effect the regioselective functionalization of aromatics.1 Historically, bases such as organolithiums and lithium dialkylamides have typically been employed for this purpose. However, either because they are highly polar or because they generate polar aryllithiums, their use for the functionalization of sensitive substrates (e.g. bearing functional groups that are prone to nucleophilic attack, rearrangement or elimination) requires strictly controlled conditions. In addition, organolithium compounds react with electrophiles, such as aroyl chlorides, through double addition, limiting synthetic possibilities by rendering the direct synthesis of ketones unlikely.Recently, the use of organic bases containing two metals has come to the fore. These so-called synergic reagents exhibit behaviour that is different from that of the corresponding monometal bases. Whilst the field is a challenging one, since its inception2 in 1999 with the development of the putative lithium zincate But2Zn(TMP)Li (TMP = 2,2,6,6-tetramethylpiperidide), a variety of complexes of the type (R)m(R′)nMM′ (M = alkali metal, M′ = lower polarity metal; R, R′ = alkyl, amido, chloro, etc.), and in particular those containing the very hindered TMP ligand, have been reported to act as efficient and selective superbases.3 These complexes – which now feature Zn, Al, Cu, Mn and Mg as the lower polarity metal – all exhibit 'ate formulations, and, as such, reveal both anionic activation4 and reduced aggregation5 when suitable reaction media are employed. Recently, more elaborate versions of these complexes have appeared; these include salt-activated organometallic compounds (e.g. “turbo”-Grignard reagents), and synergic pairs of metal amides that complement each other in deprotometallation reactions.3d,g
The present Perspective depicts the behaviour towards aromatics of lithium cuprates specifically, and the development of the concept of Directed ortho Cupration (DoC) that was first reported in 2007.6 Though they were first prepared in the 1950s, evidence for the actual nature of lithium cuprates has been slow to be forthcoming and it is only recently that solid-state structural data has appeared for examples of these complexes designed to behave as deprotonating agents. The present article is divided into five main parts. After this introduction, cuprate synthesis and structural characterisation are detailed in the second part. Understanding of the structural principles at work in lithium cuprates is important in order to fully appreciate differences in their reactivities. As is discussed below, there has been much debate into whether such differences in reactivity can be explained in terms of reagent structure. Recently, however, very interesting work has gone a long way to explaining the contrasting activities of cuprates (e.g.7 and 12, see below) that are now known to be structurally analogous.7 After this structural discussion, the scope and applications of aromatic directed cupration are presented. Discussion then shifts to the theoretical interrogation of these systems with a view to understanding their mechanistic chemistry. Finally, the concluding section seeks to draw together structural evidence and theoretical work with a view to rationalising the reported reactions and identifying future directions.
2. Advances in the solid-state structural chemistry of alkali metal amidocuprates
2.1. Lipshutz cuprates: the solid-state evidence
The most investigated method by which organocopper compounds can provide reactive organic anions is via organocuprates. First reported in 1952, Gilman noted that the addition of 1 equiv. MeLi to CuI resulted in the formation of a highly reactive and ether-insoluble yellow product thought to be (MeCu)∞. This dissolved upon addition of further MeLi8 to give (empirically) the lithium cuprate Me2CuLi 1.9 In 1981, the replacement of CuI with CuCN to give more reactive cuprates for halide substitution reactions was proposed. At this time it was argued that the cyanide anion σ-bonded to Cu,10 though while some NMR studies were interpreted in terms of the presence of such a motif in solution,11 solid-state evidence for the exact nature of so-called ‘Lipshutz cuprates’ remained elusive.Initial crystallographic evidence for the structures of lithium cuprates appeared only in the early 1980s, with publication of the structures of [Cu5Ph6]−[Li·4THF]+2 and [Li2Cu3Ph6]2[Li4Cl2·10Et2O] 3.12,13 Shortly thereafter, van Koten revealed the solid-state structure of the dimer of (C6H4{CH2NMe2}-2)2CuLi 4.14 This structure, in which an 8-membered metallocycle was supported by amine chelation of the alkali metal centres agreed well with that previously proposed based on a combination of multinuclear and variable-temperature NMR spectroscopic studies.15 This period also saw the publication of evidence suggestive of the true nature of Lipshutz cuprates in particular. Attempts to fabricate lithium cuprates from CuI and (halide-free) organolithiums in the presence of a crown ether yielded both [CuMe2]− and [CuPh2]− ions.16 These linear cuprate ions have been defined as so-called ‘lower-order’, with the possibility of ‘higher-order’ cuprate formation, in which copper is 3-coordinate, having been previously invoked to explain the enhanced reactivity of Lipshutz species.10,17 By 1990 the lack of data supporting such higher-order cuprates prompted the publication of NMR spectroscopic studies on mixtures of the bis(organyl)cuprates R2CuLi with LiX (R = Me, Ph; X = I, CN). The identical nature of the resulting data was argued to be consistent with lower-order structures in solution.18 However, the controversy continued with the presentation of contrary spectroscopic evidence, with the chemical shifts of the Me2CuLi/LiI system changing upon the addition of LiCN and the postulated formation of a Cu–CN interaction.19 That said, alternative explanations, e.g. LiCN coordination to an R group, remained, and this ambiguity led to the theoretical investigation by Snyder of possible structural candidates (see section 4). This led to the conclusion that higher-order structures were less stable than their lower-order analogues.20 Whilst in response to these finding it was proposed that CN− might σ-bond to lithium and π-bond to copper,21 further theoretical work suggested the former coordinative mode to be more likely than the latter.22
It is only since 1998 that unambiguous structural evidence for the behaviour of cyanide ligands in the presence of Cu(I) organometallics has appeared in the literature. Initial reports verified the solid-state structures of ion-separated [But2Cu]−[(μ-CN)(Li·THF·PMDTA)2]+5 (PMDTA = N,N,N′,N′′,N′′-pentamethyldiethylenetriamine) and the polymer of closely related {(2-Me2NCH2C6H4)2Cu}{(μ-CN)(Li·2THF)2} 6 (Fig. 1).23,24 In the first of these examples, the use of a strong Lewis base (PMDTA) has induced ion-separation by extruding Li+ from the cuprate moiety. Concordant with this and, as is described in section 4, based both on crystallographic and theoretical studies, the ability of the cuprate component of the structure to interact with an alkali metal is contingent not only on the Lewis basicity of the solvent but also on the nature of the Cu-bonded organyl groups. In the case of 5, the adoption of a Cu(μ-But)Li motif is not strongly favoured by the choice of potential bridging group. Accordingly, therefore, separated and linear [But2Cu]− and [(μ-CN)Li2]+ ions are noted. In contrast, the absence of PMDTA and the utilisation of bifunctional organyl groups in the cuprate ion instead lead to ion-association in 6via the formation of N(Me2)–Li interactions.
 | ||
| Fig. 1 The first solid-state evidence for lithium cyanocuprate structures was revealed by ion-separated 5 and the polymer of 6. | ||
The issue of cyanide incorporation by cuprates has only recently been revisited. This work first appeared in 2007, with attempts to fabricate and structurally elucidate the proposed Lipshutz cuprate afforded by treating TMPLi with 0.5 equiv. of CuCN (Scheme 1). The imperative for this investigation came from the first reports of Directed ortho Cupration (DoC) facilitated by the use of cuprate moieties that incorporate sterically demanding amide ligands.6 As noted in section 3.2, a reagent with the putative (Lipshutz) formulation (TMP)2Cu(CN)Li2 was found to offer significant potential for aromatic homocoupling processes. X-ray diffraction on the isolated crystals established the structure as being a dimer based on monomers of (TMP)2Cu(CN)Li2·THF 7 (Fig. 2). The core is based on a classical (NLi)2 cyclic architecture synonymous with the metallocycle formation of Snaith's vertical25 and lateral26 ring aggregation principles. Peripheral to this core are two cuprate components, based on approximately linear N–Cu–N motifs. The metal-bridging potential of the amido ligands is clearly demonstrated. Crucially, in terms of understanding the structural properties of cyanide-containing cuprates, the ion-associated species formed does not contain a bonded CuCN component in the solid-state. Instead, it closely approximates to the most stable of the structures computed by Snyder in 1994,20 with the cyanide group bridging between alkali metal ions. The manner in which it does this is different for the various metals in the aggregated structure, with each cyanide interacting with three different alkali metal centres; spanning two metal ions in near linear σ-fashion and exhibiting an offset side-on bond to the third.
 | ||
| Scheme 1 Formation of the dimer of Lipshutz bis(amido)cuprate 7. | ||
 | ||
| Fig. 2 Crystal structure (H atoms omitted) of the dimer of Lipshutz cuprate 7. | ||
2.2. Extending the principle: Lipshutz-type structures
The structural motif revealed by 7 turns out to have a great deal in common with the 6-membered metallocyclic lithiocuprate (Ph2N)2Cu(NPh2)Li2·2OEt28.27 This species revealed a solid-state monomer comprising a near linear geometry at Cu and both Cu–(μ-N)–Li and Li–(μ-N)–Li bridges (Fig. 3). Taken together, the observation of 7 and 8 suggests that there is nothing unique in the inter-lithium bridging activity of cyanide in the former complex. This idea led us to propose the creation of a series of what might be called ‘Lipshutz-type’ cuprates. These have the formulation R2Cu(X)Li2, such that for 7 X = CN. This idea has been tested by the replacement of CuCN with CuHal (Hal = Cl, Br, I) in essentially the same synthetic sequence as that which yielded 7 (Scheme 2). In the first instance, CuCl was investigated (our interest in this deriving from its ability to promote the deprotonative metallation of 2-chloropyridine as a precursor to benzoylation, see section 3.3). Initial work towards the isolation and characterisation of halide-containing reagent mixtures focused on the deployment of TMPLi and CuCl, with data quickly establishing that solvent conditions capable of promoting the retention of in situ-formed LiCl in solution (where it is capable of combining with putative R2CuLi) promoted cuprate reactivity. Importantly, attempts to replicate organic transformations using pre-formed (TMP)2CuLi (section 3) produced distinctly inferior reactivity. Overall, therefore, it could be posited that the optimal reagent adhered to the putative formulation (TMP)2Cu(Cl)Li2.28 Corroboration came from subsequent studies conducted on crystalline material isolated from reaction of TMPLi with CuCl in THF. The resulting structure, of (TMP)2Cu(Cl)Li2·THF 9 (Fig. 4), revealed a solid-state dimer based on a (ClLi)2 core.29 Superficially, the structure is highly analogous to that revealed by both 7 (albeit the Cu-containing rings are now 6- rather than 7-membered) and 8 (albeit the structure is dimeric). As with these structures, 9 incorporates lower-order, near-linear Cu centres. | ||
| Fig. 3 Monomeric structure of amidocuprate 8. | ||
 | ||
| Scheme 2 Formation of the dimers of Lipshutz-type bis(amido)cuprates 9, 10 and 12. | ||
 | ||
| Fig. 4 Structure (H atoms omitted) of the crystallographic dimer of chloride 9. | ||
Comparative research on the synthesis of diaryl ketones using lithiocuprates prepared with LiCl and LiBr has been reported and is discussed in section 3.3. As with chloride-based 9, the dimer of (TMP)2Cu(Br)Li2·THF 10 proved straightforward to isolate and characterise.30 The dimeric structure of this species contrasted with that noted previously for monomeric (C6H4{CH2NMeCH2CH2NMe2}-2)2Cu(Br)Li211 in which aggregation was inhibited by internal coordination of the alkali metals (Fig. 5).31 Finally, the iodide analogue of 9 and 10, (TMP)2Cu(I)Li2·THF 12, was also prepared (Fig. 6).32
 | ||
| Fig. 5 Monomeric structure of internally stabilized cuprate 11. | ||
 | ||
| Fig. 6 Structure of the non-centrosymmetric dimer of 12 (H atoms and iodide centre positional disorder omitted). | ||
The Cu-containing metallocycles in 7 are 7-membered and show near perpendicular geometry at the bridging nitrogen centres, there being only a slight asymmetry here (N1/2–Li2/1 1.995(4)/1.982(5) Å; Cu1–N1/2–Li2/1 91.13(13)/94.92(14)°) in spite of the fact that the two alkali metal centres in the ring have significantly different coordination environments. On moving to the chloride 9 and bromide 10, the most immediate structural change is that the cuprate moieties yield 6-membered rings which are highly asymmetric. This is revealed by the remarkably different N–Li distances and bridging angles at N. For example, in 10 the unsolvated alkali metal ion participates in a relatively short interaction with TMP (N2–Li2 1.963(4) Å compares with N1–Li1(THF) 2.002(5) Å). In a similar vein, the bridging angle at N2 is expanded relative to that at N1 (cf. 92.81(14) and 85.45(13)°, respectively). The same pattern is clearly seen for chloride 9, but moving to iodide 12, patterns in the bonding parameters depart somewhat from those seen in 9 and 10 in that the extreme asymmetry seen for the 6-membered cuprate metallocycles in the chloride and bromide dimers is present to a much reduced extent. Thus, although N–Li is significantly shorter than N–Li·THF, four very similar Cu–N–Li bond angles (range 93.11(18)–94.63(18)°) are seen. The N–Li interactions reveal a final point about the cuprate metallocycles in 12. At 1.965 Å (mean) these are shorter than those in 7 (mean 1.989 Å), consistent with the preference of hard Li+ for hard nitrogen over soft iodide.
In a similar vein to the synthesis of THF-solvates 7, 9, 10 and 12, the extension in Li–X bonding interactions as Group 17 is descended is noted also for the employment of OEt2 as Lewis base. The resulting lithiocuprates 13–16 (for X = CN, Cl, Br and I, respectively; Fig. 7) have all been found to be essentially analogous to their THF-solvated counterparts. Poor quality data prevents detailed discussion of 13. However, analysis of the bonding in 14–16 reveals that, in comparison to the THF-solvates discussed above, their 6-membered rings show a higher level of symmetry, in spite of the clearly different alkali metal environments therein.
 | ||
| Fig. 7 The solid-state formulation of diethyl ether-solvated 13–16. | ||
2.3. Gilman reagents
Of course, as noted above, the first lithium cuprates to be prepared were not Lipshutz or Lipshutz-type but rather were considered to have the formulation R2CuLi (e.g. R = Me 1). These have come to be known as Gilman reagents. In the context of deprotonative cupration, we describe elsewhere in this article (section 3.2) synthetic transformations done in hydrocarbon media that drive the removal of lithium salt from the reaction and which, therefore, promote Gilman cuprate activity. Specifically, lithium cuprates of the type RmCu(NR2)nLi (m = 1, n = 1; m = 0, n = 2) are relevant to the organic transformations described here and these have not been thoroughly studied historically. The structural possibilities for these systems have only begun to be elucidated in the last few years and this has led the field to be the subject of review.33Concerning lithium (TMP)cuprates, the treatment of TMPLi with CuCN and various CuX (X = halide) reagents in THF has been described above as yielding 7 and 9, 10 and 12 (and their OEt2-solvated analogues 13–16). Bis(amido)cuprate formulations such as these have been shown (see section 3.2) to effect DoC with the TMP ligand acting as a base, enabling homocoupling to give 2,2-biaryls. However, the solution structures of these systems are still not fully established. This study involves not only the elucidation of aggregation state but also understanding of the affinity of (TMP)2CuLi for LiX; that is, the question of whether a Lipshutz-type cuprate may interconvert with a Gilman cuprate through the inclusion/exclusion of LiX. As the discussion of recent theoretical studies in section 4 makes clear, this question is one of enormous importance in understanding DoC reactivity. By way of an example, whereas the previous sections of this structural discussion focus on Lipshutz cuprates (TMP)2Cu(CN)Li2·S 7 and 13 (S = THF, OEt2) and also Lipshutz-type reagents (TMP)2Cu(X)Li2·S 9, 10, 12 and 14–16 (X = halide; S = THF, OEt2), a bis(TMP) Gilman cuprate has been prepared using remarkably similar chemistry. Hence, the reaction of TMPLi with CuI in the presence of THF (in the ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1) has given (TMP)2Cu(I)Li2·THF 12 (see above). In contrast, similar reaction in the absence of THF represents a strategy designed to allow the precipitation and removal of lithium halide. This has borne fruit as discussed in the organic synthesis section of this review, where the efficiency with which deprotonative transformations have been achieved was found to correlate with whether or not lithium salt was abstracted from the system. In the present case, LiI precipitation facilitated the isolation of Gilman bis(amido)cuprate (TMP)2CuLi 17.32 Crystallography reveals a cyclic dimer in the solid-state (Fig. 8 and Scheme 3). In spite of the known ability of organolithium and organocopper species to exchange (see below) the metal sites in the dimer of 17 revealed hardly any substitutional disorder and the structure compares closely with the 8-membered metallocyclic dimer of MesNHCu(PhNH)Li·DME 18 (DME = 1,2-dimethoxyethane; Fig. 9).27 The isolation of 17 proved useful because it provided pure Gilman bis(TMP)cuprate for comparative tests of Lipshutz-type and Gilman cuprates in organic synthesis (section 3.3).
