 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The first ruthenium-silsesquioxyl complexes – synthesis, structure and mechanistic implications in silylative coupling†
Patrycja
Żak
,
Maciej
Kubicki
,
Bogdan
Marciniec
*,
Szymon
Rogalski
,
Cezary
Pietraszuk
and
Dawid
Frąckowiak
Faculty of Chemistry, Adam Mickiewicz University, Umultowska 89b, 61-614 Poznan, Poland. E-mail: bogdan.marciniec@amu.edu.pl; Fax: (+48)61 8291508; Tel: (+48)61 8291366
First published on 11th March 2014
Abstract
The first ruthenium-silsesquioxyl complexes have been synthesised and characterized via spectroscopic and X-ray methods. Mechanistic studies were performed and the complexes obtained were proved to be intermediates in the catalytic cycle of silylative coupling of olefins with vinylsilsesquioxane. Moreover, a mechanism for silylative coupling of styrene with vinylsilsesquioxanes was proposed.
Introduction
Polyhedral oligosilsesquioxanes (POSS) of the general formula (RSiO3/2)n and particularly those (n = 8) containing the inorganic cubic core of Si–O–Si bonds form a class of versatile building blocks for the production of inorganic–organic materials thanks to the three-dimensional highly symmetrical nature of the POSS core.1 The properties of the core permit a wide range of technological applications of silsesquioxanes that can be used as nanofillers for the preparation of nanostructured composites, catalysts,2–4 and dendrimers,5 as precursors for optoelectronic materials,6–9 as dry resists in microelectronics10 and as precursors for SiO deposition.11Mono- and octa-functionalized silsesquioxanes and spherosilicates have been synthesized by TM catalyzed reactions such as hydrosilylation by Si–H12 substituted POSS and silylative coupling13–16 by vinyl-substituted POSS with olefins, which occurs via intermediates containing a TM–silicon bond. The mechanism of silylative coupling of olefins with vinyl-substituted silicon compounds was the subject of our earlier study [see ref. 17; for reviews see ref. 18, 19]. So far only two reports have been published on the isolation and crystal structure of the complexes containing a TM–silicon bond (exactly cobalt–silicon bond).20,21
This paper reports the synthesis of the first silsesquioxyl complexes of ruthenium containing Ru–Si bonds and describes the crystal structure of one of them. Moreover, the catalytic activity of this complex in the silylative coupling of vinylheptaalkylsilsesquioxane with styrenes is analysed.
Results and discussion
When a toluene solution of the ruthenium hydride complex [RuHCl(CO)(PPh3)3] 1 was heated in the presence of 1 equiv. of vinylheptaisobutylsilsesquioxane at 110 °C the color of the solution gradually turned yellow-brown within 48 h. The 1H NMR spectrum of the post-reaction mixture revealed disappearance of signals at δ = −6.60 (dt) ppm characteristic of the Ru–H bond, and formation of a new singlet at δ = 5.25 ppm which can be assigned to ethylene. Moreover, the appearance of a new singlet at δ = 37.49 ppm was observed in the 31P NMR spectrum. On the basis of these observations, a general scheme for the synthesis of ruthenium-silsesquioxyl complexes was proposed (Scheme 1).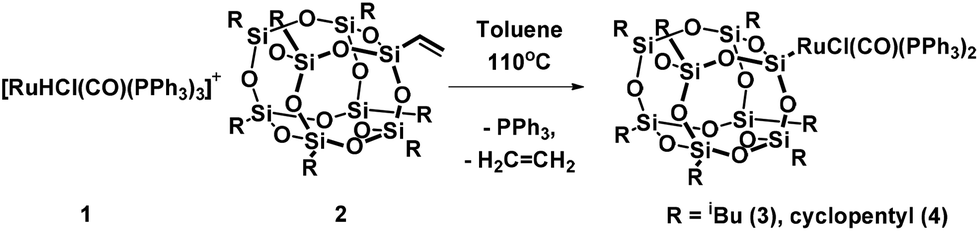 | ||
| Scheme 1 Synthesis of ruthenium-silsesquioxyl complexes. | ||
Further experiments led to the development of efficient synthetic procedures for the synthesis of silsesquioxyl complexes 3 and 4 (see ESI†). The compounds were isolated in high yields (89% and 86% respectively) as pale yellow powders. The obtained products were fully characterized by 1H, 13C, 29Si, and 31P NMR spectroscopy and high resolution mass spectrometry (see ESI†). Moreover the structure of complex (3) was confirmed via X-ray analysis (Fig. 1). Single crystals were obtained by slow evaporation of a pentane solution. Ru is 5-coordinated in a square-pyramidal fashion. Four atoms, P1, P2, Cl1 and C1, are almost coplanar (maximum deviation from the least-squares plane is 0.053(3) Å) while the fifth one, Si1, is 2.485 Å out of this plane. The Ru atom is also slightly displaced, by 0.0219 Å, from the basal plane towards the apical Si1. Because of static disorder, in approximately one of six molecules the CO and Cl ligands are interchanged (cf. Experimental part) without a significant influence on the complex geometry. The Si8O12 moiety is quite regular, with deviations from the ideal symmetry caused mainly by the presence of Ru-coordination. The Si–O distances involving the coordinated silicon atom are systematically longer than the other Si–O bonds; the mean values are 1.636(4) Å for Si1 and 1.620(5) Å for all other ones. In fact, Si1–O distances are the three longest Si–O distances in the molecule. Also, the majority (8 of 12) of Si–O–Si angles are within quite a narrow range, with the mean value of 145.4(16)°, while the other four (O4,O8, O9, O10) are significantly larger, 154°–161°.
