 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation of a Eu(III) borate solid species from a weak Eu(III) borate complex in aqueous solution†
Juliane
Schott
ab,
Jerome
Kretzschmar
a,
Margret
Acker
*b,
Sascha
Eidner
c,
Michael U.
Kumke
c,
Björn
Drobot
a,
Astrid
Barkleit
ad,
Steffen
Taut
b,
Vinzenz
Brendler
a and
Thorsten
Stumpf
ad
aHelmholtz-Zentrum Dresden-Rossendorf (HZDR), Institute of Resource Ecology, 01314 Dresden, Germany
bTechnische Universität Dresden (TUD), Central Radionuclide Laboratory, 01062 Dresden, Germany. E-mail: margret.acker@tu-dresden.de
cUniversity of Potsdam, Institute of Chemistry (Physical Chemistry), 14476 Potsdam-Golm, Germany
dTechnische Universität Dresden (TUD), Division of Radiochemistry and Radioecology, 01062 Dresden, Germany
First published on 28th April 2014
Abstract
In the presence of polyborates (detected by 11B-NMR) the formation of a weak Eu(III) borate complex (lg β11 ∼ 2, estimated) was observed by time-resolved laser-induced fluorescence spectroscopy (TRLFS). This complex is a precursor for the formation of a solid Eu(III) borate species. The formation of this solid in solution was investigated by TRLFS as a function of the total boron concentration: the lower the total boron concentration, the slower is the solid formation. The solid Eu(III) borate was characterized by IR spectroscopy, powder XRD and solid-state TRLFS. The determination of the europium to boron ratio portends the existence of pentaborate units in the amorphous solid.
Introduction
Actinides such as Am or Pu will define the long-term radiotoxicity of spent nuclear fuel. Therefore, nuclear waste has to be enclosed for a time period of up to 106 years to protect the environment. There is worldwide consensus that nuclear waste repositories should be constructed in deep geological formations (salt, argillaceous rock, and granite) to store the waste.Borates occur in significant amounts in such suitable geological formations, particularly in salt formations. For instance, the elemental analysis of brines of the WIPP site (Waste Isolation Pilot Plant near Carlsbad, New Mexico, USA) showed boron concentrations up to 160 mM.1 Accordingly, this has to be expected in other salt-based repository formations, like in Germany, too. In salt-based geological formations boron compounds can occur dissolved in evaporite enclaves or as boron containing minerals, such as sassolite, borax, ulexite and colemanite.2 Furthermore, boron containing materials from nuclear technological processes stored in a nuclear waste repository will be not negligible and have to be considered. For instance, borosilicate glass coquilles in which the high-level radioactive waste is vitrified and containers for the direct storage of spent nuclear fuel which may carry the remains of boron that stems from the cooling water of the nuclear power plant will be part of the inventory of a nuclear waste repository.
One scenario to be evaluated within the safety and risk assessment of a nuclear waste repository is the contact of the repository inventory with water. Dissolution processes of the stored inventory (container material and radioactive waste) and host rock components are to be expected, followed by various physicochemical reactions of the mobilized species (complexation, sorption, formation of solid phases, etc.). These basic processes have to be understood in order to provide a stable and safe repository for nuclear waste over the time scale of 106 years.
The corrosion of the waste containers during their storage in the repository for thousands of years could lead to the release of radionuclides and boron containing compounds. Corrosion processes are also estimated for the coquilles containing the vitrified radioactive waste. Due to the water-induced corrosion of the borosilicate glass matrix dissolved radionuclide species and locally high boron contents have to be considered.
Until now, the interactions of radionuclides, especially trivalent actinides, with boric acid and (poly)borates have been investigated only insufficiently.
Borkowski et al. were the first to investigate the complexation between Nd(III) as an analog for trivalent actinides (e.g., Am(III), Pu(III), and Cm(III)) and tetraborate under WIPP conditions.1 They found a Nd(III) borate complex (lg β11 = 3…4), which indicates that an accordant actinide(III) borate species could compete with the actinide carbonate complexation.1,3 In consequence, under WIPP conditions (up to 160 mM borate, pH = 8…9), this actinide(III) borate complex would be a predominant actinide species.1,3
Furthermore, synthetic solid actinide borate phases are known. For instance, Polinski et al. and Wang et al. synthesized different solid borates of trivalent actinides (and lanthanides) via hydrothermal syntheses.4–8 The obtained (poly)borates have complex structures, which depend strongly on the experimental conditions and the used metal.
However, more fundamental data are required for a better understanding of the actinide/lanthanide(III)–B(OH)3/(poly)borate system.
This work introduces the results of the investigated europium (poly)borate complexation (similarly to neodymium, europium was used as a chemical analog for trivalent actinides) and provides evidence for a new europium solid phase in which (poly)borates are involved. The actinide/lanthanide(III) mobilization by (poly)borates will be discussed.
Experimental
Chemicals and materials
Chemicals of analytical grade and deionized water were used for the preparation of solutions. Boric acid (Merck), B(OH)3, was used to adjust the total boron concentration. All solution samples were prepared at 0.1 M ionic strength (NaClO4 (Merck)). A 0.03 M Eu(III) stock solution was prepared by dissolving Eu2O3 (Aldrich) in 0.1 M HClO4. The pH measurements were carried out with a glass electrode, which was calibrated with buffer solutions (NIST/PTB standard buffers). The pH of the solutions was adjusted with NaOH or HClO4 (Merck). Elemental analyses were carried out with ICP-MS (Elan 9000, Perkin Elmer) and AAS (AAS-4100, Perkin Elmer). For the preparation of the solid Eu(III) borate (see below) EuCl3·6H2O (Aldrich) was used.Boron speciation studies
Samples with variable total boron concentrations (0.02 M–0.7 M) were prepared under ambient conditions (T = 22 °C, pCO2 = 10−3.5 atm) at pH 5 and pH 6. The high total boron concentrations (up to 0.7 M) were used to induce the formation of polyborates in appropriate amounts to influence the Eu(III) speciation investigated in subsequent complexation studies. After the dissolution of boric acid the samples were stored for four days to establish the boric acid/polyborate equilibrium. The samples were measured by means of 11B-NMR spectroscopy.Eu(III) borate complexation studies
Samples with variable total boron concentrations were prepared as described above at ∼pH 6. Then 2 mL of the boron solution were transferred into a quartz cuvette and the 0.03 M Eu(III) stock solution was added to adjust a total Eu(III) concentration of 3 × 10−5 M. The samples were titrated at the same day (to exclude precipitation) from ∼pH 6 down to ∼pH 2 by adding appropriate amounts of HClO4. After each titration step a stationary europium luminescence spectrum was recorded.Eu(III) borate solid formation studies
Samples with variable total boron concentrations were prepared as described above at pH 5 and pH 6. With the 0.03 M Eu(III) stock solution a total Eu(III) concentration of 3 × 10−5 M was set. Directly after the addition of the Eu(III) stock solution and then during the next 427 days, stationary and time-resolved europium luminescence spectra were recorded for each sample. Membrane filtration (1.2 μm and 0.2 μm pore size) of the samples and a subsequent determination of the europium content in the filtrates by ICP-MS were carried out to provide evidence of the solid formation.Synthesis of the Eu(III) borate solid
A solution containing 0.7 M total boron was prepared at pH 6 as described above. Then solid EuCl3·6H2O was added to adjust 10 mM total Eu(III) concentration. A white solid precipitates rapidly. The solid was stored in its solution for three weeks and then separated from the liquid phase by centrifugation. The solid was washed several times with deionized water and then dried by lyophilization.The ratio of europium, boron and sodium in the Eu(III) borate solid was determined by dissolving a defined amount of the solid in a defined volume of concentrated nitric acid and performing a subsequent analysis of the europium, boron and sodium content in this solution by ICP-MS (Eu, B) and AAS (Na). The sodium content was determined because there is evidence of a sodium pentaborate phase (see discussion of powder XRD results below) as a byproduct of the Eu(III) borate precipitation. In fact, an enhanced sodium content in the Eu(III) borate solid was detected.
