 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fac and mer isomers of Ru(II) tris(pyrazolyl-pyridine) complexes as models for the vertices of coordination cages: structural characterisation and hydrogen-bonding characteristics†
Alexander J.
Metherell
,
William
Cullen
,
Andrew
Stephenson
,
Christopher A.
Hunter
and
Michael D.
Ward
*
Department of Chemistry, University of Sheffield, Sheffield S3 7HF, UK. E-mail: m.d.ward@sheffield.ac.uk
First published on 16th October 2013
Abstract
We have prepared a series of mononuclear fac and mer isomers of Ru(II) complexes containing chelating pyrazolyl-pyridine ligands, to examine their differing ability to act as hydrogen-bond donors in MeCN. This was prompted by our earlier observation that octanuclear cube-like coordination cages that contain these types of metal vertex can bind guests such as isoquinoline-N-oxide (K = 2100 M−1 in MeCN), with a significant contribution to binding being a hydrogen-bonding interaction between the electron-rich atom of the guest and a hydrogen-bond donor site on the internal surface of the cage formed by a convergent set of CH2 protons close to a 2+ metal centre. Starting with [Ru(LH)3]2+ [LH = 3-(2-pyridyl)-1H-pyrazole] the geometric isomers were separated by virtue of the fact that the fac isomer forms a Cu(I) adduct which the mer isomer does not. Alkylation of the pyrazolyl NH group with methyl iodide or benzyl bromide afforded [Ru(LMe)3]2+ and [Ru(Lbz)3]2+ respectively, each as their fac and mer isomers; all were structurally characterised. In the fac isomers the convergent group of pendant –CH2R or –CH3 protons defines a hydrogen-bond donor pocket; in the mer isomer these protons do not converge and any hydrogen-bonding involving these protons is expected to be weaker. For both [Ru(LMe)3]2+ and [Ru(Lbz)3]2+, NMR titrations with isoquinoline-N-oxide in MeCN revealed weak 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding (K ≈ 1 M−1) between the guest and the fac isomer of the complex that was absent with the mer isomer, confirming a difference in the hydrogen-bond donor capabilities of these complexes associated with their differing geometries. The weak binding compared to the cage however occurs because of competition from the anions, which are free to form ion-pairs with the mononuclear complex cations in a way that does not happen in the cage complexes. We conclude that (i) the presence of fac tris-chelate sites in the cage to act as hydrogen-bond donors, and (ii) exclusion of counter-ions from the central cavity leaving these hydrogen-bonding sites free to interact with guests, are both important design criteria for future coordination cage hosts.
:1 binding (K ≈ 1 M−1) between the guest and the fac isomer of the complex that was absent with the mer isomer, confirming a difference in the hydrogen-bond donor capabilities of these complexes associated with their differing geometries. The weak binding compared to the cage however occurs because of competition from the anions, which are free to form ion-pairs with the mononuclear complex cations in a way that does not happen in the cage complexes. We conclude that (i) the presence of fac tris-chelate sites in the cage to act as hydrogen-bond donors, and (ii) exclusion of counter-ions from the central cavity leaving these hydrogen-bonding sites free to interact with guests, are both important design criteria for future coordination cage hosts.
Introduction
Host–guest chemistry of hollow container molecules is an increasingly important field of study because of the fundamental insights it can offer into molecular recognition processes,1,2 and because of potential applications in areas such as alterations of reactivity of bound guests which allows novel synthetic transformations;3 catalysis;4 and targeted drug delivery.5 Of these, all rely on highly selective binding of specific guests, sometimes involving hydrogen-bond based recognition between the guest and the cavity of the host.6We have recently described some detailed studies of guest binding in the cavity of a family of [M8(L15naph)12]16+ coordination cages7 (see Scheme 1 for ligand structure, and Fig. 1) which have structure with a metal ion at each vertex of a cube and a bis-bidentate ligand, containing two chelating pyrazolyl-pyridine termini, spanning each edge.8 Variation in external substituents has allowed these to be solubilised in different solvents without affecting the structure of the core cage and its central cavity. Importantly, the eight metal ions are not all in the same coordination environment: two of them (at either end of a long diagonal) have a fac tris-chelate coordination geometry whereas the other six have a mer geometry. Thus the cages are superficially ‘cubic’ due to the arrangement of metal ions, but the variation in fac or mer tris-chelate geometries at different sites results in S6 molecular symmetry in solution.
 | ||
| Scheme 1 (a) Structure of the L15naph family of ligands used to make cubic coordination cages (R′ = H, CH2OH); (b) structures of some of the simple bidentate ligands discussed in this paper. | ||
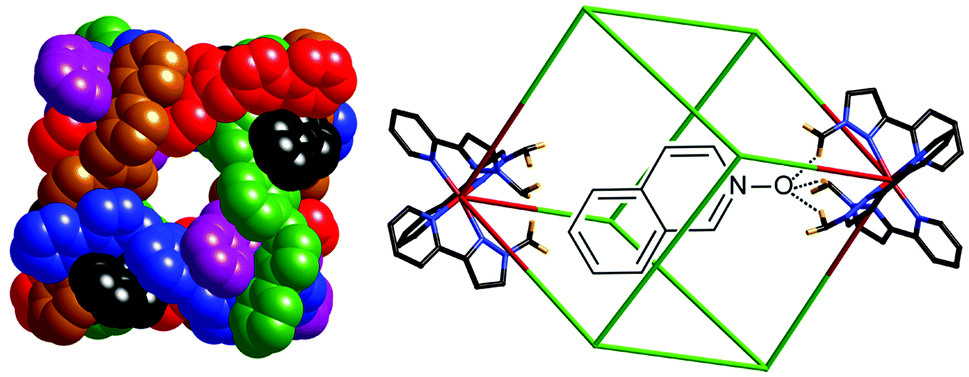 | ||
| Fig. 1 The general structure of the [M8(L15naph)12]16+ coordination cage, with ligands coloured differently for clarity (left); and a sketch showing the likely mode of binding of hydrogen-bond accepting guests at one of the two fac tris-chelate metal vertices (right). | ||
The formation of two fac tris-chelate sites results in assembly of convergent groups of inwardly-directed methylene protons, which lie quite close to the Co(II) centre and are therefore in a region of high electrostatic potential. This provides a binding pocket where electron-rich atoms can bind via a set of charge-assisted C–H⋯X hydrogen-bonds (Fig. 1). At the remaining six mer tris-chelate Co(II) centres there is no such convergent group of C–H protons, and additionally these metal ions are more sterically protected by the ligand substituents such that no close approach of an electron-rich guest atom to the metal centre is possible.7
These cages bind, with high shape and size selectivity, a range of hydrogen-bond-accepting bicyclic organic molecules such as coumarin and isoquinoline-N-oxide, in which the exocyclic O atom acts as the hydrogen-bond acceptor. 1H NMR studies on numerous host/guest combinations in MeCN showed a strong correlation between guest binding strength and the hydrogen-bond acceptor ability of the guest, confirming that a hydrogen-bonding interaction with the interior surface of the cage makes an important contribution to complex formation.7b Molecular modelling studies showed that the minimum-energy structures had the guests oriented such that their exocyclic oxygen atom did indeed lie in the pocket defined by the convergent set of methylene protons at one of the two fac tris-chelate vertices, in the regions of the cavity where electrostatic potential is most positive.
So far we have not been able to isolate good-quality crystals of a host M8L12 coordination cage containing a bound guest to confirm the binding mode. It is important for this work however to have clear proof that our hypothesis – viz. that the fac tris-chelate sites in the cages provide preorganised binding sites for recognition of hydrogen-bond acceptors7b – is correct, as this knowledge will influence design and study of future generations of host cages. We note that there are several examples of simple tris-chelate complexes in which a convergent arrangement of three polar substituents such as amides or carboxylates on one face of the complex provides a binding site for interacting with anions9 and even proteins.10 Although our cage complexes only contain inwardly directed C–H groups as the hydrogen-bonding sites we might expect to see the same geometric discrimination between fac (convergent set of hydrogen-bond donor atoms) and mer (non convergent hydrogen-bond donors) isomers.
We have therefore prepared simple, kinetically stable, mononuclear Ru(II) complexes based on pyrazolyl-pyridine type ligands, as either their fac or mer isomers, to use as models for the different metal vertices in the coordination cage. Structural characterisation of these has been used to confirm their similarity to the metal centres in the cage superstructure, and 1H NMR spectroscopic titrations have been performed to see if the coordination geometry of the metal complex does influence how well an hydrogen-bonding guest interacts with it. From this we have been able to find clear evidence that the fac tris-chelate metal complex units do act as better hydrogen-bond donors than the related mer tris-chelate complexes, and that this interaction is associated with the convergent group of methylene protons in the fac complexes. We therefore have good evidence (beyond molecular modelling) to support our understanding of how guest molecules interact with the interior surface of the cage hosts.
