 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and evaluation of new polynuclear organometallic Ru(II), Rh(III) and Ir(III) pyridyl ester complexes as in vitro antiparasitic and antitumor agents†
Prinessa
Chellan
a,
Kirkwood M.
Land
b,
Ajit
Shokar
b,
Aaron
Au
b,
Seung Hwan
An
b,
Dale
Taylor
c,
Peter J.
Smith
c,
Tina
Riedel
d,
Paul J.
Dyson
d,
Kelly
Chibale
a and
Gregory S.
Smith
*a
aDepartment of Chemistry, University of Cape Town, Private Bag, Rondebosch 7701, South Africa. E-mail: gregory.smith@uct.ac.za; Fax: +27-21-6505195; Tel: +27-21-6505279
bDepartment of Biological Sciences, University of the Pacific, Stockton, CA 95211, USA
cDivision of Pharmacology, Department of Medicine, University of Cape Town, K45, OMB, Groote Schuur Hospital, Observatory, 7925, South Africa
dInstitut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland
First published on 2nd October 2013
Abstract
New polynuclear organometallic Platinum Group Metal (PGM) complexes containing di- and tripyridyl ester ligands have been synthesised and characterised using analytical and spectroscopic techniques including 1H, 13C NMR and infrared spectroscopy. Reaction of these polypyridyl ester ligands with either [Ru(p-cymene)Cl2]2, [Rh(C5Me5)Cl2]2 or [Ir(C5Me5)Cl2]2 dimers yielded the corresponding di- or trinuclear organometallic complexes. The polyaromatic ester ligands act as monodentate donors to each metal centre and this coordination mode was confirmed upon elucidation of the molecular structures for two of the dinuclear complexes. The di- and trinuclear PGM complexes synthesized were evaluated for inhibitory effects on the human protozoal parasites Plasmodium falciparum strain NF54 (chloroquine sensitive), Trichomonas vaginalis strain G3 and the human ovarian cancer cell lines, A2780 (cisplatin-sensitive) and A2780cisR (cisplatin-resistant) cell lines. All of the complexes were observed to have moderate to high antiplasmodial activities and the compounds with the best activities were evaluated for their ability to inhibit formation of synthetic hemozoin in a cell free medium. The in vitro antitumor evaluation of these complexes revealed that the trinuclear pyridyl ester complexes demonstrated moderate activities against the two tumor cell lines and were also less toxic to model non-tumorous cells.
Introduction
The aromatic ester functionality is widely used in medicinal chemistry as aromatic esters tend to be highly lipophilic thereby enhancing the diffusion of the drug across cell membranes.1,2 There are numerous examples of aromatic esters with a variety of bioactivities described in the literature. As examples, a series of lipophilic esters prepared from 4-acetyl-2-(2-hydroxyethyl)-5,6-bis(4-chlorophenyl)-2H-pyridazin-3-one (I, Fig. 1) show in vivo efficacy as antihypertensive agents,3 a library of aromatic esters derived from cinnamic acid (II) have demonstrated anti-inflammatory effects in vitro4 and ester functionalised analogues of bezafibrate (III) have also been investigated as orally active hypolipidemic agents.5 Tyrosyl (2-(4-hydroxyphenyl)ethanol) is a known antioxidant and its ester derivatives have been assessed as antimicrobial and antileishmanial agents.6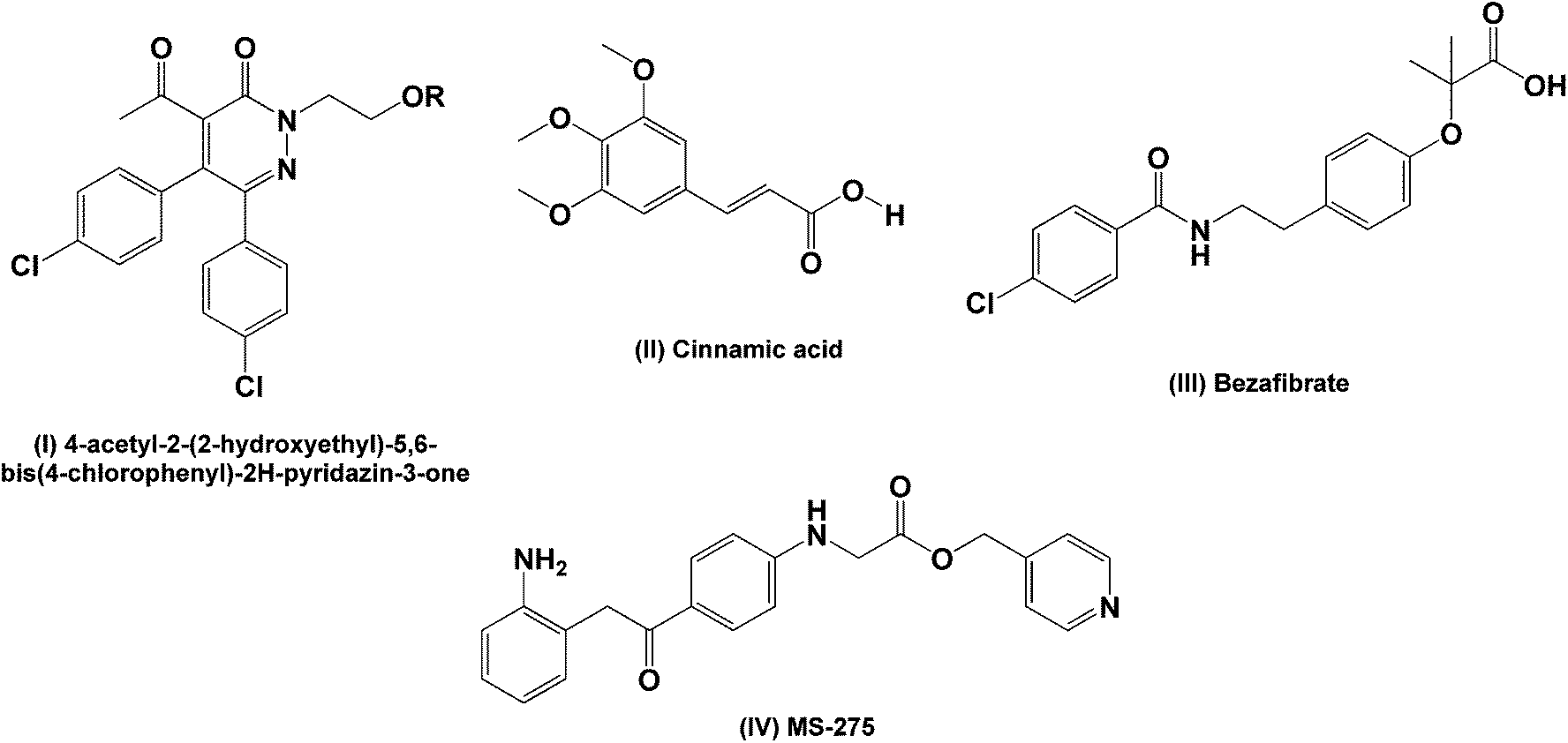 | ||
| Fig. 1 Examples of aromatic ester derivatives (I, IV) and ester precursors (II, III) studied for different bioactivities. | ||
The pyridyl ester containing compound MS-275 (IV) is currently undergoing phase II clinical trials for malignant melanomas and in combination with retinoic acid for solid tumors and lymphomas.7 Larger polyester scaffolds have also gained considerable attention for tumor targeting either as micelles or via conjugation of the potential drug molecule onto the polyester scaffold.8–12 Polyester dendrimers tend to be less toxic and exhibit better biodegradability compared to polyamine and polyamide derivatives.13 Thus, the premise of these studies is that these macromolecular scaffolds act as drug delivery vehicles, allowing preferential uptake by tumors through endocytosis. Once in the tumor cell, the ester bonds are hydrolyzed by esterases and the active drug moieties are released.
Mono- and polynuclear ruthenium–arene and pentamethylcyclopentadienyl rhodium and iridium pyridyl complexes show promising pharmacological activities, particularly as antiproliferative agents, making them viable candidates for further study.14–18 There have been several reports of the conjugation of the ruthenium arene moieties to mono- and di-aryl esters to give antitumoral agents with encouraging activities. An ethacrynic acid derivatized ruthenium–arene complex (V, Fig. 2) was found to inhibit glutathione-S-transferases with an activity that was better than the free acid while the simple p-cymene complex was completely inactive.19–21 Mono- and dipyridyl ester containing ruthenium arene complexes (VI and VII) have demonstrated similar in vitro activities against both the cisplatin sensitive (A2780) and resistant (A2780cisR) human ovarian tumor cell lines.22 The pyridyl ester containing complex, 5-fluorouracil-1-methyl isonicotinate ruthenium arene (VIII), showed a moderate increase in activity against human BEL-7402 hepatocellular carcinoma cells compared to 5-fluorouracil.14
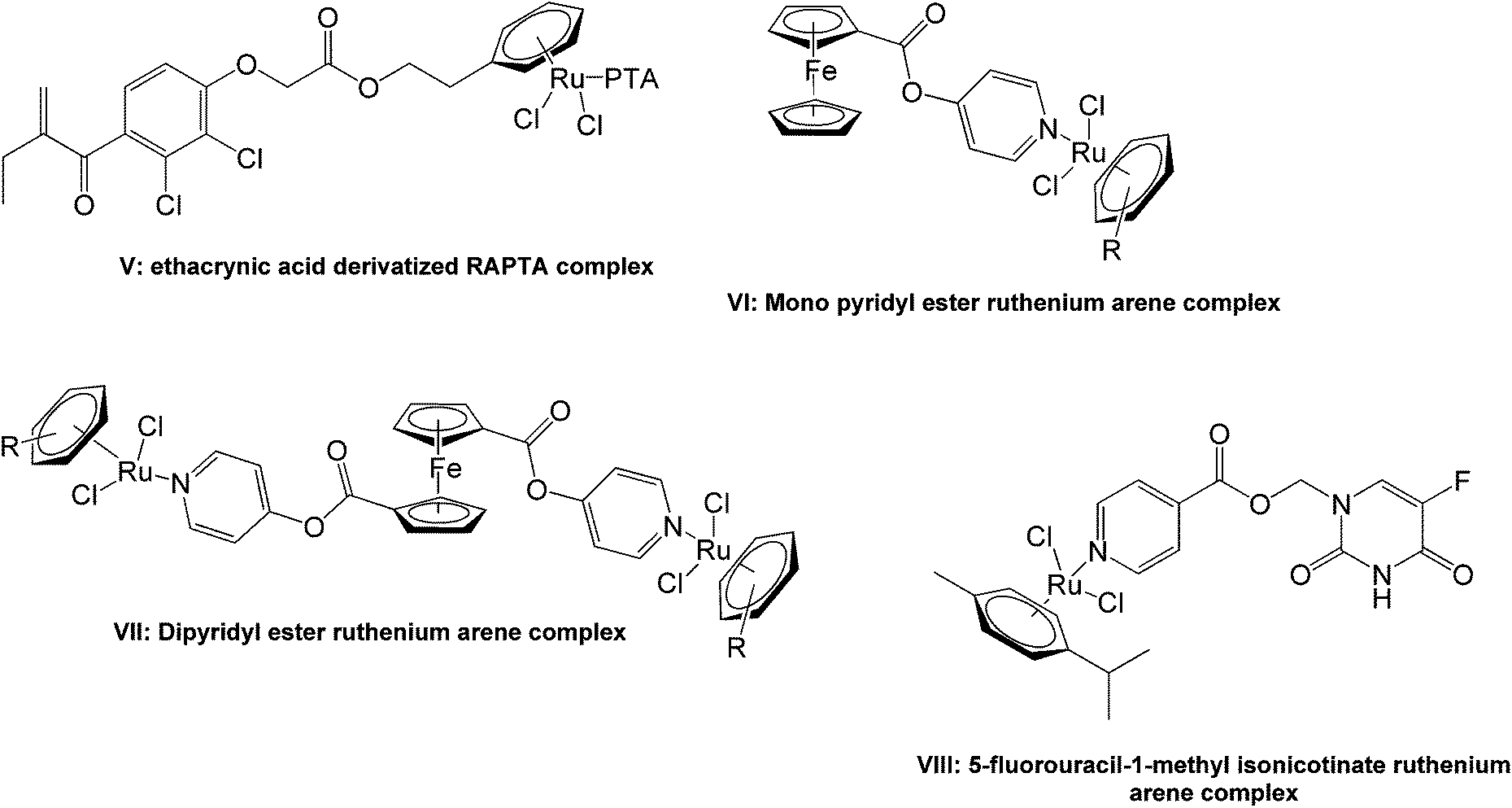 | ||
| Fig. 2 Examples of ruthenium arene ester complexes studied for activity as in vitro antitumor agents. | ||
As antiparasitics, the use of metals from the platinum group series is gaining rapid attention due to the pioneering work done by Sánchez-Delgado et al. They reported that a [Ru(II)–chloroquine]2 dimer and a mononuclear Rh(I)–chloroquine complex proved active against P. falciparum strains in vitro.23 The dinuclear ruthenium complex is 4.5 times more active than chloroquine diphosphate against two chloroquine resistant P. falciparum strains, FcB1 and FcB2. The rhodium complex exhibited activities similar to chloroquine diphosphate. Ruthenium–arene chloroquine conjugates have been extensively studied for their in vitro antiplasmodial activity against three chloroquine resistant (W2, Dd2 and K1) and four chloroquine sensitive (FcB1, 3D7, PFB and F32) P. falciparum strains.24 Iridium–chloroquine analogues have also shown activity in the nanomolar range on cultures of P. berghei.25 Since then, the application of PGM complexes as antiparasitic agents has emerged as a viable area of drug discovery.26–29
In this study, the synthesis and characterisation of a series of di- and tripyridyl aromatic ester organometallic complexes containing Ru(II), Rh(III) and Ir(III) are described. The rationale for the use of these ligands is to increase the lipophilic nature of the complexes by having multiple ester functionalities. Furthermore, these complexes couple the established in vitro pharmacological activities of half-sandwich Ru(II), Rh(III) and Ir(III) moieties with that of alkyl-pyridines. All of the compounds synthesized have been evaluated in vitro as antitumor agents against the A2780 (cisplatin-sensitive) and A2780cisR (cisplatin-resistant) human ovarian carcinoma cell lines as well as antiparasitic agents against P. falciparum strain NF54 (chloroquine sensitive) and T. vaginalis strain G3.