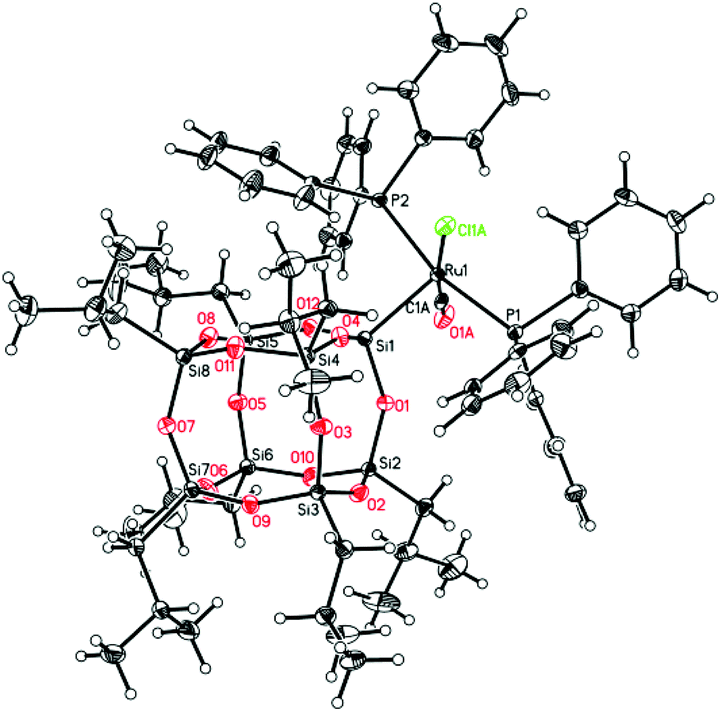 | ||
| Fig. 1 A perspective view of complex 3, the ellipsoids are drawn at the 33% probability level. Hydrogen atoms are drawn as spheres of arbitrary radii. Some relevant geometrical parameters (Å, °; values for less-occupied part are given in square brackets): Ru1–C1 1.793(4) [1.690(16)], Ru1–Cl1 2.4229(9) [2.491(5)], Ru1–Si1 2.2992(6), Ru1–P1 2.3944(5), Ru1–P2 2.3572(5), C1–O1 1.166(5) [1.19(2)], <Si–O> 1.624(11), Cl1–Ru1–C1 170.55(10) [171.1(5)], P1–Ru1–P2 165.16(2). | ||
Stoichiometric reactions of the ruthenium-silsesquioxyl complex with styrene
As we previously demonstrated for the silylative coupling mechanism of vinylsilicon compounds and olefins, complexes 3 and 4 can be treated as intermediate ones in the catalytic cycle. In order to better understand the mechanism of silylative coupling of styrenes with vinylsilsesquioxanes an equimolar reaction between ruthenium-silsesquioxyl complex 3 and styrene was performed (Scheme 2).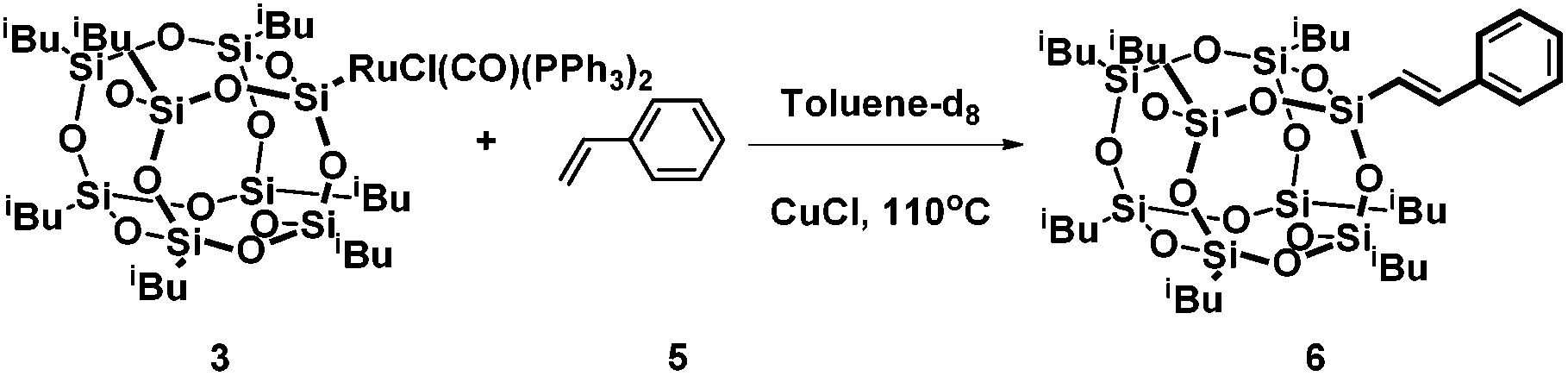 | ||
| Scheme 2 Stoichiometric reactions of styrene with ruthenium-silsesquioxyl complexes. | ||
The tests were monitored by 1H NMR spectroscopy. Addition of 1 equiv. of styrene and 5 equiv. of CuCl to the solution of complex 3 followed by heating the reacting mixture at 110 °C resulted in formation of the cross-coupling product 6. Although spectroscopic examination does not confirm the formation of a complex 9 containing a Ru–H bond, we observed signals at δ = 6.47 ppm (d, 1H, J = 19.2 Hz, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHSi) and δ = 7.56 ppm (d, 1H, J = 19.2 Hz, CHPh) characteristic of product 6. The intermediate complex containing a Ru–H bond was also not observed in the reaction of Ru–Si complexes with olefins, which was previously reported by our group.18,19 Moreover, GC-MS analysis confirmed the formation of the desired coupling product 6. Formation of compound 6 is evidence for insertion of styrene into the Ru–Si(POSS) bond in complex 3 followed by β-H elimination and evolution of E-phenyl(silyl)ethene.