Results and discussion
Initial attempts to isolate fac- and mer-[Co(Lbz)3](BF4)2 by crystallisation
Our initial targets for investigation were fac and mer tris-chelate Co(II) complexes of the benzyl-substituted pyrazolyl-pyridine ligand Lbz, which is effectively half of the bridging ligand L15naph and will provide a coordination environment around a single metal ion that is as close as possible to what is found in the [Co8(L15naph)12]16+ cages.7,11 The ligand Lbz is simply prepared by alkylation of the pyrazole ring of 3-(2-pyridyl)-1H-pyrazole with benzyl bromide. Reaction of Lbz with Co(BF4)2 afforded [Co(Lbz)3](BF4)2 as a mixture of fac and mer isomers as shown by 1H NMR spectroscopy. Statistically these are expected to form in a 1:3 ratio in the absence of other factors which might favour one geometry over the other. In the fac isomer (expected C3 symmetry) all three ligands will be equivalent. In the mer isomer (expected C1 symmetry) all three ligands will be in different chemical environments. Thus a mixture of fac and mer isomers of [Co(Lbz)3](BF4)2 is expected to show in its 1H NMR spectrum four different environment for the ligand L, and if the statistical 1:3 ratio occurs this means that all four ligand environments will be present with equal likelihood. Bulky substituents which are too close to one another in the fac isomer would skew this equilibrium in favour of the mer isomer; at the other extreme, cooperative non-covalent interactions between ligands can strongly favour the fac isomer, as shown recently by Scott and co-workers.9i,12
The 1H NMR spectrum of [Co(Lbz)3](BF4)2 (Fig. 2) shows a statistical mix of fac and mer isomers with four ligand environments in equal abundance. As we have observed before the paramagnetism of high-spin Co(II) spreads out the signals over a wide chemical shift range making identification of the mixture of isomers straightforward.7,8,11,13 Whilst all signals are not individually assigned, and some of the less shifted ones clearly overlap in the 0–10 ppm region, the presence of four independent ligand environments for the mixture of fac and mer isomers is obvious: some sets of four signals that are clearly the same proton (a–d) in four different environments are labelled on Fig. 2.
 | ||
| Fig. 2 1H NMR spectrum of [Co(Lbz)3](BF4)2 in MeCN, showing the mixture of fac and mer isomers with four independent ligand environments present in equal abundance. | ||
Crystallisation of this mixture from CHCl3 afforded X-ray quality crystals of what proved to be the mer isomer of [Co(Lbz)3](BF4)2 (Fig. 3). The quasi-octahedral coordination geometry and the Co–N distances (in the range 2.14–2.20 Å) are unremarkable and very similar to what is observed in the complete coordination cages. One of the pendant phenyl rings [C(31C)–C(36C)] lies stacked with a coordinated pyrazolyl-pyridine fragment from a different ligand within the same molecule. Importantly the three CH2 groups are not convergent and do not form a clearly-defined binding pocket, although three of them [H(26B), H(26D) and H(26F)] form close contacts with F atoms of the fluoroborate anions or the O atom of a water molecule (H⋯X separations involving these H atoms, 2.49–2.93 Å).
 | ||
| Fig. 3 Structure of the complex cation of mer-[Co(Lbz)3](BF4)2·CHCl3·H2O (thermal ellipsoids at 40% probability level). One ligand is shown with paler colours for clarity. | ||
We were unable to isolate any crystals of the fac isomer of [Co(Lbz)3](BF4)2 for comparison purposes. Only the mer isomer crystallised, and – given the kinetic lability of Co(II) – this likely resulted in re-establishment of the 3:1 mer:fac equilibrium ratio in solution, such that the amount of fac isomer present diminished as the crystallisation proceeded. This was confirmed by measuring the 1H NMR spectrum of redissolved crystals of mer-[Co(Lbz)3](BF4)2 which was exactly the same as that in Fig. 2: i.e. the pure mer isomer equilibrated to the 3:1 mer:fac equilibrium in the time it took to dissolve the crystals and record the NMR spectrum. It is clear from this that trying to isolate and study separately the fac and mer isomers of [Co(Lbz)3](BF4)2 as models for the separate types of cage vertex is not feasible, and we therefore changed strategy to the use of a more kinetically inert metal centre.
Isolation and structural characterisation of fac- and mer-[RuL3](PF6)2
We required a kinetically inert metal ion for which fac and mer tris-chelate complexes are sufficiently stable to be prepared at modest temperatures, purified by chromatography or fractional crystallisation, and studied in solution, without undergoing any significant isomerisation. To be an accurate model for the vertices of the cage, the metal ion must also have a 2+ charge and have metal–ligand bond distances similar to that of the Co(II) complexes. The obvious candidate is Ru(II), with which there are numerous well-studied examples of isolation and characterisation of fac and mer isomers of non-symmetric chelating ligands.9b,g,h,10,14,15Preparation of [Ru(Lbz)3]2+ salts as a mixture of isomers is routine and the 3:1 mer:fac ratio was confirmed by 1H NMR spectroscopy. However chromatographic separation was difficult. There are examples from Fletcher and co-workers of effective chromatographic separation of fac and mer isomers of Ru(II) complexes, but in those cases the large, polar substituents exaggerated the geometric and electronic differences between the isomers which may have facilitated the separation.14 With relatively compact and non-polar benzyl substituents in [Ru(Lbz)3]2+ salts the structural difference between the isomers did not appear to be enough to allow effective chromatographic separation under a range of conditions. Attempts at fractional crystallisation provided a few crystals of one isomer or the other but not on a sufficient scale or in a predictable way.
We therefore adopted a different strategy, which was to prepare the complex [Ru(LH)3]2+ using unsubstituted 3-(2-pyridyl)-1H-pyrazole, and then to separate the isomers according to their different ability to form adducts with other metal ions via the pendant pyrazolyl groups.16 Reaction of RuCl3 with excess LH in ethylene glycol at reflux afforded a yellow solution from which [Ru(LH)3](PF6)2 precipitated on addition of aqueous KPF6. This was purified (without separation of the isomers) by column chromatography on silica using a MeCN–water–aqueous KNO3 mixture, and the resulting sample of [Ru(LH)3](PF6)2 was shown by 1H NMR spectroscopy to be the expected 3:1 mixture of mer and fac isomers with four independent ligand environments of equal abundance being present (Fig. 4a).
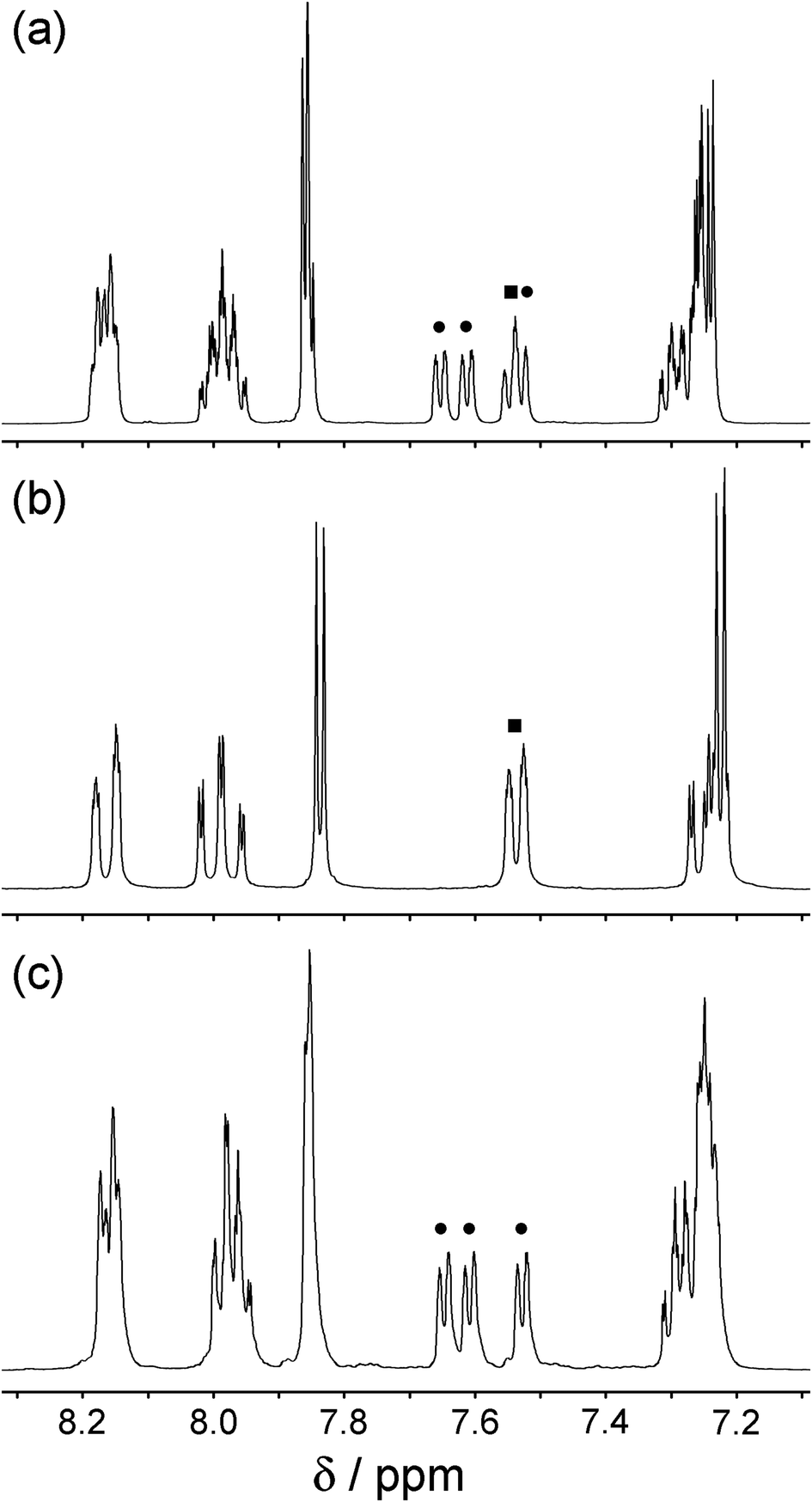 | ||
| Fig. 4 1H NMR spectra (MeCN, 400 MHz) of [Ru(LH)3](PF6)2: (a) the as-isolated statistical mixture of fac and mer isomers; (b) the pure fac isomer; (c) the pure mer isomer. | ||
Separation of this into its geometric isomers was achieved quantitatively by reaction of [Ru(LH)3](PF6)2 with Cu(BF4)2 in MeOH containing Et3N to give the pentanuclear complex [{Ru(L−)3}2Cu3](PF6) as previously reported by Lam and co-workers.16 In this complex, two fac-[Ru(LH)3]2+ units have had their pyrazolyl NH groups deprotonated by the Et3N. The resulting fac-[Ru(L−)3]− unit has an array of three anionic pyrazolyl donors on the same face of the complex, and two of these fac-[Ru(L−)3]− units sandwich a triangle of three Cu(I) ions to give a stable Ru2Cu3 complex which has a triple helical structure and with each Cu(I) ion coordinated by two pyrazole anions, one from each Ru(II) unit. This complex precipitates from MeOH as it forms and is trivially separated by filtration. In contrast mer-[Ru(LH)3]2+ cannot form a stable Cu(I) adduct in this way as the three pyrazolyl N3 atoms are not convergent; so it remains in the reaction solution from which it may be separated and purified. Treatment of the precipitated [{Ru(L−)3}2Cu3](PF6) with CF3CO2H in CH2Cl2 re-protonates the pyrazole rings, to regenerate [Ru(LH)3]2+ which is now (after a simple workup) the pure fac isomer. Fig. 4(b) and 4(c) show the 1H NMR spectra of fac- and mer-[Ru(LH)3](PF6)2 respectively, with one ligand environment and three ligand environments (6 proton environments and 18 proton environments respectively). The correspondence of these signals with the spectrum of the initially-isolated mixture of isomers in Fig. 4a is obvious; note especially the set of three doublets at around 7.6 ppm for the mer isomer (Fig. 4c, each labelled with a black circle), and the corresponding single doublet for the fac isomer (Fig. 4b, labelled with a black square), which overlap in the spectrum of the mixture of isomers (Fig. 4a). This separation via an intermediate Cu(I) adduct proved to be a simple and effective way to isolate pure fac and mer isomers.