Results and discussion
Synthesis
The ligands, diisonicotinic acid 1,4-xylylene diester (1) and benzene-1,3,5-tricarboxylic acid tripyridin-4-ylmethyl ester (2), were reacted with the dimeric precursors, [Ru(p-cymene)Cl2]2, [Rh(C5Me5)Cl2]2 or [Ir(C5Me5)Cl2]2, to yield the corresponding di- or trinuclear complexes 3–8 (Schemes 1 and 2) in moderate to high yields. All of the complexes were prepared using the same general method. The appropriate ligand and dimer were stirred in DCM at room temperature for 16 hours and the resulting product was precipitated from the reaction solution using diethyl ether.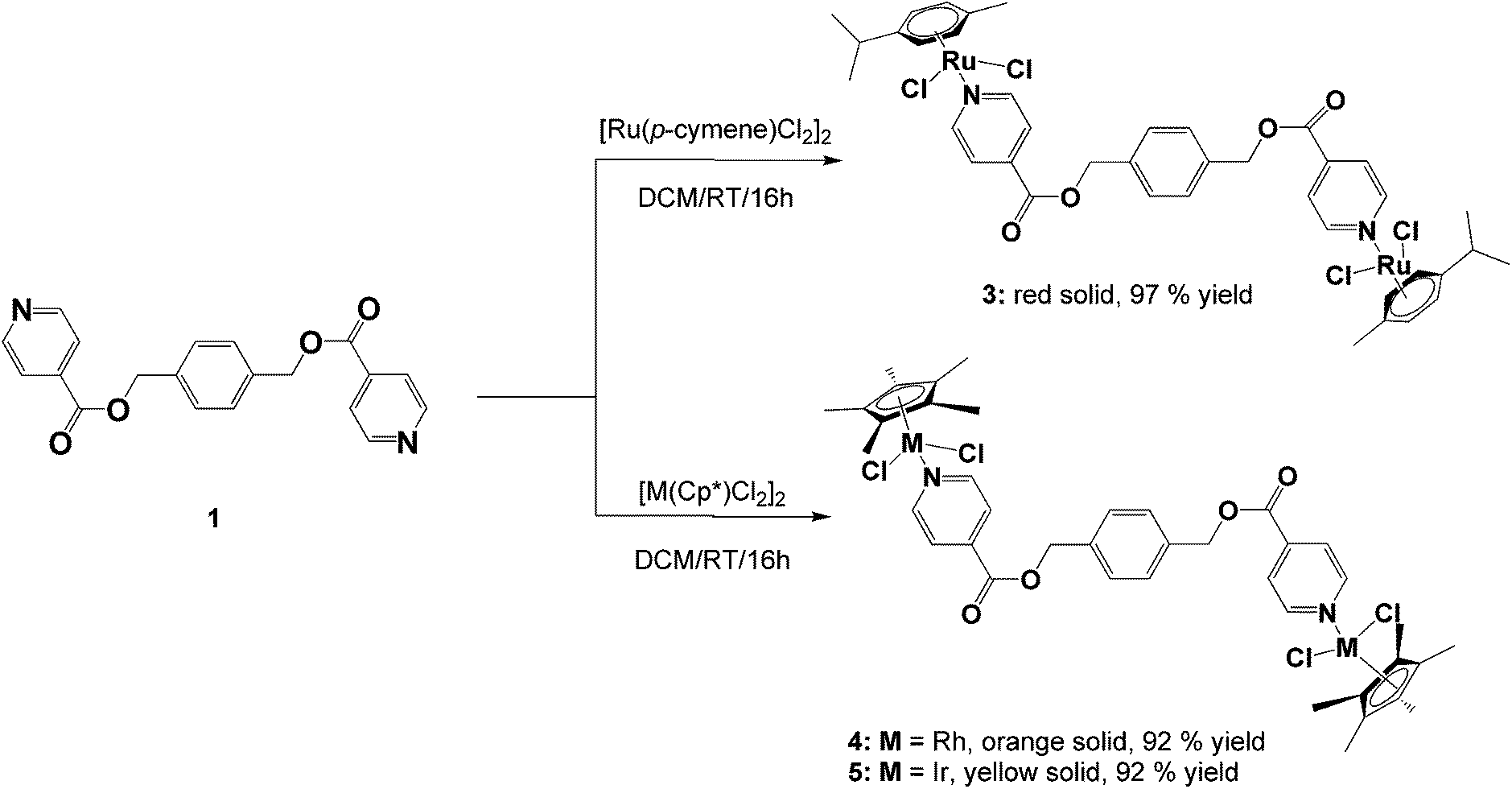 | ||
| Scheme 1 | ||
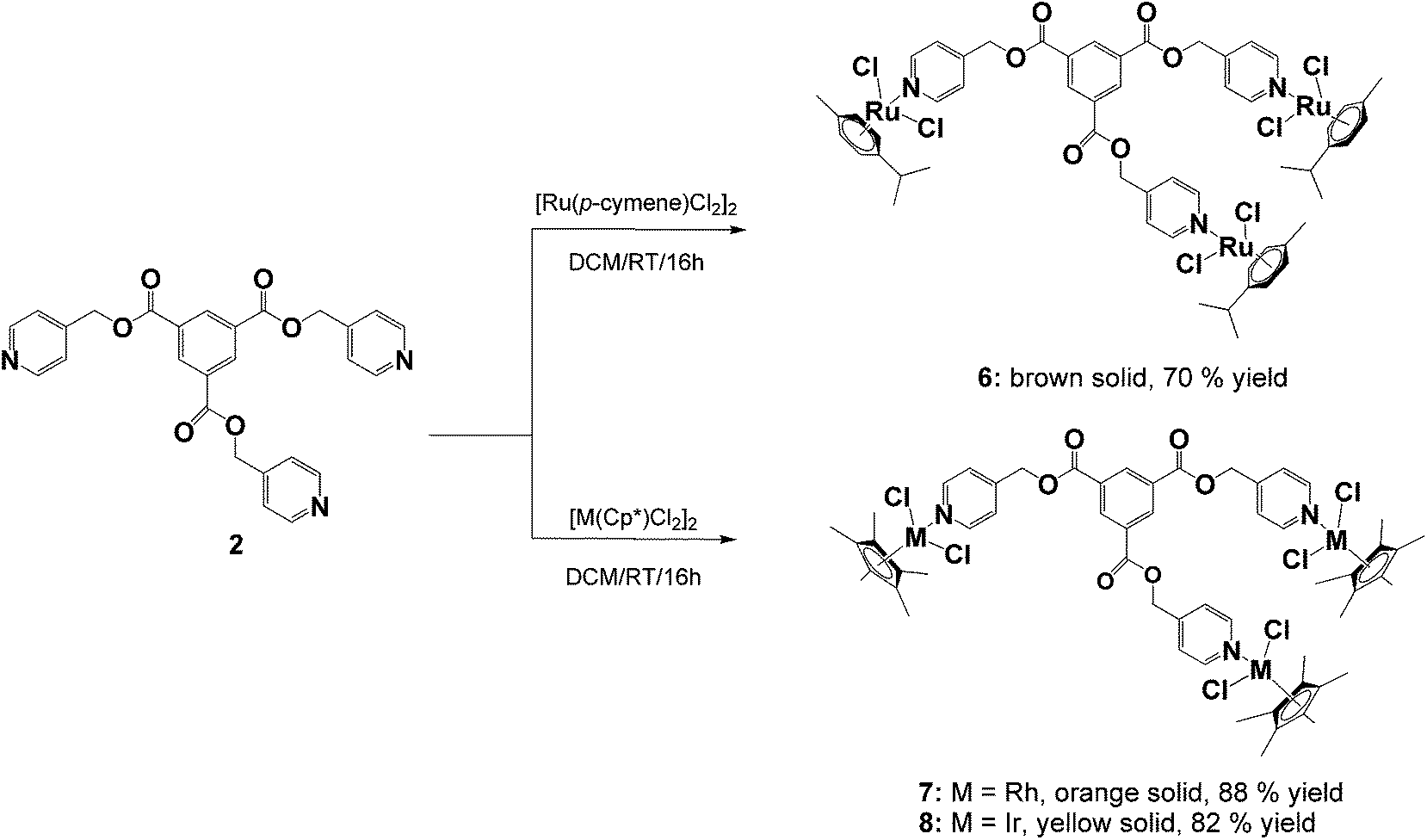 | ||
| Scheme 2 | ||
Complexes 3–8 were fully characterized using analytical and spectroscopic techniques and the structures of 4 and 5 were established in the solid state by X-ray crystallography. Spectroscopic evidence confirming metalation of ligands 1 and 2via the pyridyl nitrogens was obtained using NMR and IR spectroscopy.
