 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anion exchange in [Ni(η5-C5H4R)(Cl)(NHC)]. Counterion effect on the structure and catalytic activity†
Włodzimierz
Buchowicz
*,
Łukasz
Banach
,
Joanna
Conder
,
Piotr A.
Guńka
,
Dominik
Kubicki
and
Piotr
Buchalski
Warsaw University of Technology, Faculty of Chemistry, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: wbuch@ch.pw.edu.pl; Fax: +48 22 234 5462; Tel: +48 22 234 5150
First published on 30th January 2014
Abstract
A series of novel complexes [Ni(η5-C5H4R)(L)(NHC)]+A−2a–2j and [Ni(η5-C5H5)(A)(NHC)] 3a–3c has been obtained by anion metathesis from the corresponding chlorides 1a–1d, depending on the anion binding properties and reaction conditions. Solid-state structures of two cationic complexes (2c, 2j) and two complexes with a coordinated anion (3a, 3c) have been determined by X-ray diffraction revealing a trigonal planar geometry in all cases. Unexpectedly, 3c displayed unprecedented for this type of compounds temperature-dependent NMR spectra that were interpreted in terms of spin equilibrium. The cationic complexes 2 were less efficient in styrene polymerization than the parent chlorides 1. However, the activity of 2 and 3 in Suzuki cross-coupling did not depend considerably on the counterion.
Introduction
Transition metal N-heterocyclic carbene (NHC) complexes are a growing field of interest in organometallic chemistry, homogeneous catalysis, and other areas of chemistry.1 Nickel(0), nickel(I), and nickel(II) NHC complexes have attracted substantial attention in recent years, mainly due to their application as catalysts in a number of important organic transformations.2Abernethy et al. discovered that reaction of nickelocene with 1,3-dimesitylimidazolium chloride in refluxing THF afforded diamagnetic complex [Ni(η5-C5H5)(Cl)(IMes)]‡ (1a) in high yield.3 Following this original communication, a considerable variety of NHC complexes of the general formula [Ni(η5-C5H4R)(X)(NHC)] (1, R = H or alkyl; X = Cl, Br, or I) has been prepared in this manner (Scheme 1, path a).4–10 These Ni(II) complexes display promising catalytic activity in several reactions, including amination of aromatic compounds,4 polymerization of styrene,5,8 polymerization of methyl methacrylate,11 Suzuki–Miyaura cross-coupling,9,12 regioselective hydrothiolation of alkynes,13 and hydrosilylation of aldehydes and ketones.14
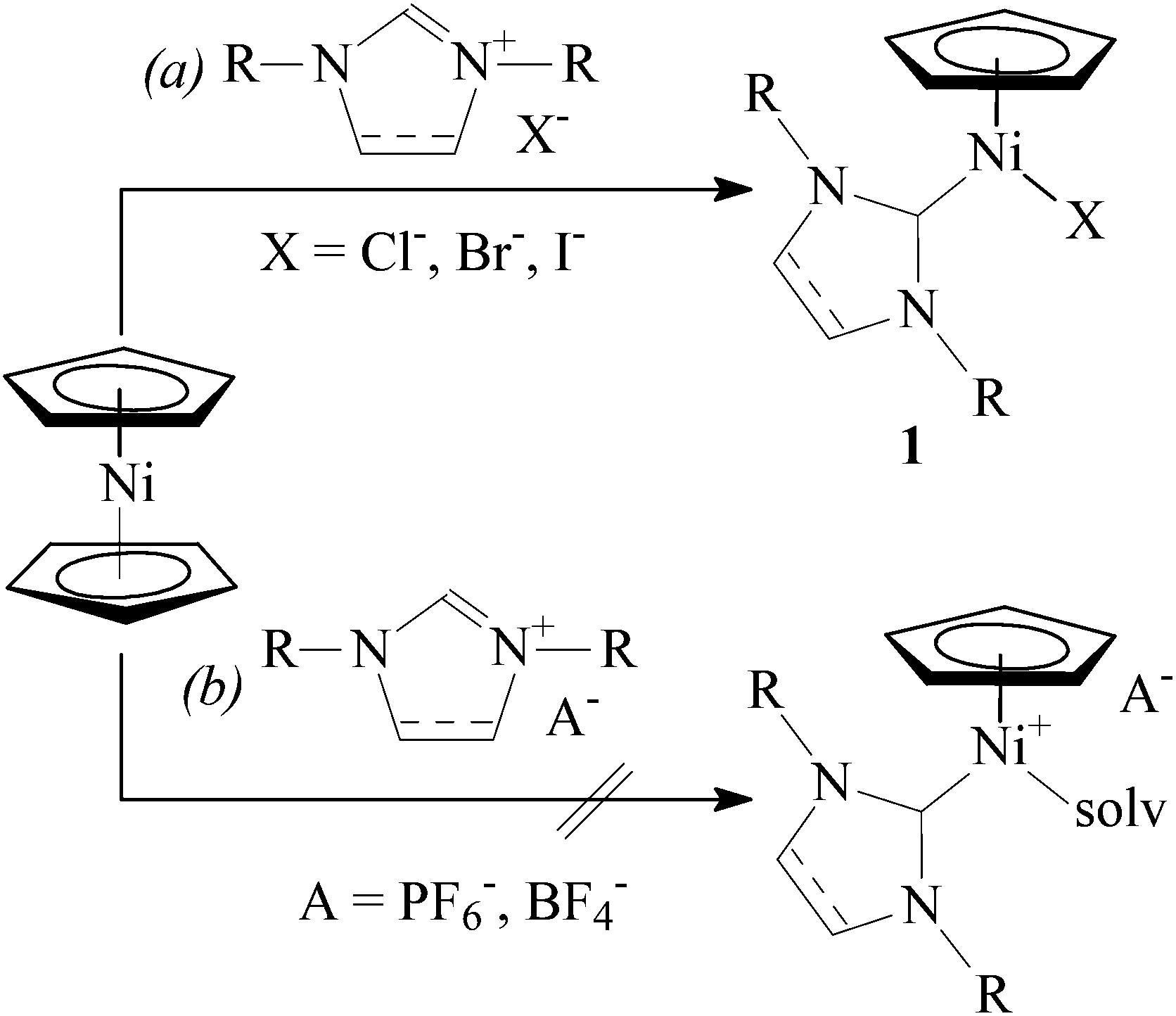 | ||
| Scheme 1 Reactions of nickelocene with NHC salts: (a) THF, heating; (b) THF or CH3CN, heating. | ||
Previously, we suggested that styrene polymerization catalysed by [Ni(η5-C5H5)(X)(NHC)] in the presence of methyl-aluminoxane (MAO) proceeded via a cationic mechanism15 involving intermediate species [Ni(η5-C5H5)(L)(NHC)]+ (L = styrene, solvent).5 In order to further elucidate this process, we sought to approach these tentative intermediates via a different route.
Halogen substitution in complexes 1 with AgBF4 or KPF6 has been recently reported. In the case of complexes bearing N-allyl functionalized NHC ligands, intramolecular cationic π-complexes [Ni(η5-C5H5)(η3-NHC)]+A− were obtained.6 However, reactions of complexes with N-aryl substituted NHCs resulted in cationic complexes [Ni(η5-C5R5)(L)(NHC)]+A− (R = H or Me, L = CH3CN or (CH3)2CO) in acetonitrile12,16 or acetone.17
In this contribution, we explore the scope of halogen substitution in complexes [Ni(η5-C5H4R)(Cl)(NHC)] with various metal salts, including non-coordinating and weakly-coordinating anions, and catalytic properties of the resulting complexes.
Results and discussion
Synthesis
Nolan and co-workers reported that nickelocene did not react with NHC tetrafluoroborates or hexafluorophosphates in refluxing THF.4a Indeed, when we attempted to synthesize [Ni(η5-C5H5)(CH3CN)(NHC)]+(BF4)− directly from nickelocene and an imidazolium tetrafluoroborate in refluxing acetonitrile, a gradual decomposition of nickelocene was observed (Scheme 1, path b).Accordingly, complexes [Ni(η5-C5H4R)(L)(NHC)]+A− (2) were obtained by the two-step route involving isolation of chlorides 1.
Complexes 1a–1e reacted cleanly with KPF6, AgClO4, AgCF3SO3, AgCF3CO2, or AgNO3 in a nitrile solution (acetonitrile, pivalonitrile) at room temperature. The expected cationic complexes 2 were obtained as yellow-brown solids in high yields for KPF6, AgClO4, and AgCF3SO3 (Scheme 2). However, reactions of 1a or 1b with AgCF3CO2 in acetonitrile or toluene afforded complexes 3a and 3b with the coordinated carboxylate (Scheme 3). Attempts to extend this methodology to other carboxylates (acetate, pivalate) were not successful.