:1:1) has given (TMP)2Cu(I)Li2·THF 12 (see above). In contrast, similar reaction in the absence of THF represents a strategy designed to allow the precipitation and removal of lithium halide. This has borne fruit as discussed in the organic synthesis section of this review, where the efficiency with which deprotonative transformations have been achieved was found to correlate with whether or not lithium salt was abstracted from the system. In the present case, LiI precipitation facilitated the isolation of Gilman bis(amido)cuprate (TMP)2CuLi 17.32 Crystallography reveals a cyclic dimer in the solid-state (Fig. 8 and Scheme 3). In spite of the known ability of organolithium and organocopper species to exchange (see below) the metal sites in the dimer of 17 revealed hardly any substitutional disorder and the structure compares closely with the 8-membered metallocyclic dimer of MesNHCu(PhNH)Li·DME 18 (DME = 1,2-dimethoxyethane; Fig. 9).27 The isolation of 17 proved useful because it provided pure Gilman bis(TMP)cuprate for comparative tests of Lipshutz-type and Gilman cuprates in organic synthesis (section 3.3).
 | ||
| Fig. 8 The dimer of 17 with H atoms omitted. | ||
 | ||
| Scheme 3 Formation of Gilman reagent 17 accompanied by the expulsion of lithium iodide, which precipitates in the absence of donor solvent and so avoids the formation of 12. | ||
 | ||
| Fig. 9 The 8-membered metallocyclic dimer of MesNHCu(PhNH)Li·DME 18. | ||
As noted above, the article in which DoC was first presented discussed the synthesis of homocoupled products.6 However, it was also suggested that the synthesis of heterocoupled products could be achieved by using the putative cuprates R(TMP)Cu(CN)Li2 (R = alkyl, aryl). To investigate, CuCN was treated with RLi and TMPLi. For R = Ph crystallography now revealed the formation of monomeric PhCu(TMP)Li·3THF 19 (Scheme 4 and Fig. 10). In a similar vein, the ability of putative cuprate Me(TMP)Cu(CN)Li2 to achieve products in which aryl substrates were methylated led to investigation of the MeLi–CuCN–TMPLi mixture. With crystallisation being aided by the introduction of TMEDA (N,N,N′,N′-tetramethylethylenediamine) it proved possible to isolate cuprate MeCu(TMP)Li·TMEDA 20.34
 | ||
| Scheme 4 The formation of Gilman cuprates 19 (R = Ph, n = 3, S = THF) and 20 (R = Me, n = 1, S = TMEDA). | ||
 | ||
| Fig. 10 Structures of 19 (left) and 20 (right) with H atoms and disorder omitted. | ||
The observation of a terminal Cu–Ph interaction in 19 is unusual. Whilst it is known from other types of organocopper compound,35 the only lithiocuprate examples are [Ph2Cu]−[Li·2(12-crown-4)]+21 and {[Ph2Cu]−}2[(Cu(·2PPh3){(CN)Li}2·5THF)2]2+22,36 whilst the remaining reported terminal Cu–Ar bonds in this area all feature bulky Mes,37 C6H3-2,6-Mes2,38 C6H3-2,6-Trip2 (Trip = C6H2-2,4,6-Pri3)39 or ferrocenyl groups.40 Moving to the similarly underpopulated field of heteroleptic organyl(amido)cuprate structural chemistry,4119 fails to demonstrate the aggregation behaviour of aryl(amido)cuprate MesCu(NBz2)Li 23 (Bz = benzyl; Fig. 11),42 the head-to-tail dimeric structure of which confirmed theoretical expectations. Even in solution, the dimer showed negligible signs of deaggregation.43 However, it was clear that 23 represented the only compound isolable from a Schlenk equilibrium involving a variety of structural isomers in solution.42 Methylcuprate ions are even less well documented, having been noted only in [Me2Cu]−[Li·nS]+ (24n = 2, S = 12-crown-4; 25n = 3, S = DME)36,44 and the alkyl(phosphido)cuprate MeCu{P(But)2}Li·3THF 26 (Fig. 12).45 The observation of a single N-atom bridge between metals in 19 and 20 is interesting in the wider context of synergic amido-reagents. Hitherto, these have frequently revealed metallocyclic M(μ-R)(μ-N)Li cores, even after isolation from (and including within their structures) Lewis base media. Hence, metallocycle formation has proved unambiguous in aluminates (where M is Group 13)46 and zincates (where M is Group 12 and therefore stands comparison with Group 2).47 However, in recently observed manganates (where M is Group 7 and therefore stands comparison with Groups 2 and 12),48 closure of the ring via the formation of an agostic bridge49 to match the amido one was much more ambiguous (Scheme 5). Finally, in the present case (where M is Group 11 and stands comparison with Group 1) the essentially linear geometry at Cu50 clearly prevents the formation of a metallocycle.
 | ||
| Fig. 11 The dimeric aggregate of MesCu(NBz2)Li 23. | ||
 | ||
| Fig. 12 Alkyl(phosphido)cuprate 26. | ||
 | ||
| Scheme 5 The cores of synergic bases containing Al, Zn, Mn and Cu. (a) and (b) metallocycle completion via short Li⋯C approaches of 2.258(4)46b and 2.374(3)47d Å, respectively; (c) the central ring fails to form, with Li⋯C relatively extended at 2.742(6) Å;47a (d) Cu exhibits a near linear geometry, precluding agostic stabilisation of the alkali metal. | ||
2.4. Amidocuprate adducts: the abstraction of lithium halide
Recent studies in the rapidly evolving field of so-called synergic base chemistry have focused on minimizing the costs of reagent preparations.51 This has led to the deployment of DMPH (= cis-2,6-dimethylpiperidine) in place of TMPH. At time of writing, the relative costs of these two amines are £2.62 g−1 (TMPH) and £0.20 g−1 (DMPH), making the potential savings of developing DMP-ligated synergic bases significant.52 The adoption of much cheaper amide ligands has gone hand-in-hand with the removal of some of the significant steric hindrance associated with TMP ligands, allowing fundamental changes to the orientation of the amide group and enabling the observation of new amidocuprate structure-types.53As noted above, the use of diethyl ether as a solvent enabled the synthesis and isolation of Lipshutz-type (TMP)2Cu(Cl)Li2·OEt214. However, upon exchanging the amide component for DMP a dramatic effect was observed. Initially, the combination of DMPLi with CuCl was attempted in limited (2 equiv.) Et2O. Reaction led to the isolation of crystalline material that analyzed as a triangulated structure based on a LiCl core and having the formulation [(DMP)4CuM(Li·OEt2)2]LiCl 27 where optimal refinement of multiple crystallographic datasets suggested a pattern of disorder consistent with M = 0.3:0.7 Cu:Li (Scheme 6).
 | ||
| Scheme 6 Synthesis of 27 (M = 30:70 Cu:Li, n = 2 equiv. wrt Cu) and 28 (M = Cu, n = bulk). | ||
The disorder at one of the metal sites complicates interpretation of the nature of 27 but is interesting because it is suggestive of the phenomenon of exchange between organocopper and organolithium species that has been directly observed spectroscopically in the past.54 Overall, two views of 27 present themselves. For M = Li the complex could be viewed as a 1:2 adduct between Lipshutz-type (DMP)2Cu(Cl)Li2 and DMPLi. However, for M = Cu it can be argued that 27 represents an adduct between Gilman cuprate (DMP)2CuLi·OEt2 and Lipshutz-type cuprate (DMP)2Cu(Cl)Li2·OEt2. To overcome this ambiguity and to remove the observed metal disorder, solvent mixtures were varied. Ultimately, the use of bulk Et2O served to reinforce the interpretation of 27 as an adduct between two cuprates, with [(DMP)2CuLi·OEt2]2LiCl 28 being isolated (Scheme 6 and Fig. 13).
 | ||
| Fig. 13 Adduct [(DMP)2CuLi·OEt2]2LiCl 28. H atoms and minor Et2O disorder omitted. | ||
It proved possible to replicate the formation of 28 using CuBr (Scheme 7 and Fig. 14). A number of iterations were required to allow the isolation of [(DMP)2CuLi·OEt2]2LiBr 29. Hence, the reaction of BunLi (1 eq. wrt amine) with DMPH to give DMPLi was initially affected in limited Lewis base (2 equiv. Et2O). However, this afforded [(DMP)2CuLi·OEt2]1.45[(DMP)2CuLi·DMPH]0.55LiBr 30. This problem could be overcome, in two ways: the use of excess BunLi (wrt amine) or the deployment of bulk Et2O as solvent. Variation in solvent conditions has therefore been shown to have an effect on the production of adducts 28 and 29, with bulk Et2O working best for X = Cl and either bulk or 2 equiv. Et2O best for X = Br. Attempts to extend the new chemistry to X = I demonstrated a need to more stringently control the quantity of Et2O, with the use of 1 equiv. wrt Cu yielding [(DMP)2CuLi·OEt2]2LiI 31 (Scheme 8 and Fig. 15).
 | ||
| Scheme 7 Synthesis of 29 (n = bulk; L = OEt2) and 30 (n = 2 equiv. wrt Cu; L = (OEt2)0.45(DMPH)0.55). | ||
 | ||
| Fig. 14 Adduct [(DMP)2CuLi·OEt2]2LiBr 29. H atoms omitted. | ||
 | ||
| Scheme 8 Synthesis of 31 (n = 1 equiv. wrt Cu). | ||
 | ||
| Fig. 15 Adduct [(DMP)2CuLi·OEt2]2LiI 31. H atoms and minor disorder omitted. | ||
The metal disorder in 27 notwithstanding, the adduct structures all approximate to C2 symmetry about a core lithium halide axis. The metal–halide bonds in adducts 28, 29 and 31 extend as Group 17 is descended. Though this is unsurprising in itself, the extension proves not to be consistent. At 1.10, the ratio between Li2–X and the mean of Li1/3–X is greatest for X = I. For both the smaller halides it is 1.04, and this can be rationalised in terms of the relative preference of the alkali metal for interaction with hard and soft donors. The extension of metal–nitrogen bonding as a function of halide hardness is also reflected in the Li2–N–Cu angles, which increase in response to the presence of a softer halide. Moreover, a persistent feature of these ether-solvated adducts is distinct asymmetry in both the N–Li bond lengths and bridging angles at nitrogen. This contrasts with the much more symmetrical rings in diethyl ether solvates 14–16 (see section 2.2) in spite of the observation of essentially the same Cu-containing 6-membered rings in which only one of the two Li+ ions is solvated by OEt2. For example, angles at either N in a 6-membered ring deviate by <3° in 14–16 whereas in adducts 28, 29 and 31 they differ by 4.2–6.6°.
The observation of a transition in structure-type from Lipshutz-type dimers to adducts is intriguing. It is also potentially important in terms of rationalising DoC. As is explained in sections 3 and 4 of this review, DoC requires reagents that are formally Lipshutz(-type) but which may generate active monomeric Gilman bases. This revelation – that generation of the active base takes place in situ – has prompted an examination of structure-types synonymous with the abstraction of LiX from Lipshutz-type dimers, and it is clear that adducts of the type now discovered can be viewed in these terms.
Since 14–16 and 28, 29 and 31 are all (mono)ether solvates it stands to reason that the choice of amide ligand must be having a significant effect on structure. This idea bears fruit if the steric constraints associated with TMP and DMP ligands are considered. In, for example, the ether-solvated Lipshutz-type chloride dimer of (TMP)2Cu(Cl)Li2·OEt214 it is clear that the two TMP ligands bonded to either Cu atom project away from one another so that the two 6-membered piperidide rings lie in a fashion best described as endo,endo relative the (ClLi)2 aggregate core (Fig. 16, top). The need for this to happen is rationalised by considering the positions of the 8 methyl groups associated with this pair of ligands. Four of these methyl groups (blue in Fig. 16) exhibit equatorial positions relative to the piperidide rings while the remaining four are axially disposed. Clearly, the axial Me-groups (red in Fig. 16) would interact with each other in the event that the piperidide ligands, instead of lying endo,endo, adopted an exo,exo (or face-on) arrangement. Such an arrangement is, however, enabled by the replacement of TMP by DMP in 28. In this case the two DMP ligands bonded to a given Cu centre can now exhibit a face-on motif that is exo,exo with respect to the structure core (the LiCl vector in 28) without there being steric interaction between the methyl groups (Fig. 16, bottom).
 | ||
| Fig. 16 Structures of the 6-membered heterocyclic Lipshutz-type components that form parts of the dimer of TMP-based chloride 14 (top) and DMP-based adduct 28 (bottom). In 14 the piperidide ligands lie flat in order that the axial Me-groups (red) do not sterically interact. In 28 only equatorial Me-groups are present (blue) and the piperidide ligands can reside face-on. | ||
The fact that, in the absence of steric interaction between methyl groups, the piperidide rings adopt a face-on configuration vexes the question: is this a preferred arrangement? This unfortunately cannot be answered by an analysis of the adduct structures reported here because the ability of the piperidide ligands to adopt a face-on orientation confers significant stability on the structure in terms of steric interactions not only within the monomeric building block but also between Lipshutz-type and Gilman components of the triangulated structure. As suggested by Fig. 17, the exo,exo orientation of the amide ligands allows steric congestion at the unsolvated Li+ (Li2 in Fig. 13–15) to be acceptable. An endo,endo structure would be precluded for obvious reasons while the inability to observe exo,exo TMP-based analogues of DMP-based adducts 28–31 may also be explicable on steric grounds.
 | ||
| Fig. 17 A schematic of the observed exo,exo (left) and less favoured endo,endo forms (middle) of DMP-based adducts akin to 28–31 clarifies why only the first option has been seen experimentally. Steric interaction at the structure apex may also explain why TMP-based adducts have not so far been seen (right). | ||
The adducts so far prepared have demonstrated a significant tendency to retain their structures in hydrocarbon solution. This has best been evidenced by 7Li NMR spectroscopy. For the chloride adduct [(DMP)2CuLi·OEt2]2LiCl 28 the bis(amide)-complexed alkali metal is seen at more positive chemical shift (δ 1.83 ppm) than the externally solvated metal ions, which resonate at δ 1.48 ppm, with the two peaks exhibiting a 1:2 ratio. Identical trends are observed for 29 and 31. The incompletely deprotonated complex [(DMP)2CuLi·OEt2]1.45[(DMP)2CuLi·DMPH]0.55LiBr 30 presents a yet more interesting spectrum. The unique bis(amide)-complexed alkali metal ion is seen at δ 1.83 ppm, while equivalent integration signals are now also seen at δ 1.66 ppm and δ 1.48 ppm, and these are attributable to Li·DMPH and Li·OEt2.
Lastly, recent work on the new class of cuprate adduct discussed here has sought to extend our understanding of the generality with which they form by varying the solvent. Reaction of DMPLi with CuBr followed by recrystallization in the presence of THF has afforded [(DMP)2CuLi·2THF]2LiBr 32 (Fig. 18 and Scheme 9). In so doing the ability of THF to furnish an adduct has been demonstrated along with the observation that much more extensive solvation of the alkali metals is possible in these structures.
 | ||
| Fig. 18 Adduct [(DMP)2CuLi·2THF]2LiBr 32. H atoms omitted. | ||
 | ||
| Scheme 9 Synthesis of adduct 32. | ||
3. Recent applications of lithium cuprates in organic synthesis
3.1. Gilman cuprates and structural interconversion
By employing a Gilman cuprate base, that is of the type R2CuLi, Gais and co-workers noted in 2008 the attack of (Z)- and (E)-alkenyl sulfoximines at the α-position and the 1,2-metallate rearrangement of the resulting copper derivatives prior to trapping. A pre-complex between the cuprate base and the sulfoximine group was proposed as a possible explanation for the reaction.55 However, with the exception of this study, other examples reported in the literature concerning deprotometallation using cuprate bases have involved aromatic substrates. Moreover, it is noteworthy that these studies have focused on Lipshutz cuprates and, as defined in section 2 of this review, Lipshutz-type cuprates. This emphasis is borne of studies on the Gilman cuprates RCu(TMP)Li·nS (R = Ph, n = 3, S = THF 19, R = Me, n = 1, S = TMEDA 20) and their tendency to include or exclude LiCN. Crucially, the same work established that 20 was relatively inactive in DoC.34 Thus, N,N-diisopropylbenzamide was treated both with pre-isolated 20 and a solution containing 20 and LiCN, prior to electrophilic quenching with I2. Results showed that iodination proceeded in both cases, but yielding only 37% 2-iodo-N,N-diisopropylbenzamide when employing pre-isolated 20. This improved to 89% using the solution containing 20 and LiCN. Clearly, these data pointed to the importance of the presence of a Lipshutz or Lipshutz-type formulation base in solution, albeit the relationship between Gilman cuprate and LiX (X = CN, halide) remained complex.3.2. Lipshutz formulations: efficient DoC reagents
The ability of several lithium cuprate bases to deprotonate aromatic compounds was first evidenced in 2007 by Usui et al.6 The efficiency of the bases was evaluated by reacting benzonitrile with a variety of amidocuprates (formally 2 equiv. of cuprate monomer wrt benzonitrile, the cuprate being prepared in situ by the treatment of amidolithium, organolithium and copper cyanide in appropriate ratios) at 0 °C in THF. This was followed by iodolysis after stirring for 3 h at room temperature (in Table 1 the cuprates are expressed formally as monomers). Whereas amidocuprates prepared from CuI failed to give the 2-iodo derivative in satisfying yields (see next section), it was clear that cuprates of Lipshutz formulation promoted DoC of the aromatic in generally high yields (70–93%). In particular, the TMP ligand, one of the bulkiest amide ligands available, was identified as being crucial for both good reactivity and chemoselectivity.
The putative cuprate MeCu(TMP)(CN)Li2, identified in Table 1 as a very efficient lithium cuprate base for the deprotonation-iodolysis of benzonitrile, was next employed in the attempted functionalization of a variety of other aromatic substrates (Table 2). In this way, the reaction was extended with success to the elaboration of N,N-diisopropylbenzamide, 3-cyanoanisole, 1,3-dimethoxybenzene and 1,3-dicyanobenzene. Notably, the presence of a bromo or iodo group on the benzene ring was tolerated provided that a lower reaction temperature was used (−78 °C, cf. 0 °C in Table 1). Lastly, five-membered aromatic heterocycles could be similarly regioselectively converted at 0 or −40 °C.











