 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermodynamics and high-pressure kinetics of a fast carbon dioxide fixation reaction by a (2,6-pyridinedicarboxamidato-hydroxo)nickel(II) complex†
O.
Troeppner
a,
D.
Huang
b,
R. H.
Holm
b and
I.
Ivanović-Burmazović
*a
aDepartment of Chemistry and Pharmacy, University of Erlangen-Nürnberg, Egerlandstr. 1, 91058, Erlangen, Germany. E-mail: ivana.ivanovic-burmazovic@fau.de
bDepartment of Chemistry and Chemical Biology, Harvard University, Cambridge, Massachusetts 02138, USA. E-mail: holm@chemistry.harvard.edu
First published on 26th February 2014
Abstract
The previously reported carbon dioxide fixation reaction by the planar terminal hydroxide complex [Ni(pyN2Me2)(OH)]1− in DMF has been further characterized by determination of the equilibrium constants K298eq = 2.4 ± 0.2 × 105 M−1 and K223eq = 1.3 ± 0.1 × 107 M−1, as well as the volume of activation for the CO2 binding (ΔV≠223on = −21 ± 3 cm3 mol−1) and back decarboxylation (ΔV≠223off = −13 ± 1 cm3 mol−1) by high-pressure kinetics. The data are consistent with an earlier DFT computation, including the probable nature of the transition state, and support designating the reaction as one of the most completely investigated carbon dioxide fixation reactions of any type.
Fixation and utilization of CO2 is an increasingly prominent problem1–4 not only because CO2 is a contributor to the greenhouse effect, but also because of its importance as a potentially cheap, nontoxic and abundant source of renewable synthetic fuels.5 There are many general approaches that can lead to fixation and utilization of CO2 to form energetically more valuable molecules like carbon monoxide, methanol or methane, ranging from photochemical6–18 to electrochemical1,5,19–34 to chemical reduction35–43 pathways. They often involve a variety of transition metal centers as redox catalysts, ranging from rather expensive and rare metals like ruthenium21 and platinum44 to cheaper and more abundant ones like nickel22,45 and copper.1 A number of metal complexes have been synthesized and investigated as possible mimics of naturally occurring carbon monoxide dehydrogenase (CODH), an enzyme catalyzing the reversible redox reaction CO2 + 2H+ + 2e− ⇌ CO + H2O and either containing Mo–Cu–S or Ni–Fe–S active sites.46–49
Besides redox methods, carbon dioxide fixation can also be achieved through non-redox processes.4 Of the latter, reactions of a terminal MII/III–OH group with CO2 to afford bridging carbonate and terminal or bridging bicarbonate products are well documented.50–54 Nature utilizes such a reaction within the catalytic cycle of carbonic anhydrases, where the terminal hydroxo group at the four-coordinate ZnII center plays a crucial role in the reversible hydration of CO2.55 Formation of a unidentate bicarbonate complex seems to be critical for the efficiency of that catalytic cycle.56 Some of us have recently observed that members of a set of planar terminal hydroxo complexes derived from N,N′-2,6-disubstituted phenyl-2,6-dicarboxamide pincer ligands react rapidly and completely with CO2 in DMF to form η1-bicarbonate products (Scheme 1).3,57 Reactions are second-order with rate constants (ca..104–106 M−1 s−1) that tend to decrease as the steric size of the phenyl R-substituents increases. The most notable feature is the second-order rate constant kon = 9.5 × 105 M−1 s−1 for reaction (1) at 298 K (Table 1), extrapolated from measurements at lower temperatures.3 [Ni(pyNMe2)(OH)]1− carries the smallest ortho-substituent in the set; the unsubstituted ligand forms octahedral [Ni(pyN2)2]2−.58
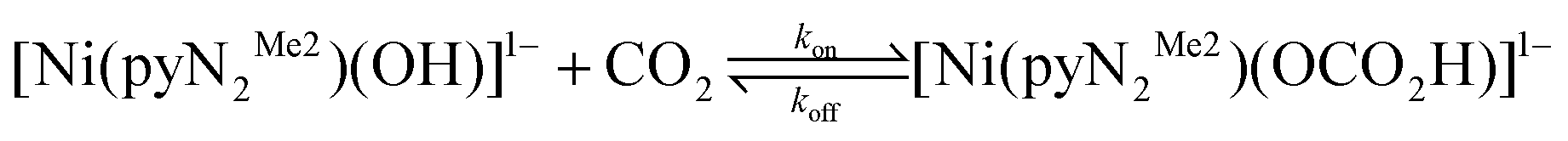 | (1) |