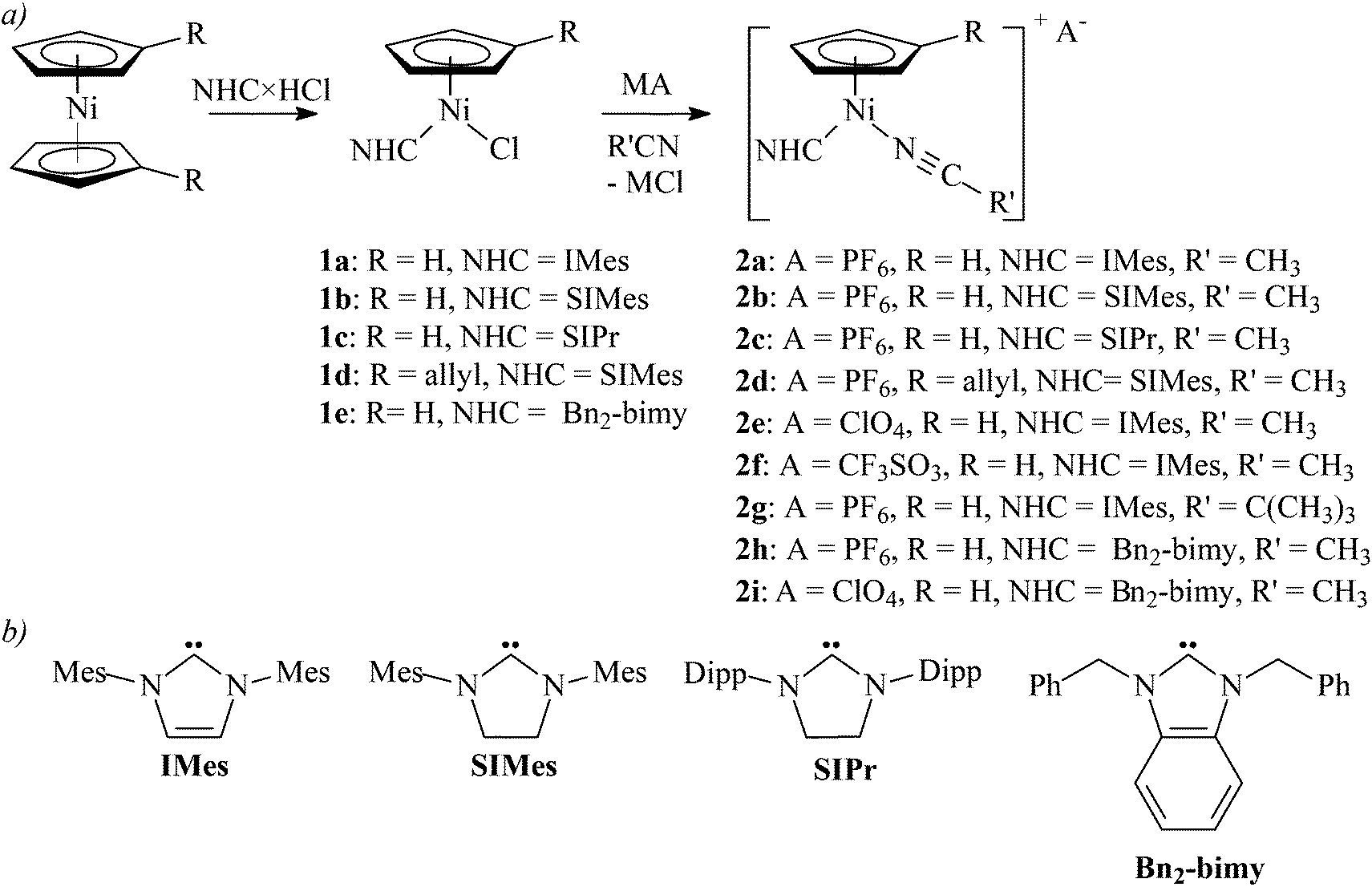 | ||
| Scheme 2 (a) The synthesis of cationic complexes 2a–2i (M = K or Ag; complex 2a has been reported by Chetcuti et al.12). (b) Structures of NHC ligands used in this work (Mes = 2,4,6-trimethylphenyl; Dipp = 2,6-diisopropylphenyl). | ||
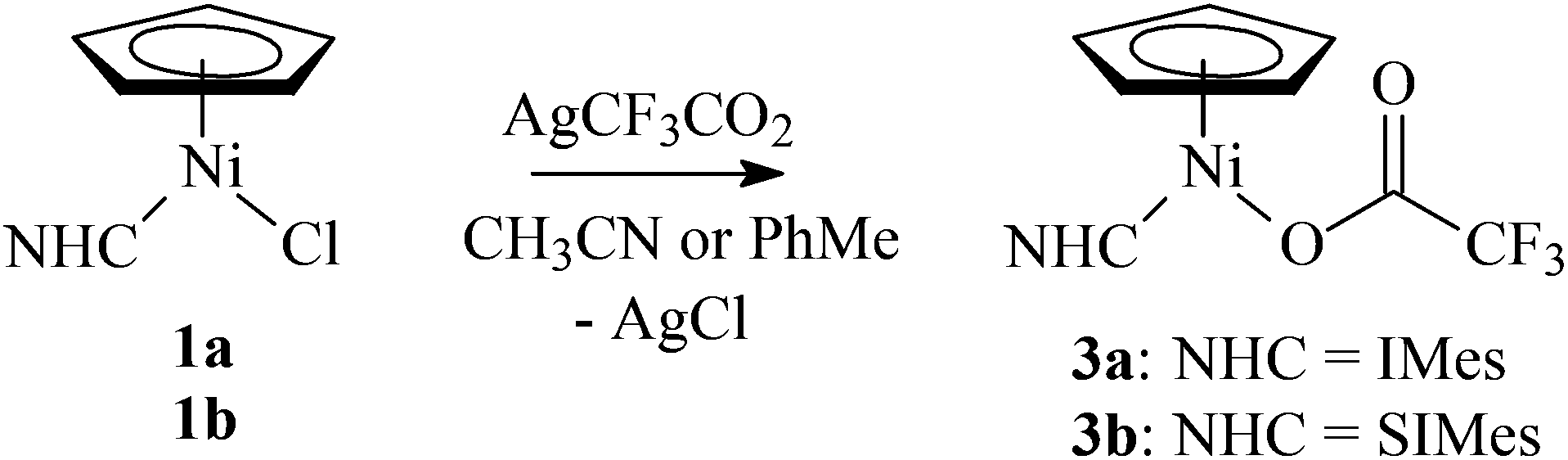 | ||
| Scheme 3 The synthesis of trifluoroacetate complexes 3a and 3b. | ||
In contrast to AgCF3CO2, reaction of 1a with AgNO3 in acetonitrile afforded cationic complex 2j. However, when this reaction was repeated in toluene/THF, neutral complex 3c with a coordinated nitrate was isolated. Moreover, complex 2j could be also obtained by dissolving 3c in acetonitrile (Scheme 4). In contrast to 2a–2i that were stable in CDCl3 solutions (see below), dissolving 2j in CDCl3 resulted in a red solution giving NMR spectra corresponding to 3c. A detailed inspection of these spectra revealed also a residual broad singlet at 4.77 ppm that could be assigned to 2j. This behaviour suggests that 2j and 3c exist in equilibrium in a polar solvent, with 3c being the major species.18
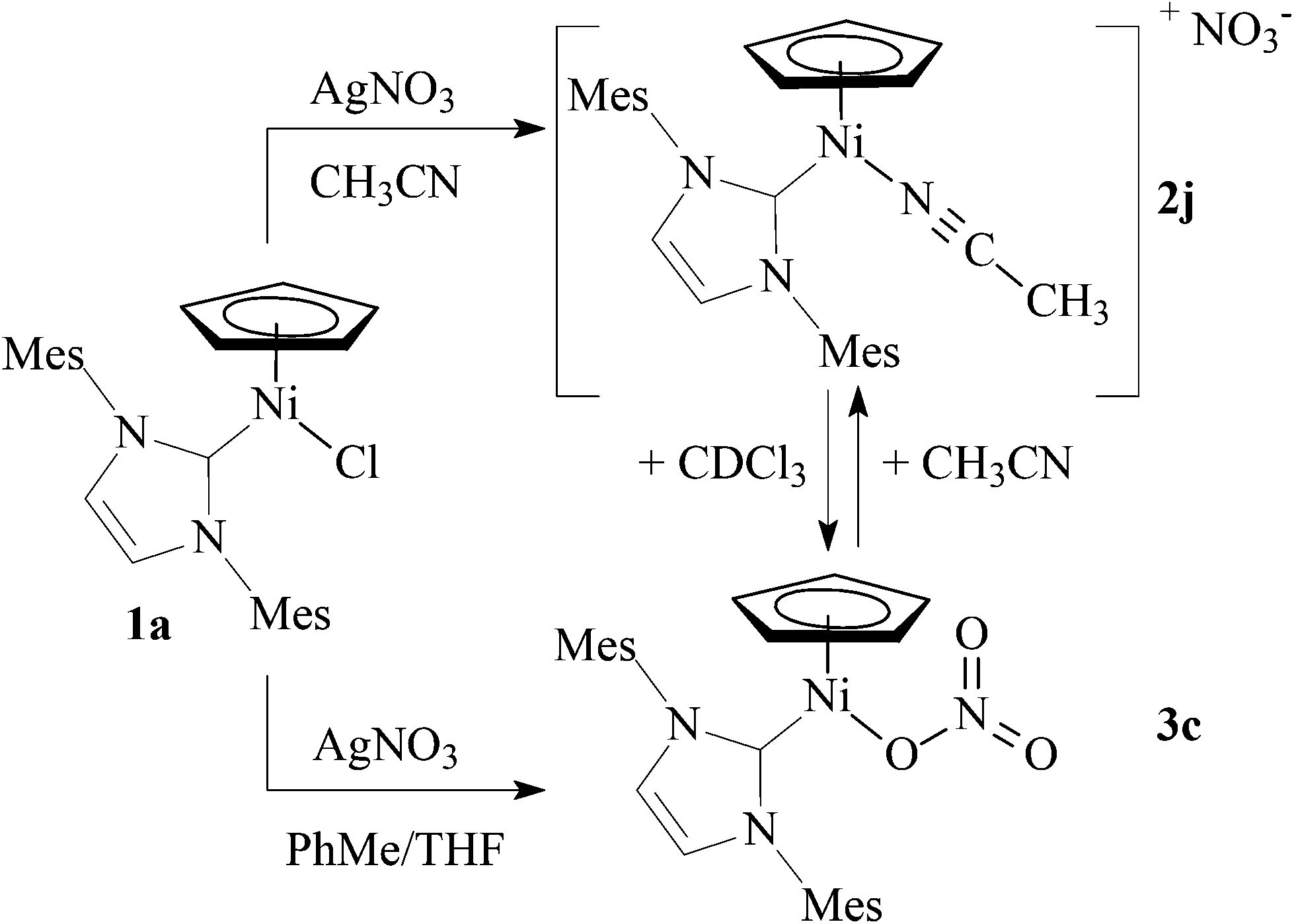 | ||
| Scheme 4 The synthesis of nitrates 2j and 3c (Mes = 2,4,6-trimethylphenyl). | ||
Characterization
The carbene carbon atom chemical shift varied from 159.9 ppm to 199.4 ppm, depending on the type of NHC ligand. For IMes complexes (2a, 2e–2g, 2j, and 3a), the carbene carbon atom signal appeared from 159.9 to 166.2 ppm. The 13C NMR spectra of Bn2-bimy complexes 2h and 2i displayed their carbene atom signals at 174.1 ppm and 174.2 ppm, respectively. The highest chemical shift of the carbene carbon atom in the range of 195.6–199.4 ppm was observed for SIMes complexes (2b, 2d and 3b). While the spectra of trifluoroacetates 3a and 3b were unexceptional, the spectra of nitrates 2j and 3c deserve a further comment.
