 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
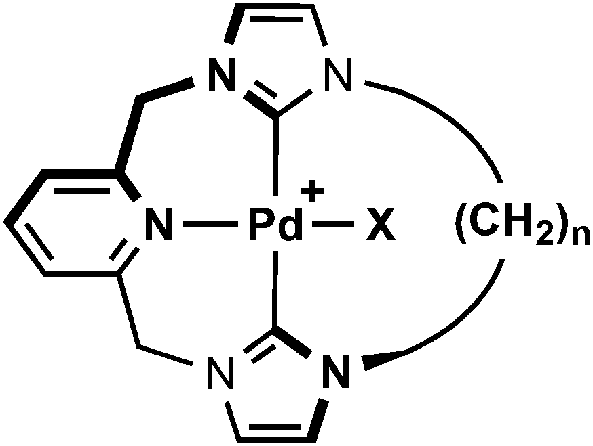
Synthesis, structure and dynamics of NHC-based palladium macrocycles†
Rhiann E.
Andrew
and
Adrian B.
Chaplin
*
Department of Chemistry, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK. E-mail: a.b.chaplin@warwick.ac.uk
First published on 31st October 2013
Abstract
A series of macrocyclic CNC pincer pro-ligands based on bis(imidazolium)lutidine salts with octa-, deca- and dodecamethylene spacers have been prepared and their coordination chemistry investigated. Using a Ag2O based transmetallation strategy, cationic palladium(II) chloride complexes [PdCl{CNC–(CH2)n}][BArF4] (n = 8, 10, 12; ArF = 3,5-C6H3(CF3)2) were prepared and fully characterised in solution, by NMR spectroscopy and ESI-MS, and in the solid-state, by X-ray crystallography. The smaller macrocyclic complexes (n = 8 and 10) exhibit dynamic behaviour in solution, involving ring flipping of the alkyl spacer across the Pd–Cl bond, which was interrogated by variable temperature NMR spectroscopy. In the solid-state, distorted coordination geometries are observed with the spacer skewed to one side of the Pd–Cl bond. In contrast, a static C2 symmetric structure is observed for the dodecamethylene based macrocycle. For comparison, palladium(II) fluoride analogues [PdF{CNC–(CH2)n}][BArF4] (n = 8, 10, 12) were also prepared and their solution and solid-state structures contrasted with those of the chlorides. Notably, these complexes exhibit very low frequency 19F chemical shifts (ca. −400 ppm) and the presence of C–H⋯F interactions (2hJFC coupling observed by 13C NMR spectroscopy). The dynamic behaviour of the fluoride complexes is largely consistent with the smaller ancillary ligand; [PdF{CNC–(CH2)8}][BArF4] exceptionally shows C2v time averaged symmetry in solution at room temperature (CD2Cl2, 500 MHz) as a consequence of dual fluxional processes of the pincer backbone and alkyl spacer.
Introduction
N-heterocyclic carbenes (NHCs) have quickly emerged as a powerful class of carbon-based ligand in organometallic chemistry and catalysis.1 With generally stronger σ-donating characteristics and orthogonal steric profiles to widely used phosphine ligands, NHCs are rapidly being established as ligands of choice for many transition metal catalysed reactions. Aided by simple and efficient synthetic protocols, intricate polydentate ligand topologies can be readily constructed based on NHC donors.2,3 Building on the flourishing chemistry of phosphine-based pincer complexes,4 analogous NHC-based tridentate architectures have received considerable attention in particular.2,5 Archetypical NHC pincer ligands are derived from bis(imidazol-2-ylidene)-benzene, pyridine, xylene or lutidine frameworks (Fig. 1) and the coordination chemistry of these CNC and CCC pincer ligands has been explored for a variety of late transition metals.2,5,6 Notably, palladium CNC based complexes have been thoroughly investigated, following seminal work by Crabtree and Piers who demonstrated the application of these species in cross coupling reactions.7–14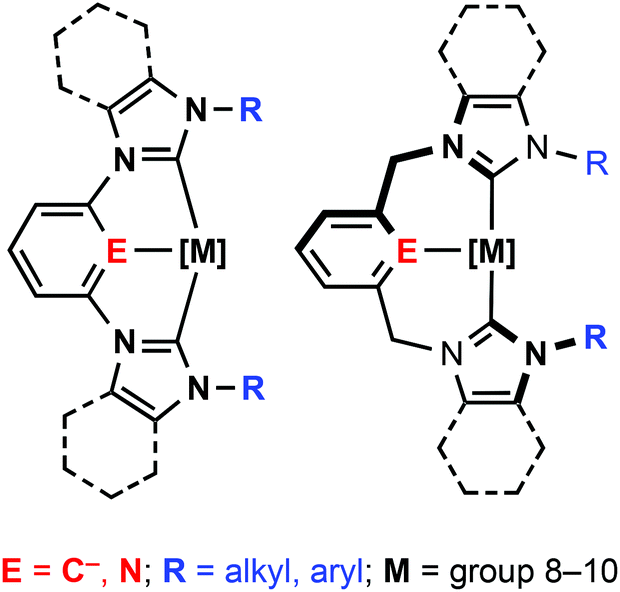 | ||
| Fig. 1 NHC-based pincer architectures. | ||
The substitution geometries of NHC pincers are well suited to the construction of macrocycles. Principally based on lutidine and xylene backbones, tridentate macrocycles find notable application in supramolecular chemistry; exploiting chemistry of the coordinated metal centre for molecular recognition15 and the construction of interlocked catenane and rotaxane systems.16 The vast majority of known CNC and CCC based pincer complexes feature ligands bearing simple alkyl (i.e. Me, nBu, tBu, Ad) and aryl (e.g. Mes, Dipp) groups. Despite facile synthetic procedures to macrocyclic imidazolium pro-ligands,17 there are very few examples of tridentate NHC-based macrocyclic complexes. Macrocyclic complexes that have been reported are limited to small (e.g.A, 15 membered, Fig. 2)18,19 or very large ring sizes (B and C; 28–38 membered).20,21 Of these systems, tridentate coordination of the macrocycle was observed within B and C, although not in A; the smaller 15 membered ring adopting a bidentate cyclophane geometry2,22 with no pyridine coordination as a consequence of the short butamethylene spacer. Although showing significantly lower catalytic activity than related non-macrocyclic variants,7,10,13,14 both xylene and lutidine based B, promote C–C bond coupling reactions.20
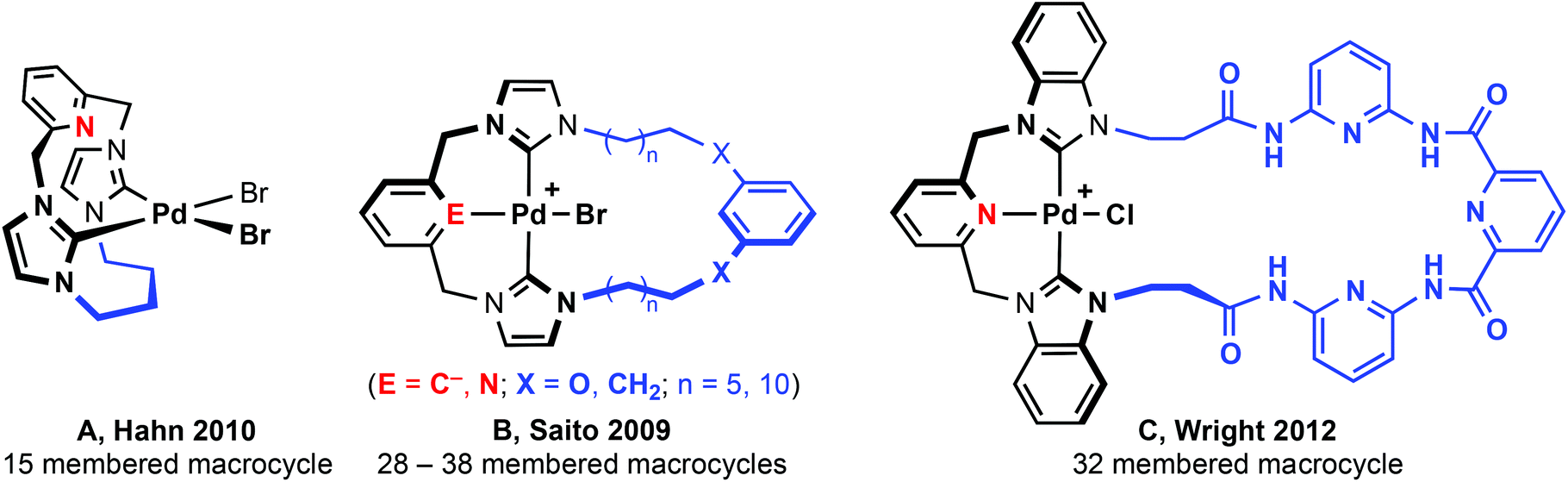 | ||
| Fig. 2 NHC-based macrocyclic complexes. | ||
Motivated by the possibility for exploiting their unique steric profile in organometallic chemistry and potential applications as building blocks in interlocking architectures, in this communication we present the synthesis of a range of medium sized (19–23 membered) macrocyclic pro-ligands consisting of bis(imidazolium)lutidine salts with octa-, deca- and dodecamethylene spacers. The coordination chemistry of the corresponding CNC pincer ligands with palladium is detailed, particularly focusing on the effect of the aliphatic chain and ancillary ligands on the structure and dynamics of the resulting macrocyclic complexes, as probed by variable temperature NMR spectroscopy and X-ray diffraction.