X-Ray quality crystals of both isomers of the complex were readily obtained and the structures are shown in Fig. 5 and 6. In the structure of mer-[Ru(LH)3](PF6)2 (Fig. 5) the Ru–N distances lie in the range 2.04–2.09 Å, slightly shorter than in the Co(II) complexes but still sufficiently similar for the Ru(II) complexes to act as credible structural analogues of the Co(II) vertices in the [Co8(L15naph)12]16+ coordination cages.7,8 It is generally unremarkable with the expected pseudo-octahedral structure.
 | ||
| Fig. 5 Structure of the complex cation of mer-[Ru(LH)3](PF6)2·2MeCN (thermal ellipsoids at 40% probability level). | ||
 | ||
| Fig. 6 Structure of the hydrogen-bonded dinuclear complex cation of fac,fac-[Ru(LH)3Ru(L−)3] (PF6)·3C7H8 (thermal ellipsoids at 40% probability level). Some of the ligands are shown in paler colours for clarity; the hydrogen-bonds between the two complex units are indicated by the striped lines. | ||
The structure of the fac isomer (Fig. 6) however reveals a surprise: the complex is partially deprotonated (at the pyrazolyl NH sites) to give the hydrogen-bonded dimer [Ru(LH)3Ru(L−)3](PF6). In this complex there are two independent complex fragments in the unit cell, both lying on a C3 axis which passes through both metal ions such that one third of each complex is in the asymmetric unit. One complex is fac-[Ru(LH)3]2+, containing Ru(1), and is the fully protonated dication, as expected. The other however is fac-[Ru(L−)3]−, containing Ru(2), in which all three pyrazolyl rings are deprotonated. The protonated and deprotonated complex units are closely associated via three NH⋯N hydrogen bonds involving the pyrazolyl NH from the Ru(1) complex [N(21A)] and the deprotonated pyrazolyl ring from the Ru(2) complex [N(21B)]. As both units have the same chirality, the result – if we take a hydrogen-bonded pair of ligands pyridyl–pz–H⋯pz–pyridyl as a single ‘strand’ spanning two metal ions – is a triple helical architecture similar to that of [{Ru(L−)3}2Cu3](PF6) reported earlier16 but with H+ ions replacing the Cu(I) ions in the centre. The N⋯N separations involved in the NH–N hydrogen bonds (all equivalent) are 2.66 Å, indicative of short, strong hydrogen-bonding interactions. In addition this close association of the two complex units results in π-stacking between the pyrazolyl rings of each, with an average interplanar separation of ca. 3.3 Å between overlapping ligand fragments. This will be facilitated by the fact that one pyrazolyl ring in each stacked pair is deprotonated and therefore electron-rich compared to the other, so the stack involves a donor–acceptor interaction. These crystals grew from a solution containing fully protonated fac-[Ru(LH)3](PF6)2; presumably the loss of three protons per two complex units is driven by the extra stability of the hydrogen-bonded and π-stacked pair of cationic and anionic complex units in the crystal. Telfer and co-workers have reported related examples of helicates formed from homochiral mononuclear units that assemble via inter-ligand hydrogen-bonding interactions in exactly the same way.17
With fac- and mer-[Ru(LH)3](PF6)2 readily available by this route, conversion to the corresponding isomers of [Ru(Lbz)3](PF6)2 was carried out by alkylation of the pyrazole groups with benzyl bromide in CH2Cl2 at reflux, using solid Cs2CO3 as base and a catalytic amount of tetrabutylammonium iodide (Finkelstein reaction). This straightforward reaction works under a range of conditions but we wanted to use conditions as mild as possible to prevent any isomerisation of the Ru(II) complex which might be facilitated by high temperatures, and by use of good donor solvents which would facilitate ligand dissociation. We found that under these conditions conversion of [Ru(LH)3](PF6)2 to [Ru(Lbz)3](PF6)2 proceeded cleanly and with no evidence (by NMR spectroscopy) of any of the alternate isomer forming. Chromatographic purification afforded good yields of fac- and mer-[Ru(Lbz)3](PF6)2. By extrapolation we now have a potentially general method to prepare fac and mer isomers of any complex-[Ru(LR)3]2+ where ‘R’ denotes a group that can be attached to a pyrazole ring by alkylation.
The X-ray crystal structure of fac-[Ru(Lbz)3](PF6)2 is shown in Fig. 7. In the complex cation (Fig. 7a) the Ru–N bond distances all lie in the range 2.07–2.09 Å. In each ligand the pendant phenyl ring lies stacked with the coordinated pyrazolyl-pyridine unit of another adjacent ligand, with a separation between near-parallel overlapping groups of ca. 3.4 Å, exactly as we see in the fac tris-chelate vertices of the metal cages. This arrangement of ligands results in the formation of a set of three methylene groups close together on the same face of the complex. The ‘inwardly’ directed member of each pair [H(26A), H(26C) and H(26E)] lies ca. 3.3 Å from the Ru(II) centre and these are the closest H atoms to the metal centre apart from the pyridyl H6 atoms (3.1–3.2 Å). This set of protons defines what we believe to be the site where hydrogen-bonding to electron-rich atoms of guests occurs inside the cage cavities.7
 | ||
| Fig. 7 (a) Structure of the complex cation of fac-[Ru(Lbz)3](PF6)2·2Me2CO (thermal ellipsoids at 40% probability level); one ligand is shown with paler colours for clarity. (b) Alternative view of the complex cation with the phenyl rings not shown, emphasising the convergent arrangement of methylene protons to form an hydrogen-bond donor site, and the presence of a hydrogen-bonded molecule of acetone at this site; the two shortest CH⋯O interactions are shown by dashed lines. | ||
Evidence for this comes from the presence of an acetone solvent molecule in the lattice (Fig. 7b) which lies such that its oxygen atom is located in this pocket, where we would expect a guest to bind. The O atom is not exactly symmetrically located in the pocket in the solid state but lies closer to C26(B) and C26(C) (O⋯C non-bonded distances are 3.11 and 3.18 Å respectively, indicative of the presence of CH⋯O hydrogen bonding; these are indicated by dashed lines in Fig. 7b) than it does to O(26C) (3.63 Å), with the result that there are four O⋯H–C hydrogen-bond separations in the range 2.51 to 2.80 Å. This solvent molecule rather nicely illustrates the presence of the binding site at the fac tris-chelate vertices of the cages.
The X-ray crystal structure of mer-[Ru(Lbz)3](PF6)2 (Fig. 8) shows that the structure of the complex cation is generally similar to that of mer-[Co(Lbz)3](BF4)2; the most obvious π-stacking interaction between ligands involves the pendant phenyl ring of ligand B (according to the numbering scheme of Fig. 8) with the coordinated pyrazolyl-pyridine unit of ligand C. Compared to the fac isomer the divergent arrangement of the methylene protons no longer results in a specific hydrogen-bonding recognition site.
 | ||
| Fig. 8 Structure of the complex cation of mer-[Ru(Lbz)3](PF6)2·Me2CO (thermal ellipsoids at 40% probability level); one ligand is shown with paler colours for clarity. | ||
Measurement of guest binding to fac- and mer-[Ru(Lbz)3](PF6)2
To measure the differential abilities of fac- and mer-[Ru(Lbz)3](PF6)2 to act as a hydrogen-bond donor we measured the equilibrium constant for adduct formation with isoquinoline-N-oxide. On the basis of previous measurements using complete cages as hosts, this was identified as one of the strongest-binding guests in MeCN (K = 2100 M−1, cf.Fig. 1).7b We suggested that this is due to the high partial negative charge on the oxygen atom (high value of the hydrogen-bond acceptor parameter β), which interacts with the hydrogen-bond donor sites of the fac tris-chelate metal vertices of the cage, which have a combined hydrogen-bond donor ability comparable to that of phenol.7bA 1H NMR titration of fac-[Ru(Lbz)3](PF6)2 with isoquinoline-N-oxide in MeCN showed a steady shift in one of the signals from the diastereotopic CH2 protons, but not the other which was essentially invariant (Fig. 9). This is emphasised in Fig. 9b which shows a series of superimposed 1H NMR spectra recorded during the titration: the methylene doublet at 4.7 ppm does not move but the other signal at around 5.4 ppm shifts by ca. 0.1 ppm. No other proton signals moved significantly during the titration. The shift of one methylene proton signal but not the other is consistent with the O atom of the guest forming an hydrogen-bonding interaction with the inwardly-directed proton from each methylene group but not with the other proton which is externally directed. Free rotation about the C–C bonds of the benzyl substituents will be hindered by the aromatic stacking (cf. the crystal structure), which would prevent the ‘inward’ and ‘outward’ H atoms of each methylene group from exchanging position on the NMR timescale. The interaction with the isoquinoline-N-oxide necessarily involves one of these two protons more than the other – as we observe. Indeed the 1H NMR spectrum confirms that the conformation observed in the crystal structure (Fig. 7) is preserved in solution, as the signals for the coordinated pyrazolyl-pyridine ligand units of fac-[Ru(Lbz)3](PF6)2 are substantially shifted compared to those of fac-[Ru(LH)3](PF6)2 and fac-[Ru(LMe)3](PF6)2 by π-stacking with the pendant phenyl rings (see Experimental section for details).
