
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic oxidation of formic acid by dioxygen with an organoiridium complex†
Tomoyoshi
Suenobu
,
Satoshi
Shibata
and
Shunichi
Fukuzumi
*
Department of Material and Life Science, Graduate School of Engineering, Osaka University and ALCA, Japan Science and Technology Agency (JST), 2-1 Yamada-oka, Suita, Osaka 565-0871, Japan. E-mail: fukuzumi@chem.eng.osaka-u.ac.jp; Fax: +81 6 6879 7370; Tel: +81 6 6879 7368
First published on 13th August 2014
Abstract
Catalytic oxidation of formic acid by dioxygen occurred efficiently using an organoiridium complex ([IrIII(Cp*)(4-(1H-pyrazol-1-yl-κN2)benzoic acid-κC3)(H2O)]2SO4, 1) as a catalyst in a water-containing organic solvent as well as in water at ambient temperature. The catalytic cycle is composed of the reduction of 1 by formate to produce the hydride complex, which reduces dioxygen to water to regenerate 1.
Formic acid (HCOOH) is liquid at room temperature1 with a relatively high volumetric density (d = 1.22 g cm−3) and is widely utilised as a preservative and an antibacterial additive for livestock feed.2 HCOOH can be formed by reduction of CO2 with H2 and the catalytic interconversion between HCOOH and H2 (eqn (1)) has been reported to be ideal for carbon-neutral storage and transportation of H2.3–6
| H2 + CO2 ⇄ HCOOH | (1) |
In a natural enzymatic system, formate oxidase7 and formate:oxygen oxidoreductase8 reduce dioxygen (O2) to reactive oxygen species, e.g., superoxide and hydrogen peroxide that would be further reduced to water. Formate is often used as an electron donor for reductive activation of O2 to conduct enzymatic oxygenation9 and for reduction of NAD+ and FAD to regenerate NADH and FADH2,10 respectively. Subsequently, NADH or FADH2 is supplied as an electron donor to either reductase or oxidase, enabling regioselective oxidation such as epoxidation.11
In addition to its importance as a renewable hydrogen source for both enzymatic and non-enzymatic useful synthetic reactions,12 formic acid is also utilised as a fuel for direct formic acid fuel cells.13,14 The theoretical output potential is 1.45 V, which is higher than those of H2 (1.23 V) and methanol (1.21 V) fuel cells.13,14 Hence, the overall reaction for the cathodic oxidation of formic acid and the anodic reduction of oxygen is expressed in eqn (2), which is largely exergonic (ΔcH0 = −255 kJ mol−1).
| 2HCOOH + O2 → 2H2O + 2CO2 | (2) |
Formic acid is the most aggressive contributor of atmospheric corrosion for indoor environments,15 being also contained as a hazardous compound in wastewaters.16,17 The best way of removing formic acid is through oxidation by O2 into H2O and CO2 (eqn (2)). Heterogeneous catalysts have been reported to act as catalysts for oxidation of HCOOH by O2.17 From an economical point of view, there is still a need to improve the catalytic activity of oxidation of HCOOH by O2 at temperatures and pressures as low as possible.17 There has been no report so far on the use of a homogeneous catalyst for efficient oxidation of HCOOH by O2 at ambient pressure and temperature or its catalytic mechanism.
We report herein the catalytic oxidation of HCOOH by O2 in water and a water-containing protic solvent, ethylene glycol, in the presence of a water-soluble iridium aqua complex ([IrIII(Cp*)(4-(1H-pyrazol-1-yl-κN2)benzoic acid-κC3)(H2O)]2SO4, [1]2·SO4; see the ESI†) acting as an efficient catalyst for the removal of HCOOH by O2. A mixture solvent of water and ethylene glycol was examined because ethylene glycol has been used to improve the energy density of electric capacitors, in which a trace of HCOOH has to be removed.181 reacts with HCOOH to produce the corresponding Ir–hydride complex (3),4,19 which can reduce O2 to H2O.