Results and discussion
The target macrocyclic pro-ligands [CNC–(CH2)n]·2(HBr) (n = 8, 2a; 10, 2b; 12, 2c) were prepared as bromide salts using a straightforward two-step synthesis, involving alkylation of 2,6-bis(bromomethyl)pyridine with bis(imidazole)alkanes 1 in dioxane (Scheme 1). All three intermediate bis-imidazoles (n = 8, 10, 12) were readily prepared following minor adaptions to literature procedures employing commercially available alkyl dibromides.23 The overall synthetic route readily afforded gram scale quantities of 2 in high purity following recrystallisation from acetonitrile–diethyl ether (19–32% overall yields). The salts are notably hygroscopic and best stored and manipulated under a dry inert atmosphere.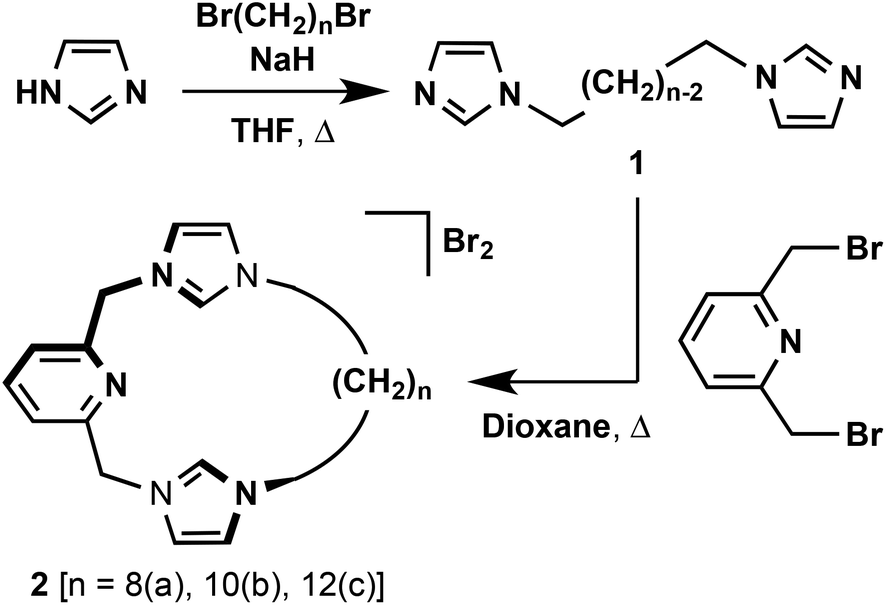 | ||
| Scheme 1 Preparation of pro-ligands 2. | ||
The structures of 2 were confirmed by a combination of NMR spectroscopy and ESI-MS, with the latter being particularly diagnostic. Strong dication parent ion signals are observed for each compound: 2a, 175.6207 (calc. 175.6206) m/z; 2b, 189.6361 (calc. 189.6362) m/z; 2c, 203.6521 (calc. 203.6519) m/z. The NMR spectra of 2 in CD2Cl2 exhibit singlet 4H resonances for the methylene bridge at ca. 5.8 ppm, with characteristic 1H and 13C signals of the pre-carbenic centre at ca. 10.7 and 138 ppm, respectively. The remaining features of the spectroscopic data fully corroborate the structures of 2 and are otherwise unremarkable. Satisfactory microanalysis was obtained for all the macrocycles, and the structure of 2b was further confirmed by X-ray crystallography (ESI†).
Typically, coordination of CNC pincers to palladium has been achieved through reaction of the corresponding imidazolium pro-ligands with [Pd(OAc)2] in DMSO at elevated temperatures.5 With a view of developing a more general procedure for the coordination of pro-ligands 2 with transition metals, we have adapted Ag2O based transmetallation strategies previously employed by Youngs, Danopoulos and Cavell for this initial work involving palladium.9–11 This procedure is outlined in Scheme 2, and involves reactions of 2 with Ag2O and Na[BArF4] (ArF = 3,5-C6H3(CF3)2) in CH2Cl2, followed by subsequent addition of [PdCl2(NCPh)2] as a source of palladium(II) halide – all carried out at room temperature. Using this convenient and mild procedure, macrocyclic palladium complexes [PdCl{CNC–(CH2)n}][BArF4] (n = 8, 3a; 10, 3b; 12, 3c) were prepared with moderate-to-good isolated yields of 31% (n = 8), 55% (n = 10) and 58% (n = 12), following purification on silica and recrystallisation. [PdCl2(NCMe)2] can also be used as the palladium(II) halide source, but gave no significant improvements in yield. Complexes 3 are air stable both in solution and the solid state; dissolving readily in CH2Cl2, CHCl3, Et2O and MeCN. No attempts were made to isolate the corresponding intermediate silver complexes, although we note that the incorporation of the [BArF4]− anion appears to plays an import role in solubilising these species throughout the procedure.
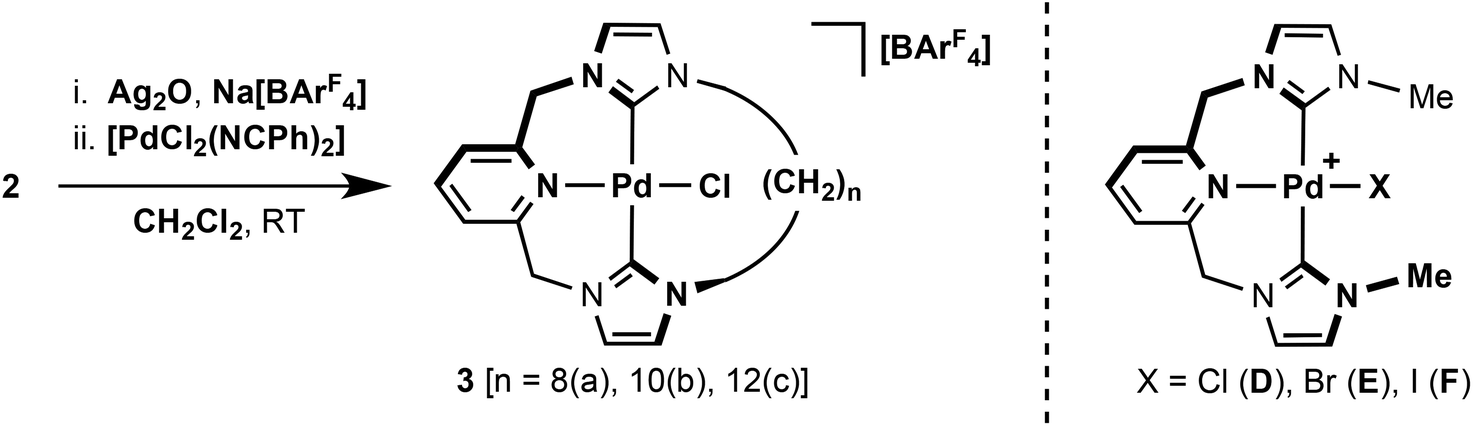 | ||
| Scheme 2 Preparation of 3. | ||
The palladium complexes 3 were fully characterised by NMR spectroscopy, ESI-MS and elemental analysis. Coordination of the ligand was confirmed through the presence of strong parent ion peaks with the expected isotope distributions in mass spectra [3a, 490.0993 (calc. 490.0989) m/z; 3b, 518.1308 (calc. 518.1303) m/z; 3c, 546.1616 (calc. 546.1617) m/z]. 1H and 13C NMR spectra further corroborate the structures of the compounds, which all exhibit approximate C2 symmetry in solution at room temperature (CDCl3 and CD2Cl2, 400 or 500 MHz). The binding of the ligand through carbene donors is established by the absence of the distinctive high frequency imidazolium proton resonances of 2 and significant 13C shifts of the carbene centre from ca. 138 to 163 ppm. These carbene resonances differ only marginally within the series of complexes, although a positive correlation with increasing frequency and macrocycle size is noted, i.e. 162.4 (3a), 163.3 (3b), 164.5 (3c) ppm. All values are in good agreement with that observed for the non-macrocyclic analogue [PdCl{CNC–Me}]+D (164 ppm, Scheme 2).13 Complexes 3 show interesting dynamic behaviour in solution and these features are discussed below in detail.