 | ||
| Fig. 9 Changes in the 1H NMR chemical shift of the two inequivalent methylene proton signals of fac-[Ru(Lbz)3](PF6)2 (0.23 mM) during titration with isoquinoline-N-oxide (up to 0.7 M) in CD3CN. (a) Stacked plots; (b) overlaid plots to emphasise how one signal moves but the other does not. | ||
It was apparent during the titration that complex formation was not complete even after a very large excess of isoquinoline-N-oxide was added, which is indicative of a low binding constant. The plot of chemical shift value for the methylene proton vs. (concentration of guest) gives a curve whose shape is consistent with 1:1 host/guest binding (Fig. 10, data points shown as circles), and the calculated value of K from this is 1(±1) M−1.‡ Thus we see an obvious interaction of fac-[Ru(Lbz)3](PF6)2 with isoquinoline-N-oxide that involves the methylene protons, even if it is weak. Importantly however no such change in chemical shift of the methylene protons could be observed using the mer isomer; the change in chemical shift of a representative 1H signal from a methylene proton is also shown in Fig. 10 (data points shown as squares). Apart from the magnitude of the Δδ for this proton during the titration being much smaller, there is no significant curvature to the line, with the result that the association constant between mer-[Ru(Lbz)3](PF6)2 and isoquinoline-N-oxide in MeCN oxide is too weak even to estimate. We therefore have clear evidence that the convergent set of methylene protons associated with a fac tris-chelate site does act as an hydrogen-bond donor to isoquinoline-N-oxide, which supports our understanding of guest binding inside the complete cage cavities.
 | ||
| Fig. 10 (a) 1H NMR binding curves showing shift of the methylene proton signals in fac-[Ru(Lbz)3](PF6)2 (circles, upper line) and mer-[Ru(Lbz)3](PF6)2 (squares, lower line) during titration with isoquinoline-N-oxide. For fac-[Ru(Lbz)3](PF6)2 the data fit a 1:1 binding isotherm with K ≈ 1 M−1; for mer-[Ru(Lbz)3](PF6)2 the binding constant is too weak to measure. (b) 19F NMR binding curve showing the change in environment of the [PF6]− ion of fac-[Ru(Lbz)3](PF6)2 as it is displaced from the hydrogen-bonding site when isoquinoline-N-oxide is added. Again the data fit a 1:1 binding isotherm with K ≈ 1 M−1. | ||
However this binding constant is surprisingly low. Based on what we observed for binding inside the host cage (K = 2100 M−1), this type of hydrogen-bonding interaction associated with mononuclear fac-[Ru(Lbz)3](PF6)2 – if it is similar to what happens inside a cage cavity – should give a much larger K value than we observed. For the [Co8(L15naph)12](BF4)16/isoquinoline-N-oxide complex that we reported earlier, the binding constant of K = 2100 M−1 gives ΔG = −19 kJ mol−1.7b Some of this arises from van der Waals interactions between the guest and the walls of the cage, and some from solvophobic effects, but even so the contribution from H-bonding alone was estimated to be several kJ mol−1 and we would expect this to be similar in the mononuclear model complex fac-[Ru(Lbz)3](PF6)2. Instead we see K ≈ 1 M−1, giving ΔG for guest binding of more or less zero despite the same type of hydrogen-bonding interaction as occurs inside the cage cavity.
One reason for this may be that competition for the hydrogen-bonding site from the hexafluorophosphate anions is occurring, which would weaken the K value for association with the neutral guest: but this competition of anions for the binding site does not occur in the complete cages for some reason. In mononuclear fac-[Ru(Lbz)3](PF6)2 the anions have free access to the relatively unhindered hydrogen-bonding site and could therefore be competing with binding of isoquinoline-N-oxide in solution. In contrast, in all structurally characterised examples of the [M8(L15naph)12]X16 cages, anions are located outside the cage cavities, with the H-bond donor sites occupied by small solvent molecules such as MeOH or water.7,11 This suggests the possibility (but does not of course prove) that the anions may also be excluded from the cage cavity in solution. This would explain the much higher binding constants for guest binding in the cage, as competition from anions would be absent.
To check for competing anion-binding in fac-[Ru(Lbz)3](PF6)2, we repeated the titration between fac-[Ru(Lbz)3](PF6)2 and isoquinoline-N-oxide in MeCN but performed 19F NMR spectra to see if there was any evidence for the hexafluorophosphate ion changing its environment. If there were no change we could say that there was no significant cation/anion association in solution. In contrast, a shift of the 19F NMR signal would suggest that the anion was being displaced from the hydrogen-bonding site by the added isoquinoline-N-oxide, and this is what we observed. The 19F NMR signal of the [PF6]− anion of fac-[Ru(Lbz)3](PF6)2 appeared as a doublet at −73.0 ppm which steadily shifted to −72.7 ppm as isoquinoline-N-oxide was titrated in. The resultant binding curve (Δδ vs. concentration of isoquinoline-N-oxide; Fig. 10b) fitted well to a 1:1 isotherm with a value of K the same within error (≈1 M−1) as that derived from the 1H NMR titration (Fig. 9 and 10). Thus, binding of isoquinoline-N-oxide to the hydrogen-bond donor site of fac-[Ru(Lbz)3](PF6)2 is accompanied by displacement of [PF6]−. As a control experiment, we also measured the change in the 19F NMR chemical shift of the [PF6]− anion of mer-[Ru(Lbz)3](PF6)2 during titration with isoquinoline-N-oxide; this resulted in a binding constant too small to measure accurately. This confirms that there is ion-pairing between cation and anion in solution with [PF6]− interacting with the hydrogen-bond donor site of the fac-[Ru(Lbz)3]2+ cation, and this competition provides one reason for the low value of K observed for binding of isoquinoline-N-oxide. An obvious experiment to try and mitigate this effect would be to use the anion tetraphenylborate which might be expected to form weaker ion pairs with the fac-[Ru(Lbz)3]2+ cation; this experiment is described in the next section.
A second contribution to the weak interaction between fac-[Ru(Lbz)3](PF6)2 and isoquinoline-N-oxide could be steric. The pendant benzyl groups of fac-[Ru(Lbz)3](PF6)2 may not be fully bent out of the way of the binding site in solution, but could move around blocking access to the hydrogen-bonding site. In the complete cages the hydrogen-bonding site is exposed because the bridging ligands have to stretch to an adjacent metal ion and are therefore stretched away from the binding site which is exposed to the cavity interior. However that may not be the case in fac-[Ru(Lbz)3](PF6)2 where steric interference from the pendant benzyl groups is possible. A control experiment to test this is to replace the pendant phenyl ring with an H atom which will remove any possible steric encumbrance, and this is also described in the next section.
Control experiment (1): measurement of guest binding to fac- and mer-[Ru(LMe)3](PF6)2
Reaction of fac- or mer-[Ru(LH)3](PF6)2 with MeI resulted in N-methylation of the pyrazolyl rings to give fac- or mer-[Ru(LMe)3](PF6)2 respectively; again, by doing this reaction under mild conditions, the individual isomers retained their structural integrity with none of the alternate isomers forming. Crystal structures are in Fig. 11 and are as expected. In fac-[Ru(LMe)3](PF6)2 there is no solvent molecule or other electron-rich atom occupying the hydrogen-bonding site, but the convergence of the methyl groups and the position of the hydrogen-bonding site is clear. Compared to fac-[Ru(Lbz)3](PF6)2, any possible steric hindrance to guest binding arising from the phenyl rings is now removed. Conversely, any possibly favourable effects – e.g. guest binding being facilitated by π-stacking with the pendant phenyl rings around the binding pocket – will also be absent. | ||
| Fig. 11 Structures of the complex cations of (a) fac-[Ru(LMe)3](PF6)2 (thermal ellipsoids at 40% probability level) and (b) mer-[Ru(LMe)3](PF6)2 (thermal ellipsoids at 30% probability level). H atoms of the methyl groups are shown in red to emphasise the binding pocket formed by convergence of the three methyl groups in the former case. | ||
1H NMR titrations of each isomer with isoquinoline-N-oxide in MeCN gave similar results to what we observed with fac- and mer-[Ru(Lbz)3](PF6)2. The fac isomer again demonstrated definite but weak binding with isoquinoline-N-oxide on the basis of a steady shift of the methyl protons (which are now all equivalent). The graph of Δδ vs. (concentration of guest) showed gentle curvature and could be fitted to a 1:1 binding isotherm (Fig. 12) with, again, K ≈ 1(±1) M−1.‡ The interaction of mer-[Ru(LMe)3](PF6)2 with isoquinoline-N-oxide in MeCN was insignificant, with much smaller Δδ for the methyl protons during the titration, and no detectable curvature in the graph. Again we see a clear difference between the behaviour of the two geometric isomers, but the important point is that the weak binding observed for fac-[Ru(Lbz)3](PF6)2 is clearly not associated with any steric blocking of the binding site by the pendant phenyl rings. This leaves ion-pairing as the other possible culprit.
 | ||
| Fig. 12 (a) 1H NMR binding curves showing shift of the methylene proton signals in fac-[Ru(LMe)3](PF6)2 (circles, upper line) and mer-[Ru(LMe)3](PF6)2 (triangles, lower line) during titration with isoquinoline-N-oxide. For fac-[Ru(LMe)3](PF6)2 the data fit a 1:1 binding isotherm with K ≈ 1 M−1; for mer-[Ru(LMe)3](PF6)2 the binding constant is too weak to measure. | ||
Control experiment (2): measurement of guest binding to fac-[Ru(LBz)3](BPh4)2
For the second control experiment we isolated fac-[Ru(Lbz)3]2+ as its tetraphenylborate salt, by simple anion metathesis starting from the hexafluorophosphate salt. The tetraphenylborate anion might be expected to result in less competition than the hexafluorophosphate ion for the hydrogen-bonding site of the cation. A 1H NMR titration with isoquinoline-N-oxide resulted in a steady shift of the signals for one of the diastereotopic methylene protons of fac-[Ru(Lbz)3]2+ but not the other, exactly as before (cf.Fig. 9); again, the resulting binding curve fitted to a 1:1 binding isotherm with K ≈ 1(±1) M−1.‡ However, during the titration the 1H NMR signals for the [BPh4]− anion also shifted steadily, consistent with it being displaced as an ion-pair breaks up, in the same way as the [PF6]− anion. The K value calculated from the shift of the most intense [BPh4]− 1H signal when it is displaced, is the same within experimental error as that derived from the shift of the methylene proton of the complex cation when the guest binds. Again, therefore, guest binding to the complex cation, and anion displacement, occur together (Fig. 13).