Synthesis and characterization of 1 were carried out according to the previous reports and are briefly described in the Experimental section in the ESI.†5,19 The carboxylate form 1-H+ is protonated to give the carboxylic acid group in 1, as shown in eqn (3), at pH 2.8 since the pKa of 1 was determined to be 4.0.5,19
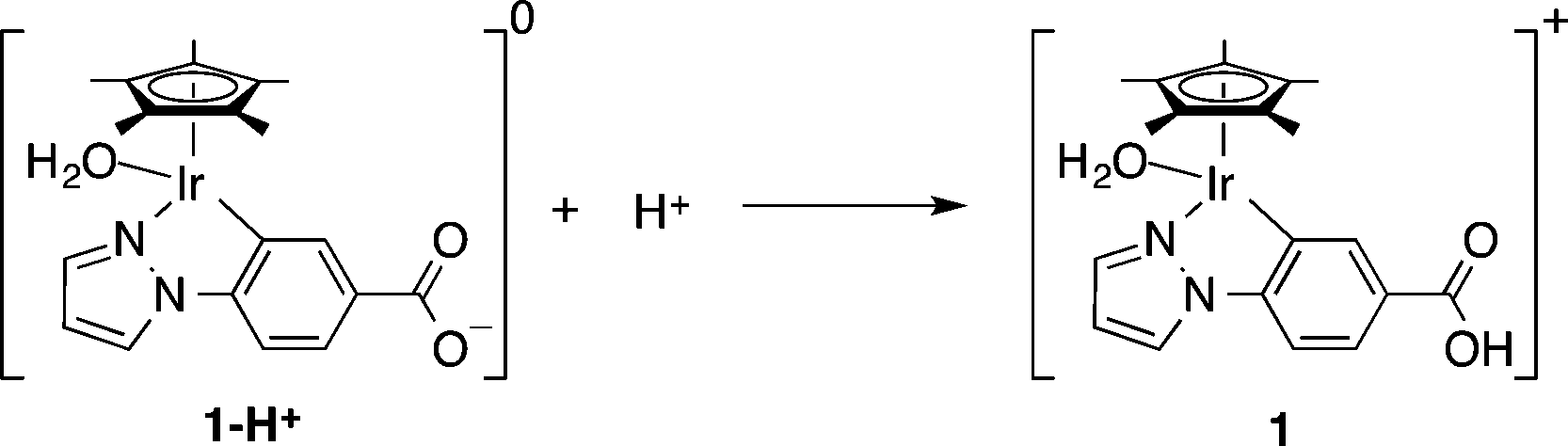 | (3) |
Under an N2 atmosphere at pH 2.8 in the presence of 1, formic acid decomposed efficiently to produce CO2 (Fig. 1a) and H2 (Fig. 1b) according to eqn (4).4 When the reaction was conducted under an O2 atmosphere,
| HCOOH → H2 + CO2 | (4) |
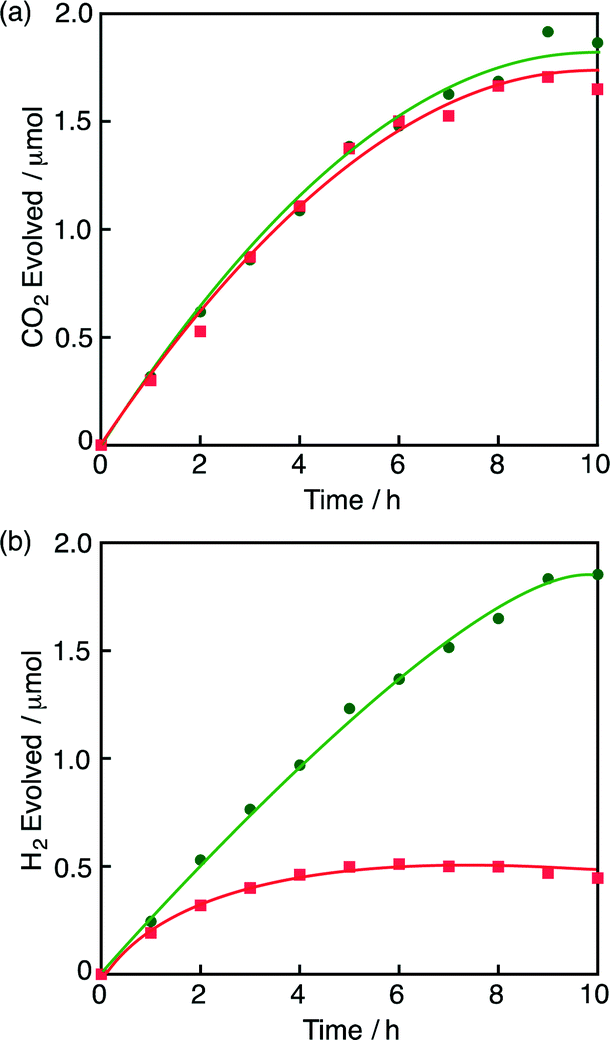 | ||
| Fig. 1 (a) Time courses of CO2 evolution from an aqueous formic acid (2.0 mM) solution (1.0 mL) in the presence of 1 (10 μM) under N2 and O2 atmosphere (green circle and red square, respectively) at pH 2.8 at 298 K. (b) Time courses of H2 evolution from a formic acid (2.0 mM) solution in the presence of 1 (10 μM) under N2 and O2 atmosphere (green circle and red square, respectively) at pH 2.8 at 298 K. The red and green lines correspond to the reactions [(eqn (2) + eqn (4)) and eqn (4), respectively]. The amounts of H2 and CO2 were analysed by GC (see the ESI†). | ||