Besides iodine, electrophiles such as D2O, methyl iodide, allyl bromide, benzoyl chloride and chlorotrimethylsilane were all successfully used to efficiently intercept the arylcuprate intermediate generated by ortho deprotometallating N,N-diisopropylbenzamide. These electrophiles afforded the expected deuterated, methylated, allylated, benzoylated and silylated derivatives in yields ranging from 84 to 100% (Table 3). The introduction of a hydroxyl group was achieved in 56% yield by exposure of the cuprate to molecular oxygen at room temperature in the presence of CuCN (1 equiv.).

When the arylcuprates prepared from N,N-diisopropylbenzamide using different putative RCu(TMP)(CN)Li2 bases were treated with nitrobenzene, selective oxidative ligand coupling took place to furnish either the 2-methylated (R = Me) or the 2-phenylated (R = Ph) cross-coupled derivative, or even the homocoupled product (R = TMP) (Scheme 10).
 | ||
| Scheme 10 Oxidation of arylcuprates coming from N,N-diisopropylbenzamide. | ||
3.3. Replacing cyanide with halide: Lipshutz-type cuprates
Since 2009, Mongin and co-workers have extensively documented the use of a cuprate of (TMP)2Cu(Cl)Li2 formulation, which has been structurally characterised as both a THF29 and a diethyl ether53 solvate (dimers of 9 and 14, respectively). In synthetic studies, the chloride-containing cuprate was generated either from air-stable CuCl2·TMEDA by reduction of CuII to CuI using butyllithium (1 equiv.) followed by the addition of TMPLi (2 equiv.)56 or, more simply, from CuCl by the addition of TMPLi (2 equiv.).57 In spite of the clear structural similarities between the chloride- (9/14) and iodide-containing (12/16) lithium cuprates, it is clear that the (TMP)2Cu(Cl)Li2 formulation base prepared in situ behaves significantly differently to the (TMP)2Cu(I)Li232 formulation base in these systems and, instead, acts rather more like the Lipshutz formulation cuprate (TMP)2Cu(CN)Li2 (see above)6 inasmuch as it is capable of deprotonating a large range of aromatic substrates. Similar results were achieved recently utilising the bromide-containing Lipshutz-type system (viz. 10 and 15 above) though with less impressive conversions than were typically attainable with the chloride.30 Taken together, these data are consistent with the view, expressed in the structural discussion above, that there is no fundamental change incurred to the cuprate on replacing X = CN (Lipshutz formulation) with X = halide (Lipshutz-type formulation) but rather, as the halide gets softer there is a general trend away from the reactivity patterns seen for the Lipshutz system. Overall, therefore, while structural evidence so far suggests the adherence of both Lipshutz and Lipshutz-type species to the general formula R2Cu(X)Li2, reactivity studies suggest that as the softness of X is increased it becomes reasonable to propose a gradual progression in solution to R2CuLi·LiX.
Reaction conditions were optimized for the chloride-containing cuprate using 2,4-dimethoxypyridine as substrate. Using THF as solvent, the best yield (60%) was recorded by carrying out the reaction with the base (1 equiv.) for 2 h at room temperature before interception with benzoyl chloride. The reaction could be efficiently carried out in toluene containing TMEDA (5 equiv.), but to the detriment of the regioselectivity. Using diethyl ether instead of THF led to the expected ketone in an improved 99% yield (Scheme 11). Meanwhile, the use of mixed organo/amido lithiocuprates resulted in lower yields due to a reduced ability to effect deprotonation and the occurrence of competitive side reactions.
 | ||
| Scheme 11 Deprotocupration–benzoylation of 2,4-dimethoxypyrimidine. | ||
The cuprate generated regioselectively from 2,4-dimethoxypyrimidine could be converted into ketones using 2- and 4-chlorobenzoyl chlorides. The 5-methyl derivative was synthesized in a reasonable yield using methyl iodide whereas a low 19% yield was noted for the 5-allylated derivative (a result that could have been due to the presence of the nucleophilic diazine nitrogen) (Table 4). Using nitrobenzene as an oxidant resulted in formation of the corresponding 5,5′-bipyrimidine in 22% yield.