11B nuclear magnetic resonance spectroscopy (11B-NMR)
11B-NMR spectra of boron containing solutions were recorded on a Varian Unity Inova 400 spectrometer with a field strength of 9.4 T and a corresponding 11B resonance frequency of 128.4 MHz with a 5 mm broadband probe. The 11B chemical shifts (δ) are referenced externally with respect to BF3 etherate in CDCl3. A 5 mm NMR tube (quartz), containing the aqueous solution and a D2O filled coaxial insert for deuterium lock, was used.Time-resolved laser-induced fluorescence spectroscopy (TRLFS)
All measurements were carried out using a time gated detection mode to avoid contributions from strayed and scattered light and to resolve the temporal characteristics of the Eu luminescence.Measurements of the europium containing solutions/suspensions were carried out with a Nd:YAG-OPO laser system (Continuum). Europium luminescence spectra of the stirred solutions/suspensions were recorded with a constant excitation wavelength of 394 nm, a time window of 1 ms, a pulse energy of 2–3 mJ and an optical multichannel analyzer (spectrograph (Oriel MS 257) and iCCD camera (Andor iStar)). Recording conditions for stationary spectra: wavelength range 565 nm–650 nm, 1200 line mm−1 grating, 0.2 nm resolution, 3000 accumulations. Recording conditions for time-resolved spectra: wavelength range 440 nm–780 nm, 300 line mm−1 grating, 0.7 nm resolution, 100 accumulations, delay time steps 15 μs–90 μs.
The solid-state TRLFS measurements at room temperature (22 °C) were performed using a Nd:YAG-OPO system as an excitation source (Nd:YAG: Quanta Ray, Spectra Physics; OPO: Flexi Scan, GWU-Lasertechnik) operated at 20 Hz repetition rate. The solid Eu(III) borate sample was placed in a self-made sample holder. The resulting luminescence emission was collected with a set of lenses and focused into a spectrograph (MS 257, LOT Oriel) equipped with an iCCD camera (iStar DH720, Andor Technology).
For the solid-state TRLFS measurements at low temperature (T < 5 K) a solid Eu(III) borate sample was placed in a copper sample holder on top of a cooling head. The low temperature was achieved using a closed cycle helium cryostate (Helium compressor unit CKW-21, Sumitomo Heavy Industries Ltd; Turbolab 80, Oerlikon Leybold Vacuum; Model 331 Temperature Controller, Lakeshore). For exciting the Eu(III) via the 5D0←7F0 transition a dye laser (Cobra Stretch, Sirah Laser- und Plasmatechnik) was used. Tuning the excitation wavelength in the spectral range 580 nm ± 5 nm was possible with Pyrromethene 597 (Sirah Laser- und Plasamatechnik) as a laser dye, which was excited by the second harmonic output of a Nd:YAG laser (Quanta Ray, Spectra Physics) operating at a repetition rate of 10 Hz and a typical pulse length of 8 ns. The excitation light was guided through an optical fiber to the Eu(III) sample. The emitted luminescence light was transferred through the same optical fiber to a spectrograph (Shamrock SR-303i, Andor Technology) attached to an iCCD camera (iStar DH 720, Andor Technology).
Infrared spectroscopy (IR)
FT-IR spectra were recorded on a Bruker Vertex 70v Fourier transform infrared spectrometer in the range 7500 cm−1–370 cm−1 with a resolution of 4 cm−1. The samples were prepared as KBr pellets.Powder X-ray diffraction (powder XRD)
The experiments were performed at the PETRA III synchrotron radiation source at DESY Hamburg, Germany (High Resolution Powder Diffraction, P02.1).9 Synchrotron radiation with an energy of 60 keV (corresponding to λ = 0.207 Å) was used. Diffraction patterns were collected in Debye–Scherrer-geometry with a PerkinElmer XRD 1621 area detector. The diffraction patterns were processed with the software FIT2D10 employing a CeO2 standard for calibration.Data analysis
Different luminescence transition bands characterize the europium luminescence spectrum. In particular the 5D0→7F0 (at ∼578 nm; forbidden for the aquo ion Eu(III)aq), 5D0→7F1 (at ∼592 nm) and 5D0→7F2 (at ∼616 nm) transition bands were analyzed. TRLFS spectra were analyzed with the software Origin™ (version 7.5G, OriginLab Corporation). Stationary and time-resolved raw spectra were baseline corrected. Stationary luminescence spectra were normalized to the 5D0→7F1 transition band because the luminescence to this transition is independent of the chemical environment of europium.11The luminescence lifetimes were determined according to the exponential decay equation, eqn (1):
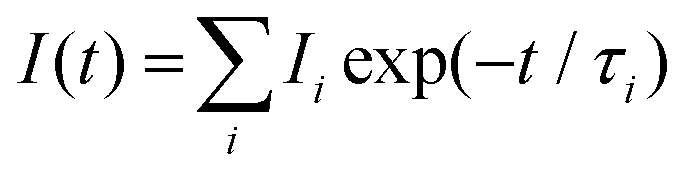 | (1) |
Depending on the characteristics of the luminescence decay, monoexponential or biexponential decay equations (see eqn (1)) were used to fit the luminescence decay curves.
The luminescence lifetime τ of europium depends on the number of water molecules in the first coordination shell of europium. They act as luminescence quenchers. In general, their substitution by other ligands leads to an increase of the luminescence lifetime τ, except for hydroxide ligands. The luminescence lifetime τ and the amount of water molecules in the first coordination shell of Eu(III) are correlated by an empirical equation,12,13eqn (2):
| nH2O ± 0.5 = 1.07/τ − 0.62 | (2) |
From nH2O, further information about the chemical environment of europium is deducible. The luminescence lifetime τ of the europium aquo ion in water is specified with 110 μs ± 5 μs corresponding to 8–9 water molecules.14–16
Speciation calculations were carried out using the program HySS (Version 4.0.31).17
The complex formation constant and the single luminescence spectrum of the Eu(III) (poly)borate complex were determined with the calculation program HypSpec.18 The application of HypSpec has been demonstrated in several studies.19–22 The data set for the fitting procedure contained as parameters the model, total metal and ligand concentrations, pH, protonation constant of the ligand, the measured europium luminescence spectra and the Eu(III) aquo ion luminescence spectrum as the known spectrum, respectively. Simplifications and approximations for the calculation of this complexation system are described in the section “Results and discussion”.