CHSi) and δ = 7.56 ppm (d, 1H, J = 19.2 Hz, CHPh) characteristic of product 6. The intermediate complex containing a Ru–H bond was also not observed in the reaction of Ru–Si complexes with olefins, which was previously reported by our group.18,19 Moreover, GC-MS analysis confirmed the formation of the desired coupling product 6. Formation of compound 6 is evidence for insertion of styrene into the Ru–Si(POSS) bond in complex 3 followed by β-H elimination and evolution of E-phenyl(silyl)ethene.
In the above stoichiometric reaction, the desired product was formed only when the reaction was conducted in the presence of CuCl. Moreover, complex 3 exhibited almost no reactivity when the reaction was performed without Cu(I) salt. To better understand the role of CuCl in the reaction mixture we performed a series of stoichiometric reactions monitored by 1H and 31P NMR spectroscopy. When a toluene solution of the ruthenium-silsesquioxyl complex [Ru(POSS)Cl(CO)(PPh3)2] 3 was heated in the presence of 5 equiv. of CuCl at 110 °C for 24 h we observed disappearance of the signals at δ = 37.49 ppm characteristic of complex 3 and formation of a new singlet at δ = 25.95 ppm in the 31P NMR spectrum (Fig. 2).
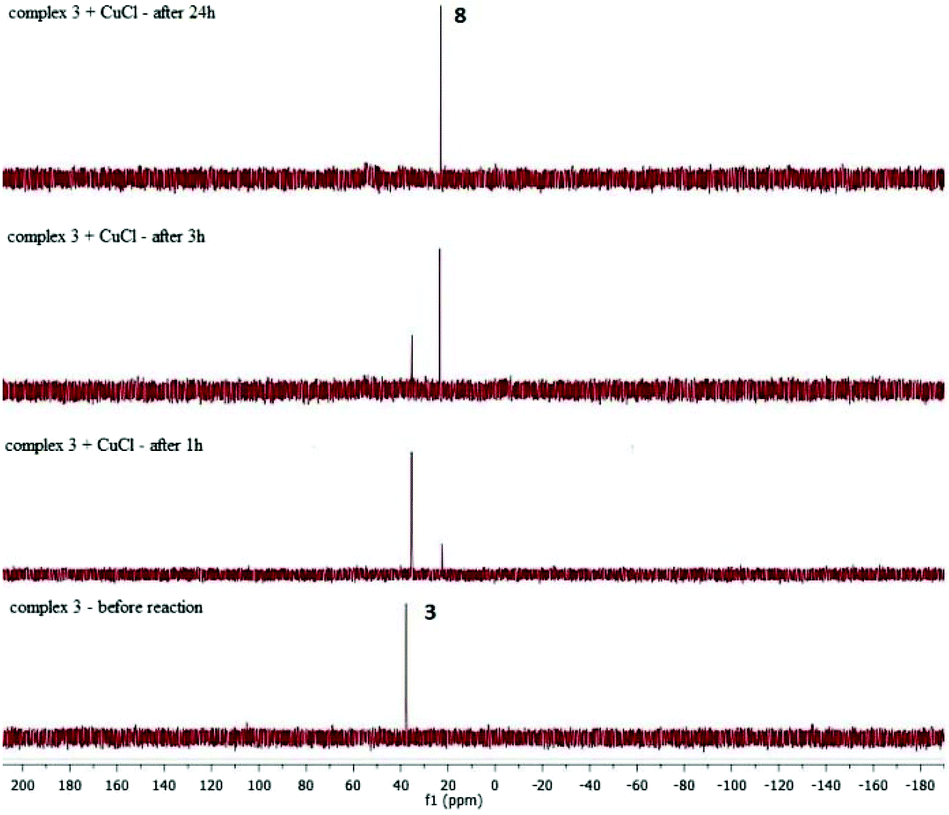 | ||
| Fig. 2 31P NMR spectra of the stoichiometric reaction of 3 with CuCl. | ||
The formation of this new complex 8 was accompanied by precipitation of an insoluble grey-brown [CuPPh3] complex, whose appearance proves the dissociation of phosphine in the proposed reaction system. The 1H NMR spectrum of the post-reaction mixture revealed changes in the aliphatic region. We observed disappearance of two multiplets at 1.85–2.06 and 2.06–2.31 and formation of a new multiplet at 2.15–2.38 ppm. All attempts to isolate this new complex 8 from the post-reaction mixture failed. However, the obtained results indicate that as a consequence of addition of CuCl to the reaction system, phosphine undergoes dissociation, as proved by [CuClPPh3] complex precipitation, and a new four-coordinated intermediate complex 8 is formed. The addition of CuCl is needed because of a large steric hindrance in the intermediate complex having the Ru–Si(POSS) bond. The presence of the steric hindrance in the intermediate complex is supported by the fact that in the analogous silyl complex [RuSiMe3Cl(CO)(PPh3)2] the insertion of styrene is effective without the addition of CuCl.17
Isotopic labelling experiments