Reaction conditions for ketone formation were carefully optimized, taking account of both steps, using 2-methoxypyridine as substrate and benzoyl chloride to intercept the intermediate arylcuprate (Table 5). Under the conditions used above for reaction of 2,4-dimethoxypyrimidine, the expected 3-benzoylpyridine was isolated in 82% yield. By using 0.5 equiv. of cuprate base, the yield was reduced by half, while reducing or extending the deprotonation time was deleterious for the yield.
|
|
|||||
|---|---|---|---|---|---|
| Entry | x | Solvent | Time 1 | Temp. 2 | Yielda (%) |
| a After purification. b The 3,6-disubstituted derivative was also formed in 10% yield. | |||||
| 1 | 1 | THF | 2 h | rt | 82 |
| 2 | 0.5 | THF | 2 h | rt | 40 |
| 3 | 1 | THF | 0.5 h | rt | 72 |
| 4 | 1 | THF | 6 h | rt | 62 |
| 5 | 1 | THF | 2 h | 60 °C | 66b |
| 6 | 1 | THF | 2 h | 0 °C | 66 |
| 7 | 1 | Et2O | 2 h | rt | 71 |
| 8 | 1 | PhMe + TMEDA (5 equiv.) | 2 h | rt | 49 |
| 9 | 1 | Et2O + TMEDA (5 equiv.) | 2 h | rt | 81 |

Interestingly, the temperature at which the quenching step was conducted in the deprotocupration–benzoylation of 2-methoxypyridine was found to have an impact on the course of the reaction. Hence, when trapping was performed at 60 °C the yield decreased on account of the competitive degradation (as observed for extended deprotonation times at room temperature). In contrast, at 0 °C the reaction proved slower. In the case of 2-methoxypyridine, switching solvent medium from THF to diethyl ether or toluene containing TMEDA did not improve the yield. A good yield was restored by using diethyl ether containing dimethyl sulfide (5 equiv.), a cosolvent which has been proven to be useful in deprotocupration reactions.58
Besides aroyl chlorides, other electrophiles were also evaluated for their room temperature-reactivity towards the pyridylcuprate intermediate (Table 6). Employing 2-phenyloxirane led to both regioisomeric alcohols. Meanwhile, the use of diphenyl disulfide afforded the expected functionalized pyridine more efficiently. Reactions involving conjugate addition were only found to be possible using alkyl propionates, affording the products as (Z) and (E) mixtures in only moderate yields. By using iodine, the expected 3-iodo derivative was formed, but again in a moderate (28%) yield on account of competitive homocoupling taking place. 4-Chlorobenzenesulfonyl chloride and, to a greater extent, dimethylsulfamoyl chloride and tetramethylthiuram behaved as oxidative agents, furnishing the 3,3′-dimer. From 2-fluoropyridine, 3,3′-dimer formation was similarly noted using chlorotrimethylsilane at 50 °C (52% yield), chloranil at 60 °C (94%) and nitrobenzene at room temperature (84%).
|
|
|||
|---|---|---|---|
| Entry | Electrophile (E) | Yield(s)a (%) | |
| a After purification. b Base prepared in the presence of TMEDA (1 equiv.). c Estimated yield. | |||
| 1b | 2-ClC6H4COCl (COC6H4-2-Cl) | 56 | — |
| 2b | 2-BrC6H4COCl (COC6H4-2-Br) | 43 | — |
| 3b |

|
39 | — |
| 4 |
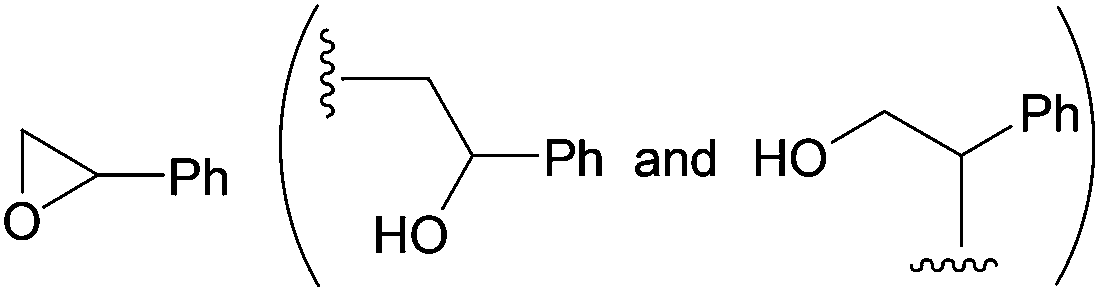
|
22 and 12 | — |
| 5 | PhSSPh (SPh) | 67 | — |
| 6 | PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCO2Et [C(Ph) CCO2Et [C(Ph)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO2Et] CHCO2Et] |
39 (Z) and 10c (E) | — |
| 7 | PeCCCO2Et [C(Pe)CHCO2Et] |
26 (E) | — |
| 8 | MeCCCO2Me [C(Me)CHCO2Me] |
30 (Z) and 17 (E) | — |
| 9 | I2 (I) | 28 | 39 |
| 10 | 4-ClC6H4SO2Cl (SO2C6H4-4-Cl) | 3 | 58 |
| 11 | Me2NSO2Cl | — | 72 |
| 12 | [Me2NC(S)S] | — | 88 |