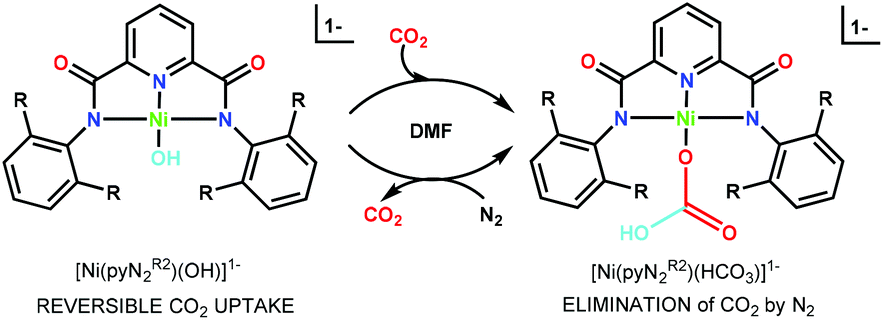 | ||
| Scheme 1 Reversible carbon dioxide fixation reaction mediated by [Ni(pyN2R2)(OH)]1−. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
Comparison of k298on for reaction (1) with second-order rate constants for other systems including model reactions for the Zn(II) enzyme carbonic anhydrase reveals an increase of ≥103 M−1 s−1. Further, under the approximation kon ≈ kcat/KM the rate constant overlaps the enzyme range of ca. 105–108 M−1 s−1.55 While precise comparisons cannot be made between synthetic or enzyme systems and reaction (1) owing to solvent differences (aqueous vs. DMF), it is nonetheless evident that this reaction is an unusually rapid CO2 fixation process. Such a rapid CO2 fixation appears to be related to several notable features of [Ni(pyN2Me2)(X)]−: presence of the uncommon terminal NiII–OH group in the reactant and unidentate η1-OCO2H bicarbonate in the product complex, despite the four-coordinate ligand environment around the NiII center.3
Although the reversible character of this process has been qualitatively demonstrated,57 quantification of this equilibrium and the back decarboxylation reaction has not yet been reported. Since the efficiency of a CO2 uptake is related to a CO2 binding constant, the equilibrium constant Keq of reaction (1) was thermodynamically and kinetically determined in this study at 298 K and 223 K, respectively, contributing to understanding of the overall solution thermodynamics. Moreover, there are no literature data for binding constants of CO2 to Ni–OH species. The only binding constant reported is for a Zn-hydroxo complex at 217 K.53 Kinetic characterization of the reaction so far has disclosed a low enthalpic barrier and a large negative entropy of activation consistent with a second-order reaction (Table 1). However, to fully comprehend the nature of this rather unique low activation pathway for CO2 insertion, here we sought to determine the pressure effect on its kinetics, applying unique high-pressure cryo-temperature stopped-flow measurements at 223 K and 0.5–70 MPa. This is important not only with respect to the determination of the volume of activation (ΔV≠) and thus visualization of a reaction transition state, but also considering an increase in solubility of CO2 under low temperature and high pressure conditions, which can have an impact on the efficiency of CO2 sequestering on a larger scale.
The compounds (Et4N)[Ni(pyN2Me2)X] (X = OH−, HCO3−) were prepared as described.46‡ To elucidate the reversible nature of reaction (1) and to determine its equilibrium constant Keq, a spectrophotometric titration was undertaken in which small aliquots of solutions of CO2 in DMF were added to a 0.1 mM solution of [Ni(pyN2Me2)(OH)]1− in DMF at 298 K in a 1 cm path-length septum-capped and gas-tight UV/visible cuvette. Saturated solutions of CO2 in DMF (0.2 M)59,60 were prepared by bubbling CO2 gas at room temperature through argon-saturated dry DMF in a 10 mL gas-tight Hamilton syringe. Because the concentration of CO2 required for the titration was not in a high excess over the complex concentration, spectrophotometric data were analyzed with eqn (2) in which [ML] is the concentration of the product as a function of the concentration of CO2 ([L0]), [M0] is the total complex concentration and Keq is the equilibrium constant. The change in the overall volume of a reaction mixture in the course of a titration is negligible because of the small amount of CO2 titrant solution added. After the addition of CO2 and prior to an absorbance measurement, the reaction mixture was allowed to equilibrate for 20 s.