In contrast to the other ionic complexes, NMR spectra of 2j could be recorded only in CD3CN since 2j appeared to easily dissociate the nitrile ligand in a polar solvent (e.g. in CDCl3) to form the neutral complex 3c (see Scheme 4). Thus, NMR spectra of 2j featured all expected signals within the usual ranges, e.g. a sharp singlet of Cp protons at 4.77 ppm.
However, NMR spectra of 3c were far from routine: we first noticed rather unusual chemical shift and linewidth of the Cp signal: in CDCl3 at ambient temperature it appeared at 3.53 ppm with ν1/2 = 5.6 Hz, and at 35 °C it appeared at higher field at 2.77 ppm with ν1/2 = 13 Hz. Moreover, the parameters of the Cp signal varied considerably also with the solvent used: it appeared at 2.34 ppm (ν1/2 = 8.9 Hz) in C6D6 at ambient temperature.20
In the 13C NMR spectrum of 3c no carbene carbon atom signal was detected; moreover, the Cp signal at 97.1 ppm was unusually broad with ν1/2 = 6.2 Hz, while for the other complexes ν1/2 was in the range 1.6–2.1 Hz. VT NMR studies in toluene-d8 in the temperature range from −55 °C to 100 °C were therefore performed (Fig. 1). The Cp signal appeared as a singlet at 4.31 ppm at −55 °C and shifted to −3.01 ppm at 100 °C. This upfield shift with increasing temperature was accompanied by signal broadening from ν1/2 = 5 Hz to ν1/2 = 36 Hz. At the same time the imidazole singlet shifted downfield slightly from 5.81 ppm to 7.33 ppm. We explain this behaviour of 3c in terms of spin equilibrium, i.e. equilibrium between a diamagnetic singlet ground state and a paramagnetic triplet excited state.21 The absence of an observable carbene carbon atom signal might be explained by its merging with the baseline as a result of the paramagnetic broadening.
 | ||
| Fig. 1 VT 1H NMR (500 MHz, toluene-d8) spectra of complex 3c (high-field range) at temperatures: (a) −55 °C, (b) −30 °C, (c) 10 °C, (d) 70 °C, and (e) 100 °C. Asterisk (*) indicates the Cp resonance. | ||
The spin equilibrium was further modelled by using a Boltzmann distribution of spins22 (for details, see the ESI†) and the thermodynamic parameters for 3c thus obtained were as follows: ΔH° = (15.15 ± 0.43) kJ mol−1, ΔS° = (16.7 ± 4.0) J (mol K)−1. The value of the high-spin species to low-spin species equilibrium constant Keq of 0.02 calculated at 298.15 K shows that at this temperature there is a large excess of the diamagnetic form of 3c. This value also explains why the magnetic susceptibility measurement by Evans’ method23 that we had attempted failed to give any significant result. At the same time such a placement of the equilibrium substantiates the observed chemical shift for Cp protons in 3c. For nickelocene (two unpaired electrons, μeff = 2.88μB)24 the magnitude of paramagnetic 1H NMR chemical shift (δ = ca. −250 ppm)25 is considerably larger than for 3c even though both in nickelocene and in 3c the distances between the nickel atom and the Cp plane are comparable (1.8177(4) Å26 and 1.765(3) Å, respectively), and therefore the paramagnetic contribution to the chemical shifts in both compounds should be of a similar order of magnitude. In the case of 3c this contribution is relatively small, creating a downfield shift of only several ppm. While it is not clear why the nitrate anion modifies the electronic properties of 3c in comparison with the other studied anions, the solid state structure of 3c (see below) revealed the expected, three-coordinate geometry.
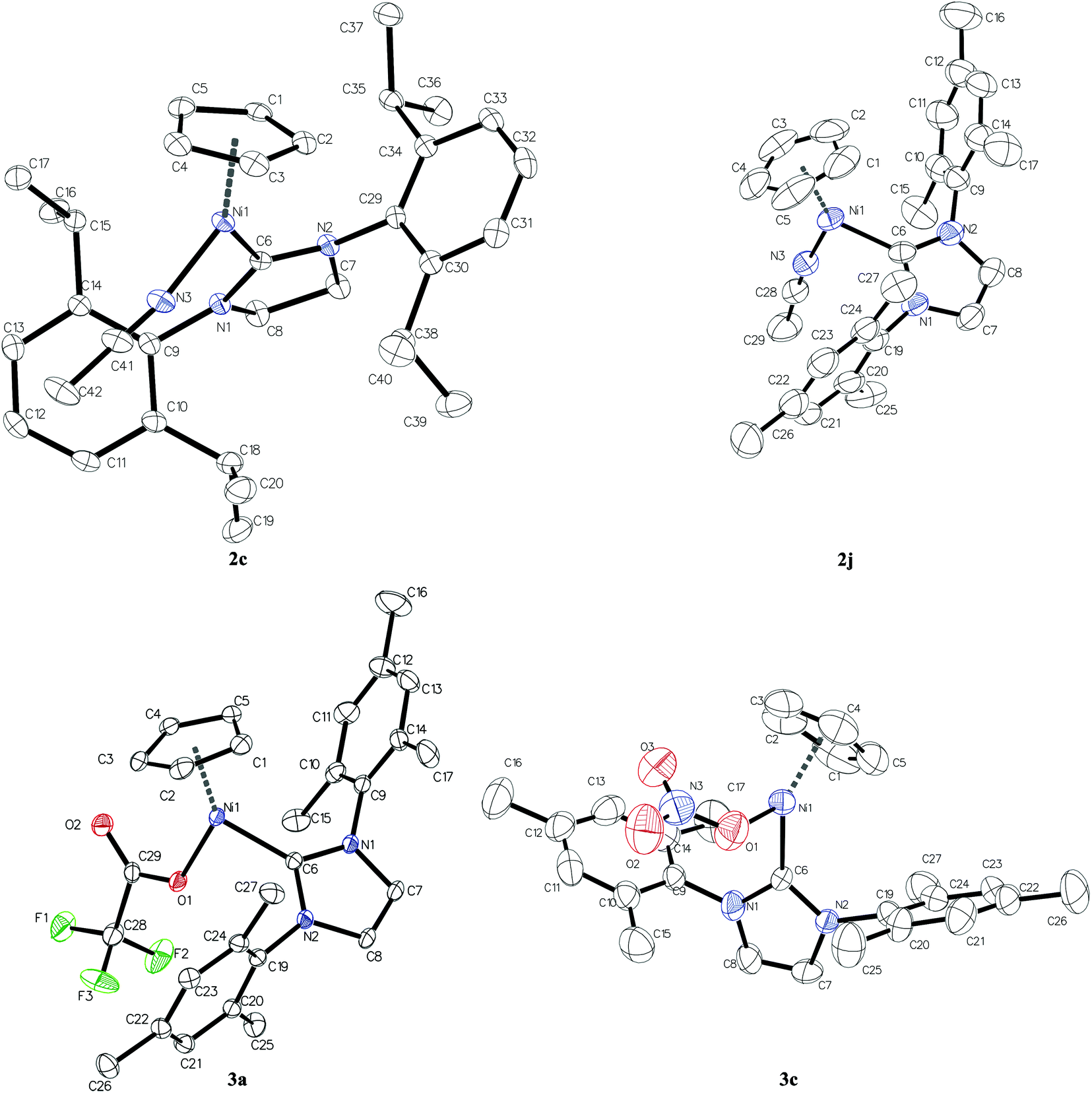 | ||
| Fig. 2 The molecular structure of cations of complexes 2c and 2j and of neutral complexes 3a and 3c. Hydrogen atoms are omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level. In the case of complexes 2c and 3c, where two independent molecules are present in the asymmetric unit, only one of them is presented. See Fig. S3 in the ESI† for the second ones of 2c and 3c. | ||
| Compound | 2c | 3a | 2j | 3c |
|---|---|---|---|---|
| Chemical formula | NiC34H46N3+·PF6− | NiC28H29F3N2O2 | NiC28H32N3+·NO3− | NiC26H29N3O3 |
| Formula mass | 700.42 | 541.24 | 531.28 | 490.23 |
| Crystal system | Triclinic | Triclinic | Triclinic | Orthorhombic |
| a/Å | 10.3048(8) | 7.9276(3) | 8.5478(3) | 17.6218(3) |
| b/Å | 18.3915(10) | 9.6611(5) | 8.8564(3) | 12.2104(3) |
| c/Å | 19.1066(10) | 20.5983(8) | 17.8908(5) | 22.7534(4) |
| α/° | 75.910(5) | 78.742(4) | 89.233(3) | 90 |
| β/° | 86.934(5) | 89.700(3) | 80.886(3) | 90 |
| γ/° | 81.371(5) | 69.643(4) | 85.015(3) | 90 |
| Unit cell volume/Å3 | 3471.8(4) | 1447.25(11) | 1332.22(7) | 4895.80(16) |
| Temperature/K | 100(2) | 100(2) | 293(2) | 293(2) |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
Pca21 |
| Z | 4 | 2 | 2 | 8 |
| No. of reflections measured | 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 217 217 |
55160 |
26005 |
53880 |
| No. of independent reflections | 17567 |
10462 |
6117 | 9968 |
| R int | 0.0708 | 0.0547 | 0.0361 | 0.0323 |
| Final R1 values (I > 2σ(I)) | 0.0468 | 0.0488 | 0.0386 | 0.0532 |
| Final wR(F2) values (I > 2σ(I)) | 0.1051 | 0.1233 | 0.0964 | 0.1444 |
| Final R1 values (all data) | 0.0646 | 0.0578 | 0.0488 | 0.0606 |
| Final wR(F2) values (all data) | 0.1151 | 0.1279 | 0.1020 | 0.1516 |
| Goodness of fit on F2 | 1.021 | 1.135 | 1.019 | 1.067 |
| d/Å | 2c | 3a | 2j | 3c |
|---|---|---|---|---|
| a N3 denotes the nitrogen atom of the coordinated acetonitrile for 2c and 2j; O1 denotes the oxygen atom of the trifluoroacetate or nitrate anions for complexes 3a and 3c, respectively (see Fig. 2 for the atom numbering scheme). | ||||