In order to confirm that the coupling process in the presence of 3 proceeds via the insertion–elimination mechanism which is characteristic of a silylative coupling reaction,17–19 we performed a series of labeling studies. In these experiments monitored by 1H NMR spectroscopy, a solution of equimolar amounts of styrene-d8 and complex 3 was heated. The insertion–elimination mechanism should afford the formation of silylstyrene-d7 and ethylene-d1 (Scheme 3), at least in the initial stage of the reaction. 1H NMR spectroscopic analysis of the products formed in the equimolar reaction carried out in the presence of 3 revealed the formation of E-1-phenyl-2-(silyl)ethene-d7 and ethylene-d1. | ||
| Scheme 3 Labeling experiments in the presence of complex 3. | ||
The results obtained in the experiments with deuterium labelled styrene clearly demonstrate that functionalisation of vinylsilsesquioxanes with styrenes proceeds according to the silylative coupling mechanism involving the activation of C–H and Si–C bonds.
Catalytic examination and mechanistic implications
The ruthenium-silsesquioxyl complex 3 was examined in the silylative coupling of selected styrenes with vinylheptaisobutylsilsesquioxane and its catalytic activity was compared to that of its parent complex [RuHCl(CO)(PPh3)3] 1 and the previously studied more reactive complex [RuHCl(CO)(PCy3)2] 7 (Scheme 4).15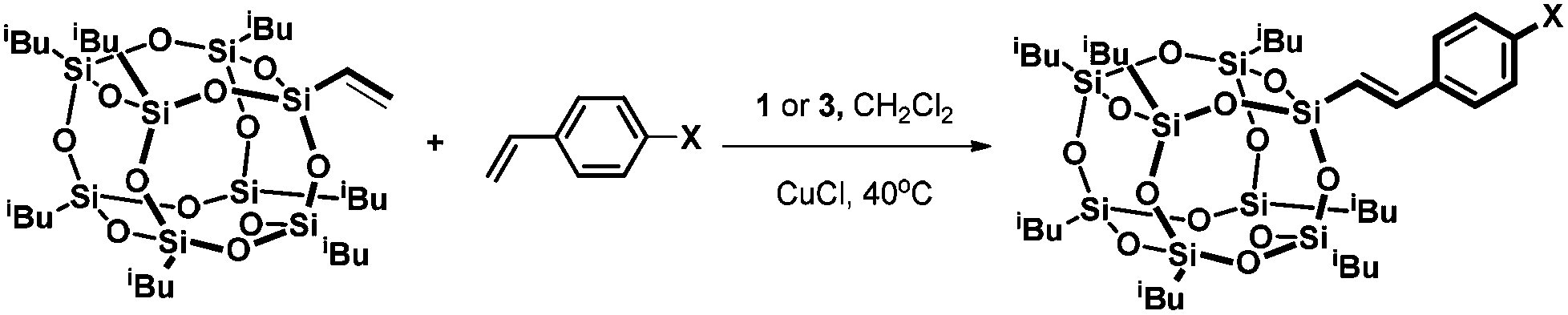 | ||
| Scheme 4 Silylative coupling of vinylheptaisobutylsilsesquioxane with styrenes. | ||
Selected data for 1 and 3 are presented in Table 1, entry 1. As indicated in this table, the silsesquioxyl catalyst 3 exhibits higher catalytic activity than catalyst 1. For example after five hours in the reaction of vinylsilsesquioxane with styrene catalysed by 3, the conversion of the silsesquioxane reached 33%, while the reaction catalysed by 1 revealed almost no conversion (6%). The activity of catalysts 1 and 3 was examined in a wide temperature range. Our experiments demonstrated that increasing the temperature of the reacting mixture from 40 °C to 110 °C did not give a higher yield of the coupling product. We compared the catalytic activity of complexes 1 and 3 with that of complex 7 bearing PCy3 ligands. In all cases, complex 7 was the most active in silylative coupling of vinylheptaisobutylsilsesquioxane with styrenes; however, because of its high reactivity we were unable to isolate ruthenium-silsesquioxyl complex bearing PCy3 ligands.