The proton NMR spectra for complexes 3–8 all show a downfield shift of the protons ortho to nitrogen compared to their corresponding free ligands, which is typical for monodentate 4-pyridyl ligands coordinated to Ru(II), Rh(III) or Ir(III) metal centers.14,16,30–37 This deshielding is expected upon metal coordination to the nitrogen as it leads to less electron density in the ortho carbon–hydrogen bond due to strong back-bonding between an empty π*-orbital of nitrogen and a filled d-orbital of the transition metal.
In the dinuclear complexes (3–5) this deshielding of the doublet associated with the ortho protons of the pyridyl ring is much more pronounced compared to the trinuclear complexes 6–8. This can be attributed to the ester functionalities that are bonded in the para position of the pyridyl rings giving rise to a much higher electron withdrawing effect on the ortho protons compared to complexes 6–8 where the coordinated ligand 2 has a methylene (CH2) spacer separating the ester functionalities from the pyridyl rings. For all of the complexes (3–8), the resonances assigned to the ester methylene protons display minimal changes compared to the free ligands confirming that no metal coordination occurs to the oxygen atoms.
For the di- and tri-ruthenium complexes, 3 and 6, the p-cymene ligand displays proton resonances characteristic of similar neutral piano-stool dichlorido ruthenium complexes.16,30,33,35–37 The methyl protons of the isopropyl group are observed at ca. 1.30 ppm, resonating as a doublet and the protons of the methyl substituent resonate as a singlet at 2.10 ppm. The aromatic protons of the p-cymene ligand resonate as two doublets between 5.00 and 5.50 ppm. The methyl protons of the pentamethylcyclopentadienyl ligands of the rhodium (4 and 7) and iridium (5 and 8) complexes are seen as a singlet between 1.50 and 1.60 ppm in the proton NMR spectra for these complexes.
Analysis of complexes 3–8 using 13C{1H} NMR spectroscopy reveals a high frequency shift of the resonance assigned to the carbon ortho to nitrogen in the pyridyl ring compared to the free ligand (observed at ca. 150.0 ppm for 1 and 2) confirming metal coordination to the pyridyl nitrogens. For complexes 3–5, a shift downfield of approximately 5.0 ppm is noted compared to 1. In comparison to the free ligand 2, a shift of approximately 5.0 ppm for the ortho carbon is observed for the tri-ruthenium complex 6 and around 3.0 ppm for the rhodium and iridium analogues (7 and 8).
Similar to the proton NMR spectra for complexes 3–8, little to no effect is observed on the other carbon resonances assigned to the metal-coordinated ester ligands. The resonances assigned to the carbonyl carbon as well as the alkyl ester carbon occur at approximately the same shift compared to the free ligand. This serves as further evidence that coordination to the ester oxygen atoms does not occur.
For the rhodium (4 and 7) and iridium (5 and 8) complexes, the carbon resonance associated with the methyl substituents of the pentamethylcyclopentadienyl ligand are observed between 8.5 and 9.0 ppm. The aromatic carbons of the ring are observed at approximately 95.0 ppm for the rhodium complexes and in the iridium complexes these carbons resonate further upfield at approximately 86.0 ppm. These shifts are similar to other piano-stool Cp* rhodium and iridium complexes.38–41 The p-cymene moieties of complexes 3 and 6 show two singlets between 82.0 and 83.0 ppm due to the resonances of the unsubstituted aromatic carbons of the ring. The isopropyl substituted aromatic carbon and the methyl substituted aromatic carbon resonates at approximately 97.0 and 104.0 ppm respectively. These resonances agree with similar mononuclear 4-pyridyl complexes.14,33,37
Further analysis of the complexes 3–8 using infrared spectroscopy reveals no shift in the absorption band associated with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond vibration of the now metalated ligands, supporting the evidence gleaned from the NMR analyses that metal coordination to the ester oxygen atoms does not occur. The CN bond vibration of the pyridyl functionalities do exhibit an expected shift to higher frequency as a consequence of metal complexation to nitrogen. A high frequency increase in the range of 15–20 cm−1 is noted and is consistent with similar examples described in the literature.42–45 For all of the complexes, a strong absorption band in the region of 1200–1300 cm−1 is observed and is assigned to the C–O bond stretching of the ester functionalities.14,33,36,46
O bond vibration of the now metalated ligands, supporting the evidence gleaned from the NMR analyses that metal coordination to the ester oxygen atoms does not occur. The CN bond vibration of the pyridyl functionalities do exhibit an expected shift to higher frequency as a consequence of metal complexation to nitrogen. A high frequency increase in the range of 15–20 cm−1 is noted and is consistent with similar examples described in the literature.42–45 For all of the complexes, a strong absorption band in the region of 1200–1300 cm−1 is observed and is assigned to the C–O bond stretching of the ester functionalities.14,33,36,46
Mass spectral analysis of complexes 3–8 using ESI-MS reveals molecular ion fragments corresponding to the molecular weights of the proposed structures. Complexes 4 and 7 exhibit base peaks corresponding to the sodium adduct and complexes 3 and 8 form adducts with methanol upon either protonation or loss of chlorido ligands. Complexes 5 and 6 show m/z peaks corresponding to the loss of a chlorido ligand. Within these complexes, there are several potential sites of ionization, thus mass fragments corresponding to multiply charged species is possible.
Single-crystal X-ray diffraction
The molecular structures of complexes 4 and 5 were elucidated using single-crystal X-ray diffraction. Crystals were grown from a solution of chloroform and hexane. Both complexes crystallize with one complex molecule and two chloroform molecules in the asymmetric unit with a monoclinic system and P21 space group. Fig. 3 and 4 show the molecular structures of 4 and 5 and Tables 1 and 2 list selected bond lengths and angles.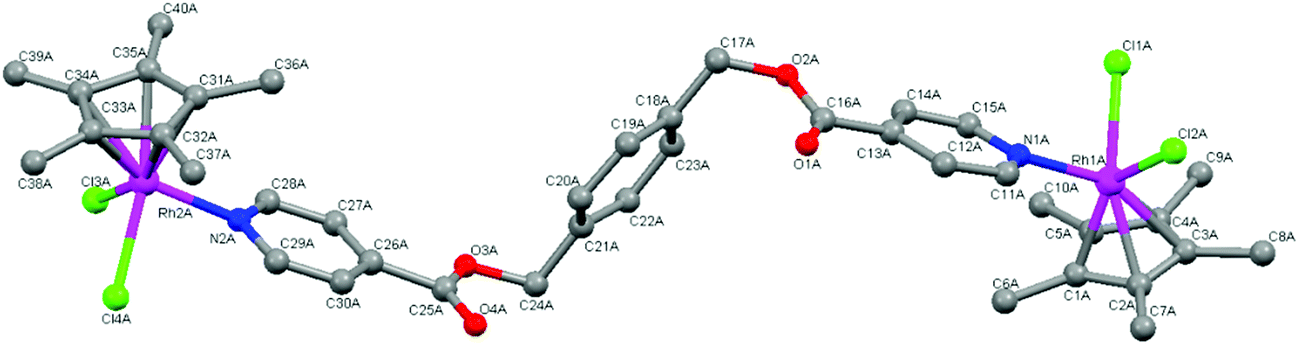 | ||
| Fig. 3 Molecular structure of complex 4 with hydrogen atoms omitted for clarity. | ||
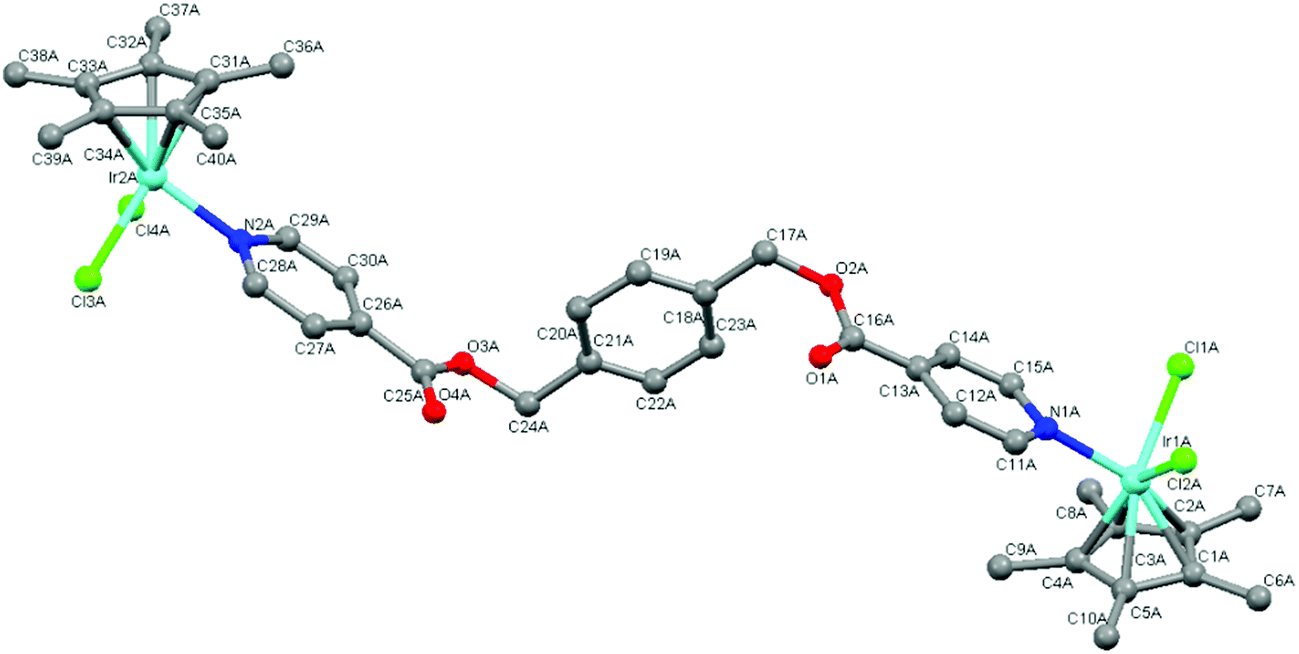 | ||
| Fig. 4 Molecular structure of complex 5 with hydrogen atoms omitted for clarity. | ||
| Complex 4·2CHCl3 M = Rh | Complex 5·2CHCl3 M = Ir | Complex 4·2CHCl3 M = Rh | Complex 5·2CHCl3 M = Ir | ||
|---|---|---|---|---|---|
| M1–N1 | 2.119(8) | 2.095(8) | M2–N2 | 2.116(8) | 2.114(8) |
| M1–C1 | 2.054(14) | 2.160(9) | M2–C31 | 2.137(9) | 2.143(10) |
| M1–C2 | 2.099(11) | 2.161(9) | M2–C32 | 2.143(8) | 2.157(10) |
| M1–C3 | 2.146(12) | 2.162(9) | M2–C33 | 2.169(9) | 2.163(11) |
| M1–C4 | 2.203(16) | 2.132(11) | M2–C34 | 2.154(9) | 2.193(12) |
| M1–C5 | 2.093(13) | 2.130(10) | M2–C35 | 2.139(8) | 2.136(11) |
| M1–Cl1 | 2.412(2) | 2.401(3) | M2–Cl3 | 2.399(2) | 2.415(3) |
| M1–Cl2 | 2.399(2) | 2.415(3) | M2–Cl4 | 2.416(2) | 2.394(3) |
| N1–C11 | 1.321(12) | 1.361(13) | N2–C28 | 1.323(12) | 1.344(13) |
| N1–C15 | 1.337(11) | 1.342(14) | N2–C29 | 1.362(12) | 1.379(13) |
| O1–C16 | 1.160(12) | 1.198(14) | O3–C25 | 1.291(12) | 1.312(13) |
| O2–C16 | 1.332(13) | 1.364(14) | O4–C25 | 1.218(12) | 1.196(14) |
| Complex 4·2CHCl3 M = Rh | Complex 5·2CHCl3 M = Ir | Complex 4·2CHCl3 M = Rh | Complex 5·2CHCl3 M = Ir | ||
|---|---|---|---|---|---|
| C4–M1–Cl1 | 92.8(4) | 136.9(4) | C34–M2–Cl3 | 94.4(3) | 93.2(3) |
| C3–M1–Cl1 | 117.0(3) | 101.8(4) | C33–M2–Cl3 | 124.8(3) | 109.6(3) |
| C1–M1–Cl1 | 138.6(4) | 121.5(3) | C31–M2–Cl3 | 135.8(3) | 152.3(4) |
| C2–M1–Cl1 | 153.0(3) | 94.7(3) | C32–M2–Cl3 | 160.4(3) | 148.2(3) |