Time-resolved emission (or luminescence) spectra of the Eu(III) borate complexation system and Eu(III) borate solid were analyzed by parallel factor analysis (PARAFAC),23 successfully applied before in a broad variety of research fields.24–26
Results and discussion
Boron speciation in aqueous solution
An understanding of the aqueous B(OH)3/(poly)borate speciation is an essential prerequisite to interpret the observed complexation and solid formation when europium is present in the borate system.Boric acid, B(OH)3, is a weak acid with a high dissociation constant (pKa = 8.98, I = 0.1 M)27 and acts as a Lewis acid (hydroxide acceptor), eqn (3):
| B(OH)3 + H2O ⇌ B(OH)4− + H+ | (3) |
Above a total boron concentration of 25 mM it forms polyborates in the pH range from 4 to 13.28 This polymerization generates tri-, tetra- and pentaborates and even higher condensed species.27,29 In the past, efforts have been made to clarify this polymerization process by potentiometric titration, IR-, Raman- and NMR-spectroscopy, showing that the aqueous chemistry of boric acid is highly complex.27,28,30–38
Ingri et al. published formation constants for the polyborates B3O3(OH)4−, B5O6(OH)4−, B4O5(OH)42− and B3O3(OH)52−.27,28,31 With these data a B(OH)3–polyborate speciation can be calculated. A speciation diagram is shown for the pH range 0 to 14 and a total boron concentration of cB,total = 0.7 M (Fig. 1).
 | ||
| Fig. 1 B(OH)3–polyborate speciation for cB,total = 0.7 M, I = 0.1 M. | ||
Up to pH 6 (the investigated pH range of this work), only the boron species B(OH)3, B3O3(OH)4− (triborate) and B5O6(OH)4− (pentaborate) are expected to be present in solution. The calculated distribution of B3O3(OH)4− and B5O6(OH)4− with the data of Ingri et al.27,28,31 as a function of cB,total and pH is shown in Fig. 2.
 | ||
| Fig. 2 Distribution of different borate species as a function of cB,total and pH, I = 0.1 M. | ||
The polyborate species B3O3(OH)4− and B5O6(OH)4− appear in significant amounts at higher cB,total between pH 4 and pH 5. At pH 6 and cB,total = 0.7 M these polyborate species represent around 3.5% of the total boron speciation. This corresponds to a maximum polyborate concentration of ∼0.025 M.
11B-NMR is well applicable to identify boron species in aqueous solution.32 Borates consist of at least one tetra-coordinated (tetrahedral) boron center [BO4] and tri-coordinated (trigonal planar) boron centers [BO3] (number depending on the (poly)borate species). Since the nucleus is sensitive to its local electronic environment, the boron atoms in [BO3] and [BO4] can easily be distinguished by their well separated NMR signals. Due to the higher coordination and, thus, negative charge, the [BO4] boron nucleus is more shielded by the increased electronic density as compared to the [BO3] boron nucleus. Its 11B chemical shift occurs around 1 ppm (for instance observable for B(OH)4−),32 whereas the NMR signal of [BO3], like in B(OH)3, occurs around 19 ppm.32 As [BO4] possesses a spherical and more symmetric electronic environment, the signal is quite narrow. Because of the non-spherical electronic environment around [BO3], the resulting electric field gradient causes a fast relaxation of the boron nucleus being quadrupolar. This leads to a considerable line broadening.
In the case of the pentaborate (at δ = 1.2 ppm; signal assignment according to Hertam et al.32) these effects may disallow the observation of the [BO3] signal. Otherwise the [BO3] signal of the pentaborate could be overlapped by the huge signal of B(OH)3. The [BO4] unit of the pentaborate is fixed by the molecular structure. This results in NMR signal properties, i.e., chemical shift and line width, quite similar to that of the monoborate anion. To interpret the position of the NMR signal of the triborate species (at δ = 13 ppm; signal assignment according to Hertam et al.32) an intramolecular OH group transfer (site exchange between [BO3] and [BO4]) has to be considered. This causes a broad signal at the population-weighted average chemical shift of the nuclei in the two sites.
At pH 5 and up to 0.5 M total boron concentration no polyborate species were detected by 11B-NMR (Fig. 3a). The only boron species found was the undissociated boric acid at δ = 19.4 ppm. Above 0.5 M B(OH)3 small amounts of polyborates occur. At pH 6 and total boron concentrations >0.4 M three boron species can be observed (Fig. 3b). The described signals at δ = 19.4 ppm, 13.3 ppm and 1.2 ppm (see above) are assigned to boric acid B(OH)3, the triborate species B3O3(OH)4− and the pentaborate species B5O6(OH)4−, respectively. With increasing total boron concentration the amount of these polyborate species increased. Besides the described species, no further polyborates were formed. Moreover, a sample with a content of 0.7 M total boron aged for six months showed no differences in the 11B-NMR spectrum compared to a freshly prepared one (Fig. 3b). Thus, the B(OH)3–(poly)borate speciation remained stable.
 | ||
| Fig. 3 11B-NMR spectra (normalized) of solutions containing variable amounts of total boron (0.2 M to 0.7 M, step size 0.1 M) (a) at pH 5 and (b) at pH 6; in each case I = 0.1 M. The insets show expansion of the polyborate region. The 11B-NMR spectrum in grey shows a six months aged solution containing 0.7 M total boron concentration at pH 6, I = 0.1 M. | ||
Eu(III) speciation in the absence and presence of boron species (B(OH)3, (poly)borates)
The experiments of this work were carried out in the acidic pH range to make the complexation studies in the Eu(III)–B(OH)3/(poly)borate system less difficult. Under these conditions strong Eu(III) hydroxo (carbonato) complexes as well as the formation of solid Eu(III) hydroxides (and carbonates) can be excluded. In the absence of boron species the Eu(III) speciation up to pH 6 is exclusively dominated by the Eu(III) aquo ion (Eu(III)aq). Hydrolysis of Eu(III) and the reaction with carbonate at pH < 6 are still negligible (at most 1% of the total europium). Furthermore, the polyborate equilibrium is still complex in the acidic pH range but simpler than in the alkaline pH range (Fig. 1 and 2).The Eu(III) speciation in aqueous solution can be investigated with high selectivity and sensitivity using the time-resolved laser-induced fluorescence spectroscopy (TRLFS). In the presence of boron (cB,total = 0…0.7 M) at pH 6 stationary and time-resolved luminescence spectra of Eu(III) were measured. With increasing total boron concentration, changes in the europium luminescence spectra and emission lifetimes τ occurred. The intensities of the 5D0→7F0 and 5D0→7F2 luminescence bands increased, pointing to a reduced overall symmetry of the Eu(III) complex(es) in comparison with the Eu(III)aq (Fig. 4a). The luminescence lifetime τ of europium rises up to ∼150 μs at the highest investigated total boron concentration (0.7 M), which corresponds to a removal of 2–3 water molecules from the first coordination shell of europium (Table 1).