Mechanistic implications
On the basis of stoichiometric experiments and labelling studies we proposed a mechanism for the coupling of styrenes with vinylsilsesquioxanes in the presence of [RuHCl(CO)(PPh3)3] 1 and CuCl as co-catalysts (Fig. 3).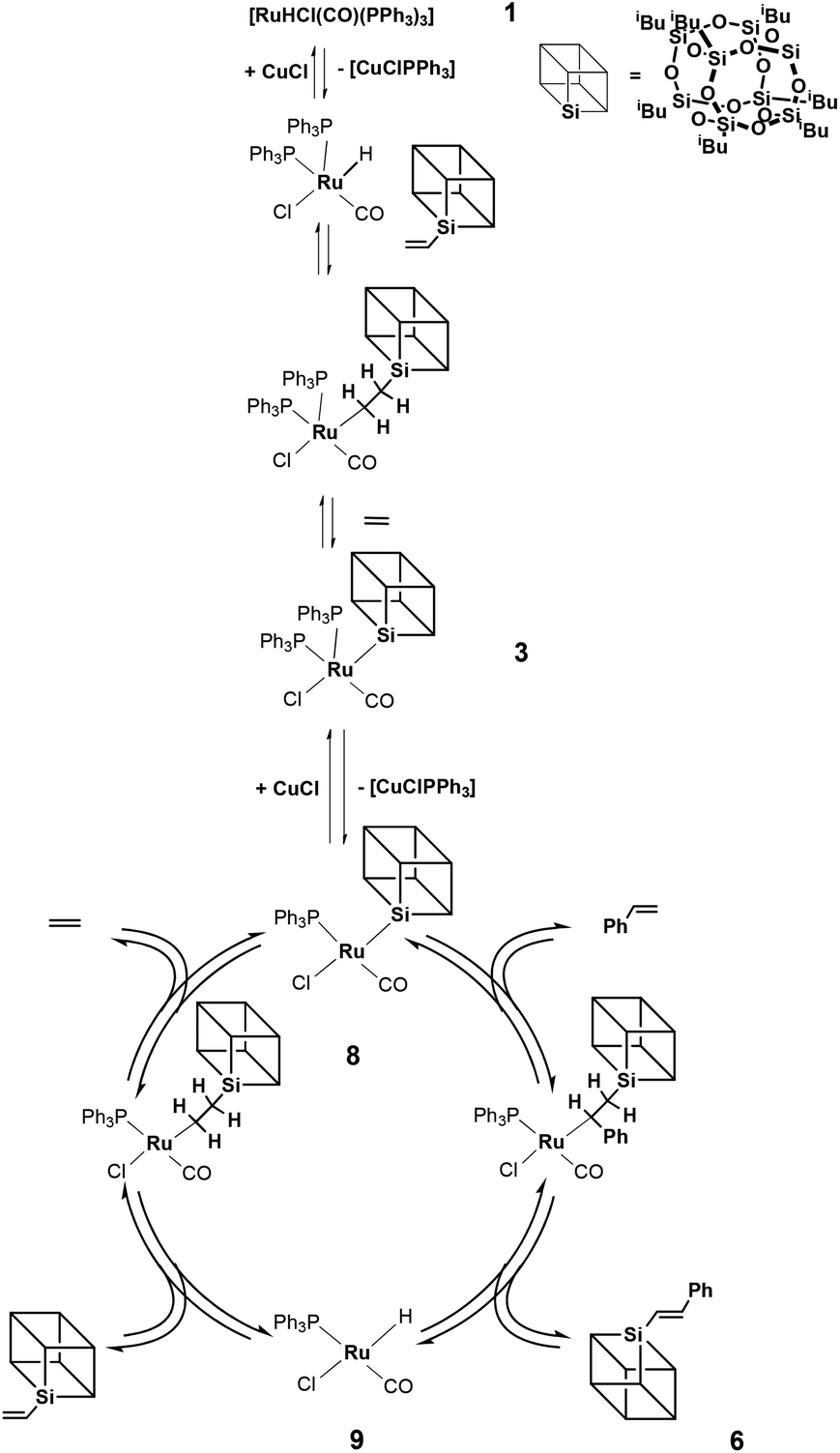 | ||
| Fig. 3 Mechanism of silylative coupling of styrene with vinylsilsesquioxane. | ||
Our studies have shown that the coupling reaction proceeds according to the well-recognized insertion–elimination mechanism17 in which, in the first step, vinylsilsesquioxane reacts with hydride complex 1 to give β-silylethyl complex. This compound decomposes via β-silsesquioxyl group migration to ruthenium and evolution of ethylene to give Ru-Si[POSS] complex 3. Addition of CuCl to complex 3 causes dissociation of phosphine to produce a less sterically hindered four-coordinate complex 8 which reacts with styrene. The next step of the catalytic cycle involves the migratory insertion of styrene into a Ru–Si bond, followed by β-H elimination to give E-phenyl(silyl)ethene 6.