| C5–M1–Cl1 | 100.9(4) | 158.8(3) | C35–M2–Cl3 | 100.3(3) | 112.5(4) |
| N1–M1–Cl1 | 88.4(2) | 87.2(3) | N2–M2–Cl3 | 89.3(2) | 86.0(2) |
| C4–M1–Cl2 | 123.7(3) | 135.8(4) | C34–M2–Cl4 | 123.7(3) | 137.9(4) |
| C3–M1–Cl2 | 96.1(3) | 161.0(3) | C33–M2–Cl4 | 95.6(3) | 103.0(3) |
| C1–M1–Cl2 | 131.7(4) | 96.7(3) | C31–M2–Cl4 | 133.9(3) | 120.7(4) |
| C2–M1–Cl2 | 97.0(3) | 125.1(3) | C32–M2–Cl4 | 100.4(3) | 94.2(3) |
| C5–M1–Cl2 | 156.5(4) | 101.2(3) | C35–M2–Cl4 | 160.3(3) | 159.3(4) |
| N1–M1–Cl2 | 88.8(2) | 87.0(2) | N2–M2–Cl4 | 87.2(2) | 86.7(2) |
| Cl2–M1–Cl1 | 89.44(8) | 87.31(10) | Cl3–M2–Cl4 | 90.28(9) | 86.94(10) |
| C25–O3–C24–C21 | −96.1(10) | −92.9(12) | C16–O2–C17–C18 | −78.5(11) | −84.3(12) |
The determined molecular structures of complexes 4 and 5 validate the structures of 3–8 evidenced by the spectroscopic and spectrometric characterisations discussed earlier. In the structures of both dinuclear complexes, each metal center adopts a typical ‘piano-stool’ conformation with the two chlorido ligands and the pyridyl ring as the three legs, giving rise to a pseudo-tetrahedral coordination geometry. The pentamethylcyclopentadienyl ring is bound to the metal in the expected η5 manner. The pyridyl ester ligand is coordinated to each metal center in a monodentate fashion as suggested by the spectroscopic data. Comparison of the Rh–C and Ir–C bond lengths between the metal and each bonded carbon of the pentamethylcyclopentadienyl ring reveal them to be similar indicating that the ring is symmetrically bound to rhodium (4) or iridium (5). The bond lengths observed in 4 and 5 between the metal and the coordinated atoms are approximately 2.40 Å (M–Cl), 2.10 Å (M–C) and 2.10 Å (M–N). These values agree with those observed for similar complexes in the literature16,30,34,40,41,44,45,47 as well as the expected length calculated from the covalent radii of iridium or rhodium, chlorine (M–Cl: 2.40 (Ir); 2.41 (Rh) Å), carbon (M–C: 2.09 (Ir); 2.10 (Rh) Å) and nitrogen (M–N: 2.09 (Ir); 2.10 (Rh) Å).48
In the coordinated pyridyl-ester ligand of both complexes 4 and 5, the C–N bond lengths in the pyridyl ring are found to be between 1.30–1.35 Å. The alkyl C–O bond lengths of the ester functionalities are between 1.30 and 1.36 Å and the CO bond lengths are shorter (1.16–1.20 Å), comparing favourably to the interatomic distances typical of pyridines and aromatic esters (CN: 1.34 Å, C–O: 1.43 Å and CO: 1.23 Å).49
With respect to the bond angles, the values observed between Cl–M–Cl and N–M–Cl are all close to 90° and the bond angles formed between the bonded carbons of the Cp* ligand, the metal and the chlorido ligand vary from 90 to 150°. This trend has also been observed within the molecular structures of similar Cp*Ir(III) and Cp*Rh(III) pyridyl complexes.16,30,34,44,45 Within the pyridyl ester ligands of both 4 and 5, the dihedral angles formed between the central phenyl ring and each ester functionality is close to 90° indicating that the pyridyl rings orientate themselves almost perpendicular to the phenyl spacer thereby minimizing electrostatic interactions within the complex molecules.
Turbidimetric assay
The solubility of a compound in aqueous-based media is an important property. Solubility has a bearing on a compound's bioavailability, which impacts on absorption, and it also validates the in vitro assay data obtained. There are three general categories that compounds can be classified based on their solubility range: (i) <10 μg mL−1 (sparingly soluble), (ii) 10–60 μg mL−1 (partially soluble) and (iii) >60 μg mL−1 (soluble). If a compound displays solubility greater than 60 μg mL−1, then it should have good absorption. One method used to validate aqueous solubility during early drug discovery is the Turbidimetric (kinetic) Solubility Assay.50–53 This assay was used to evaluate the solubility of compounds 1–8 and the [Ru(p-cymene)Cl2]2, [Rh(C5Me5)Cl2]2 and [Ir(C5Me5)Cl2]2 dimers in phosphate buffered saline at pH 7.4 (Table 3).| Compound | Determined turbidimetric solubility (μM) | Determined turbidimetric solubility (μg mL−1) |
|---|---|---|
| 1 | >200 | >70 |
| 2 | >200 | >97 |
| 3 | 160–200 | 154–192 |
| 4 | 160–200 | 155–193 |
| 5 | 80–120 | 91–137 |
| 6 | >200 | >280 |
| 7 | >200 | >282 |
| 8 | >200 | >336 |
| [Ru(p-cymene)Cl2]2 | >200 | >122 |
| [Rh(C5Me5)Cl2]2 | >200 | >104 |
| [Ir(C5Me5)Cl2]2 | >200 | >159 |
| Reserpine | 20–40 | 12–24 |
| Hydrocortisone | >200 | >72 |
The drugs, reserpine and hydrocortisone were used as controls. In micromolar concentration, ligands 1 and 2, the trinuclear complexes 6–8 and the metal dimers showed no turbidity up to the highest compound concentration tested (200 μM). All of the compounds (1–8) displayed solubility greater than 60 μg mL−1. Overall, the data ascertained suggests that the di- and tripyridyl ester compounds are good candidates for in vitro biological testing. Precipitation of these compounds from the various in vitro assays may be unlikely and thus the activities observed for these compounds are a true reflection of their in vitro activity since all of the compounds should remain in solution.
In vitro biological studies
Since metal-containing compounds have been shown to have relevant antiparasitic properties,26–29 the in vitro antiparasitic activity of all compounds was determined against the chloroquine-sensitive (CQS) NF54 P. falciparum strain and the T. vaginalis strain G3. The IC50 values obtained with standard error of the mean (SEM) values are shown in Table 4. The ruthenium, rhodium and iridium dimers, [Ru(p-cymene)Cl2]2, [Rh(C5Me5)Cl2]2 and [Ir(C5Me5)Cl2]2 and the organometallic chloroquine analogue, ferroquine, were also screened for activity at the same time as compounds 1–8. Chloroquine diphosphate was screened as a control for the in vitro experiments.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a data for compounds 1–8 against the chloroquine-sensitive P. falciparum strain NF54
a data for compounds 1–8 against the chloroquine-sensitive P. falciparum strain NF54
| Compound | Metal entities | No of metals | NF54 (μM) | SEMb (μM) |
|---|---|---|---|---|
| a Concentration inhibiting 50% of parasite growth. b SEM: standard error of the mean. c The dimers, FQ and CQDPP were screened for activity at the same time as 1–8. | ||||
| 1 | None | 0 | 48.42 | 0.62 |
| 2 | None | 0 | 22.18 | 3.46 |
| 3 | Ru(p-cymene)Cl2 | 2 | 7.04 | 0.72 |
| 4 | Rh(C5Me5)Cl2 | 2 | 25.16 | 2.23 |
| 5 | Ir(C5Me5)Cl2 | 2 | 42.63 | 1.01 |
| 6 | Ru(p-cymene)Cl2 | 3 | 5.87 | 0.58 |
| 7 | Rh(C5Me5)Cl2 | 3 | 10.84 | 2.55 |
| 8 | Ir(C5Me5)Cl2 | 3 | 10.27 | 2.04 |
|
[Ru(p-cymene)Cl2]2c |
2 | 16.80 | 2.94 | |
|
[Rh(C5Me5)Cl2]2c |
— | 2 | 20.90 | 0.87 |
|
[Ir(C5Me5)Cl2]2c |
— | 2 | 59.40 | 19.45 |
| Ferroquine c | — | 1 | 0.03 | 0.01 |
| Chloroquine diphosphate c | — | — | 0.02 | 0.01 |
With respect to the free ligands, ligand 2 showed better activity than 1. All of the transition metal complexes (3–8) displayed enhanced activity compared to the corresponding free ligands. The tripyridyl ester complex 8 (IC50 = 10.27 μM) is twice as active compared to 2 (IC50 = 22.18 μM) and the tri-ruthenium complex 6 (IC50 = 5.87 μM) is almost four times more active than 2. Consideration of the antiplasmodial data obtained for the dipyridyl ester complexes (3–5) showed that these complexes were also much more active than their corresponding free ligand 1 (IC50 = 48.42 μM). However, there was a great difference in the antiplasmodial activity as the metal fragment was varied. Complex 3 (IC50 = 7.04 μM) was the most active, followed by 4 (IC50 = 25.16 μM) and then 5 (IC50 = 42.63 μM) was the least active of the three complexes (3–5). For the dinuclear complexes it is clear that activity decreased with a change in metal fragment in the order of Ru(p-cymene)Cl2 > Rh(C5Me5)Cl2 > Ir(C5Me5)Cl2.