| Ni1–C1 | 2.033(2) | 2.0388(17) | 2.002(2) | 2.032(7) |
| Ni1–C2 | 2.157(2) | 2.1252(17) | 2.138(2) | 2.123(7) |
| Ni1–C3 | 2.181(2) | 2.1245(17) | 2.174(2) | 2.139(6) |
| Ni1–C4 | 2.122(2) | 2.1980(17) | 2.152(2) | 2.181(7) |
| Ni1–C5 | 2.134(2) | 2.1895(17) | 2.125(2) | 2.163(7) |
| Ni1–C6 (NHC) | 1.9013(19) | 1.8835(16) | 1.9020(19) | 1.874(5) |
| Ni1–X (N3, O1)a | 1.8644(18) | 1.9104(12) | 1.8693(17) | 1.910(4) |
Single crystal X-ray structure analysis reveals that compounds 2c, 3a and 2j crystallise in the triclinic P (no. 2) space group whereas complex 3c is the only one to yield non-centrosymmetric crystal structure in the orthorhombic Pca21 (no. 29) space group. While crystal structures of compounds 3a and 2j contain one molecule and a pair of cation and anion, respectively, in the asymmetric unit, there are two independent molecules or two independent pairs of cations and anions in crystal structures of complexes 3c and 2c, respectively. The sum of bond angles around Ni atoms, that is, X–Ni–C(NHC), C(NHC)–Ni–Cg and Cg–Ni–X angles, where Cg denotes the centre of gravity of Cp rings and X stands for N or O, amounts to 360° within 3 s.u.'s in all studied compounds which indicates planar trigonal coordination of nickel atoms (see Table S2 in the ESI†). The plane of the carboxylate group in complex 3a deviates considerably from the Ni coordination plane (Ni, Cg, X, C(NHC)) as evidenced by the value of C6–Ni1–O1–C29 torsion angle equal to −153.50(14)°. This is even more pronounced for nitrate anions in compound 3c where C6–Ni1–O1–N3 and C36–Ni2–O31–N33 torsion angles are −116.2(4) and 115.2(5)°, respectively. This twist results in the monodentate binding of carboxylate and nitrate ligands to nickel (Ni⋯O distances for unbound oxygen atoms amount to 3.2238(13) Å in 3a and 2.905(6) Å on average in 3c). As shown in Table 2, the distances from nickel to Cp carbon atoms differ significantly from each other within every complex. The Ni–CCp bond trans to the L ligand (acetonitrile molecule for complexes 2c and 2j, trifluoroacetate anion for 3a and nitrate anion for 3c) is shorter by ca. 0.1 Å compared to the other ones. This variation in the Ni–CCp distances can be attributed to the trans effect of the NHC ligand which leads to the elongation of Ni–CCp bonds trans to the carbene and, consequently, shortening of the Ni–CCp bonds trans to the L ligand. The C–C bond lengths in cyclopentadienyl ligands vary from 1.35 to 1.45 Å which is typical for Ni complexes comprising both Cp and NHC ligands deposited in the Cambridge Structural Database (1.35–1.46 Å).27
Catalytic activity
:Ni = 100:1) was added to a toluene suspension of complex 2 or 3. After stirring for 30 min at ambient temperature, neat styrene (styrene:Ni = 1000:1) was added and the polymerization was run in a sealed Schlenk tube for 3 h at 50 °C. The results of styrene polymerization are summarized in Table 3.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Complex (NHC) | A | L | Yieldb (%) | M n | M w/Mnc |
|
a All reactions in duplicate; conditions: [Ni] = 1.30 mmol L−1, 3 h, 50 °C, styrene:Ni = 1000:1, Al:Ni = 100:1.
b Isolated yield.
c Determined with GPC in CH2Cl2 (target Mn = 100000).
d Control experiment without MAO.
e Control experiment without Ni, styrene:Al = 10:1.
f Allyl substituted Cp.
g Bimodal distribution with low Mn oligomers.
h High molecular fraction (Mn 800000) was also present.
|
||||||
| 1 | 2a (IMes) | PF6− | CH3CN | 22 | 14000 |
2.0 |
| 1300 | 1.1 | |||||
| 2d | 2a (IMes) | PF6− | CH3CN | 0 | — | — |
| 3e | — | — | — | 0 | — | — |
| 4 | 2b (SIMes) | PF6− | CH3CN | 35 | 13000g |
2.0 |
| 800 | 1.2 | |||||
| 5 | 2c (SIPr) | PF6− | CH3CN | 13 | 10000 |
2.3 |
| 6 | 2d (SIMes)f | PF6− | CH3CN | 10 | 11000 |
2.0 |
| 7 | 2e (IMes) | ClO4− | CH3CN | 31 | 1300g | 7.0 |
| 8 | 2f (IMes) | CF3SO3− | CH3CN | 21 | 1300g,h | 11 |
| 9 | 2g (IMes) | PF6− | (CH3)3CCN | 20 | 1000g | 6.9 |
| 10 | 2h (Bn2-bimy) | PF6− | CH3CN | 14 | 14600g |
1.6 |
| 800 | 1.5 | |||||
| 11 | 2j (IMes) | NO3− | CH3CN | 42 | 1500g | 6.7 |
| 12 | 3a (IMes) | CF3CO2− | — | 94 | 11300 |
2.0 |
| 13 | 3b (SIMes) | CF3CO2− | — | 100 | 10500 |
2.0 |
| 14 | 3c (IMes) | NO3− | — | 45 | 1600g | 7.3 |

Disappointingly, hexafluorophosphates 2a and 2b (entries 1 and 4) were one order of magnitude less active than the neutral parent complexes.5 In control experiments we established that 2a without MAO gave no polymer (entry 2). Similarly, MAO itself did not yield polystyrene (entry 3). Analogously to what was observed for the chloride series, introduction of the more bulky NHC ligand (SIPr vs. SIMes, entry 5) or a substituent on the Cp ligand (entry 6) resulted in significantly lower yields than for 2b.
Other weakly- or non-coordinating anions (ClO4−, CF3SO3−, and NO3−) had no significant effect on the activity (entries 7, 8, and 11). Complex 2g with the more bulky nitrile (entry 9) provided the same efficiency as 2a. Introduction of the weaker donating benzimidazole-based NHC ligand resulted in a low yield of the polymer (entry 10). The highest activity was achieved with complexes 3a and 3b (entries 12 and 13) bearing covalently bound carboxylates; however, neutral nitrate 3c was less effective (entry 14). These findings show that strongly coordinating anions, i.e. chloride or trifluoroacetate, are the most efficient in this type of polymerization.