Experimental
General methods and chemicals
Unless mentioned otherwise, all operations were performed by using standard Schlenk techniques. 1H- and 13C-NMR spectra were recorded on a Varian 400 operating at 402.6 and 101.2 MHz, respectively. 31P NMR spectra were recorded on a Mercury 300 operating at 121.5 MHz. 29Si NMR spectra were recorded on a Varian Avance 600 operating at 119.203 MHz. All FT-IR spectra were recorded with IFS 113v FT-IR and VERTEX 70 spectrophotometers (Bruker, Karlsruhe). GC analyses were carried out on a Varian CP-3800 (column: Rtx-5, 30 m, I.D. 0.53 mm) equipped with TCD. Mass spectrometry analyses were performed using a Synapt G2-S mass spectrometer (Waters) equipped with an electrospray ion source and a quadrupole-time-of-flight mass analyzer. Acetonitrile was used as a solvent. The measurements were performed in positive ion mode with the desolvation gas flow 200 L h−1 and capillary voltage set to 5000 V with the flow rate of 20 μl min−1.The chemicals were obtained from the following sources: vinyltrichlorosilane from ABCR, dichloromethane, acetone, n-pentane, ethanol, dichloromethane-d2, benzene-d6, toluene-d8, styrene-d8, decane, dodecane, styrene, 4-chlorostyrene, 4-bromostyrene, 4-methoxystyrene, 2-methoxyethanol, triphenylphosphine, formaldehyde, copper(I) chloride, anthracene, calcium hydride and anhydrous magnesium sulphate from Aldrich, triethylamine and silica gel 60 from Fluka, ruthenium(III) chloride hydrate from Lancaster, trisilanolisobutyl POSS from Hybrid Plastics,22 and toluene and n-hexane from Chempur. [RuHCl(CO)(PPh3)3] was prepared according to the literature procedure.23 Monovinylheptaisobutylsilsesquioxane was prepared according to the literature procedure.15 Monovinylheptacyclopentylsilsesquioxane was prepared using the same procedure. All solvents were dried prior to use over CaH2 and stored under argon. CH2Cl2 was additionally passed through a column with alumina and after that it was degassed by repeated freeze–pump–thaw cycles.
Synthesis and characterization of ruthenium-silyl complexes
Analytical data: 1H NMR (C6D6, δ, ppm): 0.35–0.53 (m, 14H, CH2), 0.81–1.27 (m, 42H, CH3), 1.85–2.06 (m, 3H, CH), 2.06–2.31 (m, 4H, CH), 6.99–7.30 (m, 12H, Ph), 7.72–7.93 (m, 18H, Ph); 13C NMR (C6D6, δ, ppm): 23.05, 23.25, 23.44, 23.62, 23.81, 24.72, 24.75, 24.78, 26.15, 26.20, 26.27, 26.38, 26.43, 30.45, 128.78 (t, J = 5.0 Hz), 130.49, 131.83 (d, J = 2.5 Hz), 132.61, 132.70, 132.85, 133.07, 133.29, 135.59 (t, J = 5.8 Hz), 200.70; 29Si NMR: (C6D6, δ, ppm): −66.05 (Ru–Si), −67.32, −69.60 (core); 31P NMR: (C6D6, δ, ppm): 37.49; IR (ν, cm−1): 1931 (CO); MS (FD):m/z (%): 1231.24 (31), 1263.20 (37), 1264.20 (61), 1265.20 (99), 1267.20 (100), 1268.20 (67), 1269.20 (50), 1275.23 (30), 1287.25 (42), 1288.24 (63), 1289.25 (97), 1290.24 (80), 1292.24 (55), 1299.09 (35), 1300.09 (30), 1465.13 (33), 1466.13 (40), 1467.13 (45), 1469.13 (42), 1470.13 (30), 1525.29 (38), 1526.29 (60), 1527.29 (95), 1529.29 (100), 1530.29 (77), 1531.29 (60), 1532.29 (33), 1543.29 (31), 1551.32 (39), 1552.32 (47), 1553.32 (62), 1554.32 (51), 1555.32 (47), 1556.32 (34), 1565.34 (38), 1566.33 (61), 1567.32 (92), 1568.33 (81), 1570.34 (57), 1571.33 (37), 1715.03 (33), 1717.03 (38), 1719.03 (34), 1803.38 (34), 1804.38 (54), 1805.37 (91), 1806.38 (93), 1807.38 (100), 1808.37 (82), 1809.38 (62), 1810.37 (37); HRMS (FD) for C65H93ClO13P2RuSi8Na: calc. 1527.2875; found: 1527.2871.
Analytical data: 1H NMR (C6D6, δ, ppm): 0.64–2.2 (m, 72H, C5H9–), 6.68–7.95 (m, 30H, Ph); 13C NMR (C6D6, δ, ppm): 23.10, 23.19, 27.21, 27.45, 27.56, 27.59, 27.80, 27.83, 27.86, 28.07, 28.31, 128.47 (t, J = 4.5 Hz), 130.07, 131.59 (d, J = 2.5 Hz), 132.34, 132.44, 135.44 (t, J = 5.8 Hz), 199.82; 29Si NMR: (C6D6, δ, ppm): −48.47 (Ru–Si), −66.14, −68.75 (core); 31P NMR: (C6D6, δ, ppm): 38.47; IR (ν, cm−1): 1932 (CO); MS (FD):m/z (%):1550.32 (24), 1551.33 (65), 1553.33 (100), 1554.33 (85), 1555.33 (82), 1556.33 (60), 1557.33 (37); HRMS (FD) for C72H93O13P2RuSi8: calc. 1553.3289; found: 1553.3273.