That azines and diazines can be functionalized through deprotonative metallation at room temperature makes the method described above of interest. Indeed, such π-deficient heterocycles are sensitive to nucleophilic attacks on their CN bond due to their low LUMO levels. As a consequence, traditional lithium bases, when they can be employed, in general require very low temperature reaction conditions.59 The optimized deprotocupration–aroylation conditions were thus applied to the synthesis of different azinic and diazinic ketones (Table 7).28–30,56,57 Good results were observed using 2-substituted pyridine substrates (with the exception of 2-bromopyridine, from which elimination of metal halide to give 2,3-pyridine was suspected),60 with TMEDA favouring the reaction and regioselectivities being analogous to those observed using TMPLi.59 Except for 4-methoxypyridine and 3,5-dichloropyridine, the reaction required the presence of a substituent at the position adjacent to the ring nitrogen.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | ClCOAr′ | Product | Yielda (%) | Entry | ClCOAr′ | Product | Yielda (%) |
| a After purification. b Base prepared in the presence of TMEDA (1 equiv.). | |||||||
| 1 |

|
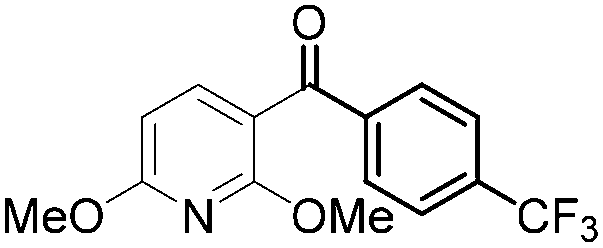
|
60 | 16 |

|

|
41b (C3), 24b (C2) |
| 2b |

|

|
91 | 17 |

|

|
42b |
| 3b |

|

|
57 | 18 |

|

|
70b |
| 4 |

|

|
85 (R = H) | 19 |

|

|
33b |
| 5 | 76 (R = 2-Cl) | ||||||
| 6 | 83 (R = 4-Cl) | ||||||
| 7 | 92 (R = 4-CF3) | ||||||
| 8 |

|
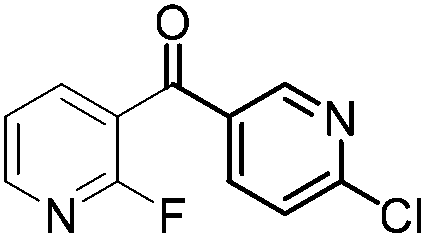
|
72 | 20 |

|

|
55b |
| 9 |

|

|
90b (R = H) | 21 |

|

|
80 |
| 10 | 59b (R = 2-Cl) | ||||||
| 11 | 80b (R = 4-Cl) | ||||||
| 12 | 80b (R = 4-OMe) | ||||||
| 13 | 66b (R = 3-OMe) | ||||||
| 14 |

|

|
65b | 22 |

|

|
21 |
| 15 |

|

|
20 | 23 |

|

|
78b |

In the framework of recent studies, the behaviour of 1,4-dimethoxybenzene and five-membered aromatic heterocycles towards (TMP)2Cu(Cl)Li2 was also investigated (Table 8). Under the reaction conditions used for the functionalization of pyridines described above, 1,4-dimethoxybenzene was converted to its 2-allylated derivative and to diarylketones in medium yields. No improvement was noted by increasing the deprotonation temperature to 40 °C.


Continuing in the same vein, N-Boc indole, furan and thiophene were similarly reacted in the deprotocupration–aroylation sequence to afford the expected ketones in approximately 50% yield. In order to reach higher yields, THF had to be replaced by either diethyl ether or diethyl ether containing dimethyl sulfide (5 equiv.). As expected, when this was undertaken, higher conversions were noted, although mixtures of 2-substituted and 2,5-disubstituted thiophenes also formed. From ethyl 2-thiophenecarboxylate and 3-thiophenecarboxylate, the reactions took place at the 5 and 2 position of the aromatic ring respectively, but with a lower chemoselectivity in the latter case. Finally, it was noted that the trapping temperature had an important impact on the efficiency on the reaction, with the best yields being obtained at 60 °C (Table 9).
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | ClCOAr′, temp. | Product | Yield(s)a (%) |
| a After purification. b After removal of the tert-butoxycarbonyl (Boc) group. c Metallation step performed at 0 °C. d Base prepared in the presence of TMEDA (1 equiv.). | ||||
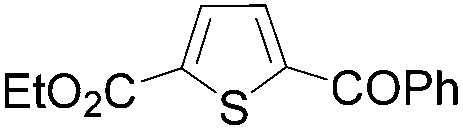
| 1 | THF | ClCOPh, 60 °C |
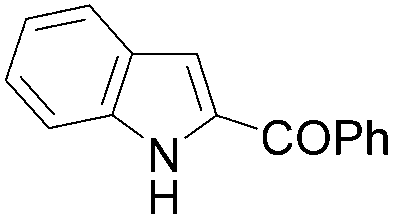
|
50b |
| 2 | THF | ClCOPh, 60 °C |

|
53 |
| 3 | THF | ClCOPh, 60 °C |

|
48 (Ar′ = Ph) |
| 4 | THF | ClCOC6H4-4-Cl, 60 °C | 58 (Ar′ = C6H4-4-Cl) | |
| 5 | Et2O | ClCOPh, rt |

|
31 and 29 |
| 6 | Et2O + Me2S (5 equiv) | ClCOPh, rt |

|
53 and 18 |
| 7 | THF | ClCOPh, rt |
|
49 |
| 8 | THF | ClCOPh, rt |

|
26, 40c |
| 9 | THF | ClCOC6H4-2-Cl, rt |

|
35d |
| 10 | THF | ClCOC6H4-2-Cl, 60 °C | 57d | |
| 11 | THF | ClCOPh, 60 °C |
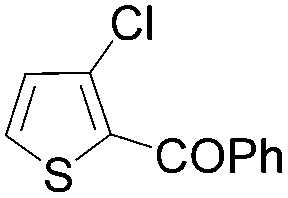
|
85 |

Whereas the arylcuprates prepared from N,N-diisopropylbenzamide using different RCu(TMP)(CN)Li2 bases were converted to the 2-methylated (R = Me) or the 2-phenylated (R = Ph) cross-coupled derivative upon treatment by nitrobenzene (Scheme 10),6 the arylcuprates obtained by deprotocupration using (TMP)2Cu(Cl)Li2 could be utilised in palladium-catalysed cross-couplings with aryl halides such as 4-iodoanisole and 2-bromopyridine in the presence of catalytic amounts of PdCl2(PPh3)2 and dppf (1,1′-bis(diphenylphosphino)ferrocene) (Table 10).57