In the catalytic reaction at pH 2.8 under an N2 atmosphere, 1 reacted with HCOO− to afford the formate complex 2, which is converted to the hydride complex 3via β-hydrogen elimination from 2. Then, 3 reacts with H3O+ to produce H2, accompanied by the regeneration of 1, as shown in Scheme 1.5 The formation of the hydride complex (3) was confirmed by comparison with the 1H NMR spectrum of the isolated hydride complex in DMSO-d6 obtained by the reaction of 1 with H2, which showed a typical hydride peak at δ = −14.74 ppm.5 Because the iridium hydride complex (3) is a neutrally charged complex, the solubility of 3 in water is too low to be detected by 1H NMR in D2O.
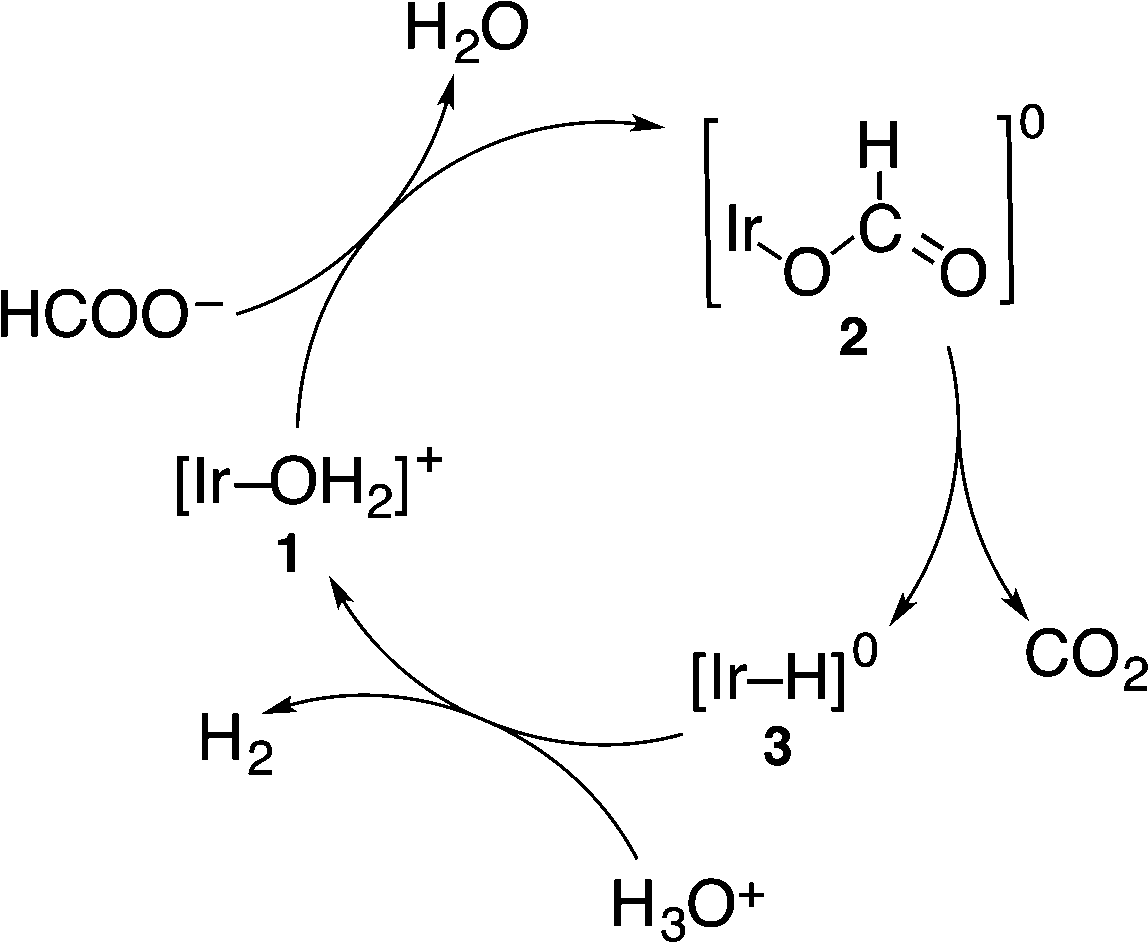 | ||
| Scheme 1 Catalytic cycle for decomposition of formic acid to form H2 and CO2 by using 1 under an N2 atmosphere.4 | ||
On the other hand, the hydride complex (3) reacts with O2 to produce H2O and reproduce 1. The overall catalytic cycle for the four-electron reduction of O2 by HCOO− with 1 in competition with H2 evolution is shown in Scheme 2. The rate-determining step of this catalytic oxidation cycle was independently examined by the deuterium kinetic isotope effect (KIE) on the catalytic oxidation of formic acid-d (DCOOH) vs. HCOOH. By comparing the time course of oxidation of HCOOH by O2 with that of DCOOH in Fig. 2 (also see Fig. S1 in the ESI†), the KIE was determined to be 4.1 ± 0.2 at pH 2.8 at 298 K. This value is nearly equal to the value (4.0) reported for