The obtained polystyrenes were examined by 13C NMR, GPC, and MALDI-TOF MS. The 13C NMR spectra were consistent with atactic microstructure of all polymers.28 GPC analyses showed that in most cases Mn was lower than that obtained with the corresponding chlorides, while Mw/Mn was higher than for the chlorides. The trifluoroacetates 3a and 3b produced polystyrenes with similar Mn and Mw/Mn to those obtained with 1a.5 MALDI-TOF MS (see the ESI†) suggested that the polystyrene chains were terminated with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds.
C double bonds.
Previously, we proposed that the initial reaction of complexes 1 with MAO resulted in cationic species [Ni(η5-C5H5)(NHC)]+ (Scheme 5, path a).29 Consequently, the efficiency of styrene polymerization with 1/MAO depended mainly on the stabilization of these intermediate species, meaning that the strongly coordinating chloride that irreversibly reacts with MAO was the most suitable counterion.5 In this study, we anticipated that the labile nitrile ligand12,30 in complexes 2 would be readily displaced with styrene (Scheme 5, path b) to produce the same intermediates as with 1. However, the low efficiency of complexes 2 in the styrene polymerization suggests that the nitrile binds to the Ni centre rather strongly and actually inhibits the polymerization. To further address this issue, we studied reactions of complexes 2 with styrene (Scheme 6).
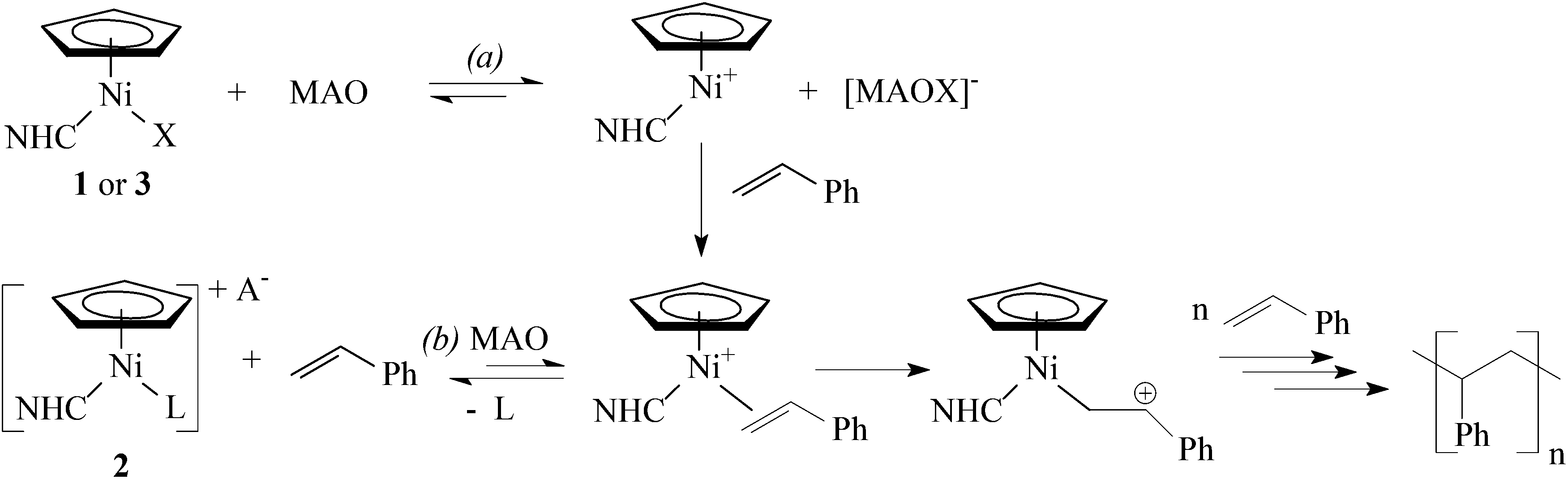 | ||
| Scheme 5 Proposed pathways of the styrene polymerization with complexes 1–3 (X = halogen or CF3CO2−, A = non- or weakly-coordinating anions used in this study, L = RCN). | ||
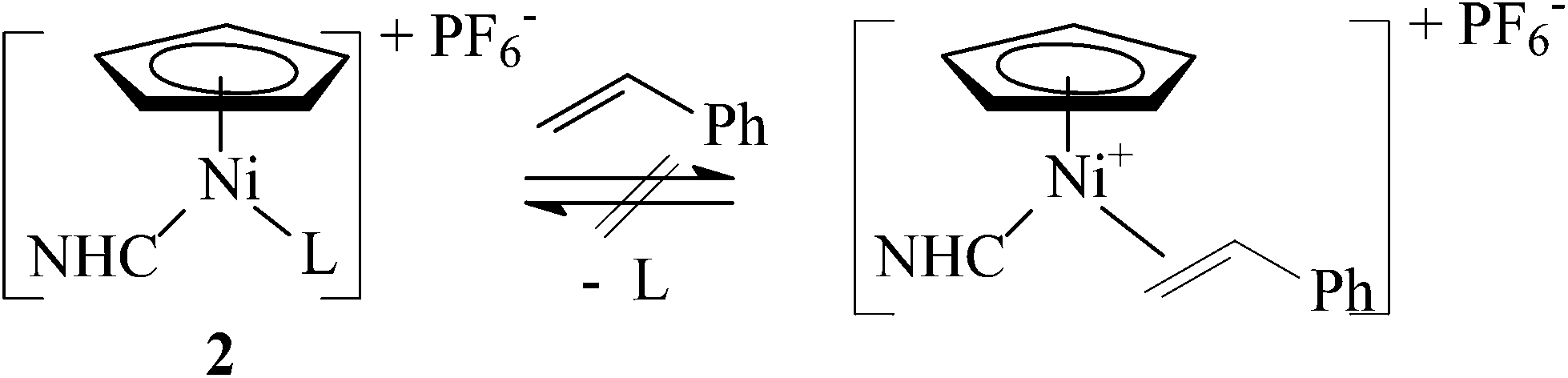 | ||
| Scheme 6 Attempted reactions of complexes 2 with styrene (L = CH3CN or (CH3)3CN). | ||
However, despite our best efforts to use as many various reaction conditions as possible (type of the nitrile ligand, solvent and temperature), the exchange of the nitrile ligand with styrene has not been accomplished. Complexes 2b and 2g remained unchanged in the presence of an excess of styrene, while 2h upon stirring in neat styrene at 35 °C partially transformed into an inseparable mixture of green nickel complexes. We note that an intramolecular analogue of [Ni(η5-C5H5)(η2-alkene)(NHC)]+ has been fully characterized by Hahn and co-workers.6,31
|
|
|||||
|---|---|---|---|---|---|
| Entry | Complex (NHC) | A | L | Yieldb (%) | Selectivityc |
|
a All runs in duplicate; reaction conditions: [Ni] = 10.2 mM (3 mol%), 90 °C, 1 h, toluene.
b Determined with GC.
c Determined with GC as the ratio A:B.
d Run with 4′-chloroacetophenone.