 | ||
| Fig. 13
1H NMR binding curves obtained from titration of [Ru(LBz)3](BPh4)2 with isoquinoline-N-oxide in MeCN: the shift of the methylene proton signal at ca. 5.4 ppm due to hydrogen-bonding with the guest (black triangles/dashed line); (b) the shift of the most intense tetraphenylborate signal at 7.3 ppm as it is displaced from the complex cation by the competing guest (black circles/solid line). Both sets of data fit a 1:1 binding isotherm with K ≈ 1 M−1. | ||
This is somewhat surprising as the tetraphenylborate anion is not a hydrogen-bond acceptor. However ion-pairing with the fac-[Ru(Lbz)3]2+ cation could occur in solution via charge-assisted π–π or CH–π interactions between electron-deficient pyrazolyl-pyridine groups that are coordinated to a 2+ metal centre, and the electron-rich phenyl rings of the anion. Examples of such ion pairs involving tetraphenylborate as the anion are known.18 Thus although hydrogen-bonding is not operative, the tetraphenylborate anion competes for binding to the complex cation just as much as does the hexafluorophosphate anion, which means that the interaction with isoquinoline-N-oxide remains weak.
The clear conclusion to be drawn from this is that the hydrogen-bonding recognition sites in the cubic coordination cage (Fig. 1) are effective at facilitating guest binding in the cavity in MeCN because anions are excluded from the cavity which is consistent with all crystal structures that we have of these cages with a variety of anions and obtained from a variety of solvents.7,11 Exclusion of anions from the binding site is therefore an important principle to take into account in design of new generations of cage hosts.
Conclusions
There are three main conclusions from this work. Firstly, we have developed a simple and general synthetic procedure which will allow access to a wide range of substituted analogues of [Ru(LH)3]2+ as their pure fac and mer isomers without tedious chromatographic separation or relying on fractional crystallisation. The mixture of fac- and mer-[Ru(LH)3]2+ may be separated by reaction with a Cu(I) salt which allows the deprotonated fac isomer to precipitate as its Cu(I) adduct whilst the mer isomer remains in solution;14 decomposition of the fac-[Ru(L−)3]−/Cu(I) adduct with acid liberates pure fac-[Ru(LH)3]2+. Alkylation of the pyrazolyl N3 positions under mild conditions allows substituted analogues to be prepared with retention of isomeric integrity.Secondly, we have shown that fac-[Ru(LBz)3]2+ and fac-[Ru(LMe)3]2+ act as better hydrogen-bond donor sites to isoquinoline-N-oxide (via formation of C–H⋯O hydrogen bonds) than do their mer isomers, because of the convergent group of weakly δ+ methylene protons in the former cases. Absolute values of binding constants are low but the clear difference between the behaviour of the fac and mer isomers confirms our earlier supposition7 that this specific recognition element – hydrogen bonding to methylene protons at a fac tris-chelate binding site – is involved in guest binding in coordination cages which incorporate metal complex vertices of this type.
Thirdly – and unexpectedly – in these simple mononuclear complexes the ability of the hydrogen-bond donor site of fac-[Ru(LBz)3]2+ or fac-[Ru(LMe)3]2+ to interact with the guest isoquinoline-N-oxide, which binds more strongly in the cavity of the cubic cage host in MeCN, is reduced by competition from anions – even those traditionally regarded as ‘weakly interacting’ such as hexafluorophosphate and tetraphenylborate. The effectiveness of the cubic cages as hosts for hydrogen-bond accepting guests therefore seems to rely not just on the presence of the two fac tris-chelate metal centres and their convergent group of CH protons, but also on the exclusion of anions from the cage cavity. All crystal structures of this family of cubic cages show that anions are located outside the cavity: although this does not prove that they cannot enter the cavity in solution, the difference in guest binding between the mononuclear model complexes reported in this paper, and the same H-bonding site inside a cage cavity, does imply that in the latter case competition from anions is prevented. Thus we have two important design principles for future generations of host cages.
Experimental
General details
3-(2-Pyridyl)pyrazole (LH) was prepared by the literature method.19 Metal salts and all organic reagents were purchased from Alfa or Sigma-Aldrich and used as received. NMR spectra were recorded on Bruker DRX 500 MHz, Bruker AV-III 400 MHz or AV-I 250 MHz instruments. Electrospray mass spectra were recorded on a Micromass LCT instrument. Calculation of 1:1 K values for guest binding from NMR titration data was performed using a program that has been described previously.20
Synthesis of LBz
A mixture of benzyl bromide (1.78 g, 10.4 mmol), 3-(2-pyridyl)pyrazole (1.51 g, 10.4 mmol), THF (25 cm3) and concentrated aqueous NaOH (20 M, 5 cm3) was stirred at 25 °C for 4 days. The organic layer was separated, dried over MgSO4 and evaporated to dryness. The clear oil was washed with Et2O resulting in precipitation of LBz as a white solid. Yield: 1.55 g, 63%. 1H NMR (250 MHz, CDCl3): δ 8.65 (1H, ddd; pyridyl H6), 7.96 (1H, d; pyridyl H3), 7.72 (1H, td; pyridyl H4), 7.42 (1H, d; pyrazolyl H5), 7.39–7.24 (5H, m; ArH), 7.20 (1H, ddd; pyridyl H5), 6.92 (1H, d; pyrazolyl H4), 5.41 (2H, s; CH2). ESMS: m/z 236.1 [M + H]+. Found: C, 76.3; H, 5.4; N, 17.6%. Required for C15H13N3: C, 76.6; H, 5.6; N, 17.9%.Synthesis of [Co(LBz)3](BF4)2
A mixture of Co(BF4)2·6H2O (0.037 g, 0.11 mmol) in MeOH (5 cm3) was added to a stirred solution of LBz (0.077 g, 0.33 mmol) in CH2Cl2 (5 cm3) and stirring was continued at temperature overnight. The solvent was removed under reduced pressure resulting in a pink residue. X-Ray quality crystals of the mer isomer were grown as orange blocks by dissolving the solid in the minimum amount of CHCl3 followed by slow evaporation of the solvent. Yield: 0.11 g, 0.10 mmol, 93%. ESMS: m/z 382 {[Co(LBz)3]}2+; 264 {[Co(LBz)2]}2+. Found: C, 57.7; H, 4.4; N, 13.2%. Required for [Co(LBz)3](BF4)2: C, 57.6; H. 4.2; N, 13.4%. For 1H NMR data, see main text.Synthesis of [Ru(LH)3](PF6)2
A mixture of RuCl3 (1.00 g, 4.82 mmol), 3-(2-pyridyl)-pyrazole (2.24 g, 15.43 mmol, 3.2 equiv.) and ethylene glycol (40 cm3) was heated to reflux with stirring for 48 h. After cooling the red mixture, excess ligand was removed by washing with chloroform. Saturated aqueous KPF6 was added to precipitate the complex which was extracted from the suspension with several portions of CH2Cl2. The combined organic extracts were dried over Na2SO4, and the solvent removed in vacuo to yield a golden yellow solid (3.71 g, 93%). ESMS: m/z 682 (M − PF6)+, 268 (M − 2PF6)2+. The 1H NMR spectrum revealed a statistical (3:1) mixture of mer and fac isomers (see main text). Slow diffusion of diisopropyl ether or toluene vapour into a solution of the complex in acetonitrile over a few weeks afforded a mixture of yellow crystals of mer-Ru(LH)3](PF6)2 and red crystals of fac,fac-[Ru(LH)3Ru(L−)3](PF6).
Separation of [Ru(LH)3](PF6)2 into fac and mer isomers
:1) and evaporation to dryness. The residue was then dissolved in dichloromethane; aqueous KPF6 was added and the mixture shaken vigorously. The organic layer was separated, dried over MgSO4, and the solvent removed in vacuo. Chromatography on silica eluting with MeCN–water–saturated KNO3(aq) (100:10:1) on a silica column resulted in a yellow band which was collected. After removing acetonitrile by rotary evaporation, excess saturated aqueous KPF6 was added and the product was extracted into CH2Cl2. The organic layer was separated, dried over MgSO4, and the solvent removed in vacuo to yield mer-[Ru(LH)3](PF6)2 as a yellow solid (yield: 0.51 g, 59%). 1H NMR (400 MHz, (CD3)2CO): δ 8.47–8.32 (3H, m; 3 × pyridyl H3), 8.18–8.01 (6H, m; 3 × pyridyl H4 and 3 × pyrazolyl H5), 7.92–7.79 (3H, m; 3 × pyridyl H6), 7.53–7.31 (6H, m; 3 × pyrazolyl H4 and 3 × pyridyl H5). ESMS: m/z 682 (M − PF6)+, 268 (M − 2PF6)2+.
:1) and evaporation to dryness. The residue was then dissolved in dichloromethane; aqueous KPF6 was added and the mixture shaken vigorously. The organic layer was separated, dried over MgSO4, and the solvent removed in vacuo to leave pure fac-[Ru(LH)3](PF6)2 as a yellow solid. Yield: 0.14 g, 99%. 1H NMR (400 MHz, (CD3)2CO): δ 8.41 (1H, ddd; pyridyl H3), 8.12 (1H, td; pyridyl H4), 8.07 (1H, d; pyrazolyl H5), 7.83 (1H, ddd; pyridyl H6), 7.44 (1H, d; pyrazolyl H4), 7.42 (1H, ddd; pyridyl H5). ESMS: m/z 682 (M − PF6)+, 268 (M − 2PF6)2+.