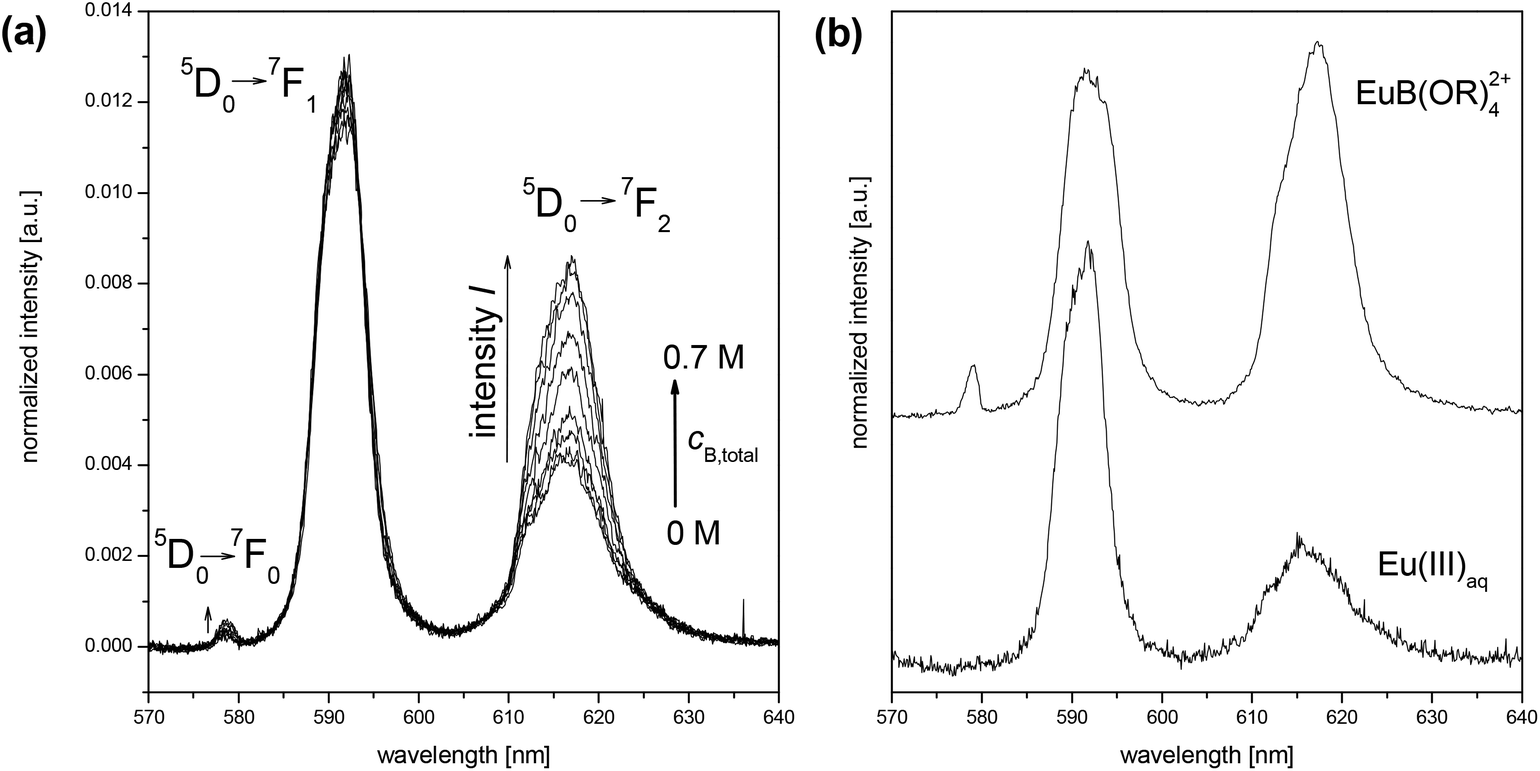 | ||
| Fig. 4 (a) Europium luminescence spectra at pH 6 as a function of cB,total, 3 × 10−5 M Eu(III), I = 0.1 M; (b) single spectra of Eu(III)aq (measured) and the Eu(III) borate complex, EuB(OR)42+ (calculated by HypSpec;18 R = H and/or [BO3] units). | ||
| c B, total [M] | τ [μs] | n H2O |
|---|---|---|
| 0 | 113.0 ± 1.0 | 8–9 |
| 0.02 | 113.5 ± 0.9 | 8–9 |
| 0.1 | 113.4 ± 0.8 | 8–9 |
| 0.2 | 115.0 ± 1.0 | 8–9 |
| 0.3 | 120.1 ± 0.5 | 8–9 |
| 0.4 | 126.8 ± 0.7 | 7–8 |
| 0.5 | 134.5 ± 0.6 | 7–8 |
| 0.6 | 142.9 ± 0.6 | 6–7 |
| 0.7 | 149.4 ± 0.7 | 6–7 |
These findings clearly show a europium complexation by the in solution existing boron species. Up to a total boron concentration of ∼0.1 M, where no or almost none polyborates exist, no changes in the luminescence parameters of europium in comparison with Eu(III)aq occurred. Hence, boric acid forms no complexes with europium. At higher total boron concentrations polyborates occur and concurrently changes in the luminescence parameters of europium were observed. Obviously, an interaction between polyborates and Eu(III) occurs which influences the Eu(III) speciation.
Additionally, the influence of polyborates was investigated for different total boron concentrations as a function of pH. For illustration the F1/F2 ratio (intensity ratio of the 5D0→7F1 and 5D0→7F2 luminescence bands) is plotted against pH (Fig. 5).
 | ||
| Fig. 5 F1/F2 ratios from TRLFS pH titration of solutions containing 3 × 10−5 M Eu(III) and variable cB,total, I = 0.1 M. | ||
The Eu(III) polyborate complexation was observed in the pH range ∼4.2 to 6 (highest investigated pH) at the highest investigated total boron concentration (0.7 M). Below pH ∼4.2, the polyborate concentration is too low to influence the europium speciation. This is in good agreement with the calculated boron speciation (Fig. 2).
We tried to determine a complexation constant of the Eu(III) borate species from the TRLFS pH titration of solutions containing Eu(III) and variable amounts of total boron (Fig. 5). This, however, proved to be difficult, because several polyborates coexist. The separation of their complexes with europium was not possible.
Therefore, a simpler model was used at least to estimate the order of magnitude of the Eu(III) (poly)borate complexation constant. It was assumed that all polyborate species with the structural unit “B(OR)4−” (R = H and/or [BO3] units) show similar complexation properties with similar values for the complexation constants. Therefore, a model species “B(OR)4−” was used for the estimation.
The PARAFAC of the time-resolved europium spectra as a function of cB,total (Table 1) showed that until 0.7 M total boron concentration, only the Eu(III)aq and one europium complex are existent. Therefore, only the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Eu(III) borate complex, EuB(OR)42+, has to be considered in the calculations. The complexation constant for this complex is expressed in eqn (4):
:1 Eu(III) borate complex, EuB(OR)42+, has to be considered in the calculations. The complexation constant for this complex is expressed in eqn (4):
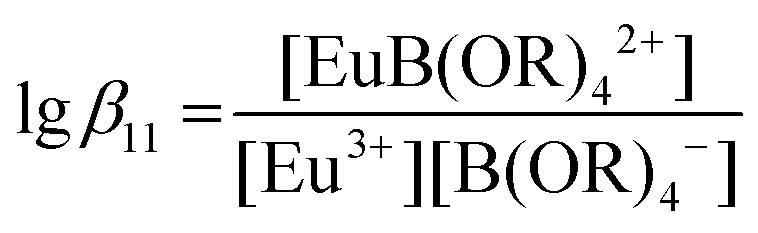 | (4) |
The equilibrium concentration of the model species “B(OR)4−” is the sum of the equilibrium concentrations of the (poly)borate species. This sum was calculated from the speciation data of Ingri et al.27,28,31
The analyses of the titration data corresponding to different total boron concentrations using the concentration of the model species “B(OR)4−” as a function of the pH yielded the complexation constants for the Eu(III) borate complex, EuB(OR)42+ (Table 2).
| Data set | c B,total [M] | lg β11 |
|---|---|---|
| 1 | 0.4 | 2.29 |
| 2 | 0.5 | 2.13 |
| 3 | 0.5 | 2.05 |
| 4 | 0.6 | 1.97 |
| 5 | 0.6 | 2.03 |
| 6 | 0.7 | 1.83 |
| 7 | 0.7 | 1.83 |
| 2.02 ± 0.33 (2σ) (averaged value) |
For the EuB(OR)42+ complex an averaged value of lg β11 = 2 was estimated. This value illustrates the order of magnitude of Eu(III) (poly)borate complexes and shows that these complexes are quite weak. The single spectrum of the EuB(OR)42+ complex extracted from the measured sum spectra is shown in Fig. 4b.