General procedure for the catalytic examination
The oven dried 5 mL glass reactor equipped with a condenser and a magnetic stirring bar was charged under argon with 2 mL of CH2Cl2, monovinylsilsesquioxane (0.1 g, 1.19 × 10−4 mol), styrene (41 μL, 3.56 × 10−4 mol) and 20 μL of an internal standard (decane or dodecane). The reaction mixture was placed in an oil bath and preheated to 40 °C. Then ruthenium complex 1 or 3 (0.0011 g (complex 1) or 0.0017 g (complex 2), 1.19 × 10−6 mol) was added under argon. After 5 min of the reaction, copper(I) chloride (0.0006 g, 5.93 × 10−6 mol) was added. The reaction mixture was heated at 40 °C under a gentle flow of argon. Reaction yields were calculated on the basis of the 1H NMR spectra of the reaction mixture. The corresponding reactions with 4-methoxystyrene, 4-bromostyrene and 4-chlorostyrene were performed using the same procedure.X-ray analysis (complex 3)
X-ray diffraction data were collected at 100(1) K by the ω-scan technique, on an Agilent Technologies four-circle Xcalibur diffractometer equipped with an Eos detector24 with an MoKα radiation source (λ = 0.71073 Å). The temperature was controlled with an Oxford Instruments Cryosystem device. The data were corrected for Lorentz-polarization effects as well as for absorption (multiscan).24 Accurate unit-cell parameters were determined by a least-squares fit of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 093 reflections of highest intensity, chosen from the whole experiment. The calculations were mainly performed within the WinGX program system.25 The structures were solved with SIR9226 and refined with the full-matrix least-squares procedure on F2 using SHELXL97.27 Scattering factors incorporated in SHELXL97 were used. The function ∑w(|Fo|2 − |Fc|2)2 was minimized, with w−1 = [σ2(Fo)2 + (0.0313P)2 + 2.2712P], where P = [Max(Fo2, 0) + 2Fc2]/3. All non-hydrogen atoms were refined anisotropically, hydrogen atoms from methyl groups were placed geometrically, in idealized positions, and refined as riding group with their Uiso's set at 1.2 (1.5 for methyl groups) times Ueq of the appropriate carrier atom. Disorder was detected during structure refinement: Cl and CO ligands are partially exchanged (site occupation factors 0.83/017), and in one iPr group the methyl groups are disordered over two positions (s.o.f.'s 0.70/0/30). Crystallographic data (excluding structure factors) for the structural analysis have been deposited with the Cambridge Crystallographic Data Centre, no. CCDC-919730.
093 reflections of highest intensity, chosen from the whole experiment. The calculations were mainly performed within the WinGX program system.25 The structures were solved with SIR9226 and refined with the full-matrix least-squares procedure on F2 using SHELXL97.27 Scattering factors incorporated in SHELXL97 were used. The function ∑w(|Fo|2 − |Fc|2)2 was minimized, with w−1 = [σ2(Fo)2 + (0.0313P)2 + 2.2712P], where P = [Max(Fo2, 0) + 2Fc2]/3. All non-hydrogen atoms were refined anisotropically, hydrogen atoms from methyl groups were placed geometrically, in idealized positions, and refined as riding group with their Uiso's set at 1.2 (1.5 for methyl groups) times Ueq of the appropriate carrier atom. Disorder was detected during structure refinement: Cl and CO ligands are partially exchanged (site occupation factors 0.83/017), and in one iPr group the methyl groups are disordered over two positions (s.o.f.'s 0.70/0/30). Crystallographic data (excluding structure factors) for the structural analysis have been deposited with the Cambridge Crystallographic Data Centre, no. CCDC-919730.
Crystal data: C65H93ClO13P2RuSi8, Mr = 1505.57, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 13.4910(3) Å, b = 14.8072(3) Å, c = 19.0764(4) Å, α = 89.338(2)°, β = 81.060(2)°, γ = 77.729(2)°, V = 3677.57(13) Å3, F(000) = 1580, dx = 1.36 g cm−3, μ(MoKα) = 0.48 cm−1, 33625 reflections measured, 14864 unique (Rint = 0.024), 12507 with I > 2σ(I). Final R(obs) = 0.031, R(all) = 0.042, wR2(obs) = 0.071, wR2(all) = 0.075, S = 1.04, max/min Δρ in the final ΔF map: 0.49/−0.62 e Å−3.
, a = 13.4910(3) Å, b = 14.8072(3) Å, c = 19.0764(4) Å, α = 89.338(2)°, β = 81.060(2)°, γ = 77.729(2)°, V = 3677.57(13) Å3, F(000) = 1580, dx = 1.36 g cm−3, μ(MoKα) = 0.48 cm−1, 33625 reflections measured, 14864 unique (Rint = 0.024), 12507 with I > 2σ(I). Final R(obs) = 0.031, R(all) = 0.042, wR2(obs) = 0.071, wR2(all) = 0.075, S = 1.04, max/min Δρ in the final ΔF map: 0.49/−0.62 e Å−3.