Crystal structures of all three palladium macrocycles were obtained, revealing significant structural differences between the complexes (Fig. 3 and Table 1). Tridentate coordination of the macrocyclic ligand is observed for each complex, with the pincer moiety adopting a characteristic twisted pseudo C2 conformation, where the pyridine (ca. 40°) and imidazolylidene rings are angled out of the coordination plane as a consequence of the methylene bridges.8–10,13 Associated Pd1–N1 (ca. 2.1 Å), Pd1–C (ca. 2.0 Å) and Pd1–Cl1 (ca. 2.3 Å) distances closely match those found in other related palladium CNC pincer complexes, as exemplified by comparison to D (Table 1).13 The principle differences between the structures are manifested in the conformation of the alkyl spacer, which is skewed to one side of the Pd–Cl bond for the shorter spacers (n = 8 and 10) as a consequence of the steric interactions between the spacer and the chloride ligand. This interaction distinctly leads to distortions of the metal coordination geometry which are most pronounced for 3a, with C7–Pd1–C11 and especially N1–Pd1–Cl1 angles deviating dramatically from 180° [168.82(7) and 163.39(4)°]. With the largest dodecamethylene spacer, 3c adopts an overall pseudo C2 geometry, with the coordinated chloride ligand fully enclosed within the macrocycle cavity; the C7–Pd1–C11 and N1–Pd1–Cl1 angles are essentially linear [172.8(6) and 176.2(3)°] as observed for non-macrocyclic D [173.60(7) and 178.19(3)°].13 Related palladium chloride CNC pincer complexes bearing bulky tBu, Mes and Dipp substituents retain N–Pd–Cl bond angles of effectively 180°.8–10 Distortion from linearity is uncommon within known palladium(II) square planar complexes (following inspection of CSD metrics), although complexes with angles down to 150° are known.24
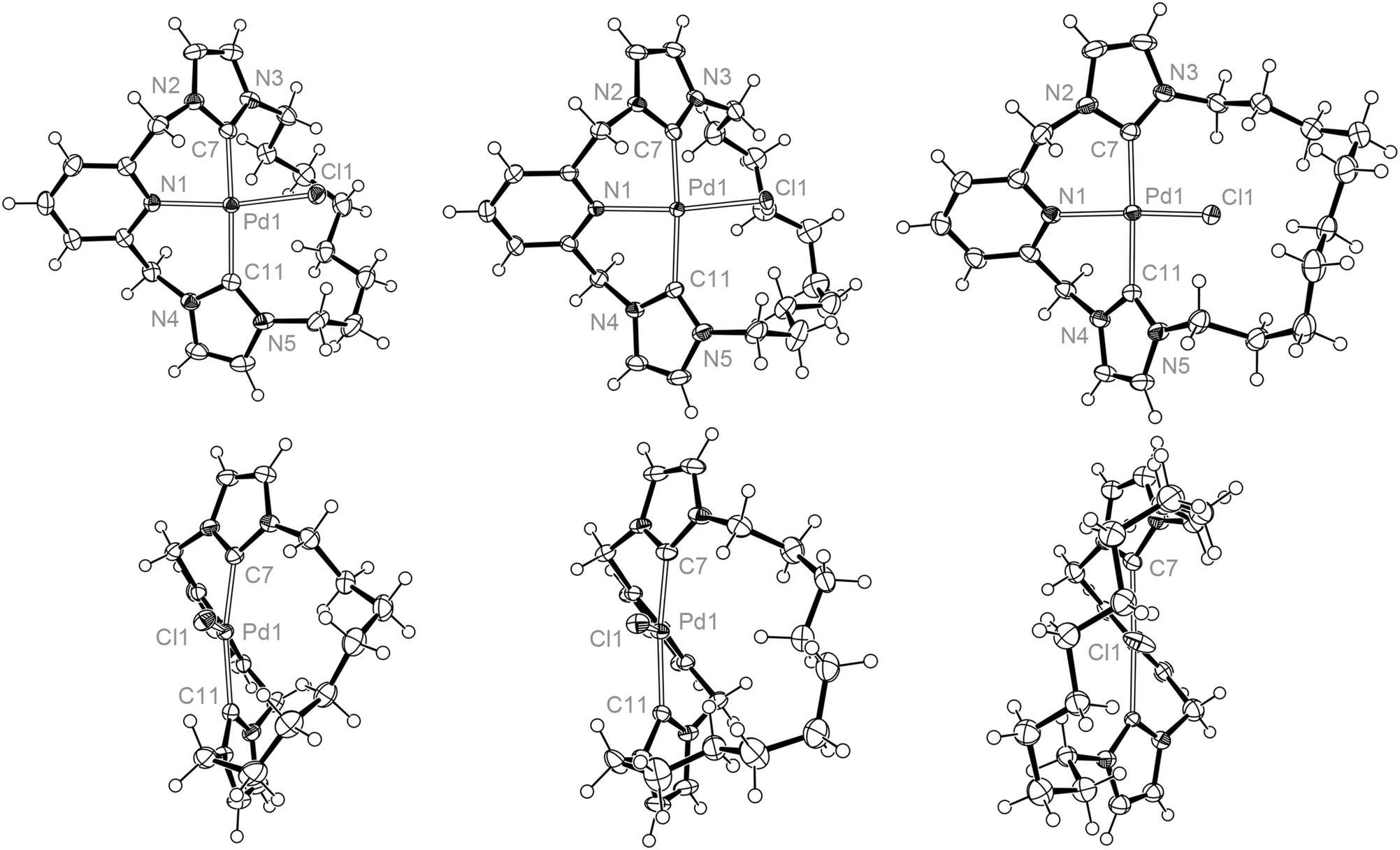 | ||
| Fig. 3 Solid-state structures of 3a, 3b and 3c viewed perpendicular (top) and along (bottom) Pd1–N1 bond vectors. Thermal ellipsoids drawn at 50%; minor disordered component (alkyl spacer) in 3b, anions and solvent molecules omitted for clarity. Selected structural metrics are listed in Table 1. | ||
| 3a | 3b | 3c | D | |
|---|---|---|---|---|
| a lspl(atoms) denotes the least squares plane through the indicated group of atoms; coord. refers to Pd1, C7, C11 and N1. b Recalculated using CIF matrix from ref. 13. | ||||
| Pd1–N1 | 2.087(2) | 2.080(2) | 2.077(10) | 2.068(1) |
| Pd1–Cl1 | 2.3051(5) | 2.3217(5) | 2.287(4) | 2.2978(5) |
| Pd1–C7 | 1.983(2) | 2.004(2) | 2.056(13) | 2.026(2) |
| Pd1–C11 | 2.073(2) | 2.078(2) | 2.036(12) | 2.029(2) |
| N1–Pd1–Cl1 | 163.39(4) | 166.29(5) | 176.2(3) | 178.19(3) |
| C7–Pd1–C11 | 168.82(7) | 171.93(8) | 172.8(6) | 173.60(7) |
| C7–Pd1–Cl1 | 86.19(5) | 87.76(6) | 95.9(4) | 93.15(5) |
| C11–Pd1–Cl1 | 98.40(5) | 98.22(6) | 91.1(4) | 93.21(5) |
| lspl(NHC) ∠lspl(NHC′) | 70.72(8) | 76.88(9) | 81.0(5) | 85.08(7)b |
| lspl(py) ∠lspl(coord.) | 33.07(6) | 39.14(7) | 42.5(4) | 41.83(5)b |
To investigate the apparent inconsistency between the solution (C2 symmetry) and solid-state (C1 symmetry) structures of 3a and 3b, we turned to variable temperature NMR measurements. The fluxional behaviour of CNC based pincer complexes [PdX{CNC–Me}]+D–F (Scheme 2) has previously been investigated in detail by Crabtree and co-workers.12,14 These complexes undergo atropisomerisation between left and right-handed C2 conformations with a free energy barrier of 60.0 kJ mol−l for [D]Cl (CD2Cl2, Tc = 303 K for the methylene bridge protons).25 The rate of this process is highly dependent on the halide–anion combination. Increased activation barriers are observed for complexes containing the weakly coordinating OTs− anion ([D]OTs, ΔG‡(CDCl3) = 66.0 kJ mol−1) and along the series: [F]I, [E]Br, [D]Cl [ΔG‡(CDCl3) = 41.9, 52.9 and 59.3 kJ mol−1]. On the basis of these observations and supporting computational work, a two-step interconversion mechanism involving dissociation of the pyridine group and coordination of the counter anion was proposed. Atropisomerisation (ΔG‡(CDCl3) = 57–59 kJ mol−1) is also observed in the macrocyclic complexes [B]Br (Fig. 2, E = N) although only very small changes in barrier height are observed in comparison to the directly related nBu substituted analogue [PdBr{CNC–nBu}]Br (ΔG‡(CDCl3) = 57.4 kJ mol−1) – indicating no significant macrocyclic effect.20 Remarkably, no dynamic behaviour of the pincer coordination geometry is observed for 3 with the methylene bridge protons diastereotopic in CD2Cl2 solution (500 MHz) up to 308 K (i.e. ΔG‡ ≫ 62 kJ mol−1). This is consistent with the non-coordinating nature of the [BArF4]− anion26 and the calculated gas-phase barrier of D of 73.2 kJ mol−1.12
In agreement with the solid-state structure, 3c retains C2 symmetry from 200–298 K in CD2Cl2 solution (500 MHz), further confirming the good fit of the coordinated chloride ligand within the cavity of the macrocyclic ligand. For complexes 3a and 3b, variable temperature NMR experiments demonstrate ring flipping of the macrocycle across the palladium chloride bond. This fluxional process mediates an atropisomerisation between skewed C1 symmetric confirmations observed in the solid-state (Fig. 3) and leads to the time averaged C2 symmetry in solution at room temperature. For 3a, the slow exchange limit for this process is reached at 200 K in CD2Cl2 (500 MHz), with the 1H NMR spectrum showing three pyridine and four imidazolylidene resonances together with pairs of diastereotopic methylene bridge (pyCH2) and N-CH2 signals (Fig. 4). Simulation27 of the 1H NMR data over the temperature range 185 to 298 K allowed rate data to be extracted for the exchange process, leading to values of ΔH‡ = 43 ± 4 kJ mol−1, ΔS‡ = −7 ± 17 J mol−1 K−1 and ΔG‡(298 K) = 45 ± 9 kJ mol−1 following an Eyring analysis (see ESI†). In line with the longer alkyl spacer, the fluxional process in 3b is much faster than 3a: the exchange could not be completely frozen out, even upon cooling to 185 K, although the barrier for this process can be approximated at ΔG‡ = 37 kJ mol−1 from signal coalescence at low temperature.28