Synthesis of fac-[Ru(LBz)3](PF6)2
A mixture of fac-[Ru(LH)3](PF6)2 (0.06 g, 0.07 mmol), benzyl bromide (0.1 cm3, 0.82 mmol), Cs2CO3 (0.33 g, 1.00 mmol), Bu4NI (0.12 g, 0.33 mmol) and CH2Cl2 (25 cm3) was heated to reflux in the dark with stirring for 24 h. After cooling to room temperature, excess Cs2CO3 was filtered off and the solvent removed by rotary evaporation, before purification of the yellow solid by column chromatography on silica. Elution with MeCN–water–saturated aqueous KNO3 (100:10:2) resulted in a single yellow band moving down the column which was collected. After removing acetonitrile by rotary evaporation, excess saturated aqueous KPF6 was added and the product was extracted from the suspension into dichloromethane. The organic layer was separated, dried over MgSO4, and the solvent removed in vacuo to yield fac-[Ru(LBz)3](PF6)2 as a yellow solid. Slow diffusion of di-isopropyl ether vapour into a solution of the complex in acetone afforded the product as yellow needles. Yield: 0.01 g, 85%. 1H NMR (400 MHz, (CD3)2CO): δ 8.10 (1H, d; pyrazolyl), 8.05 (1H, td; pyridyl H4), 7.95 (1H, d; pyridyl H3), 7.52 (1H, ddd; pyridyl H6), 7.41 (1H, ddd; pyridyl H5), 7.21 (1H, t; phenyl H4), 7.14 (1H, d; pyrazolyl), 7.05 (2H, t; phenyl H3/H5), 7.22 (2H, d; phenyl H2/H6), 5.73 (1H, d; CH2), 5.06 (1H, d; CH2). ESMS: m/z 952.2 (M − PF6)+, 403.6 (M − 2PF6)2+. Found: C, 48.1; H, 3.6; N, 11.0. C45H39F12N9P2Ru·2H2O requires C, 47.7; H, 3.8; N, 11.1%. UV/Vis in MeCN [λmax/nm (10−3ε/M−1 cm−1)]: 399 (15.4), 284 (52.0), 241 (41.6).
Synthesis of mer-[Ru(LBz)3](PF6)2
This was prepared according to the method above for fac-[Ru(LBz)3](PF6)2, but using mer-[Ru(LBz)3](PF6)2 (0.23 g, 0.27 mmol), benzyl bromide (0.5 cm3, 4.21 mmol), Cs2CO3 (0.49 g, 1.52 mmol) and acetonitrile (50 cm3). Yield: 81%. Slow diffusion of di-isopropyl ether vapour into a solution of the complex in acetone after two weeks affords the product as yellow blocks. 1H-NMR (400 MHz, (CD3)2CO): δ 8.42 (1H, ddd), 8.22 (1H, d), 8.11 (1H, td), 8.07 (1H, d), 8.02–7.97 (3H, m), 7.94 (1H, dt), 7.91 (1H, td), 7.75 (1H, d) 7.66 (1H, d), 7.63 (1H, d), 7.55 (1H, d), 7.49–7.42 (2H, m), 7.34 (1H, ddd), 7.30–7.12 (8H, m), 7.05 (2H, t), 6.96 (1H, d), 6.53 (2H, d), 6.39 (2H, d), 6.21 (2H, d), 5.44 (2H, d), 5.03 (1H, d), 4.91 (1H, d), 4.71 (2H, m). ESMS: m/z 952.2 (M − PF6)+, 403.6 (M − 2PF6)2+. Found: C, 48.8; H, 3.8; N, 10.7. C45H39F12N9P2Ru·H2O·(acetone)0.5 requires C, 48.8; H, 3.9; N, 11.0%. UV/Vis in MeCN [λmax/nm (10−3ε/M−1 cm−1)]: 396 (13.9), 282 (49.8), 241 (38.5).Synthesis of fac-[Ru(LMe)3](PF6)2
A mixture of fac-[Ru(LH)3](PF6)2 (0.06 g, 0.07 mmol), methyl iodide (0.2 cm3, 3.9 mmol), Cs2CO3 (0.16 g, 0.49 mmol) and CH2Cl2 (30 cm3) was heated to reflux in the dark with stirring for 14 h. Subsequent workup and purification was as described above for fac-[Ru(LBz)3](PF6)2. Yield: 0.05 g, 73%. Slow diffusion of di-isopropyl ether vapour into a solution of the complex in acetone afforded the product as yellow blocks. 1H NMR (400 MHz, (CD3)2CO): δ 8.43 (3H, ddd; pyridyl H3), 8.15 (1H, td; pyridyl H4), 8.10 (1H, d; pyrazolyl H5), 7.74 (1H, ddd; pyridyl H6), 7.48 (1H, d; pyrazolyl H4), 7.45 (1H, ddd; pyridyl H5), 3.52 (9H, s; CH3). ESMS: m/z 724 (M − PF6)+, 290 (M − 2PF6)2+. Found: C, 37.4; H, 3.1; N, 14.3. C27H27F12N9P2Ru requires C, 37.3; H, 3.1; N, 14.5%. UV/Vis in MeCN [λmax/nm (10−3ε/M−1 cm−1)]: 400 (15.5), 281 (49.2), 238 (40.4).Synthesis of mer-[Ru(LMe)3](PF6)2
This was prepared using mer-[Ru(LH)3](PF6)2 (0.06 g, 0.07 mmol), methyl iodide (0.2 cm3, 3.9 mmol), Cs2CO3 (0.16 g, 0.49 mmol) and CH2Cl2 (30 cm3) exactly as described above for the fac isomer. Yield: 0.05 g, 73%. Slow diffusion of di-isopropyl ether vapour into a solution of the complex in acetone afforded the product as yellow blocks. 1H NMR (400 MHz, (CD3)2CO): δ 8.47–8.35 (3H, m), 8.16–8.06 (6H, m), 8.04 (1H, ddd), 7.98 (1H, ddd), 7.79 (1H, ddd), 7.53 (1H, d), 7.51–7.40 (5H, m), 3.47 (3H, s; CH3), 3.41 (3H, s; CH3), 3.35 (3H, s; CH3). ESMS: m/z 724 (M − PF6)+, 290 (M − 2PF6)2+. Found: C, 37.4; H, 3.1; N, 14.3. C27H27F12N9P2Ru requires C, 37.3; H, 3.1; N, 14.5%. UV/Vis in MeCN [λmax/nm (10−3ε/M−1 cm−1)]: 397 (15.8), 281 (54.2), 239 (41.8).X-ray crystallography
Crystals were removed from the mother liquor, coated with oil, and transferred rapidly to a stream of cold N2 on the diffractometer (Bruker APEX-2) to prevent any decomposition due to solvent loss. In all cases, after integration of the raw data, and before merging, an empirical absorption correction was applied (SADABS)21 based on comparison of multiple symmetry-equivalent measurements. The structures were solved by direct methods and refined by full-matrix least squares on weighted F2 values for all reflections using the SHELX suite of programs.22 Pertinent crystallographic data are collected in Table 1, and coordination-sphere bond distances and angles are in Tables 2–8.| Complex | mer-[Co(Lbz)3](BF4)2·CHCl3·H2O | mer-[Ru(LH)3](PF6)2·2MeCN | fac,fac-[Ru(LH)3Ru(L−)3] (PF6)·3C7H8 |
|---|---|---|---|
| Formula | C46H42B2Cl3CoF8N9O | C28H27F12N11P2Ru | C69H63F6N18PRu2 |
| Molecular weight | 1075.79 | 908.62 | 745.74 |
| T (K) | 100(2) | 100(2) | 100(2) |
| Crystal system | Monoclinic | Monoclinic | Trigonal |
| Space group | P2(1)/c | P2(1)/c |
P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) |
| a (Å) | 12.2347(3) | 14.3036(9) | 14.1604(10) |
| b (Å) | 31.6797(9) | 13.2235(8) | 14.1604(10) |
| c (Å) | 13.5802(4) | 23.5081(16) | 19.0761(14) |
| α (°) | 90 | 90 | 90 |
| β (°) | 115.6540(10) | 121.776(4) | 90 |
| γ (°) | 90 | 90 | 120 |
| V (Å3) | 4744.7(2) | 3779.9(4) | 3312.6(4) |
| Z | 4 | 4 | 4 |
| ρ (g cm−3) | 1.503 | 1.597 | 1.495 |
| Crystal size (mm3) | 0.25 × 0.30 × 0.40 | 0.25 × 0.30 × 0.40 | 0.02 × 0.02 × 0.03 |
| μ (mm−1) | 0.610 | 0.596 | 0.555 |
| Data, restraints, parameters | 10912, 0, 619 | 5413, 327, 491 | 5090, 0, 292 |
| Final R1, wR2a | 0.0628, 0.1655 | 0.0858, 0.2955 | 0.0506, 0.1594 |
| fac-[Ru(Lbz)3](PF6)2·2Me2CO | mer-[Ru(Lbz)3](PF6)2·Me2CO | fac-[Ru(LMe)3](PF6)2 | mer-[Ru(LMe)3](PF6)2 |
|---|---|---|---|
