 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrazone-based switches, metallo-assemblies and sensors
Xin
Su
and
Ivan
Aprahamian
*
Department of Chemistry, Dartmouth College, Hanover, New Hampshire 03755, USA. E-mail: ivan.aprahamian@dartmouth.edu
First published on 16th January 2014
Abstract
The hydrazone functional group has been extensively studied and used in the context of supramolecular chemistry. Its pervasiveness and versatility can be attributed to its ease of synthesis, modularity, and most importantly unique structural properties, which enable its integration in different applications. This review provides an overview of the utilization of hydrazones in three supramolecular chemistry related areas: molecular switches, metallo-assemblies and sensors. These topics were chosen because they highlight the diversity of hydrazones, and emphasize their uniqueness vis-à-vis the imine functional group. Discussion entails (i) chemical and light activated switching of hydrazones, and how this can be used in controlling the properties of self-assembled systems, (ii) the use of hydrazones in the formation of dynamic and stimuli responsive metallogrids, and (iii) the use of hydrazones in detecting metal cations (Zn2+, Cu2+, Hg2+, etc.), anions (F−, CN−, P2O74−, etc.) and neutral molecules (amines, water, Cys, etc.).
 Xin Su | Xin Su received his BS degree in chemistry from Nankai University (2009), where he worked in the Liu group on the recognition and assembly of water-soluble calixarenes. He joined the Aprahamian group at Dartmouth College in 2009, and has since then been working on the development of novel hydrazone-based molecular switches and switching systems thereof. He received his PhD degree in December, 2013. |
 Ivan Aprahamian | Ivan Aprahamian received all his degrees (BSc – 1998, MSc – 2000, and PhD – 2005) from the Hebrew University of Jerusalem, Israel. His doctoral research was conducted under the supervision of Professors Mordecai Rabinovitz and Tuvia Sheradsky. He started his independent career at Dartmouth College in 2008, after finishing his postdoctoral research with Sir Fraser Stoddart (UCLA). His research focuses on using structurally simple, modular, and tunable hydrazone-based building blocks in the development of adaptive functional materials. |
1 Introduction
The hydrazone functional group is ubiquitous in various fields ranging from organic synthesis1 and medicinal chemistry2 to supramolecular chemistry,3 and has been used in metal and covalent organic frameworks,4 dynamic combinatorial chemistry (DCC),5 dye6 and hole-transporting materials,7 among other applications.8 The modularity, straightforward synthesis, and stability towards hydrolysis (cf. imines) of hydrazones can be cited as reasons for their popularity. But in reality it is the functional diversity of this azomethine group, which is characterized by the triatomic structure C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–N, that enables its use in various fields. A quick survey of the structure of a hydrazone (Fig. 1) reveals that it has (i) nucleophilic imine and amino-type (more reactive) nitrogens, (ii) an imine carbon that has both electrophilic and nucleophilic character, (iii) configurational isomerism stemming from the intrinsic nature of the CN double bond, and (iv) in most cases an acidic N–H proton. These structural motifs give the hydrazone group its physical and chemical properties, in addition to playing a crucial part in determining the range of applications it can be involved in.
N–N, that enables its use in various fields. A quick survey of the structure of a hydrazone (Fig. 1) reveals that it has (i) nucleophilic imine and amino-type (more reactive) nitrogens, (ii) an imine carbon that has both electrophilic and nucleophilic character, (iii) configurational isomerism stemming from the intrinsic nature of the CN double bond, and (iv) in most cases an acidic N–H proton. These structural motifs give the hydrazone group its physical and chemical properties, in addition to playing a crucial part in determining the range of applications it can be involved in.
 | ||
| Fig. 1 The structural and functional diversity of the hydrazone group. | ||
Hydrazones can be made by three main synthetic pathways (Scheme 1), (i) coupling between aryl diazonium salts and β-keto esters or acids, which is also known as the Japp–Klingemann reaction,9 (ii) coupling between hydrazines and ketones or aldehydes,10,11 and (iii) coupling between aryl halides and non-substituted hydrazones.12,13 The readily obtained compounds are usually highly crystalline and crash out of the reaction mixture, which simplifies their purification process and adds to the appeal of these systems.