To the best of our knowledge there is only one publication declaring a Ln(III) borate complexation constant. Borkowski et al. determined in their pioneering work a complexation constant for a Nd(III) tetraborate complex with lg β11 ∼ 3…4 as a function of ionic strength.1 This constant differs from the result of this work, which might be due to different reasons.
Borkowski et al. determined the complexation constant at about pH 8.6 by solubility experiments. The tetraborate species, which was the supposed complexing agent in their work, has two binding sites to interact with the metal ion. Therefore, the Ln(III) complex with tetraborate is probably stronger than the complexes with polyborates containing only one binding site, as investigated in this work. Additionally, Ln(III) hydroxoborate species could exist under alkaline conditions. For instance, in the inorganic Cm(III)–NO3 system ternary Cm(III)–OH–NO3 species were found under alkaline conditions.39 It is imaginable that the attachment of hydroxo ligands onto the metal borate complex strengthens the Ln/An(III) complexation.
Independent of the previous discussion there is a need for clarification of the supposed borate speciation published by Borkowski and co-workers. Na2B4O7·10H2O was used in their work to prepare tetraborate containing solutions. But it is known that Na2B4O7 is not stable in aqueous solution and dissociates into B(OH)3/B(OH)4−. From boric acid a new polyborate equilibrium with further polyborates is regulated. Therefore, the borate speciation would involve more complexes than the supposed tetraborate species. Even if the polyborate concentration is low, due to the dissociation of the tetraborate molecule more complexing borate molecules (B(OH)4−) are generated than expected. This would lead to an underestimation of the effective borate concentration in their experiments. Therefore, the complexation constant for a Nd(III) borate would be smaller than the reported one.
Furthermore, Hinz et al. carried out solubility experiments under the same conditions. A clear solubility decrease of Nd(III) in the presence of borate was determined.40 This is contradictory to the results of Borkowski et al. and indicates the formation of a further solid phase in which borates are involved. Therefore, the An/Ln(III)–borate system is much more difficult under alkaline conditions and a multitude of reactions occur which are not clear and actually not manageable for complexation studies.
The investigations of this work were carried out under acidic conditions. Strongly competing reactions (for instance hydrolysis of Eu(III)) can be excluded. The calculation of the equilibrium (poly)borate concentrations under acidic conditions is more reliable, because the amount of different polyborate species is reduced. The used TRLFS is a species sensitive method that allows us to observe the europium (poly)borate species even in the presence of only small amounts of complexing (poly)borates in the investigated pH range. Because the complexed europium species was observed directly by spectroscopy the derived complexation constant for the Eu(III)-(poly)borate complex seems to be reliable.
Nevertheless, the work of Borkowski et al. demonstrates the importance of studying the actinide–borate system, because borate compounds are not negligible in a nuclear waste repository.
Formation of a Eu(III) borate solid species in aqueous solution
Europium and boron containing solutions at pH 5 and pH 6 (see the Experimental section) were investigated by TRLFS as well as membrane filtration and subsequent determination of the europium content in the filtrates using ICP-MS.Some days after the preparation of the boron and europium containing solutions at pH 6 remarkable changes in the europium luminescence spectra and lifetimes τ were observed in some samples. In these spectra the luminescence bands were characteristically split (Fig. 6a) and the luminescence lifetimes τ distinctly increased (Fig. 6b) in comparison with the luminescence spectra and lifetimes measured directly after the sample preparation. These changes indicate the formation of a new europium species. In contrast, in the same investigation period at pH 5 no changes in the europium emission spectra and lifetimes of freshly prepared samples in comparison with the aged samples were observed.
 | ||
| Fig. 6 (a) Formation progress of the Eu(III) borate solid species for a solution containing 3 × 10−5 M Eu(III), cB,total = 0.7 M, I = 0.1 M at pH 6; (b) luminescence lifetime τ of europium observed with time. | ||
Membrane filtrations of the europium and boron containing solutions at pH 5 and pH 6 through different pore sizes were carried out. The investigation of the filtrates of the solutions with the characteristically split europium luminescence spectra by ICP-MS showed a removal of europium. Therefore, the formation of a solid species can be demonstrated. In solutions at pH 6 with low total boron concentrations (<0.3 M) and pH 5 with variable total boron concentrations up to 0.7 M no formation of a solid species was detectable by membrane filtration and subsequent determination of the europium content in the filtrates using ICP-MS.
No other europium solids (hydroxides, carbonates, hydroxycarbonates) are thermodynamically stable under the used experimental conditions; thus, the observed solid must be a solid Eu(III) borate species. The described Eu(III) (poly)borate complex (see above) is supposed to be the precursor of this solid. In solutions at pH 5 with variable total boron concentrations up to 0.7 M and pH 6 with low total boron concentrations (<0.3 M) no precipitations were observed. This indicates that the concentration of the polyborates and, therefore, the amount of the Eu(III) polyborate complex is too low to induce the Eu(III) borate precipitation.
The progress of the solid formation of the Eu(III) borate was investigated by TRLFS in solutions containing 3 × 10−5 M Eu(III) and variable concentrations of total boron (0.2 M…0.7 M) at pH 6. A splitting of the luminescence bands corresponding to the 5D0→7F1 and 5D0→7F2 transitions during the formation progress of the solid species occurred till a spectrum characteristic of the Eu(III) borate solid was reached (Fig. 6a). In the transition phase (see Fig. 6b) a biexponential luminescence decay curve was determined. A short luminescence lifetime (120 μs…150 μs) for the dissolved Eu(III) species and a long lifetime (600 μs…700 μs) for the solid Eu(III) species were identified (Fig. 6b, Fig. S1, S2†). After a certain time, depending on the total boron concentration, only the long europium luminescence lifetime τ (600 μs…700 μs) attributed to the Eu(III) borate solid species was observed. It shows the complete formation of the solid, Fig. S1.† The solid Eu(III) borate seems to be stable over a long time. The long lifetime of 600 μs–700 μs characteristic of this europium solid was determined even after 1.5 years, Fig. S2.†
It was shown that the lower the total boron concentration, the slower is the formation of the solid species (Fig. S2†). At 0.7 M total boron concentration the solid formation starts much earlier (between 2 and 6 days) than at 0.4 M total boron concentration (between 98 and 141 days). The solid formation was detected for total boron concentrations ≥0.3 M. Below that concentration the amount of the dissolved Eu(III) polyborate complex is too low to induce the precipitation of the solid, at least within the investigated time of 1.5 years.