|
|||||
| 1 | 2a (IMes) | PF6− | CH3CN | 78 | 99:1 |
| 2 | 2b (SIMes) | PF6− | CH3CN | 73 | 99:1 |
| 3 | 2c (SIPr) | PF6− | CH3CN | 69 | 98:2 |
| 4 | 2e (IMes) | ClO4− | CH3CN | 74 | 99:1 |
| 6 | 2f (IMes) | CF3SO3− | CH3CN | 62 | 99:1 |
| 7 | 2g (IMes) | PF6− | (CH3)3CCN | 67 | 99:1 |
| 8 | 2h (Bn2-bimy) | PF6− | CH3CN | 56 | 97:3 |
| 9 | 2i (Bn2-bimy) | ClO4− | CH3CN | 47 | 98:2 |
| 10 | 2j (IMes) | NO3− | CH3CN | 88 | 99:1 |
| 11 | 3a (IMes) | CF3CO2− | — | 57 | 99:1 |
| 12 | 3b (SIMes) | CF3CO2− | — | 60 | 99:1 |
| 13 | 3c (IMes) | NO3− | — | 38 | 99:1 |
| 14 | 2e (IMes)d | ClO4− | CH3CN | 47 | 99:1 |
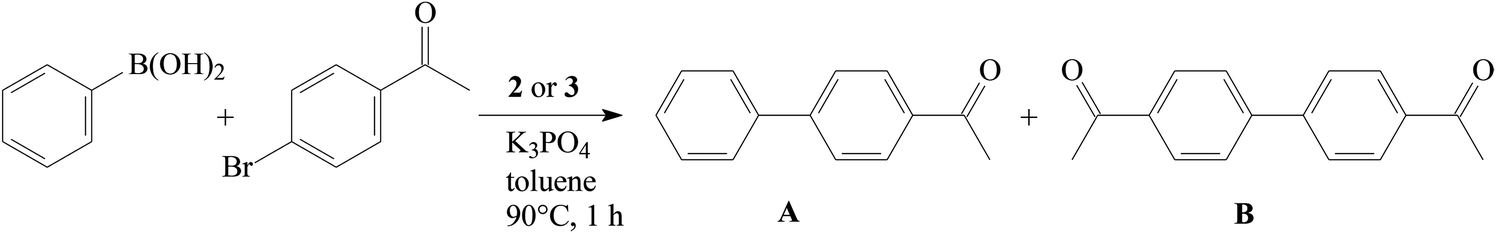
All studied complexes provided high yields of the expected cross-coupling product, i.e. 4-acetylbiphenyl (A), with excellent selectivity. The highest yield was achieved with cationic nitrate 2j (entry 10); however, the advantageous effect of this counterion was not confirmed with neutral nitrate 3c (entry 13). The weakly donating Bn2-bimy ligand was consistently less efficient (entries 8 and 9) than the other NHC ligands. With the more challenging substrate, 4′-chloroacetophenone, complex 2e was significantly less efficient than with 4′-bromoacetophenone (entry 14). The absence of a pronounced structure–activity relationship for the studied series of complexes is consistent with previous hypothesis that the Ni(II) complexes 2–3 serve as a convenient source of Ni(0) in this catalytic reaction.9,12
Conclusions
In summary, we have shown that, depending on the anion binding properties and reaction conditions, cationic 2 or neutral complexes 3 were obtained by anion metathesis in complexes 1. In the case of nitrate, both cationic complex 2j and neutral 3c could be isolated. Complexes 2j and 3c were found to easily interconvert with each other in a solution. This facile exchange of ligands opens up prospects for further optimization of electronic and catalytic properties of these complexes, in particular discovery of systems with switchable magnetic properties and plausible applications in spintronics.Experimental section
General
All manipulations (except polymer separation and purification, and work-up of the Suzuki cross-coupling reactions) were performed under an inert atmosphere of argon using Schlenk techniques. Solvents were purified with conventional methods.32 Styrene (ReagentPlus®, Aldrich) was distilled from CaH2 under reduced pressure and passed through a column with neutral Al2O3. Other reagents were purchased from commercial suppliers and were used without further purification. Complexes 1a–1e were prepared from nickelocene33 or 1,1′-bis(allyl)nickelocene34 and the appropriate imidazolium salt according to the published method with minor modifications.3NMR spectra were recorded, unless otherwise noted, at ambient temperature on a Mercury-400BB spectrometer operating at 400 MHz for 1H NMR, at 101 MHz for 13C NMR, at 376 MHz for 19F NMR, and at 162 MHz for 31P NMR. ESI MS were measured on a Mariner spectrometer. EI MS (70 eV) were measured on a AutoSpec Premier (Waters) spectrometer. MALDI-TOF MS of polystyrenes were acquired with a Bruker Daltonics ultrafleXtreme™ mass spectrometer (DCTB matrix with AgCF3CO2). The average molecular weights of PS were measured on a LabAlliance liquid chromatograph equipped with a Jordi Gel DVB Mixed Bed column (250 mm × 10 m) using CH2Cl2 as the mobile phase at 30 °C and calibrated with standard PS. Conversion and selectivity of Suzuki reactions were determined on an Agilent Technologies 7820 GC System equipped with a FID detector and an Agilent 19091J-413 column. Tetradecane was used as an internal standard.
Synthesis of cationic complexes 2a–2j
CH), 7.13 (4H, s, m-ArH), 4.76 (5H, s, C5H5), 2.43 (6H, s, p-ArCH3), 2.14 (3H, s, CH3CN), 2.11 (12H, s, o-ArCH3).121H NMR (400 MHz, CD3CN) δ(ppm): 7.43 (2H, s, HCCH), 7.21 (4H, s, m-ArH), 4.79 (5H, s, C5H5), 2.43 (6H, s, p-ArCH3), 2.16 (3H, s, CH3CN), 2.12 (12H, s, o-ArCH3).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CH2)(CH3CN)(SIMes)]+(PF6)− (2d).
Obtained from 1d (104.0 mg, 0.206 mmol) and KPF6 (37.0 mg, 0.203 mmol, 1 eq.) in acetonitrile (5.0 mL). Yield: 75%, yellow-brown solid (100.0 mg, 0.152 mmol). 1H NMR (400 MHz, CDCl3) δ(ppm): 7.07 (4H, s, m-ArH), 5.42 (1H, m, CH), 4.89, 4.91, and 4.93 (2H, m, CH2), 4.73 (2H, m, J = 2.2 Hz, C5H4), 4.30 (2H, t, J = 2.4 Hz, C5H4), 4.00 (4H, s, NCH2–CH2N), 2.38 (6H, s, p-ArCH3), 2.32 (12H, s, o-ArCH3), 2.26 (3H, s, CH3CN), 2.19 (2H, d, J = 6.8 Hz, C5H4CH2). 13C{1H} NMR (101 MHz, CDCl3) δ(ppm): 197.4 (NCN), 139.2 (ArC), 135.9 (ipso-ArC), 133.4 (ArC), 130.2 (CH), 129.7 (mArC), 128.7 (CH3CN), 116.8 (CH2), 114.8 (C5H4CH2), 97.9 (C5H4CH2), 86.7 (C5H4CH2), 51.5 (NCH2–CH2N), 31.4 (C5H4CH2), 21.1 (p-ArCH3), 18.1 (o-ArCH3), 3.9 (CH3CN). ESI MSm/z (58Ni): 510 ([M − PF6]+). 469 ([M − CH3CN − PF6−]+). Anal. Calc. for C31H38N3NiPF6: C, 56.7; H, 5.79; N, 6.40. Found: C, 56.4; H, 5.75; N, 6.28%.
CH), 7.13 (4H, s, m-ArH), 4.77 (5H, s, C5H5), 2.43 (6H, s, p-ArCH3), 2.23 (3H, s, CH3CN), 2.12 (12H, s, o-ArCH3). 13C{1H} NMR (101 MHz, CDCl3) δ(ppm): 161.5 (NCN), 140. 4 (ArC), 135.6 (ArC), 135.0 (ArC), 129.8 (ArC), 130.1 (CH3CN), 125.7 (HCCH), 93.7 (C5H5), 21.4 (p-ArCH3), 18.2 (o-ArCH3), 4.5 (CH3CN). Anal. Calc. for C28H31ClN3NiO4: C, 59.24; H, 5.50; N, 7.40. Found: C, 59.18; H, 5.64; N, 7.39%.
CH), 7.13 (4H, s, m-ArH), 4.76 (5H, s, C5H5), 2.43 (6H, s, p-ArCH3), 2.22 (3H, s, CH3CN), 2.11 (12H, s, o-ArCH3). 13C{1H} NMR (101 MHz, CDCl3) δ(ppm): 161.2 (NCN), 140.4 (ArC), 135.6 (ArC), 134.9 (ArC), 129.7 (ArC), 130.0 (CH3CN), 125.7 (HCCH), 93.7 (C5H5), 21.3 (p-ArCH3), 18.2 (o-ArCH3). 4.4 (CH3CN). 19F NMR (376 MHz, CDCl3) δ(ppm): −78.65 (s). Anal. Calc. for C29H32F3N3NiO3S: C, 56.33; H, 5.22; N, 6.80. Found: C, 56.31; H, 5.30; N, 6.79%.
CH), 7.13 (4H, bs, m-ArH), 4.75 (5H, s, C5H5), 2.44 (6H, s, p-ArCH3), 2.09 (12H, s, o-ArCH3), 1.24 (9H, s, (CH3)3CCN). 13C{1H} NMR (101 MHz, CDCl3) δ(ppm): 159.9 (NCN), 140.2 (ArC), 138.2 (CN), 135.7 (ArC), 134.8 (ArC), 129.8 (ArC), 126.1 (HCCH), 93.9 (C5H5), 30.9 ((CH3)3CCN), 27.6 ((CH3)3CCN), 21.4 (p-ArCH3), 18.2 (o-ArCH3). 19F NMR (376 MHz, CDCl3) δ(ppm): −73.55 (d, J = 709.0 Hz). 31P NMR (162 MHz, CDCl3) δ(ppm): −143.35 (sep, J = 709.0 Hz). ESI MSm/z (58Ni): 427 ([M − (CH3)3CN − PF6]+). Anal. Calc. for C31H38F6N3NiP: C, 56.73; H, 5.84; N, 6.40. Found: C, 56.70; H, 5.77; N, 6.40%.
CH), 7.19 (4H, s, m-ArH), 4.77 (5H, s, C5H5), 2.42 (6H, s, p-ArCH3), 2.21(1.6H due to exchange with CD3CN, bs, CH3CN), 2.11 (12H, s, o-ArCH3). 13C{1H} NMR (101 MHz, CD3CN) δ(ppm): 160.0 (NCN), 140.8 (ArC), 136.8 (ArC), 136.3 (ArC), 130.2 (ArC), 127.0 (HCCH), 94.24 (C5H5), 21.2 (p-ArCH3), 18.3 (o-ArCH3). Anal. Calc. for C28H32N4NiO3: C, 63.30; H, 6.07; N, 10.55. Found: C, 62.99; H, 6.07; N, 10.64%.
CH2)(CH3CN)(SIMes)]+(PF6)− (2d).