Compared with the reactivities of previously reported lithium–zinc bases, it is clear that the lithiocuprate (TMP)2Cu(Cl)Li2 behaves more like a lithium amide, as it leads to deprotonation of the most acidic hydrogen atoms and is incompatible with reactive functional groups. This, however, is offset by a satisfying reactivity towards a larger range of electrophiles.
3.4. New amidocuprate adducts and the formation of reactive species
Lastly, and in line with the investigation of less expensive amide ligands,51 the effect of replacing TMP with DMP has been studied by Harford et al.53 As noted above, attempts to fabricate Lipshutz-type cuprates analogous to 9, 10, 12 and 14–16 instead afforded the series of complexes 28, 29, 31 and 32 that have been interpreted as adducts between Lipshutz-type and Gilman cuprates. The potential importance of this new structure-type is explained fully in the final section of this review where DFT analysis has suggested that the abstraction of LiX from Lipshutz or Lipshutz-type cuprates is fundamental to incurring in situ formation of the reactive base in DoC. It is clear, therefore, that these new adducts can all be viewed in terms of the elimination of 1 equiv. LiX having occurred from the corresponding Lipshutz-type dimer (e.g.142 → 28 + LiCl). Crucial to this view of their importance, using the base prepared in situ from DMPLi (4 equiv.) and CuBr (2 equiv.) in THF, N,N-diisopropylbenzamide was readily converted to the corresponding 2-metallated derivative, as shown by subsequent trapping with iodine (80% yield). An essentially identical result (82% yield) was recorded using pre-isolated [(DMP)2CuLi(OEt2)]2LiBr 28 (1 equiv.). It is clear, therefore, that efficient DoC is achievable not only by applying bases of Lipshutz(-type) formulation but also by using newly reported adducts.4. DFT modelling of cuprate structure and DoC reaction pathways
As noted in section 2 of this review, there has been controversy in the literature for some time regarding the behaviour of cyanide ligands in so-called Lipshutz cuprates. Spectroscopic data were used in 1990 to support both the view that the structure might be lower-order (with no copper–cyanide interaction)18and that it might be higher-order (Cu–CN bonded).19 This led to theoretical studies on a variety of solvated lithium dialkylcuprates of formulation Me2Cu(CN)Li2·2OH2.20,22,61 Results suggested higher-order models to be at least 20 kcal mol−1 higher in energy than their lower-order counterparts and that spectroscopic data suggesting higher-order structures could be accommodated by metallocyclic bridged structures containing linear, lower-order copper.62 Of most interest, a tandem EXAFS/ab initio study assessed a variety of structural possibilities for the formulation Me2Cu(CN)Li2 and concluded that there was a strong likelihood that the structure was as shown in Fig. 19.61 Plainly, this corresponds closely with that recently noted for complexes 7 and 13 by X-ray diffraction in which the cyanide group bonds only to alkali metal centres.6 | ||
| Fig. 19 Preferred ab initio structure of alkylcuprate Me2Cu(CN)Li2. | ||
More recently, among the organocuprate(I) complexes receiving increased attention have been heteroleptic cuprates ([R–Cu–R′]−). Organyl(amido)cuprates represent an important class of these, particularly since they have now been harnessed to achieve a variety of organic transformations. Thus, 2007 saw publication of the first of several reports that TMP-cuprates of the type RCu(TMP)(CN)Li2 (R = alkyl, phenyl, TMP) could promote the highly chemoselective DoC of multifunctionalized aromatic compounds.6,34,57 The arylcuprate intermediates presumed to result from deprotometallation of the aromatic substrate were effectively employed not only in the trapping of electrophiles, but also in oxidative ligand coupling to form new C–C bonds with alkyl/aryl groups (see section 3.2).
The chemistry of organocuprates(I) has traditionally been dominated by two structure types: Gilman cuprates and Lipshutz(-type) cuprates. As revealed above, these have now been structurally elucidated, with the former capable of exhibiting a homodimeric structure and Lipshutz-type cuprates demonstrating heteroaggregate structures based on metallocycles that are either 6-membered (halide-containing) or 7-membered (cyanide-containing, Scheme 12). Overall, with the help of X-ray crystallography, structure–reactivity relationship studies of TMP-cuprates have revealed that (1) the cyclic dimer of Gilman cuprate (TMP)2CuLi is unreactive in DoC and (2) a variety of Lipshutz-type amidocuprates, including (TMP)2Cu(CN)Li2, exhibit enhanced deprotonative reactivity.32
 | ||
| Scheme 12 Structures of Gilman and Lipshutz-type cuprates (top) and monoGilman-type cuprates with solvent coordination (bottom, S = solvent molecule). | ||
Further insights into the natures of active cuprate species and the mechanisms by which they exhibit DoC have been gained through DFT study of the derivatisation of N,N-dimethylbenzamide. The deprotonation process using MeCu(NMe2)(CN)Li2·2OMe2 as a model Lipshutz cuprate is summarized in Scheme 13.32
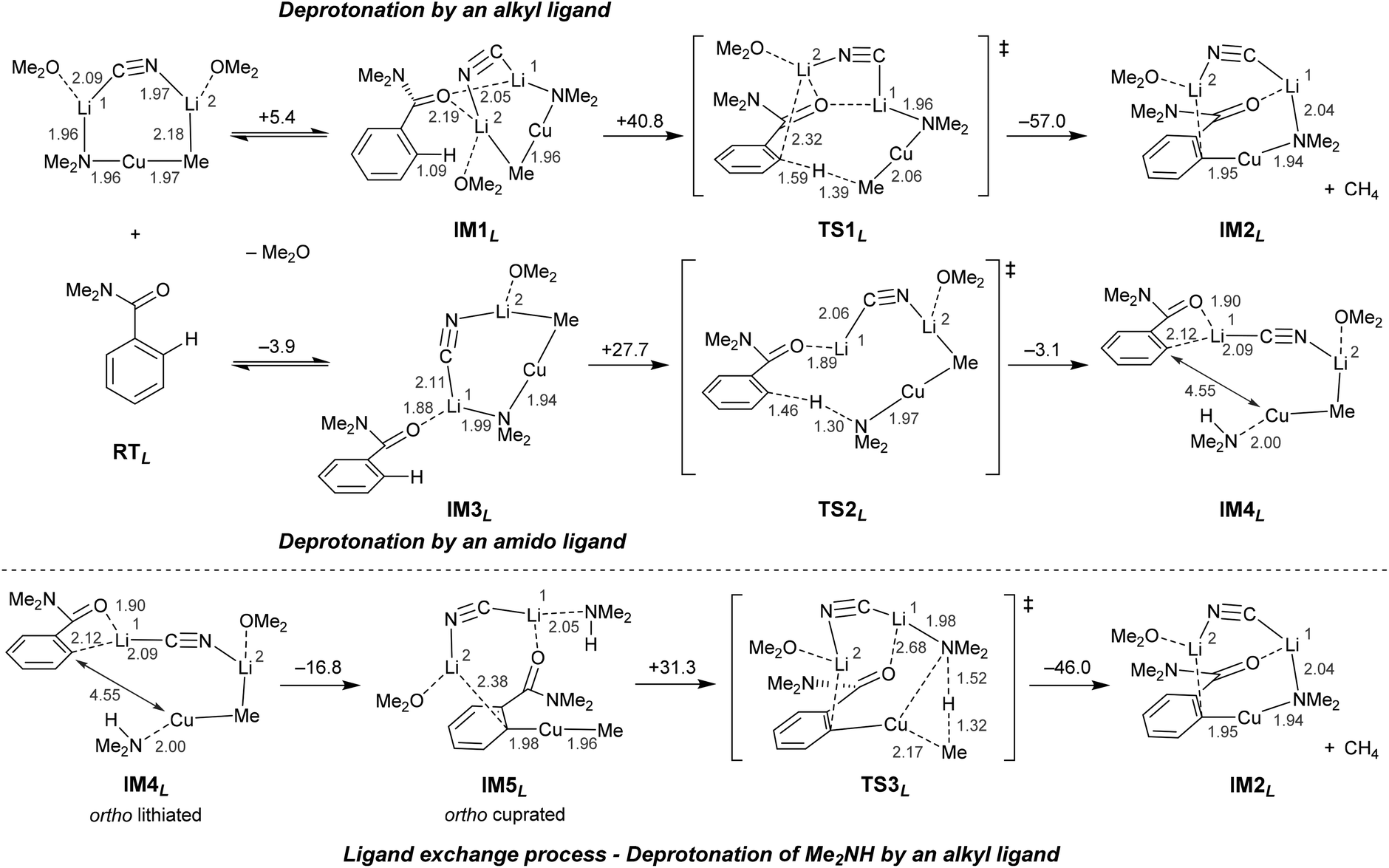 | ||
| Scheme 13 Proposed reaction pathways for the DoC of N,N-dimethylbenzamide using Lipshutz cuprate MeCu(NMe2)(CN)Li2·2OMe2. Energy changes and bond lengths at the B3LYP/6-31+G* and SVP (Cu) level of theory are shown in kcal mol−1 and Å, respectively. | ||
In common with the reaction pathways previously proposed for TMP-zincates2,47 and TMP-aluminates,46 two plausible pathways were discovered for regioselective DoM (Directed ortho Metallation). These differ in terms of whether the deprotonation step is triggered by the amido or the alkyl ligand in the cuprate. Although in Scheme 13 the formation of IM2L represents a thermodynamically favorable outcome, the pathway by which the alkyl ligand promotes the ortho deprotonation (IM1L–TS1L–IM2L) is kinetically unfavorable on account of there being a relatively high energy transition state (TS1L, ΔG‡ = 40.8 kcal mol−1). In contrast, TS2L represents the transition state immediately preceding amido ligand-promoted deprotonation (IM3L–TS2L–IM4L). While the activation barrier to TS2L formation is somewhat high (+27.7 kcal mol−1) relative to reaction pre-complex IM3L, it is still reasonably accessible and can be viewed as allowing metalated intermediate IM4L to form. The ligand exchange step (IM5L–TS3L–IM2L), a process seen in TMP-zincate chemistry47 but not in TMP-aluminate chemistry,46 has a higher activation barrier (+31.3 kcal mol−1). Taken together with the observation of heterocoupling processes achieved using organyl(amido)cuprate reagents (discussed in section 3.2) it is reasonable to conclude that the ligand exchange step would be difficult in DoC.
Interestingly, investigations into possible DoC pathways using the Lipshutz reagent MeCu(NMe2)(CN)Li2·2OMe2 have led to the identification of another ortho cupration pathway that involves an active monomeric Gilman complex (Scheme 12, bottom). Importantly, the monomeric structure-type of this so-called monoGilman species has been established experimentally using TMP-cuprates isolated from various Lewis basic media.34 Calculations that take the Lipshutz reagent MeCu(NMe2)(CN)Li2·2OMe2 as a starting point suggest that the monoGilman complexes IM1G and IM3G can be formed by the elimination of solvated LiCN (Scheme 14). Although IM1G is more stable than IM3G, its alkyl ligand fails to promote the deprotonative step in the reaction (IM1G–TS1G–IM2G), this sequence having a very high activation energy (+40.3 kcal mol−1). In contrast, the pathway IM3G–TS2G–IM4G, in which deprotonation is promoted by the amido ligand in the monoGilman base, has a very low activation barrier (+14.9 kcal mol−1 with respect to IM3G) en route to IM4G. Although the total activation energy (26.0 kcal mol−1) from RTL to TS2G is comparable to that from RTL to TS2L (23.8 kcal mol−1, in Scheme 13), it is noteworthy that themonoGilman-pathway does not require more than 15 kcal mol−1for any individual step, and this contrasts with the Lipshutz pathway (which requires more than 27 kcal mol−1 for the deprotonation step IM3L–TS2L). As with the Lipshutz system, quenching of the amine Me2NH (IM5G–TS3G–IM2G) appears to be unlikely to proceed on account of the rather high energy difference between RTL and TS3G (more than 28 kcal mol−1), though the activation barrier to reach TS3G is entropically permissible at 22.4 kcal mol−1.
 | ||
| Scheme 14 Reaction pathways for the DoC of N,N-dimethylbenzamide using Lipshutz cuprate MeCu(NMe2)(CN)Li2·2OMe2 and proceeding via a monoGilman intermediate. Energy changes and bond lengths at the B3LYP/6-31+G* and SVP (Cu) level of theory are shown in kcal mol−1 and Å, respectively. | ||
Oxidative ligand coupling involving the putative arylcuprate intermediate generated from the DoC of N,N-diisopropylbenzamide by MeCu(TMP)(CN)Li2 has been discussed in section 3.2 – with the use of nitrobenzene affording 2-methyl-N,N-diisopropylbenzamide (Scheme 15).6 This result clearly shows that the methyl ligand is retained by the copper centre in the arylcuprate intermediate and this bears out the theoretical work described here. A significant increase in the activation energy of deprotonation starting from a Gilman dimer has been found theoretically,32 again in agreement with experimental findings described in this section.
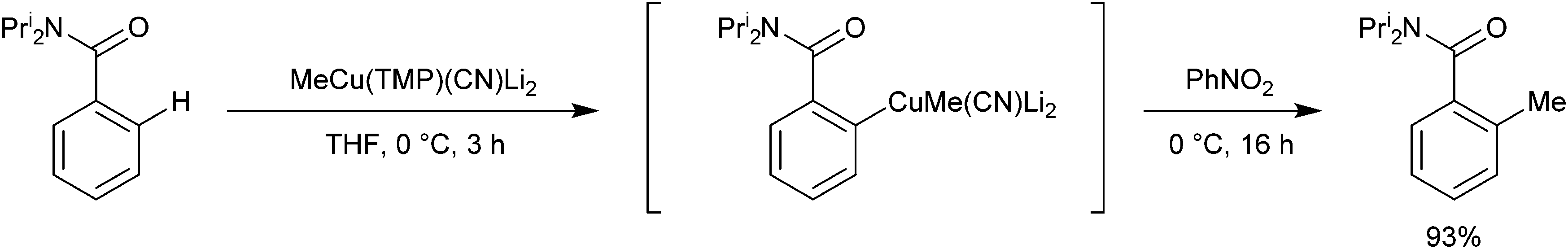 | ||
| Scheme 15 Oxidative ligand coupling employing an arylcuprate intermediate. | ||