Functionalization of the free ligands (1 and 2) with either the Ru(p-cymene)Cl2 (3 and 6) or Rh(C5Me5)Cl2 (4 and 7) moieties led to a strong increase in antiplasmodial activity suggesting that these metal moieties play a role in activity. When comparing the activities of the dimers [Ru(p-cymene)Cl2]2, [Rh(C5Me5)Cl2]2 and [Ir(C5Me5)Cl2]2, to the corresponding ester (3–8) complexes, it can be seen that the dimers were less active than the di- and trinuclear complexes with the exception of complex 4. This observation, along with the fact that the complexes are more active than the free ligands, advocates that there is a cooperative effect between metal moiety and ligand on antiplasmodial activity. The ligands (1 and 2) as well as the dimers were only moderately active individually but conjugation of the different metal moieties onto the corresponding ligand yielded a beneficial increase in activity with an increase in metal moieties leading to an increase in antiplasmodial activity in vitro. While the results obtained show that complexes 1–8 are more active than their corresponding ligands, they were not as active as ferroquine (IC50 = 0.03 μM) and chloroquine diphosphate (IC50 = 0.02 μM).
From all of the compounds (1–8) screened for in vitro antiplasmodial activity against the NF54 CQS P. falciparum strain, compounds showing cytotoxicities of approximately 10 μM or less were assayed for β-hematin inhibition (Table 5). The dimers, [Ru(p-cymene)Cl2]2, [Rh(C5Me5)Cl2]2 and [Ir(C5Me5)Cl2]2, as well as the ligands 1 and 2 were also assayed for comparison.
| Compound | IC50 (μM) | 95% Confidence interval |
|---|---|---|
| a Only compounds showing in vitro cytotoxic values of approximately 10 μM or less were screened. | ||
| 1 | No inhibition | — |
| 2 | No inhibition | — |
| 3 | 29.19 | 28.09–30.34 |
| 6 | 7.76 | 7.19–8.37 |
| 7 | 24.99 | 21.47–29.90 |
| 8 | 12.48 | 11.76–13.24 |
| [Ru(p-cymene)Cl2]2 | No inhibition | — |
| [Rh(C5Me5)Cl2]2 | No inhibition | — |
| [Ir(C5Me5)Cl2]2 | No inhibition | — |
| Ferroquine | 14.51 | 13.72–15.34 |
| Chloroquine | 18.43 | 17.56–19.34 |
| Amodiaquine | 6.83 | 6.57–7.10 |
Hemozoin (malaria pigment) formation is currently a target for antiplasmodial drug discovery. In the life cycle of the Plasmodium falciparum parasite, the ingestion and degradation of hemoglobin from the infected host provides essential amino acids for parasite growth and nutrition.54 A side product of this process is the formation of free heme that is toxic to the parasite. In order to prevent the detrimental effects of free heme, the parasite initiates a detoxification mechanism and removes the threat by conversion of the free heme into a crystalline solid known as hemozoin that is nontoxic to the parasite. Clinically used drugs, amodiaquine and chloroquine are believed to inhibit the formation of hemozoin.55 The ability of a potential drug to inhibit formation of hemozoin can be measured using the NP-40 mediated β-hematin (synthetic hemozoin) inhibition assay. NP-40 is a low cost, lipophilic detergent that can mediate the formation of synthetic hemozoin (β-hematin). The NP-40 mediated assay mimics the conditions of the acidic food vacuole in the parasite to give a better measure of hemozoin formation.
The tri-ruthenium pyridyl ester complex 6 showed the highest inhibitory effect (IC50 = 7.76 μM) that was comparable to amodiaquine (IC50 = 6.83 μM). Neither ligand 1 or 2 was able to inhibit β-hematin formation, yet their corresponding complexes (3 and 6–8) showed inhibitory effects suggesting that the overall complex structure is needed to stop formation of synthetic hemozoin. The fact that the dimers did not show activity also highlights the importance of the interaction between the metal and ligand for complexes 3 and 6–8.
The tripyridyl ester rhodium complex (7) displayed moderate inhibition (IC50 = 24.99 μM) while the iridium derivative (8) was more active (IC50 = 12.48 μM). Both these complexes were better at inhibiting formation of synthetic hemozoin than ferroquine and chloroquine; drugs that target hemozoin formation within the malaria parasite.29
All of the compounds screened using the NP-40 mediated assay contain several aromatic rings making them capable of intermolecular π–π interactions. It is possible that these compounds can inhibit formation of hemozoin through π–π stacking with hematin. All of the ester complexes (3 and 6–8) screened were able to inhibit β-hematin formation in a lipidic atmosphere to some degree. It is interesting to note that while these complexes are only moderately active against the parasite, they are better inhibitors of hemozoin formation compared to known inhibitors. This property demonstrates that PGM polyester complexes do have potential as antiparasitics. Slight structural modifications could yield complexes with better in vitro activity.
All of the compounds were evaluated for inhibitory activity against the human parasite T. vaginalis strain G3 (Fig. 5). Cells were inoculated with either 50 or 25 μM doses of compounds. All of the compounds were able to influence parasite viability in a dose-dependent manner.
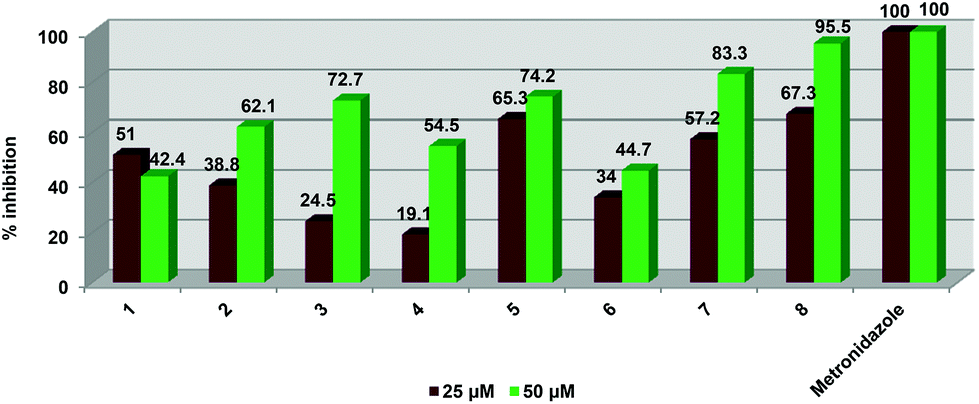 | ||
| Fig. 5 Graphical representation of % inhibition data ascertained for compounds 1–8 against T. vaginalis strain G3. | ||
At 50 μM, the tripyridyl ester ligand (2) displayed higher inhibitory activity (62.1%) than the dipyridyl ester ligand 1 (42.4%) but a decrease in compound concentration to 25 μM resulted in ligand 2 displaying a much lower activity than 1 (38.8 vs. 51.1%). At both 25 and 50 μM concentrations, the dinuclear ruthenium (3) and iridium (5) complexes exhibited better activities than the dinuclear rhodium complex 4. At 25 μM, complexes 3 and 5 were not as active (24.5 and 19.1% respectively) as their corresponding free ligand 1 (51%). The iridium derivative 5 is more active than the free ligand at both concentrations (65.3% at 25 μM and 74.2% at 50 μM). The trinuclear complexes 7 and 8 displayed better inhibitory activity compared to the free ligand 2 at both 25 and 50 μM concentrations. At 50 μM, the tri-ruthenium complex 6 showed similar inhibition (44.7%) to ligand 2 (42.4%) but is less active than 2 at 25 μM (51% vs. 34%). Complex 8 exhibited the best activity out of all the pyridyl ester complexes tested at both compound concentrations. At 25 μM, the iridium complexes (5 and 8) exhibit similar inhibitory activities of 65.3 and 67.3% respectively. Complexes 3 and 4 display the weakest activity out of all the compounds.
Overall, complex 8 displayed the best inhibitory effect out of all complexes tested. None of the compounds were as potent as metronidazole which is the current FDA approved treatment for T. vaginalis. This drug displayed 100 percent inhibition at both concentrations.
The antiproliferative activity of compounds (1–8) were evaluated on the A2780 (cisplatin-sensitive) and A2780cisR (cisplatin-resistant) human ovarian carcinoma cell lines. The toxicity of these compounds on non-tumorous cells was also assessed using the human embryonic kidney (HEK) cell line. Cells were incubated with each compound at different concentrations up to a maximum concentration of 200 μM. The IC50 determinations are shown in Table 6 and a graphical representation of the most active compounds’ is depicted in Fig. 6.
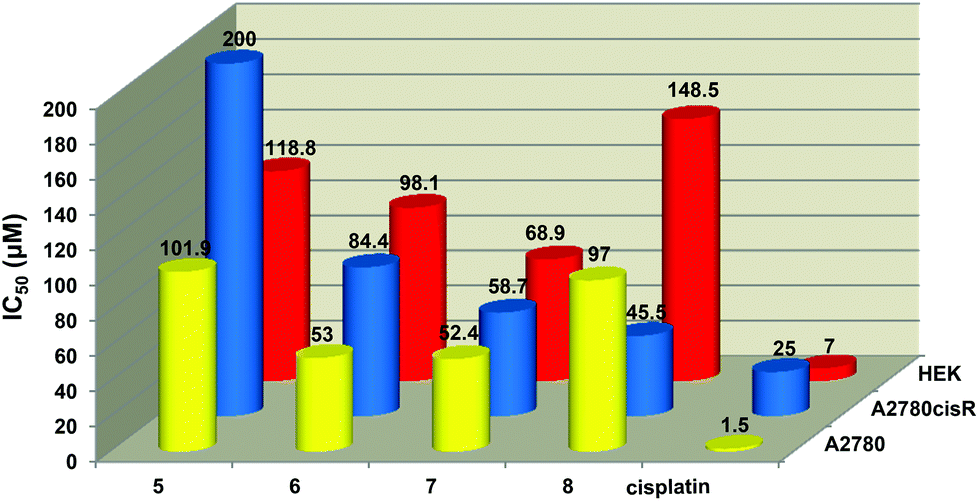 | ||
| Fig. 6 Graphical representation showing the cytotoxicity against the A2780, A2780cisR and HEK cell lines. | ||
a determinations for compounds 1–8 against cisplatin sensitive (A2780) and cisplatin resistant (A2780cisR) human ovarian carcinoma and Human Embryonic Kidney (HEK) cell linesb
| Compound | Metal moiety | No of metal moieties | A2780 (μM) | A2780 cis R (μM) | HEK (μM) |
|---|---|---|---|---|---|
| a IC50: concentration inhibiting 50% of cell growth. b Error values are given in parentheses. | |||||
| 1 | None | 0 | >200 | >200 | >200 |
| 2 | None | 0 | >200 | >200 | >200 |
| 3 | (p-Cymene)RuCl2 | 2 | >200 | >200 | >200 |
| 4 | Cp*RhCl2 | 2 | >200 | >200 | >200 |
| 5 | Cp*IrCl2 | 2 | 101.9 (8.2) | >200 | 118.8 (99.8) |
| 6 | (p-Cymene)RuCl2 | 3 | 53.0 (3.0) | 84.4 (5.2) | 98.1 (2.0) |
| 7 | Cp*RhCl2 | 3 | 52.4 (0.4) | 58.7 (4.1) | 68.9 (6.4) |
| 8 | Cp*IrCl2 | 3 | 97.0 (4.0) | 45.5 (5.7) | 148.5 (24.7) |
| Cisplatin | — | 1 | 1.5 | 25 | 7 |
The dinuclear iridium complex 5 and the trinuclear complexes (6–8) were the only compounds to show antiproliferative activity implying that the role played by the number of aryl ester groups and the type of metal moiety in the complex influences inhibitory activity. Complexes 6 and 7 showed comparable activity to each other (IC50 = 53.0 and 52.4 μM respectively) in the A2780 cell line. The di- and trinuclear iridium complexes 5 (IC50 = 101.9 μM) and 8 (IC50 = 97.0 μM) also displayed similar weak activities.