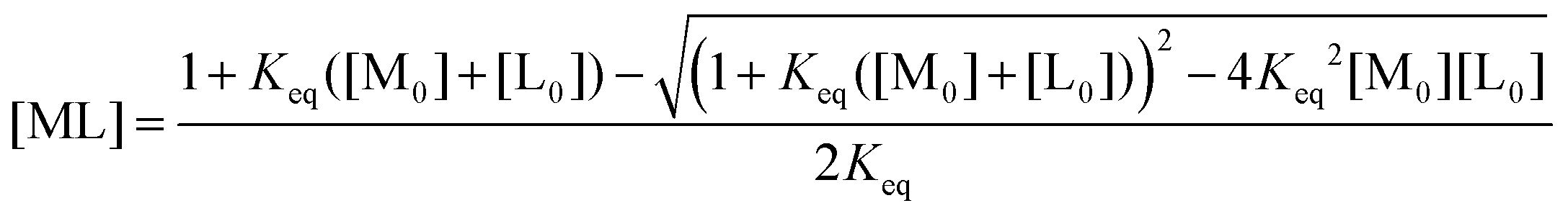 | (2) |
The UV/visible spectra obtained in the course of the titration are depicted in Fig. 1(left). Observed absorbance changes and tight isosbestic points at 279 and 395 nm are consistent with previous results3 describing the expected single-step reaction; the concentration of the product vs. [CO2] is shown in Fig. 1(right). Analysis of the global spectra data with eqn (2) leads to K298eq = 2.4 ± 0.2 × 105 M−1.
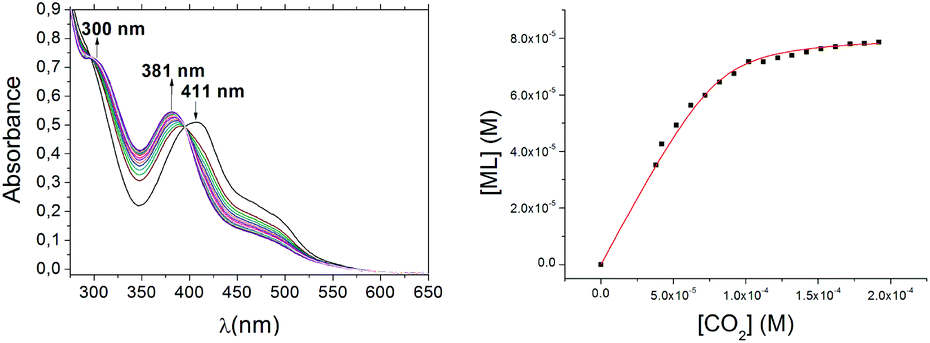 | ||
| Fig. 1 UV/visible spectral changes upon addition of small aliquots of CO2 in DMF to a 0.1 mM solution of [Ni(pyN2Me2)(OH)]1− in DMF at room temperature (left). Plot of the concentration changes of the bicarbonate reaction product ([ML], based on the absorbance change at 381 nm) as dependent on [CO2]; the red line is a fit to eqn (2) (right). | ||
Using Keq = kon/koff, we estimate the first order rate constant koff for the back decarboxylation reaction to be 4 s−1 at 298 K. We have previously demonstrated the reversibility of the binding reaction by its reversal upon passing dinitrogen through a solution of [Ni(pyN2Me2)(OCO2H)]1−.3,57
The only other quantified equilibrium for CO2 fixation is bicarbonate formation by CO2 insertion in a Zn–OH bond (Keq = 6 ± 2 × 103 M−1 in dichloromethane at 217 K, corresponding to ΔG = −3.8 ± 0.2 kcal mol−1).53 Under comparable low temperature conditions in DMF, reaction (1) is three to five orders of magnitude more efficient with estimated binding constants in the 4 × 107 to 8 × 108 M−1 range. The values were estimated from our Keq value and reaction enthalpies previously computed with BP86 and B3LYP functionals, respectively.3
In terms of the mechanism of reaction (1), one can envision a transition state that is less or more compact than the product η1-OCO2H bicarbonate complex. The former will speak in favour of a four-coordinate transition state (hydroxo attack on CO2 followed by a proton transfer),61 whereas the latter supports a five-coordinate transition state (insertion of a carbon dioxide C–O bond into the Ni–OH bond).3 This ambiguity is possible to overcome by determining the volume of activation, ΔV≠, which will offer a clearer picture of the transition state in the solution. Given the information potentially forthcoming from the effects of applied pressure on reaction rates,62–64 the high-pressure kinetics of reaction (1) was investigated. To determine the activation volume ΔV≠, pressure-dependent stopped-flow spectrophotometry at low temperature was employed.§ The decrease in absorbance of the NiII complex at 450 nm and constant temperature, during the course of CO2 binding, was monitored within a pressure range of 0.5–70 MPa (Fig. 2).