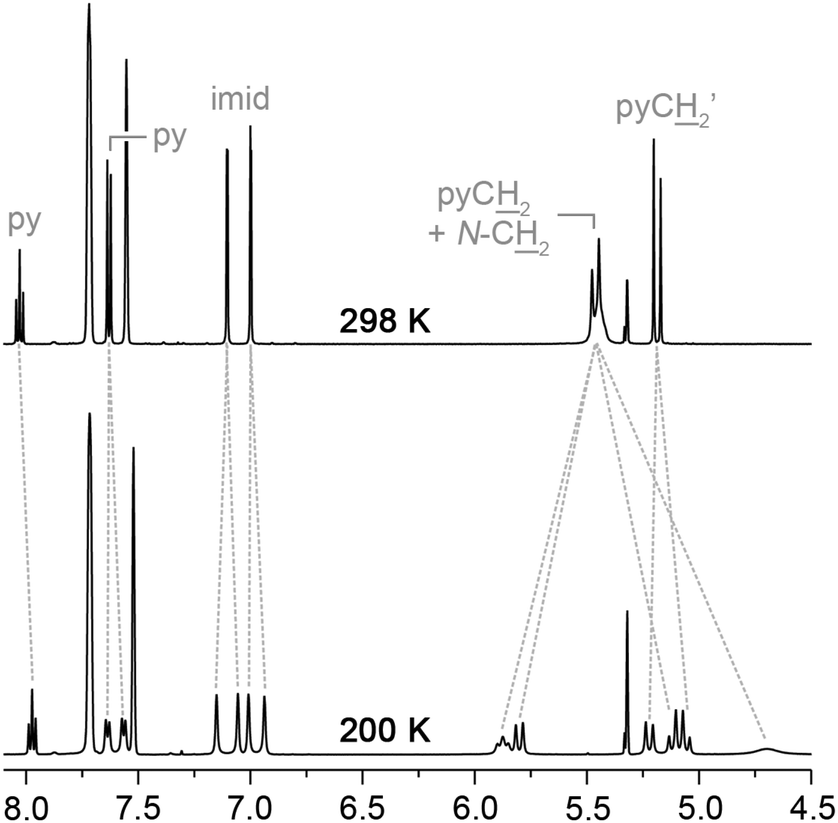 | ||
| Fig. 4 Selected 1H NMR spectra of 3a (CD2Cl2, 500 MHz). | ||
To further investigate the dynamics of the macrocyclic ligands, metathesis of the chloride ligand with the smaller fluoride ligand was targeted. This was achieved by reactions of 3 with excess AgF in acetonitrile to afford the palladium(II) fluoride complexes [PdF{CNC–(CH2)n}][BArF4] (n = 8, 4a; 10, 4b; 12, 4c) with good to moderate isolated yields of 45–88% (Scheme 3). Unlike their palladium(II) chloride analogues, the fluorides are markedly water sensitive and degrade readily on silica. Facile loss of the fluoride ligand is also apparent by ESI-MS and only fragment ion derivatives can be observed. Although many examples of palladium fluoride complexes containing phosphine and nitrogen based donor ligands are known,29 to the best of our knowledge no NHC based analogues have been reported. The formation of 4 are readily corroborated by 1H, 13C and 19F NMR spectroscopy and elemental analysis. The coordinated carbene centres are observed as broad singlets at ca. 165 ppm by 13C NMR spectroscopy to slightly higher frequency than observed in 3, while 19F NMR spectra reveal characteristically low frequency Pd(II) fluoride resonances at ca. −400 ppm. 19F chemical shifts of this magnitude are uncommon,30 although notably the palladium PNP pincer G (Scheme 3) has a 19F NMR shift of −414.3 ppm for the coordinated fluoride ligand.31
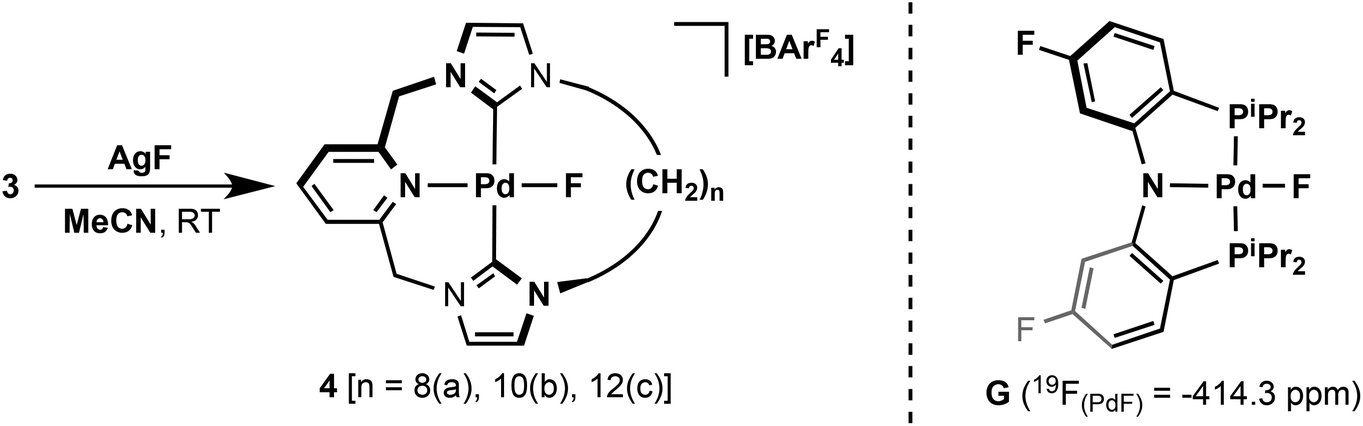 | ||
| Scheme 3 Preparation of 4. | ||
Like its chloride analogue, temperature invariant 1H NMR spectra of 4c demonstrate the adoption of C2 symmetry in solution. For the smaller 4b, a very low energy fluxional process can be detected only by cooling to 185 K, where the onset of coalescence is noted for one of the N-CH2 resonances. In comparison to 3b, the interconversion barrier for 4b must be considerably less as at 185 K the slow exchange limit is essentially reached for all signals of 3b. Given the large Δν possible for the N-CH2 shifts it is difficult to approximate the barrier, although we estimate a value of ΔG‡ ≤ 34 kJ mol−1 (based on Δν = 600 Hz from 3a). The solid-state structure of 4b shows a more symmetric structure than 3b (Fig. 5), with the N1–Pd1–F1 angle approaching 180° [175.77(7) cf. 166.29(5)° for 3b], although meaningful conclusions from this observation are difficult due to crystal packing effects. Notably the cations in 4b pack with relatively close Pd1–Pd1′ and Pd1–F1′ distances of 4.3357(7) and 3.866(2) Å, respectively. A further interesting feature of note is the presence of weak N-CH–H⋯F interactions, which were detected by carbon–fluorine coupling for both 4b and 4c (2hJFC = 6 Hz, 298 K). These 2hJFC values and the corresponding interatomic distances observed in solid-state structure of 4b [C⋯F = 2.968(3), 3.146(3) Å], are fully consistent with other C–H⋯F interactions reported in the literature.32
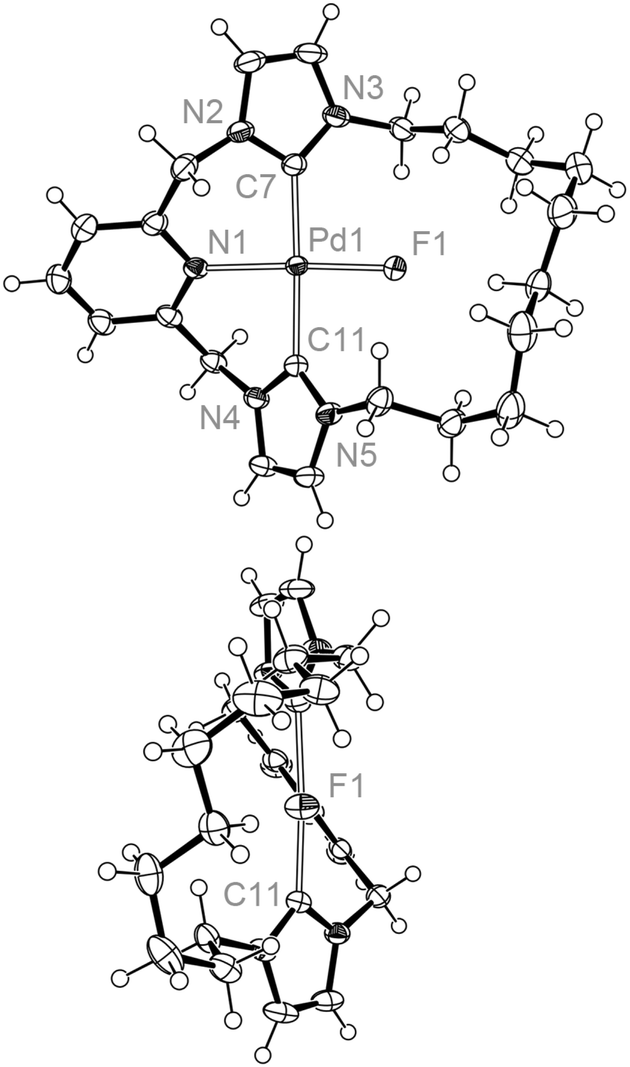 | ||
| Fig. 5 Solid-state structures of 4b viewed perpendicular (top) and along (bottom) the Pd1–N1 bond vector. Thermal ellipsoids drawn at 50%, anion omitted for clarity. Selected bond lengths (Å) and angles (°): Pd1–N1, 2.035(2); Pd1–F1, 1.9588(13); Pd1–C7, 2.018(2); Pd1–C11, 1.999(2); N1–Pd1–F, 175.77(7); C7–Pd1–C11, 175.66(10); C7–Pd1–F1, 92.96(8); C11–Pd1–F1, 88.99(7); lspl(NHC) ∠lspl(NHC′), 73.2(1); lspl(py) ∠lspl(coord.), 36.52(8). | ||
In solution, 4a is highly fluxional. At 298 K in CD2Cl2 (500 MHz), C2v symmetry is observed indicating an atropisomerisation mechanism involving dynamics of both the pincer and octamethylene spacer. Upon cooling the sample to 225 K onset of decoalescence of the methylene bridge and N-CH2 protons was observed, while those of the imidazolylidene remained relatively sharp. Further cooling to 185 K led to broadening of the later signals, although the slow exchange limit was not reached (precluding accurate simulation). These observations support a dual fluxional process involving low energy flipping of the octenyl linker (ΔG‡ ≤ 34 kJ mol−1) and higher energy pincer twisting (ΔG‡ ∼ 43 kJ mol−1). The latter process is characteristic of D–F, with the approximate barrier height of 43 kJ mol−1 being broadly similar to that calculated for dicationic [Pd{CNC–Me}]2+ (ΔG‡ = 47.8 kJ mol−1)12 and suggestive of fluoride ligand dissociation (promoted by macrocycle induced steric pressure). Although we could obtain a solid-state structure of 4a, somewhat inline with its highly fluxional solution characteristics, it has two crystallography independent cations both with extensively disordered macrocycle geometries (see ESI†). However, what is clear from the structure is that the octamethylene group is skewed to one side of the palladium(II) fluoride bond (as with 3a and 3b) and the N–Pd–F angle deviates from linearity (ca. 172°). The shorter macrocycle linker also results in further close C–H⋯F interactions, with coupling at 1,8- (2hJFC = 14 Hz) and 4,5- (2hJFC = 6 Hz) positions along the octamethylene group resolved by 13C NMR spectroscopy.