All of the active complexes (5–8) were less toxic to non-tumorous cells with the tri-iridium complex 8 showing the greatest difference in toxicities between the carcinoma cells (A2780 and A2780cisR) and non-tumorous cells (HEK). Complexes 5–8 were also less toxic than cisplatin toward the HEK cell line. Complex 8 was more active against the A2780cisR cell line (IC50 = 45.5 μM) than the A2780 cell line whereas the ruthenium complex 6 was less active on the A2780cisR cell line (IC50 = 84.4 μM) compared to the A2780 cell line (IC50 = 53.0 μM). None of the complexes are as active as cisplatin against either cancer cell line (IC50 = 1.5 μM for A2780 and 25 μM for A2780cisR cells). The ruthenium (6) and rhodium (7) complexes have a better antiproliferative effect than the iridium complex 8. Complex 7 displayed the highest cytotoxicity against both cell lines compared to 6 and 8 and it is the least cytotoxic to healthy cells.
It has been demonstrated that the dinuclear complexes (3–5) are not as active as the trinuclear complexes, implying that increasing the size and the number of metal centers of the overall complex increases antitumor activity. This effect has also been observed for other multinuclear arene–ruthenium complexes.22,35,56 The study of other di-, tri-, tetra- and hexanuclear ruthenium arene complexes for in vitro cytotoxic activity against the A2780 and A2780cisR cell lines has been reported.22,56–59 Most of these complexes demonstrated better activities than all of the complexes screened during this study (3–8). However, it is important to note that the complexes reported in the literature contain ligands that are not structurally similar to the pyridyl ester (1 and 2) ligands used to prepare 3–8 and in some cases the coordination mode of the ligands to the metal are different to that of complexes 3–8. In some of the reported complexes, the ligands chelate to the metal in a bidentate fashion.
Furthermore, all of the compounds (1–8) must be screened for anti-tumor activity on a panel of other tumor cell lines. It is possible that these compounds may selectively inhibit growth of other tumor cell lines.
Conclusions
A new series of di- (3–5) and trinuclear (6–8) complexes containing polypyridyl ester ligands have been prepared. All of the compounds were isolated as air- and moisture-stable solids and were characterised using a variety of analytical and spectroscopic techniques. The molecular structures of 4 and 5 confirm the monodentate coordination of the pyridyl ester ligands to each metal shown by the spectroscopic and spectrometric characterisations of complexes 3–8. The elucidated molecular structures of the complexes 4 and 5 revealed a typical ‘piano-stool’ geometry around the metal and analysis of the dihedral angles formed between the central phenyl ring and the ester functionalities indicate that the pyridyl metal moieties position themselves almost perpendicular to the phenyl spacer thus minimizing electrostatic interactions.All of the complexes and their free ligands (1–8) were found to exhibit moderate to high antiplasmodial activity against the NF54 CQS P. falciparum strain. The trinuclear complexes (6–8) display better activities compared to the dinuclear derivatives thus proposing that an increase in metal moieties leads to an increase in antiplasmodial activity in vitro. All of the complexes showed better activity than the corresponding free ligand. The most active compounds were assayed for inhibition of β-hematin formation and all of the complexes tested inhibited formation of synthetic hemozoin. Against Trichomonas vaginalis strain G3, complex 8 displayed the best inhibitory effect out of all complexes tested.
Antitumoral studies against the A2780 and A2780cisR cell lines revealed that the trinuclear pyridyl ester complexes were the only complexes to display activity up to the highest concentration tested. The tri-iridium complex (8) demonstrated the lowest toxicity against human embryonic kidney (HEK) cells. The moderate activity of these complexes could be improved through modification of the ligand structure and/or the metal moieties. The design of pyridyl ester ligands that can chelate to the metal in a polydentate manner may lead to the formation of complexes that are more stable and hence display better pharmacological activities. Modification of the arene or cyclopentadienyl ligands of the metal moieties to increase hydrophilicity could also enhance activity.
Experimental
Materials and equipment
1,2,3,4,5-Pentamethylcyclopentadiene, α-phellandrene, 4-bromomethylpyridine hydro-bromide, isonicotinic acid, 1,3,5-benzenetricarboxylic acid and 1,4-bis(bromomethyl)benzene were purchased from Sigma-Aldrich and used without further purification. Ruthenium trichloride trihydrate, rhodium trichloride trihydrate and iridium trichloride trihydrate were kindly donated by AngloAmerican Platinum Limited. All solvents used were analytical grade and dried over molecular sieves. All reactions were carried out in air unless otherwise stated. The precursors, [Ru(p-cymene)Cl2]2,60 [Rh(C5Me5)Cl2]261 and [Ir(C5Me5)Cl2]261 were synthesised using literature methods. The synthesis of ligands 162 and 263 has been previously reported.
Nuclear Magnetic Resonance (NMR) Spectra were recorded on a Varian Unity XR400 MHz (1H at 399.95 MHz, 13C at 100.58 MHz), Varian Mercury XR300 (1H at 300.08 MHz, 13C at 75.46 MHz) or Bruker Biospin GmbH (1H at 400.22 MHz, 13C at 100.65 MHz) spectrometer at ambient temperature. Chemical shifts for 1H and 13C{1H} NMR shifts are reported using tetramethylsilane (TMS) as the internal standard and 31P{1H} NMR spectra were measured relative to H3PO4 as the external standard. NMR spectra were recorded in deuterated chloroform (CDCl3-d1) unless otherwise stated. Infrared (IR) absorptions were measured on Perkin-Elmer Spectrum 100 FT-IR Spectrometer using a Universal Diamond Attenuated Total Reflection (ATR) accessory. Melting points were determined using a Büchi Melting Point Apparatus B-540. Mass Spectrometry determinations were carried out on all new compounds using electrospray ionisation (ESI) on a Waters API Quattro Micro instrument in either the positive or negative mode. All final compounds were analysed by HPLC using an Xbridge C18 (4.6 × 150 mm) 5 μm column; 2.0 μL injection volume; flow 0.7 mL min−1; gradient: 30–100% B in 15 min (hold 2 min) (Mobile phase A: 10 nM NH4OAc in H2O and Mobile phase B: 10 nM NH4OAc in methanol) with a Thermo Separation Products (TSP), Spectra SERIES P200 pump UV100 detector set at 254 nm.
General synthetic method for dinuclear complexes
The ligand (1) (1 molar equivalent) was dissolved in DCM (15 cm3) and the appropriate ruthenium, rhodium or iridium dimer (1.1 molar equivalent) was added and the reaction solution was stirred for 16 hours at room temperature. The reaction solvent was then reduced to approximately a third of its original volume. The product was then precipitated from solution by addition of diethyl ether and isolated by vacuum filtration, washed with diethyl ether and dried.O), 155.9 (py), 138.5 (py), 135.5 (Ar), 128.8 (Ar), 123.5 (py), 103.8 (Ar), 97.4 (Ar), 83.1 (Ar), 82.4 (Ar), 67.5 (O–CH2), 30.7 (Ar–CH(CH3)2), 22.3 (Ar–CH(CH3)2), 18.2 (Ar–CH3). IR (atr, cm−1) ν = 1719 (s, CO), 1611 (s, CN), 1271 (s, C–O–CH2–). ESI-MS: m/z 312.01 ([M − 3Cl + 3CH3OH]3+, 100%). HPLC: tR = 13.9 min.
O), 154.0 (py), 138.7 (py), 135.6 (Ar), 128.77 (Ar), 124.1 (py), 94.2 (Cp*), 67.4 (O–CH2), 8.9 (Cp*). IR (atr, cm−1) ν = 1739 (s, CO), 1613 (s, CN), 1278 (s, C–O–CH2–). ESI-MS: m/z 989.02 ([M − H + Na]+, 100%). HPLC: tR = 13.8 min.
O), 154.4 (py), 138.7 (py), 135.6 (Ar), 128.8 (Ar), 124.5 (py), 86.1 (Cp*), 67.5 (O–CH2), 8.5 (Cp*). IR (atr, cm−1) ν = 1739 (s, CO), 1613 (s, CN), 1278 (s, C–O–CH2). ESI-MS: m/z 1100.21 ([M − Cl]+, 100%). HPLC: tR = 13.8 min.
General synthetic method for trinuclear complexes
The ligand (2) (2 molar equivalent) was dissolved in DCM (15 cm3) and the appropriate ruthenium, rhodium or iridium dimer (3.1 molar equivalent) was added and the reaction solution was stirred for 16 hours at room temperature. The reaction solvent was then reduced to approximately a third of its original volume. The product was then precipitated from solution by addition of diethyl ether and isolated by vacuum filtration, washed with diethyl ether and dried.O), 155.1 (py), 146.6 (Ar), 135.1 (Ar), 130.8 (py), 123.0 (py), 103.6 (Ar), 97.2 (Ar), 82.8 (Ar), 82.2 (Ar), 64.7 (O–CH2), 30.7 (Ar–CH3), 22.3 (Ar–CH(CH3)2), 18.2 (Ar–CH(CH3)2). IR (atr, cm−1) ν = 1728 (s, CO), 1617 (s, CN), 1232 (s, C–O–CH2–). ESI-MS: m/z 1367.03 ([M − Cl]+, 20%); 312.01 ([M − 4Cl]4+, 100%). HPLC: tR = 13.5 min.
O), 153.6 (py), 146.9 (Ar), 135.2 (Ar), 130.8 (py), 123.6 (py), 94.22 (Cp*), 64.7 (O–CH2), 8.9 (Cp*). IR (KBr, cm−1) ν = 1723 (s, CO), 1616 (m, CN), 1225 (s, –C–O–CH2–). ESI-MS: m/z 1432.01 ([M − H + Na]+, 100%). HPLC: tR = 13.5 min.
O), 153.6 (py), 146.9 (Ar), 135.2 (Ar), 130.8 (py), 123.8 (py), 85.9 (Cp*), 64.6 (O–CH2), 8.6 (Cp*). IR (KBr, cm−1) ν = 1729 (s, CO), 1612 (m, CN), 1230 (s, –C–O–CH2–). ESI-MS: m/z 363.05 ([M + 5H + 4CH3OH]+, 100%). HPLC: tR = 13.3 min.