| a The value of R1 is based on ‘observed’ data with I > 2σ(I); the value of wR2 is based on all data. | |||
| C51H51F12N9O2P2Ru2 | C48H45F12N9OP2Ru | C27H27F12N9P2Ru2 | C27H27F12N9P2Ru2 |
| 1213.02 | 1154.94 | 868.59 | 868.59 |
| 100(2) | 100(2) | 100(2) | 100(2) |
| Triclinic | Triclinic | Trigonal | Monoclinic |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P3c1 | P21/c |
| 11.1447(2) | 11.7741(3) | 10.3032(3) | 13.8161(5) |
| 12.2542(3) | 12.1017(3) | 10.3032(3) | 13.2773(5) |
| 20.6452(4) | 17.9981(4) | 17.0022(5) | 20.7149(7) |
| 93.7290(10) | 99.8650(10) | 90 | 90 |
| 95.9190(10) | 96.3270(10) | 90 | 117.692(2) |
| 111.0040(10) | 107.5040(10) | 120 | 90 |
| 2602.28(9) | 2373.61(10) | 1563.08(8) | 3364.7(2) |
| 2 | 2 | 2 | 4 |
| 1.548 | 1.616 | 1.845 | 1.715 |
| 0.20 × 0.20 × 0.50 | 0.30 × 0.18 × 0.12 | 0.18 × 0.19 × 0.25 | 0.05 × 0.31 × 0.35 |
| 0.456 | 0.494 | 0.714 | 0.664 |
| 11756, 0, 698 | 10838, 0, 660 | 2421, 1, 156 | 7735, 391, 503 |
| 0.0465, 0.1346 | 0.0260, 0.0680 | 0.0464, 0.1263 | 0.0634, 0.1538 |
| Co(1)–N(11C) | 2.136(3) | Co(1)–N(22A) | 2.161(3) |
| Co(1)–N(11A) | 2.147(3) | Co(1)–N(11B) | 2.173(3) |
| Co(1)–N(22C) | 2.154(3) | Co(1)–N(22B) | 2.195(3) |
| N(11C)–Co(1)–N(11A) | 178.17(11) | N(11A)–Co(1)–N(11B) | 86.66(10) |
| N(11C)–Co(1)–N(22C) | 77.01(11) | N(22C)–Co(1)–N(11B) | 170.58(11) |
| N(11A)–Co(1)–N(22C) | 101.16(11) | N(22A)–Co(1)–N(11B) | 88.90(11) |
| N(11C)–Co(1)–N(22A) | 102.90(11) | N(11C)–Co(1)–N(22B) | 90.03(10) |
| N(11A)–Co(1)–N(22A) | 77.16(11) | N(11A)–Co(1)–N(22B) | 90.36(10) |
| N(22C)–Co(1)–N(22A) | 97.90(11) | N(22C)–Co(1)–N(22B) | 98.20(11) |
| N(11C)–Co(1)–N(11B) | 95.17(10) | N(22A)–Co(1)–N(22B) | 161.26(11) |
| N(11B)–Co(1)–N(22B) | 76.32(10) | ||
| Ru(1)–N(51A) | 2.042(10) | Ru(1)–N(42A) | 2.060(9) |
| Ru(1)–N(62A) | 2.048(11) | Ru(1)–N(22A) | 2.062(10) |
| Ru(1)–N(11A) | 2.059(10) | Ru(1)–N(31A) | 2.085(9) |
| N(51A)–Ru(1)–N(62A) | 77.0(5) | N(11A)–Ru(1)–N(22A) | 77.3(4) |
| N(51A)–Ru(1)–N(11A) | 88.2(4) | N(42A)–Ru(1)–N(22A) | 90.3(4) |
| N(62A)–Ru(1)–N(11A) | 94.8(4) | N(51A)–Ru(1)–N(31A) | 98.1(4) |
| N(51A)–Ru(1)–N(42A) | 172.6(4) | N(62A)–Ru(1)–N(31A) | 92.3(4) |
| N(62A)–Ru(1)–N(42A) | 97.8(4) | N(11A)–Ru(1)–N(31A) | 171.5(3) |
| N(11A)–Ru(1)–N(42A) | 97.6(3) | N(42A)–Ru(1)–N(31A) | 76.6(3) |
| N(51A)–Ru(1)–N(22A) | 95.5(4) | N(22A)–Ru(1)–N(31A) | 96.3(4) |
| N(62A)–Ru(1)–N(22A) | 169.4(4) | ||
| Ru(1)–N(22A) | 2.065(4) |
| Ru(1)–N(11A) | 2.077(4) |
| N(22A)–Ru(1)–N(22A)#1 | 98.03(13) |
| N(22A)–Ru(1)–N(11A)#1 | 90.84(14) |
| N(22A)–Ru(1)–N(11A) | 77.90(14) |
| N(22A)#1–Ru(1)–N(11A) | 170.72(14) |
| N(11A)#1–Ru(1)–N(11A) | 93.73(14) |
| Ru(1)–N(11A) | 2.068(2) | Ru(1)–N(22B) | 2.081(2) |
| Ru(1)–N(11B) | 2.069(2) | Ru(1)–N(22A) | 2.084(2) |
| Ru(1)–N(11C) | 2.081(2) | Ru(1)–N(22C) | 2.094(2) |
| N(11A)–Ru(1)–N(11B) | 95.77(9) | N(11C)–Ru(1)–N(22A) | 172.58(9) |
| N(11A)–Ru(1)–N(11C) | 95.08(9) | N(22B)–Ru(1)–N(22A) | 101.71(9) |
| N(11B)–Ru(1)–N(11C) | 92.35(9) | N(11A)–Ru(1)–N(22C) | 87.27(9) |
| N(11A)–Ru(1)–N(22B) | 173.28(9) | N(11B)–Ru(1)–N(22C) | 169.73(9) |
| N(11B)–Ru(1)–N(22B) | 77.52(9) | N(11C)–Ru(1)–N(22C) | 77.58(9) |
| N(11C)–Ru(1)–N(22B) | 85.70(9) | N(22B)–Ru(1)–N(22C) | 99.41(9) |
| N(11A)–Ru(1)–N(22A) | 77.60(9) | N(22A)–Ru(1)–N(22C) | 100.63(9) |
| N(11B)–Ru(1)–N(22A) | 89.62(9) | ||
| Ru(1)–N(22A) | 2.0611(12) | Ru(1)–N(22B) | 2.0814(12) |
| Ru(1)–N(11A) | 2.0711(12) | Ru(1)–N(11B) | 2.0820(12) |
| Ru(1)–N(22C) | 2.0741(12) | Ru(1)–N(11C) | 2.0885(12) |
| N(22A)–Ru(1)–N(11A) | 78.02(5) | N(22C)–Ru(1)–N(11B) | 101.43(5) |
| N(22A)–Ru(1)–N(22C) | 167.24(5) | N(22B)–Ru(1)–N(11B) | 77.26(5) |
| N(11A)–Ru(1)–N(22C) | 92.28(5) | N(22A)–Ru(1)–N(11C) | 93.61(5) |
| N(22A)–Ru(1)–N(22B) | 102.08(5) | N(11A)–Ru(1)–N(11C) | 86.55(5) |
| N(11A)–Ru(1)–N(22B) | 171.89(5) | N(22C)–Ru(1)–N(11C) | 77.37(5) |
| N(22C)–Ru(1)–N(22B) | 88.70(5) | N(22B)–Ru(1)–N(11C) | 101.51(5) |
| N(22A)–Ru(1)–N(11B) | 87.76(5) | N(11B)–Ru(1)–N(11C) | 178.34(4) |
| N(11A)–Ru(1)–N(11B) | 94.67(5) | ||
| Ru(1)–N(21) | 2.071(4) |
| Ru(1)–N(11) | 2.085(4) |
| N(21)#1–Ru(1)–N(21) | 100.54(13) |
| N(21)#1–Ru(1)–N(11) | 88.51(13) |
| N(21)–Ru(1)–N(11) | 170.94(14) |
| N(21)–Ru(1)–N(11)#1 | 77.97(15) |
| N(11)–Ru(1)–N(11)#1 | 93.03(16) |
| Ru(1)–N(11C) | 2.050(3) | Ru(1)–N(22C) | 2.056(3) |
| Ru(1)–N(22A) | 2.052(4) | Ru(1)–N(11A) | 2.069(4) |
| Ru(1)–N(22B) | 2.056(4) | Ru(1)–N(11B) | 2.084(4) |
| N(11C)–Ru(1)–N(22A) | 88.05(14) | N(22B)–Ru(1)–N(11A) | 95.21(15) |
| N(11C)–Ru(1)–N(22B) | 99.81(15) | N(22C)–Ru(1)–N(11A) | 171.66(15) |
| N(22A)–Ru(1)–N(22B) | 170.04(15) | N(11C)–Ru(1)–N(11B) | 177.13(15) |
| N(11C)–Ru(1)–N(22C) | 77.32(14) | N(22A)–Ru(1)–N(11B) | 94.60(15) |
| N(22A)–Ru(1)–N(22C) | 100.54(15) | N(22B)–Ru(1)–N(11B) | 77.67(15) |
| N(22B)–Ru(1)–N(22C) | 87.25(15) | N(22C)–Ru(1)–N(11B) | 101.08(15) |
| N(11C)–Ru(1)–N(11A) | 94.40(15) | N(11A)–Ru(1)–N(11B) | 87.23(16) |
| N(22A)–Ru(1)–N(11A) | 77.96(16) | ||
Most of the structural determination were straightforward; the only problems were that mer-[Ru(LH)3](PF6)2·2MeCN and mer-[Ru(LMe)3](PF6)2 required a large number of restraints to assist with the refinement. Crystals of mer-[Ru(LH)3](PF6)2·2MeCN diffracted weakly and only data with 2θ < 46° were used for the final refinement; global restraints (SIMU and DELU on all ligand C and N atoms) were used to keep the displacement parameters reasonable. In mer-[Ru(LMe)3](PF6)2 one of the haxafluorophosphate anions was disordered over two sites. Weak diffraction meant that global restraints (SIMU and DELU on all C, F and N atoms) were used to keep the displacement parameters reasonable, and in addition geometric restraints were applied to some of the ligand rings to prevent the geometries from becoming too eccentric.
Acknowledgements
We thank EPSRC and the University of Sheffield for financial support.References
- Reviews: (a) B. Breiner, J. K. Clegg and J. R. Nitschke, Chem. Sci., 2011, 2, 51 RSC; (b) D. Ajami and J. Rebek Jr., Acc. Chem. Res., 2013, 46, 990 CrossRef CAS PubMed; (c) L. Adriaenssens and P. Ballester, Chem. Soc. Rev., 2013, 42, 3261 RSC; (d) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418 CrossRef CAS PubMed.