Summary
A series of macrocyclic CNC pincer pro-ligands have been prepared incorporating simple alkyl spacers, resulting in 19, 21 and 23 membered ring systems (2). The coordination chemistry of these pro-ligands has been explored, and a series of cationic palladium(II) chloride (3) and fluoride (4) complexes have been synthesised and fully characterised in solution and the solid-state. The larger macrocyclic complexes, containing dodecamethylene spacers, exhibit static C2 symmetric structures with the coordinated halogen ligands lying within the macrocyclic cavity. As the ring size is reduced, increasing dynamic behaviour is observed in solution, with the spacer undergoing ring flipping across the palladium halogen bond, and is most pronounced with the larger chloride ancillary ligand (Table 2). The atropisomerisation of the smaller macrocyclic complexes is also manifested in their solid-state structures, with significant distortions from the expected square planar geometries and skewing of the alkyl spacers to one side of the palladium–halogen bonds observed. The dodecamethylene based CNC pincer ligands accommodate the coordinated metal–ligand moiety most readily and appear most suitable for subsequent applications in organometallic chemistry and as building blocks for interlocking architectures: the organometallic chemistry of other late transition complexes bearing this ligand is currently being explored.33|
|
||
|---|---|---|
| n\X | Cl | F |
| 8 | Fluxional spacer (ΔG‡ = 43 kJ mol−1) | Highly fluxional macrocycle |
| 10 | Fluxional spacer (ΔG‡ = 37 kJ mol−1) | Fluxional spacer (ΔG‡ ≤ 34 kJ mol−1) |
| 12 | C 2 symmetric (Cl enclosed) | C 2 symmetric (F enclosed) |
Experimental
General considerations
Manipulations were performed under an inert atmosphere, using Schlenk (nitrogen) and glove box (argon) techniques unless otherwise stated. Glassware was oven dried and flamed under vacuum prior to use. Anhydrous solvents (<0.005% H2O) were purchased from ACROS or Aldrich and used as supplied: MeCN, CH2Cl2, CHCl3, THF, 1,4-dioxane, pentane and Et2O. CD2Cl2 was dried over CaH2, vacuum-distilled and stored under an atmosphere of argon. CDCl3 (for NMR characterisation of 4) was dried over 3 Å molecular sieves, vacuum-distilled and stored over sieves. Na[BArF4]34 and [Rh(CO)2Cl]2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 35 were synthesised using literature procedures. Bis-imidazoles 1 were prepared by adaptions of literature procedures and are fully detailed in the ESI.†23 [PdCl2(NCPh)2], [PdCl2(NCMe)2], Ag2O, AgF, 2,6-bis(bromomethyl)pyridine, and dibromoalkanes are commercial products and were used without further purification. NMR spectra were recorded on Bruker DPX-400, AV-400 and DRX-500 spectrometers at 298 K unless otherwise stated. Chemical shirts are quoted in ppm and coupling constants in Hz. ESI-MS were recorded on a Bruker MaXis mass spectrometer. Microanalyses were performed at the London Metropolitan University by Stephen Boyer.
35 were synthesised using literature procedures. Bis-imidazoles 1 were prepared by adaptions of literature procedures and are fully detailed in the ESI.†23 [PdCl2(NCPh)2], [PdCl2(NCMe)2], Ag2O, AgF, 2,6-bis(bromomethyl)pyridine, and dibromoalkanes are commercial products and were used without further purification. NMR spectra were recorded on Bruker DPX-400, AV-400 and DRX-500 spectrometers at 298 K unless otherwise stated. Chemical shirts are quoted in ppm and coupling constants in Hz. ESI-MS were recorded on a Bruker MaXis mass spectrometer. Microanalyses were performed at the London Metropolitan University by Stephen Boyer.
Preparation of pro-ligands (2)
2a: following the general procedure using 2,6-bis(bromomethyl)pyridine (0.79 g, 2.98 mmol) and 1a (0.73 g, 2.98 mmol), the product was obtained as an off-white foam. Yield: 0.47 g (31%).
1H NMR (400 MHz, CD2Cl2) δ 10.55 (app. t, J = 2, 2H), 8.00 (app. t, J = 2, 2H), 7.82 (dd, 3JHH = 8.4, 7.0, 1H), 7.72 (app. d, J = 8, 2H), 7.40 (app. t, J = 2, 2H), 5.82 (s, 4H), 4.42 (t, 3JHH = 6.4, 4H), 1.88 (app. pentet, J = 7, 4H), 1.32–1.41 (m, 4H), 1.18–1.28 (m, 4H). 13C{1H} NMR (101 MHz, CD2Cl2) δ 153.6, 139.5, 137.9, 124.6, 124.4, 122.1, 54.2, 50.1, 29.6, 27.5, 24.7. ESI-MS (CH2Cl2, 180 °C, 3 kV) positive ion: 175.6207 m/z, [M]2+ (calc. 175.6206). Anal. Calcd for C21H29N5Br2·1.4(H2O) (536.52 [511.30] g mol−1): C, 47.01; H, 5.97; N, 13.05. Found: C, 47.17; H, 5.45; N, 13.01.
2b: following the general procedure using 2,6-bis(bromomethyl)pyridine (1.00 g, 3.77 mmol) and 1b (1.03 g, 3.77 mmol), the product was obtained as a white crystalline solid. Yield: 0.71 g (35%).
1H NMR (400 MHz, CD2Cl2) δ 10.80 (app. t, J = 2, 2H), 7.94 (app. t, J = 2, 2H), 7.82 (dd, 3JHH = 8.1, 7.3, 1H), 7.69 (app. d, J = 8, 2H), 7.23 (app. t, J = 2, 2H), 5.82 (s, 4H), 4.42 (t, 3JHH = 7.1, 4H), 1.96 (app. pentet, J = 7, 4H), 1.37–1.46 (m, 8H), 1.30–1.35 (m, 4H). 13C{1H} NMR (101 MHz, CD2Cl2) δ 153.7, 139.6, 138.5, 124.4, 123.8, 121.9, 54, 50.5, 29.6, 27.9, 27.4, 25.0. ESI-MS (CH2Cl2, 180 °C, 3 kV) positive ion: 189.6361 m/z, [M]2+ (calc. 189.6362). Anal. Calcd for C23H33N5Br2 (539.35 g mol−1): C, 51.22; H, 6.17; N, 12.98. Found: C, 51.08; H, 6.08; N, 12.89.
2c: following the general procedure using 2,6-bis(bromomethyl)pyridine (1.08 g, 4.08 mmol) and 1c (1.23 g, 4.08 mmol), the product was obtained a white crystalline solid. Yield: 0.90 g (39%).
1H NMR (400 MHz, CD2Cl2) δ 10.84 (br, 2H), 8.17 (app. t, J = 2, 2H), 7.69–7.78 (m, 3H), 7.37 (app. t, J = 2, 2H), 5.78 (s, 4H), 4.42 (t, 3JHH = 7.0, 4H), 1.93 (app. pentet, J = 7, 2H), 1.22–1.41 (m, 16H). 13C{1H} NMR (101 MHz, CD2Cl2) δ 153.8, 139.4, 138.4, 124.3, 123.9, 122.0, 53.9, 50.2, 30.0, 28.1 (2C), 27.9, 25.3. ESI-MS (CH2Cl2, 180 °C, 3 kV) positive ion: 203.6521 m/z, [M]2+ (calc. 203.6519). Anal. Calcd for C25H37N5Br2 (567.40 g mol−1): C, 52.92; H, 6.57; N, 12.34. Found: C, 52.82; H, 6.53; N, 12.17.
Preparation of [PdCl{CNC-(CH2)n}][BArF4] (3)
3a: following the general procedure using 2a (0.060 g, 0.117 mmol) and subsequent recrystallisation from CHCl3–pentane, the product was obtained as pale-yellow crystalline solid. Yield: 0.049 g (31%).
1H NMR (400 MHz, CDCl3) δ 7.71 (t, 3JHH = 7.7, 1H, py), 7.66–7.71 (m, 8H, ArF), 7.49 (br, 4H, ArF), 7.38 (d, 3JHH = 7.7, 2H, py), 6.94 (d, 3JHH = 1.9, 2H, imid), 6.90 (d, 3JHH = 1.9, 2H, imid), 5.39–5.51 (m, 2H, N-CH2), 5.34 (d, 2JHH = 15.3, 2H, pyCH2), 5.08 (d, 2JHH = 15.3, 2H, pyCH2), 3.80 (dt, 2JHH = 13.3, 3JHH = 6.6, 2H, N-CH2), 2.79 (br, 2H, CH2), 1.98–2.14 (m, 2H, CH2), 1.72–1.86 (m, 2H, CH2), 1.60–1.72 (m, 2H, CH2), 1.41–1.54 (m, 2H, CH2), 1.12–1.25 (m, 2H, CH2). 13C{1H} NMR (101 MHz, CDCl3) δ 162.4 (s, Pd-C), 161.8 (q, 1JCB = 50, ArF), 154.7 (s, py), 141.7 (s, py), 134.9 (s, ArF), 129.1 (qq, 2JFC = 32, 3JCB = 3, ArF), 125.7 (s, py), 124.6 (q, 1JFC = 273, ArF), 123.7 (s, imid), 120.4 (s, imid), 117.7 (pentet, 3JFC = 4, ArF), 56.2 (s, pyCH2), 50.0 (s, N-CH2), 31.1 (s, CH2), 26.6 (s, CH2), 26.5 (s, CH2). ESI-MS (CH3CN, 180 °C, 3 kV) positive ion: 490.0993 m/z, [M]+ (calc. 490.0989). Anal. Calcd for C53H39BClF24N5Pd (1354.56 g mol−1): C, 46.99; H, 2.90; N, 5.17. Found: C, 46.92; H, 2.87; N, 5.15.
3b: following the general procedure using 2b (0.064 g, 0.110 mmol) and subsequent recrystallization from CHCl3–pentane, the product was obtained as pale-yellow hexagonal platelets. Yield: 0.094 g (55%).