X-Ray structure analysis
Single-crystal X-ray diffraction data were collected on a Bruker KAPPA APEX II DUO diffractometer using graphite-monochromated Mo-Kα radiation (χ = 0.71073 Å). Data collection was carried out at 173(2) K. Temperature was controlled by an Oxford Cryostream cooling system (Oxford Cryostat). Cell refinement and data reduction were performed using the program SAINT.64 The data were scaled and absorption correction performed using SADABS.42 The structure was solved by direct methods using SHELXS-9765 and refined by full-matrix least-squares methods based on F2 using SHELXL-9765 and using the graphics interface program X-Seed.66,67 The programs X-Seed and POV-Ray68 were both used to prepare molecular graphic images.For complex 4, there are four chloroform molecules in the asymmetric unit. Two of them were modelled with the chlorine atoms disordered over two positions with each having site occupancy 0.50. All non-hydrogen atoms of the main molecule, except the carbon atoms C1A–C10A and C36B–C40B, were refined anisotropically. C1A–C10A and C36B–C40B were refined with isotropic temperature factors and were restrained to a reasonable geometry. All hydrogen atoms were placed in idealised positions and refined with geometrical constraints. The structure was refined to R factor of 0.072. The highest peak is 2.89 e Å−3, 0.92 Å from Rh2B and the deepest hole is −0.71 e Å−3, 0.57 Å from CL4X. The Flack x parameter was refined with BASF and TWIN commands to be 0.54936 with esd 0.03821.
For complex 5, there are four chloroform molecules in the asymmetric unit. Two of them were modelled with the chlorine atoms disordered over two positions with each having site occupancy 0.50. All non-hydrogen atoms of the main molecule, except the carbon atoms C6A–C10A and C6B–C10B, were refined anisotropically. C6A–C10A and C6B–C10B were refined with isotropic temperature factors and were restrained to a reasonable geometry. All hydrogen atoms were placed in idealised positions and refined with geometrical constraints. The structure was refined to R factor of 0.0360. The highest peak is 3.12 e Å−3, 0.86 Å from IR1A and the deepest hole is −1.30 e Å−3, 0.56 Å from CL1Z. Crystal data and structure refinement parameters are listed in Table 7.
| Complex 4·2CHCl3 | Complex 5·2CHCl3 | |
|---|---|---|
| Formula | C42H48Cl10N2O4Rh2 | C42H48Cl10Ir2N2O4 |
| Formula weight | 1205.14 | 1383.72 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21 | P21 |
| a (Å) | 11.5800(6) | 11.572(2) |
| b (Å) | 20.4163(11) | 20.505(4) |
| c (Å) | 21.4575(11) | 21.518(4) |
| β (°) | 102.7920(10) | 103.65(3) |
| V (Å3) | 4947.1(4) | 4961.9(17) |
| Z | 4 | 4 |
| D c (g cm−3) | 1.618 | 1.852 |
| μ (mm−1) | 1.249 | 5.938 |
| θ range for data collection (°) | 1.80 to 27.56 | 1.84 to 27.46 |
| Limiting indices | −15 < h < 7 | 0 < h < 14 |
| −26 < k < 26 | 0 < k < 26 | |
| −23 < l < 27 | −27 < l < 27 | |
| No. of reflns meads | 36455 |
11634 |
| No. of reflns used (Rint) | 22713 |
10862 |
| No. of params | 1001 | 1021 |
| R 1 | 0.1052 | 0.0404 |
| wR2 | 0.2009 | 0.0942 |
| Goodness of fit on F2 | 1.023 | 1.040 |
P. falciparum in vitro assay
The test samples were tested in triplicate on one occasion against chloroquine-sensitive (CQS) NF54 strain of Plasmodium falciparum. Continuous in vitro cultures of asexual erythrocyte stages of P. falciparum were maintained using a modified method.69 Quantitative assessment of antiplasmodial activity in vitro was determined via the parasite lactate dehydrogenase assay using a modified method.70 The test samples were prepared to a 20 mg cm−3 stock solution in 100% DMSO and sonicated to enhance solubility. Stock solutions were stored at −20 °C. Further dilutions were prepared on the day of the experiment. Chloroquine (CQ) was used as the reference drug in all experiments. A full dose–response was performed for all compounds to determine the concentration inhibiting 50% of parasite growth (IC50-value). Test samples were tested at a starting concentration of 100 μg cm−3, which was then serially diluted 2-fold in complete medium to give 10 concentrations; with the lowest concentration being 0.2 μg cm−3. The same dilution technique was used for all samples. CQ was tested at a starting concentration of 100 ng cm−3 against the CQR strain and 1000 ng cm−3 against the CQS strain. The highest concentration of solvent to which the parasites were exposed to had no measurable effect on the parasite viability (data not shown). The IC50-values were obtained using a non-linear dose–response curve fitting analysis via Graph Pad Prism v.4.0 software.T. vaginalis in vitro susceptibility assay
Trichomonas vaginalis strain G3 was cultured anaerobically in 15 mL conical centrifuge tubes containing 10 mL of Diamond's TYM media, pH 6.2. Dimethyl sulfoxide was used to dissolve compounds and generate 100 mM stock concentrations of each compound. Each compound was then tested at two concentrations, 25 μM and 50 μM, for in vitro inhibition of parasite growth. DMSO alone controls as well as tubes of untreated cells were carried along side-by-side. Test cultures were incubated for 24 hours at 37 °C under anaerobic conditions. Cell densities were determined using a hemacytometer. Percentage inhibition was determined by comparing cell numbers in treatment tubes with the DMSO control tube. The results of these assays (n = 6) were analyzed and standard errors were determined.A2780 and A2780cisR cancer in vitro assay
The human A2780 and A2780cisR ovarian carcinoma cells were obtained from the European Collection of Cell Cultures (Salisbury, UK). Cells in the log phase of growth were seeded at a density of 2000–2500 per well into 96-well culture treated plates in 0.1 mL of complete media in all wells except for one column reserved for the media only control. The cells were allowed to attach during an overnight incubation at 37 °C, 5% CO2 prior to treating with test compounds. Test compounds were serially diluted in complete culture media and added to each well in a volume of 0.1 mL for a total final volume of 0.2 mL per well (max 0.1% DMSO final, where used). Cells were exposed to test compounds for 72 hours at 37 °C, 5% CO2. Then, 0.02 mL of MTT reagent was added to each well and the plates were returned to the incubator for 2 h. After the incubation period, the culture supernatant was carefully removed from all wells of each plate and DMSO (0.1 mL) was added to all wells to dissolve the formazan crystals. The plates were allowed to shake on a table shaker at room temperature for 15 min and the absorbance at 590 nm was measured using a SpectraMAX e5 plate reader (Molecular Devices). The percentage of surviving cells was calculated from the ratio of absorbance of treated to untreated cells. The IC50 values for the inhibition of cell growth were determined by fitting the plot of the log of the ratio between the percentages of surviving cells, divided by the amount of dead cells, against the log of drug concentration using a linear threshold function.Detergent mediated assay for β-hematin inhibitors
The β-hematin formation assay method described by Carter et al.55 was modified for manual liquid delivery. Stock solutions of the test compounds were prepared at 10 mM, 2 mM and 0.4 mM by dissolving each sample in DMSO with sonication. Test compounds were delivered to a 96 well plate in triplicate from 0–500 μM (final concentration) with a total DMSO volume of 10 μL in each well. Deionized H2O (70 μL) and NP-40 (20 μL; 30.55 μM) were then added. A 25 mM hematin stock solution was prepared by sonicating hemin in DMSO, for complete dissolution, and then suspending 177.76 μL of this in a 2 M acetate buffer (pH 4.8). The homogenous suspension (100 μL) was then added to the wells to give final buffer and hematin concentrations of 1 M and 100 μM respectively. The plate was covered and incubated at 37 °C for 16 hours in a water bath. Analysis of the assay was carried out using the pyridine-ferrichrome method developed by Ncokazi and Egan.71 A solution of 50% (v/v) pyridine, 30% (v/v) H2O, 20% (v/v) acetone and 0.2 M HEPES buffer (pH 7.4) was prepared and 32 μL added to each well to give a final pyridine concentration of ±5% (v/v). Acetone (60 μL) was then added to assist with hematin dispersion. The UV-vis absorbance of the plate wells was read on a SpectraMax plate reader. Sigmoidal dose–response curves were fitted to the absorbance data using GraphPad Prism v5.00.Turbidimetric solubility assay
Preparation of 0.01 M pH 7.4 Phosphate Buffered Saline (PBS) involved dissolving 1 intact PBS buffer tablet (EC Diagnostics AB, Sweden) in sufficient distilled water to make 1000 mL of a solution comprising 0.14 M NaCl, 0.003 M KCl and 0.01 M phosphate buffer. This solution was filtered through a 0.22 μm nylon filter to remove any particulate contaminants and the pH ascertained using a pH meter. The test compounds were prepared by making 10 mM stock solutions in DMSO from which, dilutions of the compounds in both DMSO and 0.01 M pH 7.4 PBS were prepared in a 96-well plate, in triplicate. Wells in columns 1–6 contained compounds in DMSO and columns 7–12 contained the compounds in PBS at similar nominal concentrations as those in DMSO. The final volume of solvent in each assay plate well was 200 μL, prepared by pipetting 4 μL each of solution from the pre-dilution plate to the corresponding well into both DMSO and PBS (both 196 μL). This ensured that the final concentration of DMSO in the PBS aqueous buffer did not exceed 2% v/v. The different concentrations in DMSO were prepared to serve as controls to determine potential false turbidimetric absorbance readings arising from the compounds in solution absorbing incident radiation at the analysis wavelength. After making the assay plate preparation, the plate was covered and left to equilibrate for 2 hours at ambient temperature. After incubation, UV-vis absorbance readings from the plate were measured at 620 nm. Corrected absorbance readings at different concentrations of test compounds were calculated by subtracting absorbance of the blank (DMSO and 1% DMSO in PBS) from each subsequent concentration absorbance.Acknowledgements
Financial support from the University of Cape Town, the National Research Foundation (NRF) of South Africa and Sasol South Africa is gratefully acknowledged. Johnson-Matthey/Anglo American Platinum Limited is kindly acknowledged for donation of metal salts. KML was supported by the Department of Biological Sciences, and the Office of Sponsored Programs and Research at the University of the Pacific.References
- T. J. Ritchie and S. J. F. Macdonald, Drug Discovery Today, 2009, 14(21–22), 1011 CrossRef CAS PubMed.
- T. J. Ritchie, S. J. F. Macdonald, S. Peace, S. D. Pickett and C. N. Luscombe, Med. Chem. Commun., 2012, 3, 1062 RSC.
- S. W. Fogt, J. A. Scozzie, R. D. Heilman and L. J. Powers, J. Med. Chem., 1980, 23, 1445 CrossRef CAS.
- S. Kumar, P. Arya, C. Mukherjee, B. K. Singh, N. Singh, V. S. Parmar, A. K. Prasad and B. Ghosh, Biochemistry, 2005, 44, 15944 CrossRef CAS PubMed.
- B. P. Bandgar, R. J. Sarangdhar, K. Fruthous, J. Mookkan, S. Chaudhary, H. V. Chavan, S. B. Bandgar and V. Y. Kshirsagar, Eur. J. Med. Chem., 2012, 57, 217 CrossRef CAS PubMed.