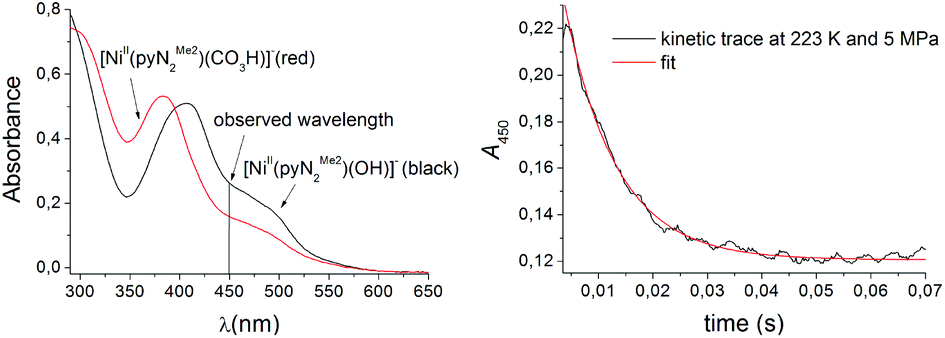 | ||
| Fig. 2 Spectral changes upon the reaction of 1 mM CO2 and 0.1 mM [Ni(pyN2Me2)(OH)]1− in DMF at room temperature (left). The kinetics trace (black) is overlaid with the one-exponential fit to the data (red) for the same reaction system at 223 K and 0.5 MPa (right). | ||
Reactions were performed under pseudo-first-order conditions, with CO2 concentrations ranging between 0.5 and 1.5 mM and a NiII concentration of 0.1 mM. The temperature of the reaction cell and the solutions was maintained at 223 ± 0.1 K. Table S1† contains the resulting second order rate constants (kon) derived from the slopes of plots of the observed rate constants as a function of CO2 concentration at variable pressures (Fig. 3(left)). At least five kinetic runs were performed per data point up to 70 MPa at 223 K. Under these conditions, the volume of activation is independent of pressure and can be evaluated from the slope of the linear plot (ln(kon) vs. pressure)62 in Fig. 3(right).35
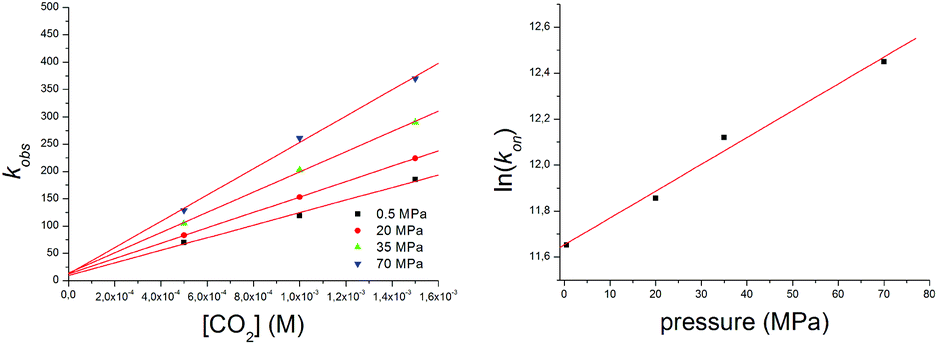 | ||
| Fig. 3 Plots of the observed rate constants (kobs) as a function of CO2 concentration at different pressures: 0.1 mM [Ni(pyN2Me2)(OH)]1− in DMF at 223 K (left). Corresponding plot of ln(kon) vs. pressure (right). | ||
Analysis of the data leads to ΔV≠on = –21 ± 3 cm3 mol−1 for reaction (1). This value is entirely consistent with the negative value of ΔS≠ for this reaction (Table 1) and supports an associative mechanism where bond breaking does not have a significant role in the rate-determining step. Unlike many classical substitution reactions, there is neither a leaving ligand nor a change in charge. Thus, the observed ΔV≠ corresponds to the intrinsic volume change and directly reflects the transition state at the molecular level. Activation volumes for the same type of reaction are reported only for CO2 uptake by [M(NH3)5(OH)]2+ complexes in aqueous solution (M = CoIII, –10.1 ± 0.6; RhIII, –4.7 ± 0.8; IrIII, –4 ± 1 cm3 M−1).61 These values are significantly less negative than that of reaction (1), suggesting a more compact transition state in that reaction. By way of comparison, the partial molar volume of CO2 in DMF is ca. 43 cm3 M−1.65 By a dilution experiment (stopped-flow mixing of [Ni(pyN2Me2)(OCO2H)]1− with degassed DMF at 223 K) at variable pressures we have directly determined the activation volume for the back decarboxylation reaction ΔV≠off = –13 ± 1 cm3 mol−1 (Fig. 4, Table S1†). This enabled us to construct a volume profile (Fig. 5), which clearly indicates that the {[Ni(pyN2Me2)(OH)]1−⋯CO2}§ transition state is more compact than the product bicarbonate complex. In addition, based on the kinetic parameters at 223 K (Table S1†) the corresponding binding constant could be obtained as K223eq = kon/koff = 1.3 ± 0.1 × 107 M−1, which is in accord with the estimations based on the thermodynamic parameters (vide supra).