Conclusions
New silsesquioxyl ruthenium complexes (3 and 4) have been synthesized and their structures were confirmed by spectroscopic and X-ray methods. These complexes were proved to be intermediates in the silylative coupling of vinylsilsesquioxane with styrene. The reaction between vinylsilsesquioxanes and styrenes in the presence of ruthenium hydride complex 1 was confirmed to proceed via the insertion–elimination mechanism. Moreover, the obtained complex 3 exhibits higher catalytic activity than its parent hydride complex 1.Acknowledgements
The authors gratefully acknowledge the financial support from the European Regional Development Fund, Operational Programme Innovative Economy, 2007–2013, project no. UDA-POIG.01.03.01-30-173/09 and from the National Science Centre (Poland) (project ‘Maestro’ no. UMO-2011/0/A/ST5/00472).References
- (a) D. B. Cordes, P. D. Lickiss and F. Rataboul, Chem. Rev., 2010, 110, 2081 CrossRef CAS PubMed; (b) S. Yamaguchi and K. Tamao, in Silicon-containing polymers, ed. R. G. Jones, W. Ando and J. Chojnowski, Kluwer, 2000, ch. 17 Search PubMed; (c) Materials Research Society Symposia Proceedings, ed. R. M. Laine, C. Sanchez, E. Giannelis and C. J. Brinker, Organic/Inorganic Hybrid Materials, Materials Research Society, Warrendale, 2000, vol. 628 Search PubMed; (d) Materials Research Society Symposia Proceedings, ed. C. Sanchez, R. M. Laine, S. Yang and C. J. Brinker, Organic/Inorganic Hybrid Materials, Materials Research Society, Warrendale, 2002, vol. 726 Search PubMed.
- B. Marciniec, I. Kownacki, A. Franczyk and M. Kubicki, Dalton Trans., 2011, 40, 5073 RSC.
- R. Duchateau, Chem. Rev., 2002, 102, 3626 CrossRef PubMed.
- E. A. Quadrelli and J. M. Basset, Coord. Chem. Rev., 2000, 254, 707 CrossRef PubMed.
- N. Mintcheva, M. Tanabe and K. Osakada, Organometallics, 2011, 30, 187 CrossRef CAS.
- A. R. Bassindale and T. E. Gentle, J. Mater. Chem., 1993, 3, 1319 RSC.
- M. A. Wahab, K. Y. Mya and C. He, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5887 CrossRef PubMed.
- M. Y. Lo, C. Zhen, M. Lauters, G. E. Jabbour and A. Sellinger, J. Am. Chem. Soc., 2007, 129, 5808 CrossRef CAS PubMed.
- K. I. Chan, P. Sonar and A. Sellinger, J. Mater. Chem., 2009, 19, 9103 RSC.
- A. Schmidt, S. Babin, K. Böhmer and H. W. P. Koops, Microelectron. Eng., 1997, 35, 129 CrossRef CAS.
- S. B. Desu, C. H. Peng, T. Shi and P. A. Agaskar, J. Electrochem. Soc., 1992, 139, 2682 CrossRef CAS PubMed.
- D. B. Cordes, P. D. Lickiss and F. Rataboul, Chem. Rev., 2010, 110, 2081 CrossRef CAS PubMed.
- Y. Itami, B. Marciniec and M. Kubicki, Chem. – Eur. J., 2004, 10, 1239 CrossRef CAS PubMed.
- J. Waehner, B. Marciniec and P. Pawluć, Eur. J. Inorg. Chem., 2007, 18, 2975 CrossRef PubMed.
- P. Żak, C. Pietraszuk, B. Marciniec, G. Spólnik and W. Danikiewicz, Adv. Synth. Catal., 2009, 351, 2675 CrossRef PubMed.
- P. Żak, B. Marciniec, M. Majchrzak and C. Pietraszuk, J. Organomet. Chem., 2011, 696, 887 CrossRef PubMed.
- B. Marciniec and C. Pietraszuk, Organometallics, 1997, 16, 4320–4326 CrossRef CAS.
- B. Marciniec, Coord. Chem. Rev., 2005, 249, 2374–2390 CrossRef CAS PubMed.
- B. Marciniec, Acc. Chem. Res., 2007, 40, 943–952 CrossRef CAS PubMed.
- G. Calzaferri, R. Imhofa and K. W. Tornroosb, J. Chem. Soc., Dalton Trans., 1993, 3741 RSC.
- M. Rattay, D. Fenske and P. Jutzi, Organometallics, 1998, 17, 2930 CrossRef CAS.
- Vinylsilsesquioxanes purchased from Hybrid Plastics Inc. and used as received do not undergo silylative coupling. Successful reactivity was achieved by preliminary purification by extraction of pentane solution of the reagent with water followed by drying of the organic phase with magnesium sulphate and solvent evaporation.
- J. Levison and S. D. Robinson, J. Chem. Soc. A, 1970, 2947 RSC.
- CrysAlis PRO, Agilent Technologies, 2010 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † CCDC 919730. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt00553h |
| This journal is © The Royal Society of Chemistry 2014 |