The B(OH)3–polyborate equilibrium is dynamic. If polyborates of the aqueous phase are removed from the equilibrium due to the Eu(III) borate solid formation there is still a sufficient amount of boric acid in solution to regulate the B(OH)3–polyborate equilibrium. Thus, the dissolved Eu(III) is almost completely converted into the solid if enough polyborate species are reproduced. This was verified by the quantitative analysis of europium in the filtrates (see above).
Characterization of the solid Eu(III) borate
 | ||
| Fig. 7 IR spectrum of the solid Eu(III) borate (below) and for comparison of boric acid (top) in the range of (a) 4000 cm−1 to 380 cm−1 and (b) 700 cm−1 to 480 cm−1 (range of characteristic pulse vibrations of polyborates). | ||
| Boric acid, solid | Eu(III) borate, solid | Assignment |
|---|---|---|
| b = broad, m = middle, s = strong, w = weak, [BO3] = trigonal planar boron center, [BO4] = tetrahedral boron center, ν = stretching vibration, δ = in-plane bending, γ = out-of-plane bending. | ||
| 450 (w) | δ([BO4]–O)45,48,50 | |
| 548 (s) | δ([BO3]–O)45 | |
| δ(B–O), in-plane O–B–O angle deformation mode43,44 | ||
| 550 (w) | δ([BO3]–O)/δ([BO4]–O)45 | |
| Potentially γp(pentaborate)45,47,50 | ||
| 645 (m) | γ([BO3]–O)31 | |
| γ(O–H), out-of-plane OH deformation mode44 | ||
| δ(B–O)43 | ||
| 680 (w) | γ([BO3]–O)47,48,50 | |
| 756 (m) | γ([BO3]–O)45,47,48,50 | |
| 800–1200 (s) | ν s([BO3]–O), νs([BO4]–O), νas([BO4]–O)45,47,48,50 | |
| 806 (m) | γ(B–O), out-of-plane BO3 angle deformation mode44,45 | |
| γ(O–H), twisting43 | ||
| 883 (s, b) | ν s(B–O)43–45 | |
| 1196 (m) | ν as([BO3]–O)45 | |
| δ(O–H), in-plane B–O–H angle deformation mode43,44 | ||
| 1269 (m) | δ(B–O–H)45–47,50 | |
| 1379 (s) | ν as([BO3]–O)45,47,50 | |
| 1450 (s) | ν as(B–O)43–45 | |
| 1631 (w) | δ(H–O–H), structural water45,50 | |
| 2262 (m) | No B–O modes (adsorbed gaseous CO2), B2O3 impurities42 | |
| 2363 (m) | Combination frequencies of ν(O–H), ν(B–O), δ(O–H), δ(B–O)43,44 | |
| 2517 (m) | ||
| 3217 (s, b) | ν(O–H)43–45 | |
| 3400 (s, b) | ν(O–H)45,50 | |
Besides the vibration modes of trigonal planar boron [BO3] units the solid Eu(III) borate showed vibration modes of tetrahedral boron [BO4] units (Fig. 7a, Table 3). The assignment of these modes (Table 3) was based on literature data.45–50 Furthermore, in the literature so-called pulse vibrations are described being typical of different polyborate structures.45 Pulse vibrations of tri-, tetra-, penta- and hexaborates occur in the wavenumber range of 650 cm−1 to 530 cm−1.45 In this range one vibration band at 550 cm−1 in the measured IR spectrum of the solid Eu(III) borate occurred (Fig. 7b). This could be an indication of a pentaborate structure in the solid Eu(III) borate. Unfortunately, this is not unambiguous, because this band can be assigned to the bending mode of the [BO3] and [BO4] units, too (Table 3). Nevertheless, the borate structure as the main structure in the isolated Eu(III) solid can be clearly confirmed by IR spectroscopy.
In addition, the vibration band at 1631 cm−1 is assigned to structural water, which obviously exists in the solid Eu(III) borate.
Solid-state TRLFS
The europium luminescence spectrum of the solid Eu(III) borate at room temperature (λex = 394.0 nm) is shown in Fig. 8a. This spectrum is comparable to that of the solid Eu(III) borate in suspension (see Fig. 6a). An asymmetrically shaped excitation spectrum of the solid Eu(III) borate has to be noted (Fig. 8b). From this asymmetry the presence of more than one Eu(III) species in the isolated Eu(III) borate solid is indicated. At λex = 394.0 nm all Eu(III) species present in the sample were excited. In order to discriminate the contributions of different species to the overall luminescence, site-selective measurements of the solid Eu(III) borate were performed. The excitation was carried out at λex = 579.45 nm (peak maximum in the excitation spectrum, see Fig. 8b) and at lower excitation wavelengths (λex = 578.50 nm and 578.0 nm, respectively) to measure further possible europium species and to minimize the contribution of the strongly luminescent species excited at λex = 579.45 nm. At room temperature the emission spectra (not shown) and the luminescence lifetimes τ (Table 4) recorded at different excitation wavelengths are comparable. The determined τ ∼ 620 μs of the europium solid at room temperature (see Table 4) is comparable to that of the Eu(III) borate solid in suspension (see Fig. 6b). | ||
| Fig. 8 Solid-state TRLFS of the solid Eu(III) borate. (a) Emission spectrum at room temperature (T = 22 °C) excited at λex = 394 nm, (b) excitation spectrum at low temperature (T < 5 K), (c) emission spectra excited at λex = 578.0 nm, 578.50 nm and 579.45 nm, respectively, and (d) emission spectra determined from the PARAFAC of the time-resolved spectra at low temperature (T < 5 K) excited at λex = 578.50 nm and 579.45 nm, respectively. | ||
| Excitation wavelength λex [nm] | ||
|---|---|---|
| 578.50 | 579.45 | |
| τ (measured) [μs], at room temperature (22 °C) | 605 | 636 |
| τ (measured) [μs], at low temperature (<5 K) | 263 | 771 |
| 619 | ||
| τ (from PARAFAC) [μs], at low temperature (<5 K) | 335 (species 2b) | 717 (species 2a) |
| 717 (species 2a) | 882 (species 1) | |
To obtain more information about the species in the solid Eu(III) borate, emission spectra were recorded at low temperature (T < 5 K). Compared to the emission spectra at room temperature the spectra at low temperature are much better resolved and a potential splitting of the luminescence bands is more clearly visible (compare Fig. 8a and 8c). Consequently, more information, for instance about the species and the molecular environment of the Eu(III) in the solid, can be extracted.
At low temperature (T < 5 K) the excitation of europium was carried out at λex = 579.45 nm, 578.50 nm, and 578.0 nm, respectively. Depending on the excitation wavelength λex two different europium species were identified (Fig. 8c). At λex = 579.45 nm the obtained emission spectrum can be assigned to the Eu(III) borate (named as species 1) as the major species in the solid, because the spectrum shape is comparable to the spectrum of the solid Eu(III) borate in suspension (Fig. 6a). In contrast to the excitation at λex = 579.45 nm distinctly different emission spectra were recorded at λex = 578.0 nm and 578.50 nm (Fig. 8c). Both spectra obtained at λex = 578.0 nm and 578.50 nm are identical and are assigned to at least one further europium solid species (named species 2). Identical spectra resulting from different excitation wavelengths are expected for amorphous materials.51
In addition, the luminescence decay kinetic at different excitation wavelengths and low temperatures (T < 5 K) was measured. At λex = 579.45 nm one luminescence lifetime with τ = 771 μs was determined (Table 4), which is slightly higher than its value at room temperature. This effect was observed elsewhere before,52 and can be ascribed to a reduction of radiationless deactivation processes at low temperature. Moreover, at λex = 578.50 nm two luminescence lifetimes were found (τ1 = 619 μs and τ2 = 263 μs, Table 4). This indicates that more than one species are observed with λex = 578.0 nm and 578.50 nm. The luminescence of species 1 and species 2 could spectrally overlap or even a third species may have to be considered.