the hydrogen evolution reaction under an N2 atmosphere under otherwise the same experimental conditions.19 This indicates that the rate-determining step in the overall catalytic cycle for oxidation of HCOOH by O2 is the β-hydrogen elimination of the formate complex (2) to form the hydride complex (3).
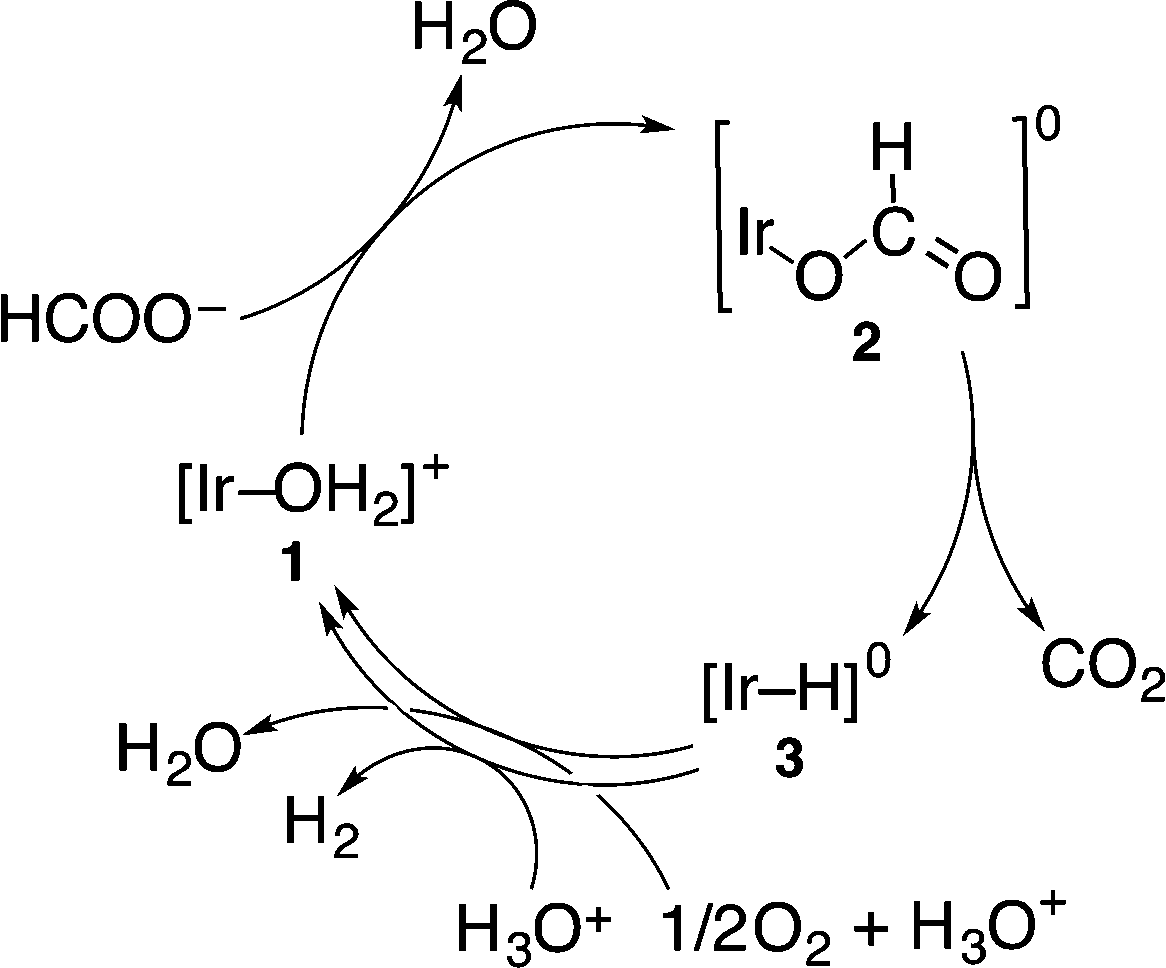 | ||
| Scheme 2 Catalytic cycle for the formation of H2, CO2 and H2O from formic acid in the presence of 1 under an O2 atmosphere. | ||
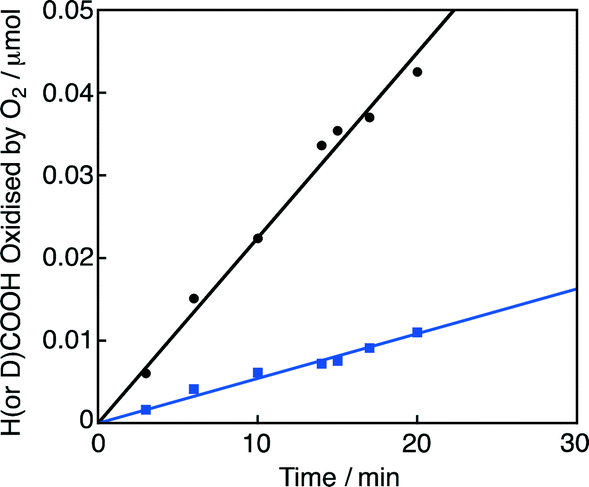 | ||
| Fig. 2 Time courses of oxidation of HCOOH (2.0 mM; black circle) and DCOOH (2.0 mM; blue square) by O2 in the presence of 1 (10 μM) under an O2 atmosphere in water (1.0 mL) at pH 2.8 at 298 K. R2 = 0.99 and 0.98 for the linear correlations (black and blue, respectively). | ||
The catalytic oxidation of HCOOH by O2 also occurred in a mixed solution (3.0 mL) of ethylene glycol and water [4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v)] and the yield of H2 was decreased as compared with that under an N2 atmosphere (Fig. 3). In the same manner, various concentrations of HCOOH were oxidised by O2 under an O2 atmosphere by using 1 in water-containing ethylene glycol at various pH values (Fig. S2 and S3 in the ESI†). The amount of the remaining HCOOH was quantified by 1H NMR, in which no oxidized product of ethylene