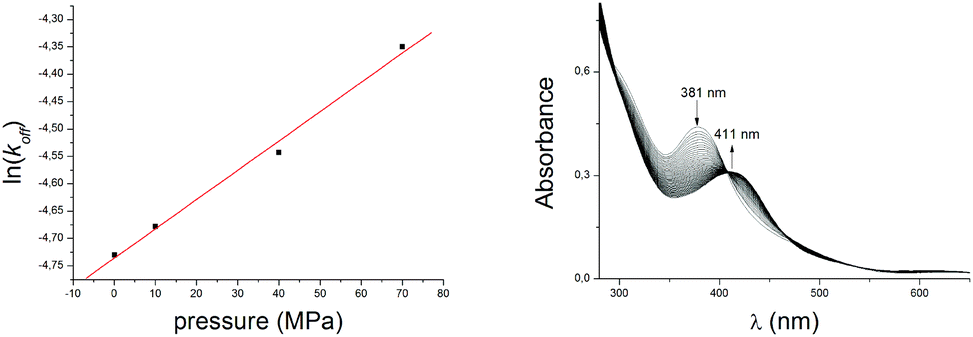 | ||
| Fig. 4 Plot of ln(koff) vs. pressure for the reaction of 0.08 mM [Ni(pyN2Me2)(OH)]1− with CO2 in DMF at 223 K (left). Corresponding time resolved UV/visible spectra in the course of the back decarboxylation reaction; 100 s observation time (right). | ||
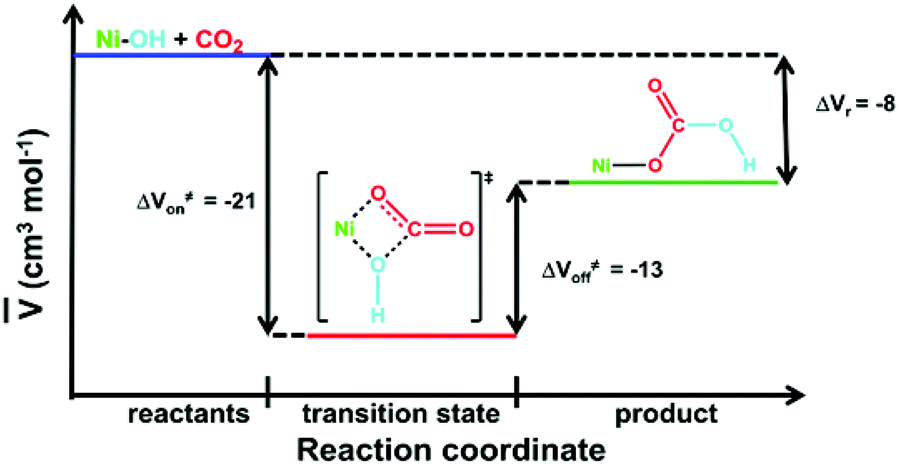 | ||
| Fig. 5 Volume profile for CO2 fixation in reaction (1). | ||
Such a volume profile supports a five-coordinate transition state with bidentate bicarbonate coordination on the way to the four-coordinate product.