From the PARAFAC of the time-resolved spectra of the solid Eu(III) borate at low temperature three europium species were clearly determined (Fig. 8d). One species is quite similar to the measured species 1 (compare Fig. 8c and 8d). It seems that the measured spectrum of species 2 (Fig. 8c) is a spectrum composed of two species. The PARAFAC was able to separate these two species (named species 2a and 2b, Fig. 8d). Furthermore, it is possible to estimate the contribution of each species to the luminescence signal (Fig. S4†). The PARAFAC of the time-resolved data at λex = 579.45 nm showed that species 2a exists as a minor component in addition to species 1. Species 2b does not contribute to the luminescence signal. Because species 1 dominates the luminescence at λex = 579.45 nm, only a monoexponential decay was measured (see above). With λex = 578.5 nm, species 2b contributes to the luminescence in comparable amounts like species 2a (result of the PARAFAC, Fig. S4†), which leads to the observed biexponential luminescence decay (see above). Species 1 does not contribute to the luminescence at λex = 578.5 nm (Fig. S4†).
From the splitting pattern of the luminescence bands of the europium emission spectra, structural information on the europium environment can be obtained.51,53–56
The spectrum of species 1 (measured and determined by PARAFAC, Fig. 8c and 8d, respectively) shows that the 5D0→7F1 luminescence band split into three peaks and the 5D0→7F2 luminescence band split into five peaks. The 5D0→7F1 and 5D0→7F2 luminescence bands in the by PARAFAC determined spectrum of species 2a show similar splitting patterns to those described for species 1. This indicates a local europium environment with low symmetry (C1, C2 or CS)53 for species 1 and species 2a. The by PARAFAC separated spectrum of species 2b does not show a clear splitting pattern and the symmetry determination of the local europium environment is difficult. It seems that the 5D0→7F1 luminescence band is split into three peaks and the 5D0→7F2 luminescence band into four peaks. This also indicates the low symmetry of the local europium environment (C2v).53
Species 2a is likely a borate species, too. The spectrum shape and luminescence lifetime of species 2a are comparable to that of species 1 (Table 4, Fig. 8d). The shape of the europium emission spectrum and the luminescence lifetime of species 2b point to a Eu2(CO3)3·nH2O species.57 It is assumed that species 2a and 2b are byproducts of the Eu(III) borate precipitation.
Europium to boron ratio in the solid
The boron–europium–sodium ratio was 14.15:2.55:1. Considering the solid sodium borate phase (see discussion of the powder XRD results above) and minor europium species (see discussion of the solid-state TRLFS results above) the major Eu(III) borate solid species is assumed to be a Eu(III) pentaborate species.
Conclusion
This work deals with the very interesting but complex Eu(III)–B(OH)3/(poly)borate system at pH ≤ 6, I = 0.1 M. Some of the main results shall be summarized.Complexation in aqueous solution
Polyborates, more precisely tri- and pentaborates with one binding site, which occur under the investigated conditions, show a weak complexation of Eu(III) (lg β11 ∼ 2).Solid formation
The formation of a Eu(III) solid phase involving polyborates was observed. To the best of our knowledge this is the first time that such a phase formation starting from a weak Ln(III) polyborate complex has been described at pH 6. Recently, a similar effect was observed at alkaline pH (see above).40Possible impact of (poly)borates on the mobilization of An(III) in a nuclear waste repository
Interpreting the complexation results, borates should have a minor mobilization potential for trivalent actinides at least in the slightly acidic to neutral pH range because the complexation is very weak in comparison with other complexation reactions, e.g., with carbonate and hydroxide ligands. Furthermore, the formation of solid (poly)borate phases already at slightly acidic pH should support the immobilization of trivalent actinides.Future research is definitely required for explaining the actinide–borate interactions, particularly in the alkaline pH range. Furthermore, other actinides such as uranium and oxidation states of actinides above +3 have to be considered in this context.
Acknowledgements
This work was funded by the Federal Ministry of Economics and Energy (BMWi) under contract numbers 02E11021 and 02E11011. The authors would like to thank A. Ritter (HZDR) for ICP-MS and AAS analyses, K. Heim (HZDR) for IR measurements, and S. Labs (FZ Jülich) and Ch. Hennig (HZDR) for XRD measurements and analysis. Powder XRD measurements were conducted at the PETRA III facility, beamline P02.1, of the Deutsches Elektronen Synchrotron (DESY) Hamburg, Germany under proposal I-20130337.References
- M. Borkowski, M. Richmann, D. T. Reed and Y. Xiong, Radiochim. Acta, 2010, 98, 577–582 CrossRef CAS.
- W. G. Woods, Environ. Health Perspect., 1994, 102(Suppl 7), 5–11 CrossRef CAS.
- J. F. Lucchini, M. Borkowski, M. K. Richmann, S. Ballard and D. T. Reed, J. Alloys Compd., 2007, 444/445, 506–511 CrossRef PubMed.
- M. J. Polinski, D. J. Grant, S. Wang, E. V. Alekseev, J. N. Cross, E. M. Villa, W. Depmeier, L. Gagliardi and T. E. Albrecht-Schmitt, J. Am. Chem. Soc., 2012, 134, 10682–10692 CrossRef CAS PubMed.
- M. J. Polinski, S. Wang, E. V. Alekseev, W. Depmeier and T. E. Albrecht-Schmitt, Angew. Chem., Int. Ed., 2011, 50, 8891–8894 CrossRef CAS PubMed.
- M. J. Polinski, S. Wang, E. V. Alekseev, W. Depmeier, G. Liu, R. G. Haire and T. E. Albrecht-Schmitt, Angew. Chem., Int. Ed., 2012, 51, 1869–1872 CrossRef CAS PubMed.
- S. Wang, E. V. Alekseev, W. Depmeier and T. E. Albrecht-Schmitt, Inorg. Chem., 2011, 50, 2079–2081 CrossRef CAS PubMed.
- M. J. Polinski, E. M. Villa and T. E. Albrecht-Schmitt, Coord. Chem. Rev., 2014, 266–267, 16–27 CrossRef CAS PubMed.
- T. Straaso, A.-C. Dippel, J. Becker and J. Als-Nielsen, J. Synchrotron Radiat., 2014, 21, 119–126 Search PubMed.
- A. P. Hammersley, S. O. Svensson, M. Hanfland, A. N. Fitch and D. Hausermann, High Press. Res., 1996, 14, 235–248 CrossRef.
- Lanthanide Probes in Life, Chemical and Earth Sciences: Theory and Practice, ed. J.-C. G. Bünzli and G. R. Choppin, Elsevier Science B.V., Amsterdam, 1989 Search PubMed.
- W. D. Horrocks and D. R. Sudnick, J. Am. Chem. Soc., 1979, 101, 334–340 CrossRef CAS.
- T. Kimura and G. R. Choppin, J. Alloys Compd., 1994, 213/214, 313–317 CrossRef CAS.
- C. Moulin, J. Wei, P. Van Iseghem, I. Laszak, G. Plancque and V. Moulin, Anal. Chim. Acta, 1999, 396, 253–261 CrossRef CAS.