glycol was detected. The amount of H2O2 produced was analysed by spectral titration using the oxo[5,10,15,20-tetra(4-pyridyl)porphyrinato]titanium(IV) complex in water.20 Neither H2O2 nor H2 was formed in the absence of HCOOH. The amount of H2O2 generated in the reaction of 2.0 mM HCOOH was 13 μM at 5 h, which is also negligible as in the case of the reaction in water. The TON reached 1300 at 22 h (Fig. 3). The amount of H2O2 was significantly increased by adding flavin mononucleotide (FMN), as shown in Fig. S4 in the ESI.† In the presence of FMN, the hydride complex (3) reacts with FMN in competition with the four-electron reduction of O2 and the reduced FMN reacts with O2 to produce H2O2.19c
:1 (v/v)] and the yield of H2 was decreased as compared with that under an N2 atmosphere (Fig. 3). In the same manner, various concentrations of HCOOH were oxidised by O2 under an O2 atmosphere by using 1 in water-containing ethylene glycol at various pH values (Fig. S2 and S3 in the ESI†). The amount of the remaining HCOOH was quantified by 1H NMR, in which no oxidized product of ethylene glycol was detected. The amount of H2O2 produced was analysed by spectral titration using the oxo[5,10,15,20-tetra(4-pyridyl)porphyrinato]titanium(IV) complex in water.20 Neither H2O2 nor H2 was formed in the absence of HCOOH. The amount of H2O2 generated in the