Obtained from 1d (104.0 mg, 0.206 mmol) and KPF6 (37.0 mg, 0.203 mmol, 1 eq.) in acetonitrile (5.0 mL). Yield: 75%, yellow-brown solid (100.0 mg, 0.152 mmol). 1H NMR (400 MHz, CDCl3) δ(ppm): 7.07 (4H, s, m-ArH), 5.42 (1H, m, CH), 4.89, 4.91, and 4.93 (2H, m, CH2), 4.73 (2H, m, J = 2.2 Hz, C5H4), 4.30 (2H, t, J = 2.4 Hz, C5H4), 4.00 (4H, s, NCH2–CH2N), 2.38 (6H, s, p-ArCH3), 2.32 (12H, s, o-ArCH3), 2.26 (3H, s, CH3CN), 2.19 (2H, d, J = 6.8 Hz, C5H4CH2). 13C{1H} NMR (101 MHz, CDCl3) δ(ppm): 197.4 (NCN), 139.2 (ArC), 135.9 (ipso-ArC), 133.4 (ArC), 130.2 (CH), 129.7 (mArC), 128.7 (CH3CN), 116.8 (CH2), 114.8 (C5H4CH2), 97.9 (C5H4CH2), 86.7 (C5H4CH2), 51.5 (NCH2–CH2N), 31.4 (C5H4CH2), 21.1 (p-ArCH3), 18.1 (o-ArCH3), 3.9 (CH3CN). ESI MSm/z (58Ni): 510 ([M − PF6]+). 469 ([M − CH3CN − PF6−]+). Anal. Calc. for C31H38N3NiPF6: C, 56.7; H, 5.79; N, 6.40. Found: C, 56.4; H, 5.75; N, 6.28%.
CH), 7.13 (4H, s, m-ArH), 4.77 (5H, s, C5H5), 2.43 (6H, s, p-ArCH3), 2.23 (3H, s, CH3CN), 2.12 (12H, s, o-ArCH3). 13C{1H} NMR (101 MHz, CDCl3) δ(ppm): 161.5 (NCN), 140. 4 (ArC), 135.6 (ArC), 135.0 (ArC), 129.8 (ArC), 130.1 (CH3CN), 125.7 (HCCH), 93.7 (C5H5), 21.4 (p-ArCH3), 18.2 (o-ArCH3), 4.5 (CH3CN). Anal. Calc. for C28H31ClN3NiO4: C, 59.24; H, 5.50; N, 7.40. Found: C, 59.18; H, 5.64; N, 7.39%.
CH), 7.13 (4H, s, m-ArH), 4.76 (5H, s, C5H5), 2.43 (6H, s, p-ArCH3), 2.22 (3H, s, CH3CN), 2.11 (12H, s, o-ArCH3). 13C{1H} NMR (101 MHz, CDCl3) δ(ppm): 161.2 (NCN), 140.4 (ArC), 135.6 (ArC), 134.9 (ArC), 129.7 (ArC), 130.0 (CH3CN), 125.7 (HCCH), 93.7 (C5H5), 21.3 (p-ArCH3), 18.2 (o-ArCH3). 4.4 (CH3CN). 19F NMR (376 MHz, CDCl3) δ(ppm): −78.65 (s). Anal. Calc. for C29H32F3N3NiO3S: C, 56.33; H, 5.22; N, 6.80. Found: C, 56.31; H, 5.30; N, 6.79%.
CH), 7.13 (4H, bs, m-ArH), 4.75 (5H, s, C5H5), 2.44 (6H, s, p-ArCH3), 2.09 (12H, s, o-ArCH3), 1.24 (9H, s, (CH3)3CCN). 13C{1H} NMR (101 MHz, CDCl3) δ(ppm): 159.9 (NCN), 140.2 (ArC), 138.2 (CN), 135.7 (ArC), 134.8 (ArC), 129.8 (ArC), 126.1 (HCCH), 93.9 (C5H5), 30.9 ((CH3)3CCN), 27.6 ((CH3)3CCN), 21.4 (p-ArCH3), 18.2 (o-ArCH3). 19F NMR (376 MHz, CDCl3) δ(ppm): −73.55 (d, J = 709.0 Hz). 31P NMR (162 MHz, CDCl3) δ(ppm): −143.35 (sep, J = 709.0 Hz). ESI MSm/z (58Ni): 427 ([M − (CH3)3CN − PF6]+). Anal. Calc. for C31H38F6N3NiP: C, 56.73; H, 5.84; N, 6.40. Found: C, 56.70; H, 5.77; N, 6.40%.
CH), 7.19 (4H, s, m-ArH), 4.77 (5H, s, C5H5), 2.42 (6H, s, p-ArCH3), 2.21(1.6H due to exchange with CD3CN, bs, CH3CN), 2.11 (12H, s, o-ArCH3). 13C{1H} NMR (101 MHz, CD3CN) δ(ppm): 160.0 (NCN), 140.8 (ArC), 136.8 (ArC), 136.3 (ArC), 130.2 (ArC), 127.0 (HCCH), 94.24 (C5H5), 21.2 (p-ArCH3), 18.3 (o-ArCH3). Anal. Calc. for C28H32N4NiO3: C, 63.30; H, 6.07; N, 10.55. Found: C, 62.99; H, 6.07; N, 10.64%.
1H NMR (400 MHz, CDCl3) δ(ppm): 7.11 (6H, s, m-ArH and HCCH overlapping), 4.62 (5H, s, C5H5), 2.44 (6H, s, p-ArCH3), 2.09 (12H, s, o-ArCH3). 13C{1H} NMR (101 MHz, CDCl3) δ(ppm): 166.2 (NCN), 163.8 (q, J = 35.6 Hz, CO), 139.34 (ArC), 136.3 (ArC), 135.6 (ArC), 129.3 (ArC), 124.6 (HCCH), 114.1 (q, J = 291.4 Hz, CF3), 91.3 (C5H5), 21.5 (p-ArCH3), 18.0 (o-ArCH3). 19F NMR (376 MHz, CDCl3) δ(ppm): −74.73 (s). EI MS (70 eV) m/z (58Ni): 540 (M+, 24%), 475 ([M − Cp]+, 29), 427 ([M − CF3CO2]+, 29), 303 (IMes+, 100). Anal. Calc. for C28H29F3N2NiO2: C, 62.14; H, 5.40; N, 5.18. Found: C, 62.18; H, 5.51; N, 5.20%.
CH), 7.12 (4H, s, m-ArH), 3.53 (5H, s, C5H5), 2.43 (6H, s, p-ArCH3), 2.16 (12H, s, o-ArCH3). 1H NMR (400 MHz, C6D6) δ(ppm): 6.86 (4H, s, m-ArH), 6.40 (2H, s, HCCH), 2.34 (5H, s, C5H5), 2.15 (6H, s, p-ArCH3), 2.06 (12H, s, o-ArCH3). 13C{1H} NMR (101 MHz, CDCl3) δ(ppm): 139.5 (ArC), 136.5 (ArC), 135.5 (ArC), 129.4 (ArC), 126.8 (HCCH), 97.1 (C5H5), 21.4 (p-ArCH3), 18.1 (o-ArCH3). 13C NMR (101 MHz, CDCl3) δ(ppm): 139.5 (s, ArC), 136.5 (s, ArC), 135.5 (s, ArC), 129.4 (d, J = 151.8 Hz, Mes), 126.6 (d, J = 195.1 Hz, HCCH), 96.9 (d, J = 169.3 Hz, C5H5), 21.4 (q, J = 127.3 Hz, p-ArCH3), 18.1 (q, J = 127.5 Hz, o-ArCH3). EI MS (70 eV) m/z (58Ni): 424 ([M − Cp]+, 80%), 378 ([M − Cp − NO2]+, 19), 320 (100), 302 (84). Anal. Calc. for C26H29N3NiO3·H2O: C, 61.44; H, 6.15; N, 8.27. Found: C, 61.85; H, 5. 92; N, 8.52%. Magnetic susceptibility measurements by Evans’ method in toluene-d8 up to 100 °C gave μ ≈ 0.
Attempted exchange of nitrile with styrene in 2
Complex [Ni(η5-C5H5)(SIMes)(CH3CN)]+PF6− (2b) (40.0 mg, 65.0 μmol) was dissolved in THF (1.0 mL) in a Schlenk tube and styrene (neat, 0.3 mL, 2.6 mmol, 40 eq.) was added. This solution was stirred at 50 °C for 4 h. The reaction mixture was evaporated in vacuo and the solid residue was washed with hexane (2.0 mL) and diethyl ether (2.0 mL), and dried in vacuo at room temperature. The solid thus obtained, according to 1H NMR analysis, appeared to be substrate 2b. When CH2Cl2 (0.3 mL) was used instead of THF at 35 °C, no reaction occurred. Stirring of 2b (107.0 mg, 173.9 μmol) in styrene (neat, 1.5 mL, 13.0 mmol, 74.8 eq.) at 50 °C gave the same result. Attempts to exchange the pivalonitrile ligand in 2g under conditions analogous to those for 2b in THF also failed. Complex 2h (20.0 mg, 32. 9 μmol) stirred in styrene (neat, 1.0 mL, 8.7 mmol, 967 eq.) at 35 °C underwent transformation to an inseparable mixture of green nickel complexes (by 1H NMR).Catalytic activity
X-ray diffraction studies
Single crystals of 2c suitable for X-ray studies were obtained from a CH2Cl2–hexanes (1:2) solution; single crystals of 3a and 3c were obtained from saturated toluene–hexane solutions at 4 °C; compound 2j was crystallized from a mixture of acetonitrile and diethyl ether at 4 °C. Diffraction data of suitable single crystals were measured on an Agilent κ-CCD Gemini A Ultra diffractometer with graphite-monochromated Mo-Kα radiation at 100(2) K for compounds 2c and 3a, and at 293(2) K for 2j and 3c. Cell refinement and data collection as well as data reduction and analysis were performed with the CrysAlisPRO software.35 The structures were solved by direct methods and subsequent Fourier-difference synthesis with ShelXS and refined by full-matrix least-squares against F2 with ShelXL within the Olex2 program suite.36,37 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were introduced at calculated positions and refined as riding atoms with isotropic displacement parameters related to that of the parent atoms. Asymmetric unit of complex 3a contained one half of a severely disordered toluene molecule which was treated with the SQUEEZE procedure implemented in PLATON.38 The structure model of compound 3c was refined as an inversion twin with the twin ratio refined to 43:57. Attempts to refine the crystal structure in centrosymmetric space group failed giving unphysical ADPs. Data analysis was carried out using Olex2 and PLATON. Crystal data and structure refinement parameters are given in Table 1 and CCDC 972867–972870.