- I. Aissa, R. M. Sghair, M. Bouaziz, D. Laouini, S. Sayadi and Y. Gargouri, Lipids Health Dis., 2012, 11, 13 CrossRef CAS PubMed.
- H. Hess-Stumpp, T. U. Bracker, D. Henderson and O. Politz, Int. J. Biochem. Cell Biol., 2007, 39, 1388 CrossRef CAS PubMed.
- O. L. P. De Jesús, H. R. Ihre, L. Gagne, J. M. J. Fréchet and F. C. Szoka Jr., Bioconjugate Chem., 2002, 13, 453 CrossRef PubMed.
- A. L. Acton, C. Fante, B. Flatley, S. Burattini, I. W. Hamley, Z. Wang, F. Greco and W. Hayes, Biomacromolecules, 2013, 14(2), 564 CrossRef CAS PubMed.
- X. Ma, Z. Zhou, E. Jin, Q. Sun, B. Zhang, J. Tang and Y. Shen, Macromolecules, 2013, 46(1), 37 CrossRef CAS.
- C. Gong, S. Deng, Q. Wu, M. Xiang, X. Wei, L. Li, X. Gao, B. Wang, L. Sun, Y. Chen, Y. Li, L. Liu, Z. Qian and Y. Wei, Biomaterials, 2013, 34, 1413 CrossRef CAS PubMed.
- Y. Xiao, H. Hong, A. Javadi, J. W. Engle, W. Xu, Y. Yang, Y. Zhang, T. E. Barnhart, W. Cai and S. Gong, Biomaterials, 2012, 33, 3071 CrossRef CAS PubMed.
- N. Feliu, M. V. Walter, M. I. Montañez, A. Kunzmann, A. Hult, A. Nyström, M. Malkoch and B. Fadeel, Biomaterials, 2012, 33, 1970 CrossRef CAS PubMed.
- K.-G. Liu, X.-Q. Cai, X.-C. Li, D.-A. Qin and M.-L. Hu, Inorg. Chim. Acta, 2012, 388, 78 CrossRef CAS PubMed.
- F. Schmitt, P. Govindaswamy, O. Zava, G. Suss-Fink, L. Juillerat-Jeanneret and B. Therrien, J. Biol. Inorg. Chem., 2009, 14, 101 CrossRef CAS PubMed.
- F. Schmitt, P. Govindaswamy, G. Süss-Fink, W. H. Ang, P. J. Dyson, L. Juillerat-Jeanneret and B. Therrien, J. Med. Chem., 2008, 51, 1811 CrossRef CAS PubMed.
- P. Govender, A. K. Renfrew, C. M. Clavel, P. J. Dyson, B. Therrien and G. S. Smith, Dalton Trans., 2011, 40, 1158 RSC.
- R. Payne, P. Govender, B. Therrien, C. M. Clavel, P. J. Dyson and G. S. Smith, J. Organomet. Chem., 2013, 729, 20 CrossRef CAS PubMed.
- W. H. Ang, E. Daldini, L. Juillerat-Jeanneret and P. J. Dyson, Inorg. Chem., 2007, 46(22), 9048 CrossRef CAS PubMed.
- W. H. Ang, A. De Luca, C. Chapuis-Bernasconi, L. Juillerat-Jeanneret, M. Lo Bello and P. J. Dyson, ChemMedChem, 2007, 2(12), 1799 CrossRef CAS PubMed.
- W. H. Ang, L. J. Parker, A. De Luca, L. Juillerat-Jeanneret, C. J. Morton, M. Lo Bello, M. W. Parker and P. J. Dyson, Angew. Chem., Int. Ed., 2009, 48(21), 3854 CrossRef CAS PubMed.
- M. Auzias, B. Therrien, G. Süss-Fink, P. Štěpnička, W. H. Ang and P. J. Dyson, Inorg. Chem., 2008, 47, 578 CrossRef CAS PubMed.
- R. A. Sánchez-Delgado, M. Navarro, H. Perez and J. A. Urbina, J. Med. Chem., 1996, 39, 1095 CrossRef PubMed.
- C. S. K. Rajapakse, A. Martınez, B. Naoulou, A. A. Jarzecki, L. Suarez, C. Deregnaucourt, V. Sinou, J. Schrevel, E. Musi, G. Ambrosini, G. K. Schwartz and R. A. Sanchez-Delgado, Inorg. Chem., 2009, 48, 1122–1131 CrossRef CAS PubMed.
- M. Navarro, S. Pekerar and H. A. Perez, Polyhedron, 2007, 26, 2420 CrossRef CAS PubMed.
- D. Gambino and L. Otero, Inorg. Chim. Acta, 2012, 393, 103 CrossRef CAS PubMed.
- P. F. Salas, C. Herrmann and C. Orvig, Chem. Rev., 2013, 113(5), 3450 CrossRef CAS PubMed.
- C. Biot, W. Castro, C. Y. Botte and M. Navarro, Dalton Trans., 2012, 41, 6335 RSC.
- M. Navarro, W. Castro and C. Biot, Organometallics, 2012, 31, 5715 CrossRef CAS.
- Y. Yamamotoa, H. Suzuki, N. Tajima and K. Tatsumi, Chem.–Eur. J., 2002, 8(2), 372 CrossRef CAS.
- P. Govindaswamy, G. Süss-Fink and B. Therrien, Organometallics, 2006, 26, 915 Search PubMed.
- A. Bacchi, G. Cantoni, P. Pelagatti and S. Rizzato, J. Organomet. Chem., 2012, 714, 81 CAS.
- J. Grau, V. Noe, C. Ciudad, M. J. Prieto, M. Font-Bardia, T. Calvet and V. Moreno, J. Inorg. Biochem., 2012, 109, 72 CAS.
- M. Yadav, A. K. Singh, R. Pandey and D. S. Pandey, J. Organomet. Chem., 2010, 695, 841 CAS.
- P. Govender, N. C. Antonels, J. Mattsson, A. K. Renfrew, P. J. Dyson, J. R. Moss, B. Therrien and G. S. Smith, J. Organomet. Chem., 2009, 694, 3470 CAS.
- G. Süss-Fink, F.-A. Khan, L. Juillerat-Jeanneret, P. J. Dyson and A. K. Renfrew, J. Cluster Sci., 2010, 21, 313 Search PubMed.
- S. Grgurić-Šipka, I. Ivanović, G. Rakić, N. Todorović, N. Gligorijević, S. Radulović, V. B. Arion, B. K. Keppler and Ž. L. Teši, Eur. J. Med. Chem., 2010, 45, 1051 Search PubMed.
- N. Mézailles, P. E. Fanwick and C. P. Kubiak, Organometallics, 1997, 16(8), 1526 Search PubMed.
- S. K. Singh, M. Trivedi, M. Chandra and D. S. Pandey, J. Organomet. Chem., 2005, 690, 647 CAS.
- P. Govindaswamy, Y. A. Mozharivskyj and M. R. Kollipara, Polyhedron, 2005, 24, 1710 CAS.
- M. Chandra, A. N. Sahay, S. M. Mobin and D. S. Pandey, J. Organomet. Chem., 2002, 658, 43 CAS.
- G. M. Sheldrick, SADABS version 2.05, University of Göttingen, Germany, 1997 Search PubMed.
- J. G. Małecki, M. Jaworska and R. Kruszynski, Polyhedron, 2006, 25, 2519 Search PubMed.
- Y.-F. Han, J.-S. Zhang, Y.-J. Lin, J. Dai and G.-X. Jin, J. Organomet. Chem., 2007, 692, 4545 CAS.
- J.-Q. Wang, C.-X. Ren and G.-X. Jin, Organometallics, 2006, 25, 74 CAS.
- R. Schobert, S. Seibt, K. Effenberger-Neidnicht, C. Underhill, B. Biersack and G. L. Hammond, Steroids, 2011, 76, 393 CAS.
- M. Gras, B. Therrien, G. Süss-Fink, A. Casini, F. Edafe and P. J. Dyson, J. Organomet. Chem., 2010, 695(8), 1119 CAS.
- L. Pauling, The Nature of the Chemical Bond, Cornell University Press, New York, 3rd edn, 1960 Search PubMed.
- L. E. Shutton, Tables of Interatomic Distances and Configurations in Molecules and Ions (Supplement), The Chemical Society, London, 1965 Search PubMed.
- E. H. Kerns and L. Di, Drug-like Properties: Concepts, Structure Design and Methods from ADME to Toxicity Optimization, Elsevier Academic Press, California, 1st edn, 2008 Search PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Duminy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3 CAS.
- J. Alsenz and M. Kansy, Adv. Drug Delivery Rev., 2007, 59, 546 CAS.
- L. Pan, Q. Ho, K. Tsutsui and L. Takahashi, J. Pharm. Sci., 2001, 4, 521 Search PubMed.
- D. E. Goldberg, A. F. Slater, A. Cerami and G. B. Henderson, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 2931 CAS.
- M. D. Carter, V. V. Phelan, R. D. Sandlin, B. O. Bachmann and D. W. Wright, Comb. Chem. High Throughput Screening, 2010, 13(3), 285 CAS.
- M. G. Mendoza-Ferri, C. G. Hartinger, A. A. Nazarov, R. E. Eichinger, M. A. Jakupec, K. Severin and B. K. Keppler, Organometallics, 2009, 28, 6260 CAS.
- F. Linares, M. A. Galindo, S. Galli, M. A. Romero, J. A. R. Navarro and E. Barea, Inorg. Chem., 2009, 48, 7413 CAS.
- L. E. H. Paul, J. Furrer and B. Therrien, J. Organomet. Chem., 2013, 734, 45 CAS.
- N. P. E. Barry, F. Edafe and B. Therrien, Dalton Trans., 2011, 40, 7172 CAS.
- M. A. Bennett, T. N. Huang, T. W. Matheson and A. K. Smith, Inorg. Synth., 1982, 21, 74 CAS.
- C. White, A. Yates and P. M. Maitlis, Inorg. Synth., 1992, 29, 228 CAS.
- X.-B. Shao, X.-K. Jiang, X. Zhao, C.-X. Zhao, Y. Chen and Z.-T. Li, J. Org. Chem., 2004, 69, 899 CAS.
- S. Ghosh and P. S. Mukherjee, J. Org. Chem., 2006, 71, 8412 CAS.
- SAINT Version 7.60a, Bruker AXS Inc., Madison, WI, USA, 2006 Search PubMed.
- G. M. Sheldrick, SHELXS-97 and SHELXL-97, University of Göttingen, Germany, 1997 Search PubMed.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189 CAS.
- J. L. Atwood and L. J. Barbour, Cryst. Growth Des., 2003, 3, 3 CAS.
- E. Mas-Marza, E. Peris, I. Castro-Rodriguez and K. Meyer, Organometallics, 2005, 24, 3158 CAS.
- W. Trager and J. B. Jensen, Science, 1976, 193(4254), 673 CAS.
- M. T. Makler, J. M. Ries, J. A. Williams, J. E. Bancroft, R. C. Piper, B. L. Gibbins and D. J. Hinrichs, Am. J. Trop. Med. Hyg., 1993, 48, 739 CAS.
- K. K. Ncokazi and T. J. Egan, Anal. Biochem., 2005, 338(2), 306 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 934524 and 934525. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt52090k |
| This journal is © The Royal Society of Chemistry 2014 |