reaction of 2.0 mM HCOOH was 13 μM at 5 h, which is also negligible as in the case of the reaction in water. The TON reached 1300 at 22 h (Fig. 3). The amount of H2O2 was significantly increased by adding flavin mononucleotide (FMN), as shown in Fig. S4 in the ESI.† In the presence of FMN, the hydride complex (3) reacts with FMN in competition with the four-electron reduction of O2 and the reduced FMN reacts with O2 to produce H2O2.19c
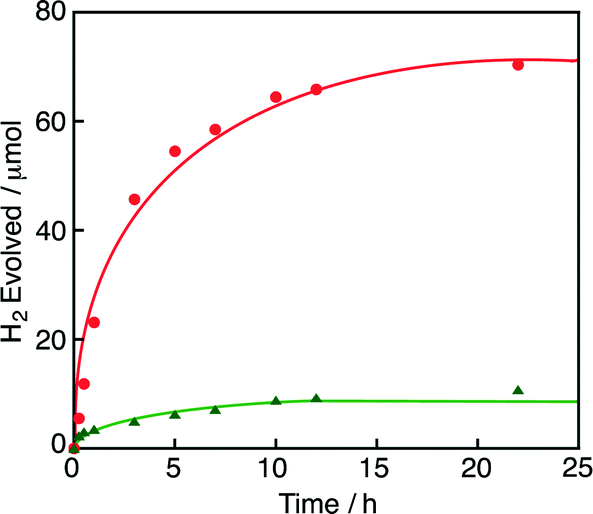 | ||
| Fig. 3 Time courses of H2 evolution from formic acid (0.50 M) in an ethylene glycol and water [4:1 (v/v)] mixed solution (3.0 mL) in the presence of 1 (18 μM) under N2 and O2 atmosphere (red circle and green triangle, respectively) at pH 5.9 at 298 K. | ||
In conclusion, a water-soluble iridium(III) complex (1) can efficiently catalyse the oxidation of HCOOH by O2 to mainly generate water with evolution of a little amount of H2 under acidic conditions at 298 K. This reaction occurred in both water and water-containing ethylene glycol. The rate-determining step of the catalytic cycle is the β-hydrogen elimination of the formate complex (2) to form the hydride complex (3) in the same manner as the hydrogen evolution from HCOOH catalysed by 1. This study provides an efficient way to remove undesired formic acid in water as well as in water-containing ethylene glycol.