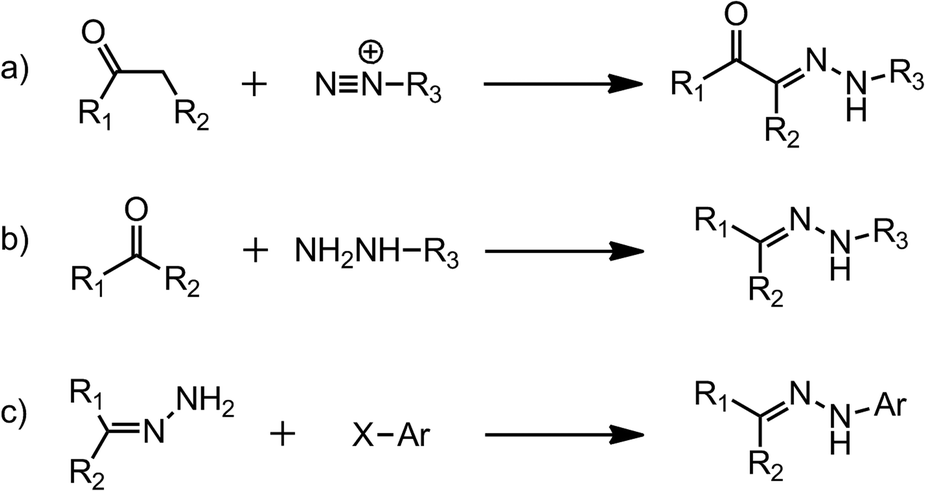 | ||
| Scheme 1 The synthesis of hydrazones via (a) the Japp–Klingemann reaction; (b) hydrazine–ketone/aldehyde condensation; and (c) aryl halide substitution. | ||
In this review we will focus on the use of the hydrazone group in: (i) molecular switches,14 (ii) metallo-assemblies,15 and (iii) sensing applications,16 in order to showcase the diversity of this functional group in the context of supramolecular chemistry. In the first section we will focus on the use of light and chemical inputs in activating the E/Z isomerization in these systems. We will also discuss how the acidic N–H proton in hydrazones differentiates it from imines, by leading for example to a new isomerization mechanism that is not present in the latter. Moreover, we will demonstrate how these structurally simple systems can be used for complex functions, such as controlling the properties of liquid crystals and the morphology of macroscopic assemblies. In the second part we will show how different metallo-assemblies can be formed using different combinations of hydrazone derivatives and metals. Moreover, we will demonstrate how such systems can be stimulated to undergo dynamic shape changes, and be used as metallo-dynamers and gels. Finally, we will show how the nucleophilicity of the hydrazone nitrogens, electrophilicity of the imine carbon, and acidity of the N–H proton can be utilized in highly sensitive and selective sensing of cations, anions and even neutral species. A common theme in all these research directions is the acidic N–H proton, which as discussed below, enables the switching, metal coordination, and sensing applications. This functional group also distinguishes and differentiates the hydrazone from the imine group.
2 Configurational switching
The control of molecular level motion is the primary objective of the field of molecular switches and machines.14 With time, there will be a need for the incorporation of more and more complicated functions into such systems. This requirement will necessitate the expansion of the currently available tool box in the field, as far as it relates to the type of intricate motions that we can control externally. The hydrazone functional group is perfectly suited to address this challenge as its E/Z isomerization (i.e., configurational switching) can be activated by both light and chemical inputs. This dual control over the rotary motion of a molecular system showcases the uniqueness of the hydrazone functional group. Here we will summarize some of the recent advances in the use of hydrazones in modulating the motion of molecules and supramolecular systems.2.1 Photoswitching
The intrinsic nature of the CN bond leads to configurational isomerism in hydrazones, which can exist in either the E or Z forms in solution.17 It has long been established that UV light can provide enough energy for the E → Z isomerization of the CN bond,18,19 although in most cases the obtained Z isomers are ephemeral species. Courtot et al.20 studied the photoisomerization of a series of 1,2,3-tricarbonyl-2-arylhydrazones (TCAHs) derived from β-diketones and β-ketoesters, and found that the less favored Z configuration can be stabilized through intramolecular H-bonding, making it kinetically stable (Scheme 2a). These studies have opened the way for the use of hydrazones as photochemically controlled configurational switches. It was proposed by Courtot et al.20 that these systems isomerize through a photo- or thermally-activated rotational mechanism. This finding prompted Tamaki et al.21 to try and use these systems as command molecules for the alignment of liquid crystals (LCs) on surfaces (Scheme 2b). These efforts showed that the configurational switching in hydrazones can indeed be used in switching the LC alignment between homeotropic and parallel modes using light and heat as inputs.