1H NMR (400 MHz, CDCl3) δ 7.66–7.71 (m, 8H, ArF), 7.64 (t, 3JHH = 7.4, 1H, py), 7.49 (br, 4H, ArF), 7.35 (d, 3JHH = 7.4, 2H, py), 6.97 (d, 3JHH = 1.7, 2H, imid), 6.87 (d, 3JHH = 1.7, 2H, imid), 5.57 (d, 3JHH = 15.3, 2H, pyCH2), 4.98 (d, 3JHH = 15.3, 2H, pyCH2), 4.59–4.73 (m, 2H, N-CH2), 4.33–4.46 (m, 2H, N-CH2), 2.30–2.43 (m, 2H, CH2), 1.77–1.92 (m, 2H, CH2), 1.57–1.70 (m, 2H, CH2), 1.08–1.48 (m, 10H, CH2). 13C{1H} NMR (101 MHz, CDCl3) δ 163.3 (s, Pd-C), 161.8 (q, 1JCB = 50, ArF), 155.0 (s, py), 141.6 (s, py), 134.9 (s, ArF), 129.1 (qq, 2JFC = 32, 3JCB = 3, ArF), 125.3 (s, py), 124.6 (q, 1JFC = 273, ArF), 123.2 (s, imid), 120.4 (s, imid), 117.7 (pentet, 3JFC = 4, ArF), 56.0 (s, pyCH2), 49.9 (s, N-CH2), 30.3 (s, CH2), 27.8 (s, CH2), 27.4 (s, CH2), 25.5 (s, CH2). ESI-MS (CH3CN, 180 °C, 3 kV) positive ion: 518.1308 m/z, [M]+ (calc. 518.1303). Anal: Calcd for C55H43BClF24N5Pd (1382.61 g mol−1): C, 47.78; H, 3.13; N, 5.07. Found: C, 47.85; H, 3.11; N, 5.07.
3c: following the general procedure using 2c (0.100 g, 0.176 mmol) and subsequent recrystallization from Et2O–hexane, the product was obtained as a white crystalline solid. Yield: 0.151 g (58%).
1H NMR (400 MHz, CDCl3) δ 7.66–7.70 (m, 8H, ArF), 7.58 (t, 3JHH = 7.6, 1H, py), 7.48 (br, 4H, ArF), 7.32 (d, 3JHH = 7.6, 2H, py), 7.02 (d, 3JHH = 1.7, 2H, imid), 6.89 (d, 3JHH = 1.7, 2H, imid), 5.63 (d, 2JHH = 15.0, 2H, pyCH2), 4.97 (d, 2JHH = 15.0, 2H, pyCH2), 4.72 (td, JHH = 12.4, 3JHH = 3.6, 2H, N-CH2), 3.75 (td, JHH = 12.4, 3JHH = 5.7, 2H, N-CH2), 2.09–2.22 (m, 2H, CH2), 1.64–1.77 (m, 2H, CH2), 1.31–1.57 (m, 14H, CH2), 1.08–1.20 (m, 2H, CH2). 13C{1H} NMR (101 MHz, CDCl3) δ 164.5 (s, Pd-C), 161.8 (q, 1JCB = 50, ArF), 154.8 (s, py), 141.5 (s, py), 134.9 (s, ArF), 129.1 (qq, 2JFC = 32, 3JCB = 3, ArF), 125.4 (s, py), 124.6 (q, 1JFC = 273, ArF), 122.4 (s, imid), 121.0 (s, imid), 117.7 (pentet, 3JFC = 4, ArF), 55.7 (s, pyCH2), 51.6 (s, N-CH2), 31.0 (s, CH2), 28.3 (s, CH2), 27.1 (s, CH2), 27.0 (s, CH2), 23.2 (s, CH2). ESI-MS (CH3CN, 180 °C, 3 kV) positive ion: 546.1616 m/z, [M]+ (calc. 546.1617). Anal. Calcd for C57H47BClF24N5Pd (1410.66 g mol−1); C, 48.53; H, 3.36; N, 4.96. Found: C, 48.41; H, 3.29; N, 4.98.
Preparation of [PdF{CNC–(CH2)n}][BArF4] (4)
4a: following the general procedure using AgF (0.023 g, 0.181 mmol) and 3a (0.050 g, 0.037 mmol) – stirred for 1 hour, the product was obtained as an off-white solid. Yield: 0.030 g (61%).
1H NMR (400 MHz, CDCl3) δ 7.67–7.72 (m, 9H, py + ArF), 7.49 (br, 4H, ArF), 7.35 (d, 3JHH = 7.8, 2H, py), 6.95 (d, 3JHH = 1.8, 2H, imid), 6.90 (d, 3JHH = 1.8, 2H, imid), 5.23 (s, 4H, pyCH2), 4.31 (app. t, JHH = 7, 4H, N-CH2), 2.04–2.15 (m, 4H, CH2), 1.42–1.53 (s, 8H, CH2). 19F NMR (377 MHz, CDCl3) δ −62.38 (s, ArF), −392.08 (s, Pd-F). 13C{1H} NMR (101 MHz, CDCl3) δ 164.8 (s, Pd-C), 161.8 (q, 1JCB = 50, ArF), 155.4 (s, py), 141.2 (s, py), 134.9 (s, ArF), 129.1 (qq, 2JFC = 32, 3JCB = 3, ArF), 126.2 (s, py), 124.6 (q, 1JFC = 273, ArF), 123.2 (s, imid), 119.7 (s, imid), 117.7 (pentet, 3JFC = 4, ArF), 55.8 (s, pyCH2), 50.0 (d, 2hJFC = 14, N-CH2), 30.2 (s, CH2), 26.0 (s, CH2), 25.5 (d, 2hJFC = 6, CH2). ESI-MS (CH2Cl2, 180 °C, 3 kV) positive ion: 500.1283 m/z (100%), [M − F]2+[HCOO]− (calc. 500.1278); 456.1377 m/z (35%), [M − F]2+H− (calc. 456.1382). Anal. Calcd for C53H39BClF25N5Pd·0.5(CHCl3) (1397.79 [1338.10] g mol−1): C, 45.97; H, 2.85; N, 5.01. Found: C, 45.77; H, 2.86; N, 4.93.
4b: following the general procedure using AgF (0.019 g, 0.150 mmol) and 3b (0.056 g, 0.041 mmol) – stirred for 3 hours, the product was obtained as a white solid (0.049 g, 88%).
1H NMR (400 MHz, CDCl3) δ 7.67–7.71 (m, 8H, ArF), 7.66 (t, 3JHH = 7.8, 1H, py), 7.49 (br, 4H, ArF), 7.35 (d, 3JHH = 7.8, 2H, py), 6.98 (d, 3JHH = 1.8, 2H, imid), 6.90 (d, 3JHH = 1.8, 2H, imid), 5.60 (d, 2JHH = 15.2, 2H, pyCH2), 4.97 (d, 2JHH = 15.2, 2H, pyCH2), 4.66 (td, JHH = 12.5, 3JHH = 3.9, 2H, N-CH2), 3.71 (td, JHH = 12.5, 3JHH = 5, 2H, N-CH2), 2.00–2.15 (m, 2H, CH2), 1.34–1.81 (m, 12H, CH2), 0.97–1.12 (m, 2H, CH2). 19F NMR (377 MHz, CDCl3) δ −62.38 (s, ArF), −399.89 (s, Pd-F). 13C{1H} NMR (101 MHz, CDCl3) δ 165.0 (s, Pd-C), 161.8 (q, 1JCB = 50, ArF), 155.6 (s, py), 141.2 (s, py), 134.9 (s, ArF), 129.1 (qq, 2JFC = 32, 3JCB = 3, ArF), 126.0 (s, py), 124.6 (q, 1JFC = 273, ArF), 122.3 (s, imid), 120.3 (s, imid), 117.7 (pentet, 3JFC = 4, ArF), 55.6 (s, pyCH2), 50.6 (d, 2hJFC = 6, N-CH2), 30.4 (s, CH2), 27.0 (s, CH2), 25.6 (s, CH2), 24.5 (s, CH2). ESI-MS (CH2Cl2, 180 °C, 3 kV) positive ion: 542.1762 m/z (100%), [M − F]2+[CH3COO]− (calc. 542.1747); 528.1604 m/z (10%), [M − F]2+[HCOO]− (calc. 528.1591). Anal. Calcd for C55H43BClF25N5Pd (1366.15 g mol−1): C, 48.35; H, 3.17; N, 5.13. Found: C, 48.45; H, 3.11; N, 5.17.
4c: following the general procedure using AgF (0.022 g, 0.173 mmol) and 3c (0.050 g, 0.035 mmol) – stirred for 1 hour, the product was obtained as an oil that was washed with pentane and dried to give a white foam (0.022 g, 45%).
1H NMR (400 MHz, CDCl3) δ 7.64–7.72 (m, 8H, ArF), 7.56 (t, 3JHH = 7.7, 1H, py), 7.48 (br, 4H, ArF), 7.29 (d, 3JHH = 7.7, 2H, py), 7.01 (d, 3JHH = 1.9, 2H, imid), 6.92 (d, 3JHH = 1.9, 2H, imid), 5.67 (d, 2JHH = 15.1, 2H, pyCH2), 4.94 (d, 2JHH = 15.1, 2H, pyCH2), 4.75 (dt, JHH = 12.1, 3JHH = 5.2, 2H, N-CH2), 3.83 (dt, JHH = 11.8, 3JHH = 5.2, 2H, N-CH2), 1.73–1.97 (m, 4H, CH2), 1.15–1.53 (m, 16H, CH2). 19F NMR (377 MHz, CDCl3) δ −62.39 (s, ArF), −399.91 (s, Pd-F). 13C{1H} NMR (101 MHz, CDCl3) δ 164.7 (s, Pd-C), 161.9 (q, 1JCB = 50, ArF), 155.8 (s, py), 141.3 (s, py), 134.9 (s, ArF), 129.1 (qq, 2JFC = 32, 3JCB = 3, ArF), 125.8 (s, py), 124.6 (q, 1JFC = 273, ArF), 121.7 (s, imid), 120.6 (s, imid), 117.6 (pentet, 3JFC = 4, ArF), 55.6 (s, pyCH2), 50.0 (d, 2hJFC = 6, N-CH2), 31.0 (s, CH2), 27.4 (s, CH2), 27.3 (s, CH2), 27.2 (s, CH2), 24.4 (s, CH2). ESI-MS (CH2Cl2, 180 °C, 3 kV) positive ion: 570.2068 m/z (100%), [M − F]2+[CH3COO]− (calc. 570.2060); 556.1902 m/z (5%), [M − F]2+[HCOO]− (calc. 556.1903). Anal. Calcd for C57H47BClF25N5Pd (1394.21 g mol−1): C, 49.10; H, 3.40; N, 5.02. Found: C, 49.10; H, 3.28; N, 5.13.