The four-coordinate structure of the starting complex and the binding of the dinegative pincer ligand enhance the nucleophilicity of a bound hydroxide. This favors an initial nucleophilic attack of a hydroxide and a cycloaddition pathway that should promote fast kinetics. The analogous reactions for the [M(NH3)5(OH)]2+ systems (M = CoIII, RhIII or IrIII)61 have three orders of magnitude smaller rate constants than that in this study. This parallels a significantly lower nucleophilicity of OH− coordinated to the +3 metal centers within the positively charged [M(NH3)5(OH)]2+ complexes in comparison to the nucleophilicity of OH− within negatively charged [Ni(pyN2Me2)(OH)]1−. The steric protection of the activated hydroxide and the increased electron density caused by the pincer ligand control the reaction thermodynamics and leads to a stable η1-bound product. These collective features render CO2 fixation by [Ni(pyN2Me2)(OH)]1− kinetically and thermodynamically favorable. In addition to reaction (1),3 the enthalpic reaction coordinate has also been computed by DFT methods for the slower reaction of CO2 with [Ni(pyN2iPr2)(OH)]1− (kon = 5.1 × 104 M−1 s−1, ΔS≠ = −27 cal mol−1 K−1 in DMF).57 Results for both systems indicate a mechanism proceeding by a nucleophilic attack at the carbon atom of CO2 by a hydroxide, formation of a cyclic transition state as depicted in Fig. 5, rupture of one Ni–O bond, and reorientation of the resulting monodentate bicarbonate by a Ni–O–C pivot motion. The predicted final perpendicular orientation of the bicarbonate relative to the NiN3 plane is observed in crystalline salt of the reaction products.46 The herewith visualized mechanism differs from that reported for [M(NH3)5(OH)]2+ (M = CoIII, RhIII or IrIII),61 where the rate-determining step in decarboxylation proceeds according to a dissociative mechanism that involves breakage of the oxygen–carbon bond on the carbonate ligand. In contrast, our back reaction has a clearly associative character, involving a Ni–O bond formation and closure of a chelate bicarbonate ring. We should point out that the reactions between CO2 and [M(NH3)5(OH)]2+ operate in aqueous solutions, where a hydrogen-bonding network assists in proton transfer from the bound hydroxide to one of the CO2 oxygen atoms. On the other hand, our reaction operates in DMF, within a hydrophobic environment of the sterically bulky ligand, and there is no possibility that the CO2 oxygen atoms can be involved in some prominent hydrogen-bonding interactions. This makes the CO2 oxygen atoms even more nucleophilic and prone to bind to the NiII center, resulting in a five-coordinate transition state. This may also explain the fact that our interpretation of the overall volume profile is consistent with, and may be said to support, the computed reaction pathway, although these computational models do not address hydrogen bonding.3
Conclusions
Here, we have revealed the thermodynamics of CO2 binding to [Ni(pyN2Me2)(OH)]−, which was previously reported as the fastest CO2 fixation reaction by a metal-bound hydroxide.3 Based on the determined equilibrium constant of 2.4 ± 0.2 × 105 M−1 at 298 K and 1.3 ± 0.1 × 107 M−1 at 223 K, it seems that the thermodynamics of this process is also highly efficient, though there is limited literature information for comparisons.53Our high-pressure kinetic studies revealed a large negative activation volume for both the CO2 uptake and the back decarboxylation reaction. These results support a very compact transition state, which can be visualized as a five-coordinate species with a bicarbonate bound in a bidentate fashion. The observed character of the transition state, in accord with computational results,3 explains a very low activation enthalpy and a negative activation entropy consistent with rapid CO2 fixation. Reaction (1) is currently the most thoroughly characterized carbon dioxide fixation reaction by a metal hydroxide complex, and among the more thoroughly examined carbon dioxide fixation reactions of any type.
Acknowledgements
I.I.-B. and O.T. gratefully acknowledge support from the “Solar Technologies Go Hybrid” initiative of the State of Bavaria. Research at Harvard University was supported by NIH grant GM-28856.Notes and references
- R. Angamuthu, P. Byers, M. Lutz, A. L. Spek and E. Bouwman, Science, 2010, 327, 313–315 CrossRef CAS PubMed.
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- D. Huang, O. V. Makhlynets, L. L. Tan, S. C. Lee, E. V. Rybak-Akimova and R. H. Holm, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1222–1227 CrossRef CAS PubMed.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. Dubois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. Kenis, C. A. Kerfeld, R. H. Morris, C. H. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- M. Jitaru, J. Univ. Chem. Technol. Metall., 2007, 42, 333–334 CAS.
- H. Takeda, K. Koike, H. Inoue and O. Ishitani, J. Am. Chem. Soc., 2008, 130, 2023–2031 CrossRef CAS PubMed.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- K. R. Thampi, J. Kiwi and M. Grätzel, Nature, 1987, 327, 506–508 CrossRef CAS.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536 RSC.
- K. Ikeue, H. Yamashita, M. Anpo and T. Takewaki, J. Phys. Chem. B, 2001, 105, 8350–8355 CrossRef CAS.
- H. Fujiwara, H. Hosokawa, K. Murakoshi, Y. Wada, S. Yanagida, T. Okada and H. Kobayashi, J. Phys. Chem. B, 1997, 101, 8270–8278 CrossRef CAS.
- J. Grodkowski, T. Dhanasekaran, P. Neta, P. Hambright, B. S. Brunschwig, K. Shinozaki and E. Fujita, J. Phys. Chem. A, 2000, 104, 11332–11339 CrossRef CAS.
- T. Ogata, S. Yanagida, B. S. Brunschwig and E. Fujita, J. Am. Chem. Soc., 1995, 117, 6708–6716 CrossRef CAS.