Acknowledgements
This work was supported by the Advanced Low Carbon Technology Research and Development (ALCA) program of Japan Science Technology Agency (JST) (to S.F.) and Grants-in-Aid (no. 24550077 to T.S.) from MEXT, Japan.Notes and references
-
(a) D. L. Royer, R. A. Berner and J. Park, Nature, 2007, 446, 530 CrossRef CAS PubMed
; (b) R. Masel, Nature, 2006, 442, 521 CrossRef CAS PubMed
-
W. Reutemann and H. Kieczka, Formic acid in Ullmann's Encyclopedia of Industrial Chemistry, online edition, Wiley VCH, Weinheim, 7th edn, 2011, DOI:10.1002/14356007.a12_013.pub2
-
(a) S. Enthaler, J. von Langermann and T. Schmidt, Energy Environ. Sci., 2010, 3, 1207 RSC
-
(a) M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171 RSC
- Y. Maenaka, T. Suenobu and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 7360 CAS
-
(a) J. F. Hull, Y. Himeda, W.-H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383 CrossRef CAS PubMed
-
(a) D. Doubayashi, T. Ootake, Y. Maeda, M. Oki, Y. Tokunaga, A. Sakurai, Y. Nagaosa, B. Mikami and H. Uchida, Biosci., Biotechnol., Biochem., 2011, 75, 1662 CrossRef CAS PubMed
-
(a) P. M. F. Sousa, M. A. M. Videira and A. M. P. Melo, FEBS Lett., 2013, 587, 2559 CrossRef CAS PubMed
- V. Kohler, Y. M. Wilson, M. Durrenberger, D. Ghislieri, E. Churakova, T. Quinto, L. Knorr, D. Haussinger, F. Hollmann, N. J. Turner and T. R. Ward, Nat. Chem., 2013, 5, 93 CrossRef CAS PubMed
-
(a) H. C. Lo, O. Buriez, J. B. Kerr and R. H. Fish, Angew. Chem., Int. Ed., 1999, 38, 1429 CrossRef CAS
-
(a) C. E. Paul, S. Gargiulo, D. J. Opperman, I. Lavandera, V. Gotor-Fernandez, V. Gotor, A. Taglieber, I. Arends and F. Hollmann, Org. Lett., 2013, 15, 180 CrossRef CAS PubMed
-
(a) K. Hofstetter, J. Lutz, I. Lang, B. Witholt and A. Schmid, Angew. Chem., Int. Ed., 2004, 43, 2163 CrossRef CAS PubMed
-
(a) R. Wang, J. Liu, P. Liu, X. Bi, X. Yan, W. Wang, X. Ge, M. Chen and Y. Ding, Chem. Sci., 2014, 5, 403 RSC
-
(a) X. Ji, K. T. Lee, R. Holden, L. Zhang, J. Zhang, G. A. Botton, M. Couillard and L. F. Nazar, Nat. Chem., 2010, 2, 286 CrossRef CAS PubMed
- M. Forslund, C. Leygraf, P. M. Claesson, C. Lin and J. Pana, J. Electrochem. Soc., 2013, 160, C423 CrossRef CAS PubMed
- (a) Hazardous Substances Data Bank (HSDB) database, United States National Library of Medicine (NLM), CASRN: 64-18-6, http://toxnet.nlm.nih.gov/ Search PubMed; (b) ACROS Organics Material Safety Data Sheet (MSDS) for formic acid (#66394) Search PubMed.
-
(a) A. Pintar, J. Batista and T. Tišler, Appl. Catal., B, 2008, 84, 30 CrossRef CAS PubMed
-
(a) C. Ramasany, J. P. del Val and M. Anderson, Electrochim. Acta, 2014, 135, 181 CrossRef PubMed
-
(a) Y. Maenaka, T. Suenobu and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 367 CrossRef CAS PubMed
- C. Matsubara, N. Kawamoto and K. Takamura, Analyst, 1992, 117, 1781 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and kinetic details. See DOI: 10.1039/c4cy00957f |
| This journal is © The Royal Society of Chemistry 2014 |