The most recent structural studies into bis(amido)cuprates have revealed the formation of triangulated complexes best viewed as adducts between Gilman and Lipshutz-type monomers. These are discussed in detail in section 2.4. Moreover, the adduct [(DMP)2CuLi·OEt2]2LiBr 29 has been shown (see section 3.4) to be an effective DoC reagent.53 In the wake of recent DFT work, this has led to speculation that the adduct can deaggregate to give an active monoGilman base. In this vein, preliminary calculations on the simplified Lipshutz-type dimer of (Me2N)2Cu(Cl)Li2·OMe2 (LD) have revealed an enthalpic preference for eliminating a solvated lithium halide to form Gilman dimer GDvia adduct LMGM, though the effects of entropy mean that the system is a finely balanced one, as reflected by the very small values of ΔG (Scheme 16).
 | ||
| Scheme 16 The interconversion of a Lipshutz-type and a Gilman dimer (LD and GD) at B3LYP/631SVP level (ΔE [ΔG] are in kcal mol−1). “LiCl” = 1/4[LiCl(OMe2)]4 − OMe2. | ||
A further important step that it is necessary for LMGM to participate in if it is to be viewed as a contender for DoC is its dissociation to generate LM and the participation of this in a pre-reaction complex with, for example, N,N-dimethylbenzamide. Data point to this sequence increasing ΔG by 6.1 kcal mol−1 with the initial dissociation of LMGM raising ΔG by 15.4 kcal mol−1 (Scheme 17). In comparison to the IM3G–TS2G–IM4G pathway modelled above (see Scheme 14) for sourcing monoGilman agents from a Lipshutz cuprate starting material, it is clear that the recently reported cuprate adducts can also be viewed as viable sources of monoGilman active species. This is consistent with their demonstrated ability (discussed in section 3.4) as DoC reagents.53
 | ||
| Scheme 17 Conversion of adduct LMGM to a monoGilman-(N,N-dimethylbenzamide) complex (IM1G) at B3LYP/631SVP level. | ||
To summarize, DoM using Lipshutz-type amidocuprates or amidocuprate adducts can be viewed as proceeding via the formation of a monoGilman intermediate, and in organyl(amido)cuprate systems the alkyl ligand is retained by the copper centre in the intermediate arylcuprate species, indicating that ligand scrambling does not occur.
5. Conclusions
The relatively new field of directed aromatic ortho metallation using synergic reagents has been significantly expanded through the deployment of lithium cuprates. Though these systems are not new in themselves it is only recently that unambiguous structural evidence for their nature has been forthcoming. Subsequent to spectroscopic and theoretical investigations on cyanide-containing lithium cuprates, it was only in 1998 that the first clear evidence for the behaviour of the cyanide ion was presented.23,24 More recently, the crystallographic elucidation of 7 revealed that the classic Lipshutz system (TMP)2Cu(CN)Li2·THF incorporated bonds between cyanide and lithium, with the lower polarity metal being ligated only by two diametrically opposed amide ligands.6 Gratifyingly, this observation was concordant with calculations reported as long ago as 1995!61 Subsequent to this, it rapidly became apparent that the replacement of cyanide with halide (= X) in (TMP)2Cu(X)Li2·THF (X = Cl 9, Br 10, I 12) had no very significant effect on lithium cuprate structure. This allows cyanide- and halide-containing TMP-cuprates to be discussed in terms of the existence of a series of cuprates that can be regarded as, to a greater or lesser extent, Lipshutz-type. This can be interpreted in terms of the elimination of LiX to give a Gilman cuprate (which is highly active, see below), though we have also seen that if X is very soft then unreactive Gilman dimers may be isolated. It should also be noted, though, that the observation of a common Lipshutz-type cuprate dimer for various X groups is also consistent with the recent suggestion that differing reactivities of cyanide and iodide complexes may be defined by the structures of reaction intermediates rather than those of the substrates themselves.7 That said, the general inefficiency of Gilman reagents in DoC is clear. The implication of these data is that for DoC to proceed with optimum efficiency, a Lipshutz-type cuprate is necessary as the starting point.Recent calculations have added a remarkable new insight to the structural and synthetic data noted above. Namely, that whereas the employment of a Lipshutz-type cuprate is necessary for efficient DoC to proceed (as evidenced synthetically and also proposed on the basis of reaction coordinates plotted in the previous section), the active species is a Gilman monomer created in situ by the expulsion of LiX from the Lipshutz-type substrate. DFT analysis reveals a compelling reaction pathway for the elimination of LiX prior to formation of a pre-reaction complex between aromatic substrate and Gilman monomer and, interestingly, also suggests that the formation of a reactive Gilman monomer from the corresponding dimer is, in fact, relatively unlikely. Again, this is borne out by structural and synthetic work.
Most recently, the isolation and characterisation of a new class of lithium cuprate that is best viewed as representing an adduct between Lipshutz-type and Gilman monomers has presented a new way of envisaging the in situ generation of reactive cuprates for DoC. The abstraction of LiX from a Lipshutz-type dimer turns out to be enthalpically plausible. Moreover, calculations suggest that deaggregation of the resulting adduct and coordination of the formed (active) Gilman monomer by an aromatic substrate provides another compelling pathway for DoC. Overall, it is clear that, like Lipshutz-type dimers, the triangulated adducts reported here can be interpreted as plausible sources of the active cuprate species required for DoC.
The interpretation of theoretical data in terms of solid-state structures has afforded significant new insights into routes towards active monoGilman species. For example, adducts such as 28 have introduced new possibilities regarding the elimination of LiX from Lipshutz-type cuprates. Of course, these data have implications for our view of the solution behaviour of lithium cuprates and direct monitoring of the aggregation behaviour of these systems in solution, examples of which have been reported recently,42 will represent an increasingly significant part of forthcoming work. Attention is also turning to heteroleptic cuprates. Though these are reported for 19 and 20, heteroleptic bis(amido)cuprates represent the next target for structural elucidation and it is expected that they will be able to take two forms: the first will be mixed dimers or adducts such as (AM)2Cu(X)Li2·(AM′)2Cu(X)Li2 and (AM)2Cu(X)Li2·(AM′)2CuLi while the second will take the form of structures that incorporate heteroleptic monomers such as AMCu(AM′)(X)Li2 (where AM and AM′ are different amides). This work is rendered particularly important by the observation of distinctly contrasting amide orientations (cf. TMP in 142 and DMP in 28) and attempts to fabricate mixed DMP/TMP-cuprates are ongoing. Moreover, the ability of DMP to result in relatively inexpensive cuprating agents opens up new possibilities in terms of varying the amide components of cuprate bases (e.g., different ring size, substitution pattern, introducing chirality) in order to develop a full understanding of the steric requirements necessary for effective DoC.
Acknowledgements
Much of this work was supported by the U.K. EPSRC (EP/J500380/1). A.W. would like to acknowledge the graduate students who have contributed to the work in the past (Drs James Morey and Joanna Haywood) and also the GB Sasakawa and Daiwa Foundations and the Royal Society for support with travel and international collaboration. A.W. and M.U. thank the Japan Society for the Promotion of Science. M.U. and R.T. acknowledge Drs Shinsuke Komagawa, Shinya Usui, Ching-Yuan Liu, Yotaro Matsumoto and Keiichi Hirano, and Professors Yuichi Hashimoto, Shuji Yasuike and Jyoji Kurita for their contributions to the work. M.U. also thanks JSPS KAKENHI (S) (No. 24229011), Takeda Science Foundation, The Asahi Glass Foundation, Daiichi-Sankyo Foundation of Life Sciences, Mochida Memorial Foundation, Tokyo Biochemical Research Foundation, Foundation NAGASE Science Technology Development, Yamada Science Foundation and Sumitomo Foundation. R.T. acknowledges JSPS Grant-in-Aid for Young Scientists (A) (No. 25713001). F.C. and F.M. would like to acknowledge the graduate students who have contributed to the work (Dr Tan Tai Nguyen and Ms Nada Marquise) and the Agence Nationale de la Recherche (ACTIVATE program) for financial support. F.M. also thanks the Institut Universitaire de France and Rennes Métropole. Thanks go to Professor Paul Raithby and Dr Tom Robinson (Bath) for X-ray crystallographic studies. Calculations were performed on the RIKEN Super Combined Cluster (RSCC) and RIKEN Integrated Cluster of Clusters (RICC).References
- (a) H. W. Gschwend and H. R. Rodriguez, Org. React., 1979, 26, 1 CAS; (b) P. Beak and V. Snieckus, Acc. Chem. Res., 1982, 15, 306 CrossRef CAS; (c) V. Snieckus, Chem. Rev., 1990, 90, 879 CrossRef CAS; (d) T. G. Gant and A. I. Meyers, Tetrahedron, 1994, 50, 2297 CrossRef CAS; (e) M. Schlosser, Organometallics in Synthesis, ed. M. Schlosser, Wiley, New York, 2nd edn, 2002, ch. 1 Search PubMed.
- Y. Kondo, M. Shilai, M. Uchiyama and T. Sakamoto, J. Am. Chem. Soc., 1999, 121, 3539 CrossRef CAS.