- G. Plancque, V. Moulin, P. Toulhoat and C. Moulin, Anal. Chim. Acta, 2003, 478, 11–22 CrossRef CAS.
- B. Marmodée, K. Jahn, F. Ariese, C. Gooijer and M. U. Kumke, J. Phys. Chem. A, 2010, 114, 13050–13054 CrossRef PubMed.
- P. Gans, A. Sabatini and A. Vacca, Hyperquad Simulation and Speciation, Protonic Software, Leeds, 2009 Search PubMed.
- P. Gans, A. Sabatini and A. Vacca, Hyperquad suite of programs, Protonic Software, Leeds, 2008 Search PubMed.
- A. Heller, A. Barkleit, H. Foerstendorf, S. Tsushima, K. Heim and G. Bernhard, Dalton Trans., 2012, 41, 13969–13983 RSC.
- A. Shokrollahi, M. Montazerozohori, T. Mehrpour, H. Tavallali, B. Z. Khafri and Z. Montaseri, Quim. Nova, 2013, 36, 1354–1359 CrossRef CAS PubMed.
- L. E. Santos-Figueroa, M. E. Moragues, M. M. M. Raposo, R. M. F. Batista, R. C. M. Ferreira, S. P. G. Costa, F. Sancenon, R. Martinez-Manez, J. Soto and J. V. Ros-Lis, Tetrahedron, 2012, 68, 7179–7186 CrossRef CAS PubMed.
- A. Günther and G. Bernhard, HZDR-IRE Annual Report 2012, HZDR-030, ISSN 2191-8716, Dresden, 2013.
- C. A. Andersson and R. Bro, Chemom. Intell. Lab. Syst., 2000, 52, 1–4 CrossRef CAS.
- T. Saito, H. Sao, K. Ishida, N. Aoyagi, T. Kimura, S. Nagasaki and S. Tanaka, Environ. Sci. Technol., 2010, 44, 5055–5060 CrossRef CAS PubMed.
- R. M. Callejón, J. M. Amigo, E. Pairo, S. Garmón, J. A. Ocana and M. L. Morales, Talanta, 2012, 88, 456–462 CrossRef PubMed.
- K. Ishida, T. Saito, N. Aoyagi, T. Kimura, R. Nagaishi, S. Nagasaki and S. Tanaka, J. Colloid Interface Sci., 2012, 374, 258–266 CrossRef CAS PubMed.
- N. Ingri, Acta Chem. Scand., 1962, 16, 439–448 CrossRef CAS PubMed.
- N. Ingri, G. Lagerström, M. Frydman and L. G. Sillén, Acta Chem. Scand., 1957, 11, 1034–1058 CrossRef CAS PubMed.
- I. Tsuyumoto, T. Oshio and K. Katayama, Inorg. Chem. Commun., 2007, 10, 20–22 CrossRef CAS PubMed.
- J. L. Anderson, E. M. Eyring and M. P. Whittaker, J. Phys. Chem., 1964, 68, 1128–1132 CrossRef CAS.
- N. Ingri, Acta Chem. Scand., 1963, 17, 573–580 CrossRef CAS PubMed.
- A. Hertam, Thesis, Freiberg University of Mining and Technology, 2011.
- T. Hirao, M. Kotaka and H. Kakihana, J. Inorg. Nucl. Chem., 1979, 41, 1217–1220 CrossRef.
- L. Maya, Inorg. Chem., 1976, 15, 2179–2184 CrossRef CAS.
- R. K. Momii and N. H. Nachtrieb, Inorg. Chem., 1967, 6, 1189–1192 CrossRef CAS.
- Y. Zhou, C. Fang, Y. Fang and F. Zhu, Spectrochim. Acta, Part A, 2011, 83, 82–87 CrossRef CAS PubMed.
- J. E. Spessard, J. Inorg. Nucl. Chem., 1970, 32, 2607–2613 CrossRef CAS.
- R. Janda and G. Heller, Z. Naturforsch., B: J. Chem. Sci., 1979, 34, 1078–1083 Search PubMed.
- M. Herm, X. Gaona, T. Rabung, C. Crepin, V. Metz, M. Altmaier and H. Geckeis, Book of Abstracts of the Migration 2013, Brighton (UK), 2013 Search PubMed.
- K. Hinz, M. Altmaier, X. Gaona, T. Rabung, D. Schild, C. Adam and H. Geckeis, Book of Abstracts of the Migration 2013, Brighton (UK), 2013 Search PubMed.
- S. Menchetti, C. Sabelli, A. Stoppioni and R. Trosti-Ferroni, Neues Jahrb. Mineral., Abh., 1983, 148, 163–180 CAS.
- E. F. Medvedev and A. S. Komarevskaya, Glass Ceram., 2007, 64, 42–46 CrossRef CAS.
- J. L. Parsons and M. E. Milberg, J. Am. Ceram. Soc., 1960, 43, 326–330 CrossRef CAS.
- D. E. Bethell and N. Sheppard, Trans. Faraday Soc., 1955, 51, 9–15 RSC.
- L. Jun, X. Shuping and G. Shiyang, Spectrochim. Acta, Part A, 1995, 51, 519–532 CrossRef.
- E. L. Belokoneva, A. G. Ivanova, S. Y. Stefanovich, O. V. Dimitrova and V. S. Kurazhkovskaya, Crystallogr. Rep., 2004, 49, 603–613 CrossRef CAS.
- S. Hua-Yu, Z. Yan, H. Ya-Xi, S. Wei and M. Jin-Xiao, Chin. J. Struct. Chem., 2010, 29, 1387–1393 Search PubMed.
- R. Janda and G. Heller, Spectrochim. Acta, Part A, 1980, 36, 997–1001 CrossRef.
- S. Lemanceau, G. Bertrand-Chadeyron, R. Mahiou, M. El-Ghozzi, J. C. Cousseins, P. Conflant and R. N. Vannier, J. Solid State Chem., 1999, 148, 229–235 CrossRef CAS.
- Z. Lixia, Y. Tao, W. Jiang and G. Shiyang, Russ. J. Inorg. Chem., 2007, 52, 1786–1792 CrossRef.
- K. S. Holliday and T. Stumpf, Environ. Radiochem. Anal., 2011, IV, 30–39 Search PubMed.
- S. Kuke, B. Marmodée, S. Eidner, U. Schilde and M. U. Kumke, Spectrochim. Acta, Part A, 2010, 75, 1333–1340 CrossRef CAS PubMed.
- C. Görller-Walrand and K. Binnemans, Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner Jr. and L. Eyring, Elsevier Science B. V., Amsterdam, 1996, vol. 23 Search PubMed.
- M. Schmidt, T. Stumpf, C. Walther, H. Geckeis and T. Fanghänel, Dalton Trans., 2009, 6645–6650 RSC.
- R. Ternane, M. Ferid, G. Panczer, M. Trabelsi-Ayadi and G. Boulon, Opt. Mater., 2005, 27, 1832–1838 CrossRef CAS PubMed.
- B. Piriou, A. Elfakir and M. Quarton, J. Lumin., 2001, 93, 17–26 CrossRef CAS.
- W. Runde, C. Van Pelt and P. G. Allen, J. Alloys Compd., 2000, 303/304, 182–190 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4dt00843j |
| This journal is © The Royal Society of Chemistry 2014 |