- S. Matsuoka, K. Yamamoto, T. Ogata, M. Kusaba, N. Nakashima, E. Fujita and S. Yanagida, J. Am. Chem. Soc., 1993, 115, 601–609 CrossRef CAS.
- J. Hawecker, J.-M. Lehn and R. Ziessel, Helv. Chim. Acta, 1986, 69, 1990–2012 CrossRef CAS.
- H. Hori, F. P. A. Johnson, K. Koike, O. Ishitani and T. Ibusuki, J. Photochem. Photobiol., A., 1996, 96, 171–174 CrossRef CAS.
- K. Koike, H. Hori, M. Ishizuka, J. R. Westwell, K. Takeuchi, T. Ibusuki, K. Enjouji, H. Konno, K. Sakamoto and O. Ishitani, Organometallics, 1997, 16, 5724–5729 CrossRef CAS.
- P. Kurz, B. Probst, B. Spingler and R. Alberto, Eur. J. Inorg. Chem., 2006, 2006, 2966–2974 CrossRef.
- M. Jitaru, D. A. Lowy, M. Toma, B. C. Toma and L. Oniciu, J. Appl. Electrochem., 1997, 27, 875–889 CrossRef CAS.
- R. Aydin and F. Köleli, J. Electroanal. Chem., 2002, 535, 107–112 CrossRef CAS.
- A. Begum and P. G. Pickup, Electrochem. Commun., 2007, 9, 2525–2528 CrossRef CAS PubMed.
- C. I. Smith, J. A. Crayston and R. W. Hay, J. Chem. Soc., Dalton Trans., 1993, 3267 RSC.
- C. Dealwis, Electrochim. Acta, 2000, 45, 2061–2074 CrossRef CAS.
- M. Gattrell, N. Gupta and A. Co, J. Electroanal. Chem., 2006, 594, 1–19 CrossRef CAS PubMed.
- S. Ikeda, T. Takagi and K. Ito, Bull. Chem. Soc. Jpn., 1987, 60, 2517–2522 CrossRef CAS.
- M. Isaacs, J. C. Canales, A. Riquelme, M. Lucero, M. J. Aguirre and J. Costamagna, J. Coord. Chem., 2003, 56, 1193–1201 CrossRef CAS.
- S. Kaneco, N.-h. Hiei, Y. Xing, H. Katsumata, H. Ohnishi, T. Suzuki and K. Ohta, Electrochim. Acta, 2002, 48, 51–55 CrossRef CAS.
- S. Kaneco, K. Iiba, N.-h. Hiei, K. Ohta, T. Mizuno and T. Suzuki, Electrochim. Acta, 1999, 44, 4701–4706 CrossRef CAS.
- S. Kaneco, R. Iwao, K. Iiba, K. Ohta and T. Mizuno, Energy, 1998, 23, 1107–1112 CrossRef CAS.
- J. Lee, Y. Kwon, R. L. Machunda and H. J. Lee, Chem.–Asian J., 2009, 4, 1516–1523 CrossRef CAS PubMed.
- J. Lee and Y. Tak, Electrochim. Acta, 2001, 46, 3015–3022 CrossRef CAS.
- R. Schrebler, P. Cury, F. Herrera, H. Gómez and R. Córdova, J. Electroanal. Chem., 2001, 516, 23–30 CrossRef CAS.
- R. Schrebler, P. Cury, C. Suárez, E. Muñoz, H. Gómez and R. Córdova, J. Electroanal. Chem., 2002, 533, 167–175 CrossRef CAS.
- P. J. Welford, B. A. Brookes, J. D. Wadhawan, H. B. McPeak, C. E. W. Hahn and R. G. Compton, J. Phys. Chem. B, 2001, 105, 5253–5261 CrossRef CAS.
- S. Kern and R. van Eldik, Inorg. Chem, 2012, 51, 7340–7345 CrossRef CAS PubMed.
- C. Creutz and M. H. Chou, J. Am. Chem. Soc., 2007, 129, 10108–9 CrossRef CAS PubMed.
- J. Dietrich and S. Schindler, Z. Anorg. Allg. Chem., 2008, 634, 2487–2494 CrossRef CAS.
- Y. Inoue, H. Izumida, Y. Sasaki and H. Hashimoto, Chem. Lett., 1976, 863–864 CrossRef CAS.
- W. Leitner, Angew. Chem., Int. Ed. Engl., 1995, 107, 2391–2405 Search PubMed.
- P. G. Jessop, Y. Hsiao, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 344–355 CrossRef CAS.
- P. G. Jessop, T. Ikariya and R. Noyori, Nature, 1994, 368, 231–233 CrossRef CAS.
- Y. Himeda, Eur. J. Inorg. Chem., 2007, 2007, 3927–3941 CrossRef.