- (a) R. E. Mulvey, Organometallics, 2006, 25, 1060 CrossRef CAS; (b) R. E. Mulvey, F. Mongin, M. Uchiyama and Y. Kondo, Angew. Chem., Int. Ed., 2007, 46, 3802 CrossRef CAS PubMed; (c) R. E. Mulvey, Acc. Chem. Res., 2009, 42, 743 CrossRef CAS PubMed; (d) B. Haag, M. Mosrin, H. Ila, V. Malakhov and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 9794 CrossRef CAS PubMed; (e) F. Mongin and M. Uchiyama, Curr. Org. Chem., 2011, 15, 2340 CrossRef CAS; (f) R. E. Mulvey, Dalton Trans., 2013, 42, 6676 RSC; (g) F. Mongin and A. Harrison-Marchand, Chem. Rev., 2013, 113, 7563 CrossRef CAS PubMed.
- (a) G. Wittig, Angew. Chem., 1958, 70, 65 CrossRef; (b) W. Tochtermann, Angew. Chem., Int. Ed. Engl., 1966, 5, 351 CrossRef CAS.
- A. Harrison-Marchand and F. Mongin, Chem. Rev., 2013, 113, 7470 CrossRef CAS PubMed.
- S. Usui, Y. Hashimoto, J. V. Morey, A. E. H. Wheatley and M. Uchiyama, J. Am. Chem. Soc., 2007, 129, 15102 CrossRef CAS PubMed.
- M. Neumeier and R. M. Gschwind, J. Am. Chem. Soc., 2014, 136, 5765 CrossRef CAS PubMed.
- H. Gilman, R. Jones and L. Woods, J. Org. Chem., 1952, 17, 1630 CrossRef CAS.
- H. House, W. Respess and G. Whitesides, J. Org. Chem., 1966, 31, 3128 CrossRef CAS.
- B. H. Lipshutz, R. S. Wilhelm and D. M. Floyd, J. Am. Chem. Soc., 1981, 103, 7672 CrossRef CAS.
- G. Whitesides, W. Fisher, J. San Filipo, R. Bashe and H. House, J. Am. Chem. Soc., 1969, 91, 4871 CrossRef CAS.
- P. G. Edwards, R. W. Gellert, M. W. Marks and R. Bau, J. Am. Chem. Soc., 1982, 104, 2072 CrossRef CAS.
- H. Hope, D. Oram and P. P. Power, J. Am. Chem. Soc., 1984, 106, 1149 CrossRef CAS.
- (a) G. van Koten and J. G. Noltes, J. Chem. Soc., Chem. Commun., 1972, 940 RSC; (b) G. van Koten, J. T. B. H. Jastrzebski, F. Muller and C. H. Stam, J. Am. Chem. Soc., 1985, 107, 697 CrossRef CAS.
- G. van Koten and J. G. Noltes, J. Am. Chem. Soc., 1979, 101, 6593 CrossRef CAS.
- H. Hope, M. Olmstead, P. Power, J. Sandell and X. Xu, J. Am. Chem. Soc., 1985, 107, 4337 CrossRef CAS.
- H. O. House, D. G. Koepsell and W. J. Campbell, J. Org. Chem., 1972, 37, 1003 CrossRef CAS.
- S. Bertz, J. Am. Chem. Soc., 1990, 112, 4031 CrossRef CAS.
- B. H. Lipshutz, S. Sharma and E. L. Ellsworth, J. Am. Chem. Soc., 1990, 112, 4032 CrossRef CAS.
- J. Snyder, D. Spangler, J. Behling and B. Rossiter, J. Org. Chem., 1994, 59, 2665 CrossRef CAS.
- B. Lipshutz and B. James, J. Org. Chem., 1994, 59, 7585 CrossRef CAS.
- J. Snyder and S. Bertz, J. Org. Chem., 1995, 60, 4312 CrossRef CAS.
- C. Kronenberg, J. Jastrzebski, A. Spek and G. van Koten, J. Am. Chem. Soc., 1998, 120, 9688 CrossRef.
- G. Boche, F. Bosold, M. Marsch and K. Harms, Angew. Chem., Int. Ed., 1998, 37, 1684 CrossRef CAS.
- D. R. Armstrong, D. Barr, R. Snaith, W. Clegg, R. E. Mulvey, K. Wade and D. Reed, J. Chem. Soc., Dalton Trans., 1987, 1071 RSC.
- D. R. Armstrong, D. Barr, W. Clegg, S. M. Hodgeson, R. E. Mulvey, D. Reed, R. Snaith and D. S. Wright, J. Am. Chem. Soc., 1989, 111, 4719 CrossRef CAS.
- P. Reiss and D. Fenske, Z. Anorg. Allg. Chem., 2000, 626, 1317 CrossRef CAS.
- K. Snégaroff, T. T. Nguyen, N. Marquise, Y. S. Halauko, P. J. Harford, T. Roisnel, V. E. Matulis, O. A. Ivashkevich, F. Chevallier, A. E. H. Wheatley, P. C. Gros and F. Mongin, Chem. – Eur. J., 2011, 17, 13284 CrossRef PubMed.
- N. Marquise, P. J. Harford, F. Chevallier, T. Roisnel, A. E. H. Wheatley, P. C. Gros and F. Mongin, Tetrahedron Lett., 2013, 54, 3154 CrossRef CAS PubMed.
- N. Marquise, P. J. Harford, F. Chevallier, T. Roisnel, V. Dorcet, A.-L. Gagez, S. Sablé, L. Picot, V. Thiéry, A. E. H. Wheatley, P. C. Gros and F. Mongin, Tetrahedron, 2013, 69, 10123 CrossRef CAS PubMed.
- C. M. P. Kronenburg, C. H. M. Amijs, J. T. B. H. Jastrzebski, M. Lutz, A. L. Spek and G. van Koten, Organometallics, 2002, 21, 4662 CrossRef CAS.
- S. Komagawa, S. Usui, J. Haywood, P. J. Harford, A. E. H. Wheatley, Y. Matsumoto, K. Hirano, R. Takita and M. Uchiyama, Angew. Chem., Int. Ed., 2012, 51, 12081 CrossRef CAS PubMed.
- R. P. Davies, Coord. Chem. Rev., 2011, 255, 1226 CrossRef CAS PubMed.
- J. Haywood, J. V. Morey, A. E. H. Wheatley, C.-Y. Liu, S. Yasuike, J. Kurita, M. Uchiyama and P. R. Raithby, Organometallics, 2009, 28, 38 CrossRef CAS.
- (a) S. Gambarotta, S. Strologo, C. Floriani and A. Chiesi-Villa, Organometallics, 1984, 3, 1444 CrossRef CAS; (b) A. M. Dattelbaum and J. D. Martin, Polyhedron, 2006, 25, 349 CrossRef CAS PubMed; (c) R. Fischer, H. Gorls and M. Westerhausen, Organometallics, 2007, 26, 3269 CrossRef CAS.
- (a) H. Hope, M. M. Olmstead, P. P. Power, J. Sandell and X. Xu, J. Am. Chem. Soc., 1985, 107, 4337 CrossRef CAS; (b) R. P. Davies and S. Hornauer, Eur. J. Inorg. Chem., 2005, 51 CAS.
- R. P. Davies and S. Hornauer, Chem. Commun., 2007, 304 RSC.
- (a) M. Niemeyer, Organometallics, 1998, 17, 4649 CrossRef CAS; (b) C.-S. Hwang and P. P. Power, Bull. Korean Chem. Soc., 2003, 24, 605 CrossRef CAS.
- (a) C.-S. Hwang and P. P. Power, J. Am. Chem. Soc., 1998, 120, 6409 CrossRef CAS; (b) C.-S. Hwang and P. P. Power, J. Am. Chem. Soc., 1999, 18, 697 CAS.
- R. Bomparola, R. P. Davies, T. Gray and A. J. P. White, Organometallics, 2009, 28, 4632 CrossRef CAS.
- R. M. Gschwind, Chem. Rev., 2008, 108, 3029 CrossRef CAS PubMed.
- R. P. Davies, S. Hornauer and P. B. Hitchcock, Angew. Chem., Int. Ed., 2007, 46, 5191 CrossRef CAS PubMed.
- (a) R. K. Dieter and M. Tokles, J. Am. Chem. Soc., 1987, 109, 2040 CrossRef CAS; (b) R. K. Dieter and T. W. Hanks, Organometallics, 1992, 11, 3549 CrossRef CAS; (c) B. E. Rossiter, M. Eguchi, G. Miao, N. M. Swingle, A. E. Hernandez, D. Vickers, E. Fluckiger, R. G. Patterson and K. V. Reddy, Tetrahedron, 1993, 49, 965 CrossRef CAS.
- M. John, C. Auel, C. Behrens, M. Marsch, K. Harms, F. Bosold, R. M. Gschwind, P. R. Rajamohanan and G. Boche, Chem. – Eur. J., 2000, 6, 3060 CrossRef CAS.
- S. F. Martin, J. R. Fishpaugh, J. M. Power, D. M. Giolando, R. A. Jones, C. M. Nunn and A. H. Cowley, J. Am. Chem. Soc., 1988, 110, 7226 CrossRef CAS.
- (a) M. Uchiyama, H. Naka, Y. Matsumoto and T. Ohwada, J. Am. Chem. Soc., 2004, 126, 10526 CrossRef CAS PubMed; (b) H. Naka, M. Uchiyama, Y. Matsumoto, A. E. H. Wheatley, M. McPartlin, J. V. Morey and Y. Kondo, J. Am. Chem. Soc., 2007, 129, 1921 CrossRef CAS PubMed; (c) J. García-Álvarez, E. Hevia, A. R. Kennedy, J. Klett and R. E. Mulvey, Chem. Commun., 2007, 2402 RSC; (d) H. Naka, J. V. Morey, J. Haywood, D. J. Eisler, M. McPartlin, F. García, H. Kudo, Y. Kondo, M. Uchiyama and A. E. H. Wheatley, J. Am. Chem. Soc., 2008, 130, 16193 CrossRef CAS PubMed.
- (a) M. Uchiyama, T. Miyoshi, Y. Kajihara, T. Sakamoto, Y. Otani, T. Ohwada and Y. Kondo, J. Am. Chem. Soc., 2002, 124, 8514 CrossRef CAS PubMed; (b) W. Clegg, S. H. Dale, E. Hevia, G. W. Honeyman and R. E. Mulvey, Angew. Chem., Int. Ed., 2006, 45, 2370 CrossRef CAS PubMed; (c) M. Uchiyama, Y. Matsumoto, D. Nobuto, T. Furuyama, K. Yamaguchi and K. Morokuma, J. Am. Chem. Soc., 2006, 128, 8748 CrossRef CAS PubMed; (d) Y. Kondo, J. V. Morey, J. M. Morgan, P. R. Raithby, D. Nobuto, M. Uchiyama and A. E. H. Wheatley, J. Am. Chem. Soc., 2007, 129, 12734 CrossRef CAS PubMed; (e) W. Clegg, J. Garcia-Álvarez, P. Garcia-Álvarez, D. V. Graham, R. W. Harrington, E. Hevia, A. R. Kennedy, R. E. Mulvey and L. Russo, Organometallics, 2008, 27, 2654 CrossRef CAS.
- J. Garcia-Álvarez, A. R. Kennedy, J. Klett and R. E. Mulvey, Angew. Chem., Int. Ed., 2007, 46, 1105 CrossRef PubMed.
- J. M. Cole, P. G. Waddell, A. E. H. Wheatley, G. J. McIntyre, A. J. Peel, C. W. Tate and D. J. Linton, Organometallics, 2014 DOI:10.1021/om500271p.
- P. P. Power, K. Ruhlandt-Senge and S. C. Shoner, Inorg. Chem., 1991, 30, 5013 CrossRef CAS.
- (a) R. Campbell, B. Conway, G. S. Fairweather, P. García-Álvarez, A. R. Kennedy, J. Klett, R. E. Mulvey, C. T. O'Hara and G. M. Robertson, Dalton Trans., 2010, 511 RSC; (b) B. Conway, P. García-Álvarez, A. R. Kennedy, J. Klett, R. E. Mulvey and S. D. Robertson, New J. Chem., 2010, 34, 1707 RSC.
- D. R. Armstrong, J. A. Garden, A. R. Kennedy, S. A. Leenhouts, R. E. Mulvey, P. O'Keefe, C. T. O'Hara and A. Steven, Chem. – Eur. J., 2013, 19, 13492 CrossRef CAS PubMed.
- P. J. Harford, A. J. Peel, J. P. Taylor, S. Komagawa, P. R. Raithby, T. P. Robinson, M. Uchiyama and A. E. H. Wheatley, Chem. – Eur. J., 2014, 20, 3908 CrossRef CAS PubMed.
- C. M. P. Kronenburg, J. T. B. H. Jastrzebski, J. Boersma, M. Lutz, A. L. Spek and G. van Koten, J. Am. Chem. Soc., 2002, 124, 11675 CrossRef CAS PubMed.
- H.-J. Gais, C. V. Rao and R. Loo, Chem. – Eur. J., 2008, 14, 6510 CrossRef CAS PubMed.
- T. T. Nguyen, F. Chevallier, V. Jouikov and F. Mongin, Tetrahedron Lett., 2009, 50, 6787 CrossRef CAS PubMed.
- T. T. Nguyen, N. Marquise, F. Chevallier and F. Mongin, Chem. – Eur. J., 2011, 17, 10405 CrossRef CAS PubMed.
- (a) T. Fujisawa, Y. Kurita, M. Kawashima and T. Sato, Chem. Lett., 1982, 1641 CrossRef CAS; (b) P. Kocienski and C. Barber, Pure Appl. Chem., 1990, 62, 1933 CrossRef CAS; (c) P. Ashworth, B. Broadbelt, P. Jankowski, P. Kocienski, A. Pimm and R. Bell, Synthesis, 1995, 199 CrossRef CAS PubMed.
- (a) G. Queguiner, F. Marsais, V. Snieckus and J. Epsztajn, Adv. Heterocycl. Chem., 1991, 52, 187 CrossRef CAS; (b) F. Mongin and G. Queguiner, Tetrahedron, 2001, 57, 4059 CrossRef CAS; (c) A. Turck, N. Plé, F. Mongin and G. Queguiner, Tetrahedron, 2001, 57, 4489 CrossRef CAS; (d) M. Schlosser and F. Mongin, Chem. Soc. Rev., 2007, 36, 1161 RSC; (e) F. Chevallier and F. Mongin, Chem. Soc. Rev., 2008, 37, 595 RSC; (f) P. C. Gros and Y. Fort, Eur. J. Org. Chem., 2009, 4199 CrossRef CAS.
- G. Queguiner, F. Marsais, V. Snieckus and J. Epsztajn, Adv. Heterocycl. Chem., 1991, 52, 187 CrossRef CAS.
- T. L. Stemmler, T. M. Barnhart, J. E. Penner-Hahn, C. E. Tucker, P. Knochel, M. Bohme and G. Frenking, J. Am. Chem. Soc., 1995, 117, 12489 CrossRef CAS.
- S. H. Bertz, G. Miao and M. Eriksson, Chem. Commun., 1996, 815 RSC.
| This journal is © The Royal Society of Chemistry 2014 |