Acknowledgements
The authors thank the National Science Centre for financial support of this work (grant DEC-2011/01/B/ST5/06297). Financial support was also provided by the Warsaw University of Technology (P.A.G.).Notes and references
- (a) W. A. Hermann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef; (b) L. Cavallo, A. Correa, C. Costabile and H. Jacobsen, J. Organomet. Chem., 2005, 690, 5407 CrossRef CAS PubMed; (c) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122 CrossRef CAS PubMed; (d) S. Diez-Gonzales, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612 CrossRef PubMed.
- For selected recent examples, see: (a) C. J. E. Davies, M. J. Page, C. E. Ellul, M. F. Mahhon and M. K. Whittlesey, Chem. Commun., 2010, 46, 5151 RSC; (b) S. Miyazaki, Y. Koga, T. Matsumoto and K. Matsubara, Chem. Commun., 2010, 46, 1932 RSC; (c) K. Zhang, M. Conda-Sheridan, S. R. Cooke and J. Louie, Organometallics, 2011, 30, 2546 CrossRef CAS PubMed; (d) C. Lohre, T. Dröge, C. Wang and F. Glorius, Chem.–Eur. J., 2011, 17, 6052 CrossRef CAS PubMed; (e) B. Liu, X. Liu, C. Chen, C. Chen and W. Chen, Organometallics, 2012, 31, 282 CrossRef CAS; (f) H. Song, D. Fan, Y. Liu, G. Hou and G. Zi, J. Organomet. Chem., 2013, 729, 40 CrossRef CAS PubMed; (g) K. Vin Tan, J. L. Dutton, B. W. Skelton, D. J. D. Wilsom and P. J. Barnard, Organometallics, 2013, 32, 1913 CrossRef; (h) E. A. Bielinski, W. Dai, L. M. Guard, N. Hazari and M. K. Takase, Organometallics, 2013, 32, 4025 CrossRef CAS.
- C. D. Abernethy, A. H. Cowley and R. A. Jones, J. Organomet. Chem., 2000, 596, 3 CrossRef CAS.
- (a) R. A. Kelly, N. M. Scott, S. Diez-Gonzalez, E. D. Stevens and S. P. Nolan, Organometallics, 2005, 24, 3442 CrossRef CAS; (b) A. R. Martin, Y. Makida, S. Meiries, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2013, 32, 6265 CrossRef CAS.
- W. Buchowicz, A. Kozioł, L. B. Jerzykiewicz, T. Lis, S. Pasynkiewicz, A. Pęcherzewska and A. Pietrzykowski, J. Mol. Catal. A: Chem., 2006, 257, 118 CrossRef CAS PubMed.
- E. F. Hahn, B. Heidrich, A. Hepp and T. Pape, J. Organomet. Chem., 2007, 692, 4630 CrossRef CAS PubMed.
- V. Ritleng, C. Barth, E. Brenner, S. Milosevic and M. J. Chetcuti, Organometallics, 2008, 27, 4223 CrossRef CAS.
- W. Buchowicz, W. Wojtczak, A. Pietrzykowski, A. Lupa, L. B. Jerzykiewicz, A. Makal and K. Woźniak, Eur. J. Inorg. Chem., 2010, 648 CrossRef CAS.
- A. M. Oertel, V. Ritleng and M. J. Chetcuti, Organometallics, 2012, 31, 2829 CrossRef CAS.
- O. R. Luca, B. A. Thompson, M. K. Takase and R. H. Crabtree, J. Organomet. Chem., 2013, 730, 79 CrossRef CAS PubMed.
- W. Buchowicz, J. Conder, D. Hryciuk and J. Zachara, J. Mol. Catal. A: Chem., 2014, 381, 16 CrossRef CAS PubMed.
- V. Ritleng, A. M. Oertel and M. J. Chetcuti, Dalton Trans., 2010, 39, 8153 RSC.
- D. A. Malyshev, N. M. Scott, N. Marion, E. D. Stevens, V. P. Ananikov, I. P. Beletskaya and S. P. Nolan, Organometallics, 2006, 25, 4462 CrossRef CAS.
- (a) L. Postigo and B. Royo, Adv. Synth. Catal., 2012, 354, 2613 CrossRef CAS; (b) L. P. Bheeter, M. Henrion, L. Brelot, C. Darcel, M. J. Chetcuti, J.-B. Sortais and V. Ritleng, Adv. Synth. Catal., 2012, 354, 2619 CrossRef CAS.
- D. Gareau, C. Sui-Seng, L. F. Groux, F. Brisse and D. Zargarian, Organometallics, 2005, 24, 4003 CrossRef CAS.
- A. M. Oertel, J. Freudenreich, J. Gein, V. Ritleng, L. F. Veiros and M. J. Chetcuti, Organometallics, 2011, 30, 3400 CrossRef CAS.
- A. M. Oertel, V. Ritleng, A. Busiah, L. F. Veiros and M. J. Chetcuti, Organometallics, 2011, 30, 6495 CrossRef CAS.
- As suggested by a referee, we tried to observe both 2j and 3c in coexistence; in a mixed solvent CDCl3–CD3CN both species were observed by 1H NMR in a ca. 1:1 ratio (according to Cp integration).
- H. V. Huynh, Y. Han, R. Jothibasu and J. A. Yang, Organometallics, 2009, 28, 5395 CrossRef CAS.
- Typical values of 1H NMR chemical shifts for η5-Cp ligands in reported [Ni(Cp)(X)(NHC)] complexes are in the range from 4.5 to 4.8 ppm. See ref. 4–10.
- For an example of spin equilibrium in a half-sandwich nickel complex [Cp*Ni(acac)] see: M. E. Smith and R. A. Andersen, J. Am. Chem. Soc., 1996, 118, 11119 CrossRef CAS ; for spin equilibria in square-planar/tetrahedral complexes [NiX2L2] see: R. G. Hayter and F. S. Humiec, Inorg. Chem., 1963, 2, 1701 Search PubMed; G. R. van Hecke and W. D. Horrocks, Inorg. Chem., 1966, 5, 1968 CrossRef; G. N. La Mar and E. O. Sherman, J. Am. Chem. Soc., 1970, 92, 2691 CrossRef.
- P. Gütlich, B. R. McGarvey and W. Kläui, Inorg. Chem., 1980, 19, 3704 CrossRef.
- D. F. Evans, J. Chem. Soc., 1959, 2003 RSC.
- G. Wilkinson, P. L. Pauson, J. M. Birmingham and F. A. Cotton, J. Am. Chem. Soc., 1953, 75, 1011 CrossRef CAS.
- F. H. Köhler, J. Organomet. Chem., 1976, 110, 235 CrossRef.
- P. Seiler; and J. D. Dunitz, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 2255 CrossRef.
- The CSD (version 5.34, May 2013) search was restricted to complexes containing Ni cations with both Cp and NHC ligands. Only structures with R factor lower than 0.05 and containing neither errors nor disorder were considered. The resulting data set comprises 20 entries.
- (a) L. F. Johnson, F. Heatley and F. A. Bovey, Macromolecules, 1970, 3, 175 CrossRef CAS; (b) N. Ishihara, T. Seimiya, M. Kuramoto and M. Uoi, Macromolecules, 1986, 19, 2464 CrossRef CAS.
- In this “classical” cationic polymerization propagation occurs through addition of a monomer to the carbocation.
- A. M. Oertel, V. Ritleng, L. Burr and M. J. Chetcuti, Organometallics, 2011, 30, 6685 CrossRef CAS.
- A related Ni(0)–NHC–(styrene)2 complex has been also reported; see M. J. Iglesias, J. F. Blandez, M. R. Fructos, A. Prieto, E. Álvarez, T. R. Belderrain and M. C. Nicasio, Organometallics, 2012, 31, 6312 CrossRef CAS.
- D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, Pergamon Press, New York, 1988 Search PubMed.
- K. W. Barnett, J. Chem. Educ., 1974, 51, 422 CrossRef CAS.
- W. Buchowicz, L. B. Jerzykiewicz, A. Krasińska, S. Losi, A. Pietrzykowski and P. Zanello, Organometallics, 2006, 25, 5076 CrossRef CAS.
- CrysAlisPro Software system, Agilent Technologies UK Ltd, Oxford, UK, 2011 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional tables and figures. CCDC 972867–972870. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt53352b |
| ‡ Standard abbreviations for NHC ligands are used throughout this manuscript: IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene, SIMes = 1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidene, SIPr = 1,3-bis(2,6-diisopropylphenyl)-4,5-dihydroimidazol-2-ylidene, and Bn2-bimy = 1,3-dibenzylbenzimidazolin-2-ylidene. |
| This journal is © The Royal Society of Chemistry 2014 |