Variable temperature NMR experiments
All measurements were performed using a DRX-500 spectrometer. Samples (0.014 M in CD2Cl2) were prepared in J. Youngs NMR tubes under inert atmosphere and equilibrated at each temperature inside the spectrometer for 10 minutes prior to data acquisition. Spectra and selected data are compiled in the ESI.†Crystallography
Crystallographic data for 2b, 3 and 4 are summarised in Table 3. Full details about the collection, solution and refinement are documented in the CIF, which have been deposited with the Cambridge Crystallographic Data Centre under CCDC 961289–961294.| 2b·MeCN | 3a | 3b·0.5(pentane) | 3c·(OEt2) | 4a | 4b | |
|---|---|---|---|---|---|---|
| CCDC | 961289 | 961290 | 961291 | 961292 | 961293 | 961294 |
| Figure | S-31 | 3 | 3/SI-32 | 3 | SI-33 + 34 | 5 |
| Formula | C25H36Br2N6 | C53H39BClF24N5Pd | C57.5H49BClF24N5Pd | C61H57BClF24N5OPd | C53H39BF25N5Pd | C55H43BF25N5Pd |
| M | 580.42 | 1354.55 | 1418.67 | 1484.77 | 1338.10 | 1366.15 |
| Crystal system | Monoclinic | Triclinic | Monoclinic | Orthorhombic | Triclinic | Monoclinic |
| Space group | P21 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c | P212121 |
P |
C2/c |
| T [K] | 150(2) | 150(2) | 150(2) | 150(2) | 150(2) | 150(2) |
| a [Å] | 8.6112(4) | 13.8039(3) | 12.92329(18) | 9.7194(4) | 12.7226(2) | 35.3011(14) |
| b [Å] | 36.1437(11) | 14.2173(3) | 25.0749(3) | 15.1274(5) | 18.5028(3) | 16.1372(3) |
| c [Å] | 8.7973(3) | 15.6408(3) | 19.5371(2) | 43.507(2) | 25.0641(5) | 27.1414(11) |
| α [°] | 90 | 93.0635(17) | 90 | 90 | 83.7891(16) | 90 |
| β [°] | 91.596(4) | 102.8239(17) | 108.9205(15) | 90 | 78.0669(17) | 133.337(7) |
| γ [°] | 90 | 110.5834(19) | 90 | 90 | 82.1611(16) | 90 |
| V [Å3] | 2737.01(17) | 2772.73(10) | 5988.95(13) | 6396.7(5) | 5699.2(2) | 11245.5(12) |
| Z (Z′) | 4 (2) | 2 | 4 | 4 | 4 (2) | 8 |
| Density [g cm−3] | 1.409 | 1.622 | 1.573 | 1.542 | 1.559 | 1.614 |
| μ (mm−1) | 2.985 | 0.506 | 0.472 | 0.447 | 0.448 | 0.456 |
| θ range [°] | 3.2 ≤ θ ≤ 25.0 | 2.9 ≤ θ ≤ 29.6 | 3.0 ≤ θ ≤ 27.9 | 3.1 ≤ θ ≤ 25.0 | 2.9 ≤ θ ≤ 25.7 | 2.9 ≤ θ ≤ 27.9 |
| Reflns collected | 19071 |
78258 |
88555 |
28972 |
50701 |
34662 |
| R int | 0.0327 | 0.0360 | 0.0292 | 0.0820 | 0.0325 | 0.0319 |
| Completeness | 99.8 | 99.9% | 99.9% | 99.7 | 99.8 | 99.8 |
| No. of data/restr/param | 9360/223/619 | 15538/605/850 |
14249/964/968 |
11264/753/934 |
21631/3975/2076 |
13379/710/836 |
| R 1 [I > 2σ(I)] | 0.0495 | 0.0358 | 0.0392 | 0.0937 | 0.0720 | 0.0412 |
| wR2 [all data] | 0.1238 | 0.0862 | 0.0975 | 0.2105 | 0.1988 | 0.0988 |
| GoF | 1.101 | 1.021 | 1.058 | 0.2105 | 1.022 | 1.016 |
| Largest diff. pk and hole [e Å−3] | 1.52/−0.47 | 0.82/−0.56 | 1.03/−0.57 | 2.11/−1.24 | 2.60/−1.27 | 0.93/−0.75 |
| Flack (x) | 0.024(15) | 0.47(7) |
Acknowledgements
We thank the University of Warwick (R.E.A.) and the Royal Society (A.B.C.) for financial support.References
- (a) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed; (b) C. Samojłowicz, M. Bieniek and K. Grela, Chem. Rev., 2009, 109, 3708–3742 CrossRef PubMed; (c) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed; (d) S. Würtz and F. Glorius, Acc. Chem. Res., 2008, 41, 1523–1533 CrossRef PubMed.
- M. Poyatos, J. A. Mata and E. Peris, Chem. Rev., 2009, 109, 3677–3707 CrossRef CAS PubMed.
- (a) P. G. Edwards and F. E. Hahn, Dalton Trans., 2011, 40, 10278–10288 RSC; (b) L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz and V. Cesar, Chem. Rev., 2011, 111, 2705–2733 CrossRef CAS PubMed.
- (a) M. Albrecht and M. M. Lindner, Dalton Trans., 2011, 40, 8733–8744 RSC; (b) J. Choi, A. H. R. MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761–1779 CrossRef CAS PubMed; (c) W. H. Bernskoetter, C. K. Schauer, K. I. Goldberg and M. Brookhart, Science, 2009, 326, 553–556 CrossRef CAS PubMed; (d) M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759–1792 CrossRef CAS PubMed; (e) M. Albrecht and G. van Koten, Angew. Chem., Int. Ed., 2001, 40, 3750–3781 CrossRef CAS.
- (a) J. A. Mata, M. Poyatos and E. Peris, Coord. Chem. Rev., 2007, 251, 841–859 CrossRef CAS PubMed; (b) D. Pugh and A. A. Danopoulos, Coord. Chem. Rev., 2007, 251, 610–641 CrossRef CAS PubMed; (c) E. Peris and R. H. Crabtree, Coord. Chem. Rev., 2004, 248, 2239–2246 CrossRef CAS PubMed.
- For representative examples see: (a) T. R. Helgert, T. K. Hollis and E. J. Valente, Organometallics, 2012, 31, 3002–3009 CrossRef CAS; (b) X. Zhang, A. M. Wright, N. J. DeYonker, T. K. Hollis, N. I. Hammer, C. E. Webster and E. J. Valente, Organometallics, 2012, 31, 1664–1672 CrossRef CAS; (c) D. Serra, P. Cao, J. Cabrera, R. Padilla, F. Rominger and M. Limbach, Organometallics, 2011, 30, 1885–1895 CrossRef CAS; (d) K. M. Schultz, K. I. Goldberg, D. G. Gusev and D. M. Heinekey, Organometallics, 2011, 30, 1429–1437 CrossRef CAS; (e) K. Inamoto, J.-I. Kuroda, E. Kwon, K. Hiroya and T. Doi, J. Organomet. Chem., 2009, 694, 389–396 CrossRef CAS PubMed; (f) A. A. Danopoulos, D. Pugh and J. A. Wright, Angew. Chem., Int. Ed., 2008, 47, 9765–9767 CrossRef PubMed; (g) J. A. Wright, A. A. Danopoulos, W. B. Motherwell, R. J. Carroll, S. Ellwood and J. Saßmannshausen, Chem. Ber., 2006, 2006, 4857–4865 Search PubMed; (h) A. A. Danopoulos, J. A. Wright, W. B. Motherwell and S. Ellwood, Organometallics, 2004, 23, 4807–4810 CrossRef CAS.
- (a) C. Gao, H. Zhou, S. Wei, Y. Zhao, J. You and G. Gao, Chem. Commun., 2013, 49, 1127–1129 RSC; (b) F. E. Hahn, M. C. Jahnke and T. Pape, Organometallics, 2007, 26, 150–154 CrossRef CAS; (c) F. E. Hahn, M. C. Jahnke, V. Gomez-Benitez, D. Morales-Morales and T. Pape, Organometallics, 2005, 24, 6458–6463 CrossRef CAS; (d) M. Poyatos, F. Márquez, E. Peris, C. Claver and E. Fernandez, New J. Chem., 2002, 27, 425–431 RSC; (e) J. A. Loch, M. Albrecht, E. Peris, J. Mata, J. W. Faller and R. H. Crabtree, Organometallics, 2002, 21, 700–706 CrossRef CAS; (f) E. Peris, J. Mata, J. A. Loch and R. H. Crabtree, Chem. Commun., 2001, 201–202 RSC.
- A. A. D. Tulloch, A. A. Danopoulos, G. J. Tizzard, S. J. Coles, M. B. Hursthouse, R. S. Hay-Motherwell and W. B. Motherwell, Chem. Commun., 2001, 1270–1271 RSC.
- A. A. Danopoulos, A. A. D. Tulloch, S. Winston, G. Eastham and M. B. Hursthouse, Dalton Trans., 2003, 1009–1015 RSC.
- D. J. Nielsen, K. J. Cavell, B. W. Skelton and A. H. White, Inorg. Chim. Acta, 2006, 359, 1855–1869 CrossRef CAS PubMed.
- R. S. Simons, P. Custer, C. A. Tessier and W. J. Youngs, Organometallics, 2003, 22, 1979–1982 CrossRef CAS.
- J. R. Miecznikowski, S. Gründemann, M. Albrecht, C. Mégret, E. Clot, J. W. Faller, O. Eisenstein and R. H. Crabtree, Dalton Trans., 2003, 831–838 RSC.
- D. J. Nielsen, K. J. Cavell, B. W. Skelton and A. H. White, Inorg. Chim. Acta, 2002, 327, 116–125 CrossRef CAS.
- S. Gründemann, M. Albrecht, J. A. Loch, J. W. Faller and R. H. Crabtree, Organometallics, 2001, 20, 5485–5488 CrossRef.