- Representative recent examples: (a) M. M. J. Smulders, S. Zarra and J. R. Nitschke, J. Am. Chem. Soc., 2013, 135, 7039 CrossRef CAS PubMed; (b) T. K. Ronson, C. Giri, N. K. Beyeh, A. Minkkinen, F. Topić, J. J. Holstein, K. Rissanen and J. R. Nitschke, Chem.–Eur. J., 2013, 19, 3374 CrossRef CAS PubMed; (c) J. S. Mugridge, A. Zahl, R. van Eldik, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2013, 135, 4299 CrossRef CAS PubMed; (d) A. Asadi, D. Ajami and J. Rebek Jr., Chem. Sci., 2013, 4, 1212 RSC; (e) Y. Fang, T. Murase, S. Sato and M. Fujita, J. Am. Chem. Soc., 2013, 135, 613 CrossRef CAS PubMed; (f) P. Jagadesan, B. Mondal, A. Parthsarathy, V. J. Rao and V. Ramamurthy, Org. Lett., 2013, 15, 1326 CrossRef CAS PubMed; (g) S. Liu, D. H. Russell, N. F. Zinnel and B. C. Gibb, J. Am. Chem. Soc., 2013, 135, 4314 CrossRef CAS PubMed.
- (a) P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697 CrossRef CAS PubMed; (b) M. Yoshizawa, T. Kusukawa, M. Fujita, S. Sakamoto and K. Yamaguchi, J. Am. Chem. Soc., 2001, 123, 10454 CrossRef CAS PubMed; (c) S. Horiuchi, T. Murase and M. Fujita, Angew. Chem., Int. Ed., 2012, 51, 12029 CrossRef CAS PubMed; (d) T. Murase, Y. Nishijima and M. Fujita, Chem.–Asian J., 2012, 7, 826 CrossRef CAS PubMed.
- (a) M. D. Pluth, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2009, 42, 1650 CrossRef CAS PubMed; (b) Z. J. Wang, K. N. Clary, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Chem., 2013, 5, 100 CrossRef CAS PubMed; (c) C. J. Brown, G. M. Miller, M. W. Johnson, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2011, 133, 11964 CrossRef CAS PubMed; (d) S. R. Shenoy, F. R. P. Crisóstomo, T. Iwasawa and J. Rebek Jr., J. Am. Chem. Soc., 2008, 130, 5658 CrossRef CAS PubMed.
- (a) J. E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Chem. Sci., 2012, 3, 778 RSC; (b) J. W. Yi, N. P. E. Barry, M. A. Furrer, O. Zava, P. J. Dyson, B. Therrien and B. H. Kim, Bioconjugate Chem., 2012, 23, 461 CrossRef CAS PubMed.
- (a) R. Custelcean, Chem. Commun., 2013, 49, 2173 RSC; (b) R. Custelcean, J. Bosano, P. V. Bonnesen, V. Kertesz and B. P. Hay, Angew. Chem., Int. Ed., 2009, 48, 4025 CrossRef CAS PubMed; (c) R. Custelcean, P. V. Bonnesen, N. C. Duncan, X. Zhang, L. A. Watson, G. Van Berkel, W. B. Parson and B. P. Hay, J. Am. Chem. Soc., 2012, 134, 8525 CrossRef CAS PubMed; (d) B. P. Hay, Chem. Soc. Rev., 2010, 39, 3700 RSC; (e) S. Yi, V. Brega, B. Captain and A. E. Kaifer, Chem. Commun., 2012, 48, 10295 RSC; (f) J.-F. Ayme, J. E. Beves, C. J. Campbell and D. A. Leigh, Chem. Soc. Rev., 2013, 42, 1700 RSC; (g) B. Dietrich, B. Dilworth, J.-M. Lehn, J.-P. Souchez, M. Cesario, J. Guilhem and C. Pascard, Helv. Chim. Acta, 1996, 79, 569 CrossRef CAS; (h) J. K. Clegg, J. Cremers, A. J. Hogben, B. Breiner, M. M. J. Smulders, J. D. Thorburn and J. R. Nitschke, Chem. Sci., 2013, 4, 68 RSC.
- (a) S. Turega, M. Whitehead, B. R. Hall, M. F. Haddow, C. A. Hunter and M. D. Ward, Chem. Commun., 2012, 48, 2752 RSC; (b) S. Turega, M. Whitehead, B. R. Hall, A. J. H. M. Meijer, C. A. Hunter and M. D. Ward, Inorg. Chem., 2013, 52, 1122 CrossRef CAS PubMed; (c) M. Whitehead, S. Turega, A. Stephenson, C. A. Hunter and M. D. Ward, Chem. Sci., 2013, 4, 2744 RSC.
- M. D. Ward, Chem. Commun., 2009, 4487 RSC.
- (a) S. G. Telfer, G. Bernardinelli and A. F. Williams, Dalton Trans., 2003, 435 RSC; (b) L. H. Uppadine, M. G. B. Drew and P. D. Beer, Chem. Commun., 2001, 291 RSC; (c) S. Goetz and P. E. Kruger, Dalton Trans., 2006, 1277 RSC; (d) L. P. Harding, J. C. Jeffery, T. Riis-Johannessen, C. R. Rice and Z. T. Zeng, Chem. Commun., 2004, 654 RSC; (e) L. P. Harding, J. C. Jeffery, T. Riis-Johannessen, C. R. Rice and Z. T. Zeng, Dalton Trans., 2004, 2396 RSC; (f) S. G. Telfer, G. Bernardinelli and A. F. Williams, Chem. Commun., 2001, 1498 RSC; (g) B. Wu, J. Yang, X. Huang, S. Li, C. Jia, X.-J. Yang, N. Tang and C. Janiak, Dalton Trans., 2011, 40, 5687 RSC; (h) N. C. A. Baker, N. McGaughey, N. C. Fletcher, A. V. Chernikov, P. N. Horton and M. B. Hursthouse, Dalton Trans., 2009, 965 RSC; (i) C. P. Sebli, S. E. Howson, D. J. Clarkson and P. Scott, Dalton Trans., 2010, 39, 4447 RSC; (j) C. R. K. Glasson, G. V. Meehan, C. A. Motti, J. K. Clegg, M. S. Davies and L. F. Lindoy, Aust. J. Chem., 2012, 65, 1371 CrossRef CAS.
- M. H. Filby, J. Muldoon, S. Dabb, N. C. Fletcher, A. E. Ashcroft and A. J. Wilson, Chem. Commun., 2011, 47, 559 RSC.
- I. S. Tidmarsh, T. B. Faust, H. Adams, L. P. Harding, L. Russo, W. Clegg and M. D. Ward, J. Am. Chem. Soc., 2008, 130, 15167 CrossRef CAS PubMed.
- (a) S. E. Howson, L. E. N. Allan, N. P. Chmel, G. J. Clarkson, R. J. Deeth, A. D. Faulkner, D. H. Simpson and P. Scott, Dalton Trans., 2011, 40, 10416 RSC; (b) S. E. Howson, L. E. N. Allan, N. P. Chmel, G. J. Clarkson, R. van Gorkum and P. Scott, Chem. Commun., 2009, 1727 RSC.
- I. S. Tidmarsh, B. F. Taylor, M. J. Hardie, L. Russo, W. Clegg and M. D. Ward, New J. Chem., 2009, 33, 366 RSC.
- R. T. Brown, N. C. Fletcher, M. Nieuwenhuyzen and T. E. Keyes, Inorg. Chim. Acta, 2005, 358, 1079 CrossRef CAS PubMed.
- (a) N. C. Fletcher, M. Nieuwenhuyzen, R. Prabarahan and A. Wilson, Chem. Commun., 2002, 1188 RSC; (b) N. C. Fletcher, R. T. Brown and A. P. Doherty, Inorg. Chem., 2006, 45, 6132 CrossRef CAS PubMed.
- M. H. W. Lam, S. T. C. Cheung, K.-M. Fung and W.-T. Wong, Inorg. Chem., 1997, 36, 4618 CrossRef CAS.
- S. G. Telfer, T. Sato and R. Kuroda, Angew. Chem., Int. Ed., 2004, 43, 581 CrossRef CAS PubMed.
- (a) E. Solari, M. Musso, E. Gallo, C. Floriani, N. Re, A. Chiesi-Villa and C. Rizzoli, Organometallics, 1995, 14, 2265 CrossRef CAS; (b) P. A. W. Dean, M. Jennings, U. Rajalingam, D. C. Craig, M. L. Scudder and I. G. Dance, CrystEngComm, 2002, 4, 46 RSC.
- (a) A. J. Amoroso, A. M. W. Cargill Thompson, J. C. Jeffery, P. L. Jones, J. A. McCleverty and M. D. Ward, J. Chem. Soc., Chem. Commun., 1994, 2751 RSC; (b) H. Brunner and T. Scheck, Chem. Ber., 1992, 125, 701 CrossRef CAS; (c) Y. Lin and S. A. Lang, J. Heterocycl. Chem., 1977, 14, 345 CrossRef CAS.
- A. P. Bisson, C. A. Hunter, J. C. Morales and K. Young, Chem.–Eur. J., 1998, 4, 845 CrossRef CAS.
- G. M. Sheldrick, SADABS: A program for absorption correction with the Siemens SMART system, University of Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnotes |
| † CCDC 959996–960002. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt52479e |
| ‡ Actually the curve-fitting software generates more precise values than these, of 0.8(±0.1) M−1 and 0.4(±0.1) M−1 for binding of isoquinoline-N-oxide to fac-[Ru(LBz)3](PF6)2 and fac-[Ru(LMe)3](PF6)2 respectively. However, as the binding is so weak, the curve fitting is based only on the early part of the binding curve, with <50% complex formation even in the presence of guest concentrations of up to 1 M. This means in practice that the errors are underestimated. Accordingly we prefer to quote a value of K ≈ 1(±1) for both cases. Any difference in K between fac-[Ru(LBz)3](PF6)2 and fac-[Ru(LMe)3](PF6)2 is not significant for the purposes of this work; the important point is that the behaviour of the fac isomers is clearly distinct from that of the mer isomers. |
| This journal is © The Royal Society of Chemistry 2014 |