- G. Laurenczy, F. Joó and L. Nádasdi, Inorg. Chem., 2000, 39, 5083–5088 CrossRef CAS.
- T. Smolinka, M. Heinen, Y. Chen, Z. Jusys, W. Lehnert and R. Behm, Electrochim. Acta, 2005, 50, 5189–5199 CrossRef CAS PubMed.
- E. Simón-Manso and C. P. Kubiak, Organometallics, 2005, 24, 96–102 CrossRef.
- D. Huang and R. H. Holm, J. Am. Chem. Soc., 2010, 132, 4693–4701 CrossRef CAS PubMed.
- S. W. Ragsdale, Crit. Rev. Biochem. Mol. Biol., 2004, 39, 165–195 CrossRef CAS PubMed.
- S. W. Ragsdale and M. Kumar, Chem. Rev., 1996, 96, 2515–2540 CrossRef CAS PubMed.
- P. A. Lindahl, Biochemistry, 2002, 41, 2097–2105 CrossRef CAS.
- D. A. Palmer and R. Van Eldik, Chem. Rev., 1983, 83, 651–731 CrossRef CAS.
- A. Looney, R. Han, K. McNeill and G. Parkin, J. Am. Chem. Soc., 1993, 115, 4690–4697 CrossRef CAS.
- N. Kitajima, S. Hikichi, M. Tanaka and Y. Morooka, J. Am. Chem. Soc., 1993, 115, 5496–5508 CrossRef CAS.
- W. Sattler and G. Parkin, Polyhedron, 2012, 32, 41–48 CrossRef CAS PubMed.
- C. Bergquist, T. Fillebeen, M. M. Morlok and G. Parkin, J. Am. Chem. Soc., 2003, 125, 6189–6199 CrossRef CAS PubMed.
- X. Zhang and R. van Eldik, Inorg. Chem., 1995, 34, 5606–5614 CrossRef CAS.
- X. Zhang, R. van Eldik, T. Koike and E. Kimura, Inorg. Chem., 1993, 32, 5749–5755 CrossRef CAS.
- D. Huang, O. V. Makhlynets, L. L. Tan, S. C. Lee, E. V. Rybak-Akimova and R. H. Holm, Inorg. Chem., 2011, 50, 10070–10081 CrossRef CAS PubMed.
- A. K. Patra and R. Mukherjee, Inorg. Chem., 1999, 38, 1388–1393 CrossRef CAS.
- A. Gennaro, A. A. Isse and E. Vianello, J. Electroanal. Chem. Interfacial Electrochemistry, 1990, 289, 203–215 CrossRef CAS.
- M. Tezuka and M. Iwasaki, Chem. Lett., 1993, 427–430 CrossRef CAS.
- U. Spitzer, R. Van Eldik and H. Kelm, Inorg. Chem., 1982, 21, 2821–2823 CrossRef CAS.
- R. Van Eldik, T. Asano and W. J. Le Noble, Chem. Rev., 1989, 89, 549–688 CrossRef CAS.
- C. D. Hubbard and R. van Eldik, Inorg. Chim. Acta, 2010, 363, 2357–2374 CrossRef CAS PubMed.
- R. Van Eldik, C. Dücker-Benfer and F. Thaler, Adv. Inorg. Chem., 1999, 49, 1–58 CrossRef.
- M. Jödecke, Á. Pérez-Salado Kamps and G. Maurer, J. Chem. Eng. Data, 2012, 57, 1249–1266 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Rate constants for the CO2 uptake (kon) and decarboxylation (koff) at elevated pressures. See DOI: 10.1039/c3dt53004c |
| ‡ All manipulations and measurements were performed under a pure argon atmosphere. All reagents and solvents were purchased from commercial sources (Sigma-Aldrich, Acros Organics) and were of p.a. grade and used without purification. DMF was purchased as an extra dry solvent and was stored in a sure-seal bottle under argon and molecular sieves. All solutions were prepared in dry DMF in an MBraun inert atmosphere glove box. |
| § Measurements were performed using a Hi-Tech Scientific HPSF 56 stopped-flow instrument externally coupled to a Huber CC905 thermostat. UV/visible spectra were recorded using an Energetiq LDLS ENQ EQ-99 FC laser-driven light source coupled to an Applied Photo Physics photomultiplier. Solutions in DMF were prepared immediately before measurement. Lower concentrations of CO2 were obtained by dilution of a saturated solution in graduated gas-tight syringes. Neither the solution of [Ni(pyN2Me2)(OH)]1− nor the CO2 solution was exposed to the atmosphere. |
| This journal is © The Royal Society of Chemistry 2014 |