- (a) X. Zhang, D. Huang, Y.-S. Chen and R. H. Holm, Inorg. Chem., 2012, 51, 11017–11029 CrossRef CAS PubMed; (b) J. Kickham, S. Loeb and S. Murphy, Chem.–Eur. J., 1997, 3, 1203–1213 CrossRef CAS; (c) B. R. Cameron, S. J. Loeb and G. P. A. Yap, Inorg. Chem., 1997, 36, 5498–5504 CrossRef CAS; (d) J. Kickham and S. Loeb, Inorg. Chem., 1995, 34, 5656–5665 CrossRef CAS; (e) J. Kickham and S. Loeb, Inorg. Chem., 1994, 33, 4351–4359 CrossRef CAS; (f) J. Kickham, S. Loeb and S. Murphy, J. Am. Chem. Soc., 1993, 115, 7031–7032 CrossRef CAS.
- (a) G. De Bo, J. De Winter, P. Gerbaux and C.-A. Fustin, Angew. Chem., Int. Ed., 2011, 50, 9093–9096 CrossRef CAS PubMed; (b) S. M. Goldup, D. A. Leigh, R. T. McBurney, P. R. McGonigal and A. Plant, Chem. Sci., 2010, 1, 383 RSC; (c) S. M. Goldup, D. A. Leigh, P. J. Lusby, R. T. McBurney and A. M. Z. Slawin, Angew. Chem., Int. Ed., 2008, 47, 3381–3384 CrossRef CAS PubMed; (d) V. Aucagne, J. Berná, J. D. Crowley, S. M. Goldup, K. D. Hänni, D. A. Leigh, P. J. Lusby, V. E. Ronaldson, A. M. Z. Slawin, A. Viterisi and D. B. Walker, J. Am. Chem. Soc., 2007, 129, 11950–11963 CrossRef CAS PubMed; (e) L. Hogg, D. A. Leigh, P. J. Lusby, A. Morelli, S. Parsons and J. K. Y. Wong, Angew. Chem., Int. Ed., 2004, 43, 1218–1221 CAS.
- Such species have notably found application in anion sensing: (a) H.-Y. Gong, B. M. Rambo, V. M. Lynch, K. M. Keller and J. L. Sessler, J. Am. Chem. Soc., 2013, 135, 6330–6337 CrossRef CAS PubMed; (b) H.-T. Niu, Z. Yin, D. Su, D. Niu, Y. Ao, J. He and J.-P. Cheng, Tetrahedron, 2008, 64, 6300–6306 CrossRef CAS PubMed; (c) V. K. Khatri, M. Chahar, K. Pavani and P. S. Pandey, J. Org. Chem., 2007, 72, 10224–10226 CrossRef CAS PubMed; (d) J. Yoon, S. K. Kim, N. J. Singh and K. S. Kim, Chem. Soc. Rev., 2006, 35, 355 RSC; (e) M. V. Baker, M. J. Bosnich, D. H. Brown, L. T. Byrne, V. J. Hesler, B. W. Skelton, A. H. White and C. C. Williams, J. Org. Chem., 2004, 69, 7640–7652 CrossRef CAS PubMed.
- C. Radloff, H.-Y. Gong, C. Schulte to Brinke, T. Pape, V. M. Lynch, J. L. Sessler and F. E. Hahn, Chem.–Eur. J., 2010, 16, 13077–13081 CrossRef CAS PubMed.
- A range of pyridine based bis-NHC cyclophanes complexes are known, although they adopt dinuclear coordination geometries: (a) M. V. Baker, D. H. Brown, R. A. Haque, P. V. Simpson, B. W. Skelton, A. H. White and C. C. Williams, Organometallics, 2009, 28, 3793–3803 CrossRef CAS; (b) A. Melaiye, Z. Sun, K. Hindi, A. Milsted, D. Ely, D. H. Reneker, C. A. Tessier and W. J. Youngs, J. Am. Chem. Soc., 2005, 127, 2285–2291 CrossRef CAS PubMed; (c) P. J. Barnard, M. V. Baker, S. J. Berners-Price, B. W. Skelton and A. H. White, Dalton Trans., 2004, 1038 RSC; (d) J. C. Garrison, R. S. Simons, J. M. Talley, C. Wesdemiotis, C. A. Tessier and W. J. Youngs, Organometallics, 2001, 20, 1276–1278 CrossRef CAS; (e) J. C. Garrison, R. S. Simons, W. G. Kofron, C. A. Tessier and W. J. Youngs, Chem. Commun., 2001, 1780–1781 RSC . For a unique exception see: M. V. Baker, B. W. Skelton, A. H. White and C. C. Williams, Organometallics, 2002, 21, 2674–2678 Search PubMed.
- S. Saito, I. Azumaya, N. Watarai, H. Kawasaki and R. Yamasaki, Heterocycles, 2009, 79, 531–548 CrossRef PubMed.
- K. Meyer, A. F. Dalebrook and L. J. Wright, Dalton Trans., 2012, 41, 14059–14067 RSC.
- (a) M. V. Baker, S. K. Brayshaw, B. W. Skelton, A. H. White and C. C. Williams, J. Organomet. Chem., 2005, 690, 2312–2322 CrossRef CAS PubMed; (b) M. V. Baker, B. W. Skelton, A. H. White and C. C. Williams, J. Chem. Soc., Dalton Trans., 2001, 111–120 RSC.
- (a) O. Shoji, S. Okada, A. Satake and Y. Kobuke, J. Am. Chem. Soc., 2005, 127, 2201–2210 CrossRef CAS PubMed; (b) J. Z. Vlahakis, S. Mitu, G. Roman, E. Patricia Rodriguez, I. E. Crandall and W. A. Szarek, Bioorg. Med. Chem., 2011, 19, 6525–6542 CrossRef CAS PubMed.
- For example see: (a) B. Szyszko, L. Latos-Grazyński and L. Szterenberg, Angew. Chem., Int. Ed., 2011, 50, 6587–6591 CrossRef CAS PubMed; (b) P. Štěpnička, H. Solařová and I. Císařová, J. Organomet. Chem., 2011, 696, 3727–3740 CrossRef PubMed; (c) M. Bröring and C. Kleeberg, Z. Anorg. Allg. Chem., 2007, 633, 2210–2216 CrossRef.
- The conditions under which this coalescence temperature was measured are unclear: either a 400 or 500 MHz spectrometer was used.
- See for example: (a) T. M. Douglas, E. Molinos, S. K. Brayshaw and A. S. Weller, Organometallics, 2007, 26, 463–465 CrossRef CAS; (b) I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef CAS PubMed.
- gNMR (Adept Scientific, Herts, UK), v 4.1.2.
- W. Kemp, NMR in Chemistry: A multinuclear introduction, Macmillan, Hampshire, UK, 1986 Search PubMed.
- (a) N. D. Ball, J. W. Kampf and M. S. Sanford, Dalton Trans., 2010, 632–640 RSC; (b) V. V. Grushin and W. J. Marshall, J. Am. Chem. Soc., 2009, 131, 918–919 CrossRef CAS PubMed; (c) D. A. Watson, M. Su, G. Teverovskiy, Y. Zhang, J. García-Fortanet, T. Kinzel and S. L. Buchwald, Science, 2009, 325, 1661–1664 CrossRef CAS PubMed; (d) N. D. Ball and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 3796–3797 CrossRef CAS PubMed; (e) V. V. Grushin, Chem.–Eur. J., 2002, 8, 1006–1014 CrossRef CAS; (f) M. C. Pilon and V. V. Grushin, Organometallics, 1998, 17, 1774–1781 CrossRef CAS; (g) S. L. Fraser, M. Y. Antipin, V. N. Khroustalyov and V. V. Grushin, J. Am. Chem. Soc., 1997, 119, 4769–4770 CrossRef CAS.
- (a) S. R. Caskey, M. H. Stewart, Y. J. Ahn, M. J. A. Johnson, J. L. C. Rowsell and J. W. Kampf, Organometallics, 2007, 26, 1912–1923 CrossRef CAS; (b) C. J. Bourgeois, S. A. Garratt, R. P. Hughes, R. B. Larichev, J. M. Smith, A. J. Ward, S. Willemsen, D. Zhang, A. G. DiPasquale, L. N. Zakharov and A. L. Rheingold, Organometallics, 2006, 25, 3474–3480 CrossRef CAS; (c) A. Yahav, I. Goldberg and A. Vigalok, Inorg. Chem., 2005, 44, 1547–1553 CrossRef CAS PubMed; (d) J. E. Veltheer, P. Burger and R. G. Bergman, J. Am. Chem. Soc., 1995, 117, 12478–12488 CrossRef CAS; (e) S. K. Agbossou, C. Roger, A. Igau and J. A. Gladysz, Inorg. Chem., 1992, 31, 419–424 CrossRef CAS; (f) S. A. Brewer, J. H. Holloway, E. G. Hope and P. G. Watson, J. Chem. Soc., Chem. Commun., 1992, 1577 RSC.
- R. Huacuja, D. E. Herbert, C. M. Fafard and O. V. Ozerov, J. Fluorine Chem., 2010, 131, 1257–1261 CrossRef CAS PubMed.
- (a) S. C. F. Kui, N. Zhu and M. C. W. Chan, Angew. Chem., Int. Ed., 2003, 42, 1628–1632 CrossRef CAS PubMed; (b) D. L. Bryce and R. E. Wasylishen, J. Mol. Struct., 2002, 602, 463–472 CrossRef; (c) G. R. Desiraju, Acc. Chem. Res., 2002, 35, 565–573 CrossRef CAS PubMed; (d) G. W. Gribble, E. R. Olson, J. H. Brown and C. H. Bushweller, J. Org. Chem., 1993, 58, 1631–1634 CrossRef CAS.
- Preliminary catalytic data for 3c (Heck reaction) is detailed in the ESI.†.
- W. E. Buschmann, J. S. Miller, K. Bowman-James and C. N. Miller, Inorg. Synth., 2002, 33, 83–91 CAS.
- J. A. McCleverty, G. Wilkinson, L. G. Lipson, M. L. Maddox and H. D. Kaesz, Inorg. Synth., 1990, 28, 84–86 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 961289–961294. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt52578c |
| This journal is © The Royal Society of Chemistry 2014 |