 | ||
| Scheme 2 The photo- and thermal isomerization of TCAHs (a) in solution and (b) on a surface. | ||
On the other hand, Smirnov et al.22 observed that acyl hydrazones can also undergo photoisomerization, which proceeds through the weakening of the CN bond by the electronic transition from the π-orbital of the amine moiety to the π*-orbital of the whole molecule, namely, the π2–π1* transition. This photoisomerization was observed in the solid state as well.23
Lehn et al.24 demonstrated that some pyridyl-2-aldehyde phenylhydrazones (1–3) and acyl hydrazones (7–10, Scheme 3) can also undergo E → Z photoisomerization upon UV light irradiation with up to 94% quantum yield (1-Z) in deuterated methanol (CD3OD). The Z isomers are highly resistant to isomerization because of the intramolecular H-bond; for example, no 1-E can be detected in the CD3OD solution of 1-Z kept at 45 °C for five days, and only refluxing in CD3OD or the addition of 20 mol% TFA can regenerate 1-E from 1-Z.
 | ||
| Scheme 3 Photo-induced E → Z configurational switching in hydrazones 1–3 and 7–10. | ||
On the other hand aryl hydrazones 4–6 (Scheme 3), which are structurally similar to 1–3, but cannot form intramolecular H-bonds nor can take advantage of their stabilization, do not undergo appreciable E ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) Z photoisomerization. Interestingly, the E isomers of switches 1–3 and 7–10, are fluorescent, whereas the Z isomers are not. This difference in photophysical properties allows for the toggling of the readout signal using the E → Z isomerization process.
Z photoisomerization. Interestingly, the E isomers of switches 1–3 and 7–10, are fluorescent, whereas the Z isomers are not. This difference in photophysical properties allows for the toggling of the readout signal using the E → Z isomerization process.
As tridentate ligands, the E isomers of hydrazones 1 and 7–10 can coordinate to transition metal cations via the NNN and NNO binding sites, respectively. This process locks the E configuration in place. Hence, the binding of 1-E with Zn2+ yields the metal complex Zn(1-E) that cannot undergo photoisomerization. The addition of a free ligand such as tris(2-aminoethyl)amine (Tren) to the mixture regenerates 1-E by removing Zn2+, and unlocks the switch and allows it to photoisomerize again.
In contrast to the stable coordination complex formed between Zn2+ and 7–10, potassium thiocyanate (KSCN) forms a weak 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex with 7 (K(7-E)), with a binding constant of 24.8 M−1 in acetonitrile (Scheme 4).25 When subjected to UV irradiation, K(7-E) can be converted into 7-Z, accompanied by the simultaneous release of K+. Upon heating at 40 °C, the isomerization from 7-Z to 7-E is accelerated by a factor of 6 in the presence of 1 equiv. of KSCN, most likely driven by the formation of K(7-E). More importantly, complex K(7-E) forms a crystalline metallosupramolecular assembly in the presence of SCN−, which deforms to amorphous stains upon photoisomerization. This process is reversible as the crystals can be regenerated through recrystallization. This study demonstrates how molecular switches can be used in controlling the morphology of macroscopic assemblies.
:1 complex with 7 (K(7-E)), with a binding constant of 24.8 M−1 in acetonitrile (Scheme 4).25 When subjected to UV irradiation, K(7-E) can be converted into 7-Z, accompanied by the simultaneous release of K+. Upon heating at 40 °C, the isomerization from 7-Z to 7-E is accelerated by a factor of 6 in the presence of 1 equiv. of KSCN, most likely driven by the formation of K(7-E). More importantly, complex K(7-E) forms a crystalline metallosupramolecular assembly in the presence of SCN−, which deforms to amorphous stains upon photoisomerization. This process is reversible as the crystals can be regenerated through recrystallization. This study demonstrates how molecular switches can be used in controlling the morphology of macroscopic assemblies.
 | ||
| Scheme 4 Photo-induced E → Z configurational switching in K(7-E) with simultaneous release of the K+ cation. | ||
2.2 pH-activated switching
As mentioned above, TCAHs20 exist in solution as an equilibrated mixture of intramolecularly H-bonded E and Z isomers (Scheme 5a). The E → Z (or vice versa) isomerization proceeds via hydrazone-azo tautomerization followed by rotation about a C–N single bond. The position of the equilibrium can be altered by catalytic amounts of acid and base, albeit the process does not proceed to full completion, because the H-bond accepting capability of the two carbonyl groups is similar, providing insufficient impetus to drive the equilibrium to full extent in either direction. This property is of course not ideal for molecular switch related applications. | ||
| Scheme 5 (a) Possible mechanism for the acid-catalyzed tautomerization in TCAHs (a similar mechanism applies to the base catalyzed process); (b) the proposed tautomerization followed by rotation mechanism in switches 11 and 12, with intermediates and the transition state (TS) structure. | ||
The replacement of one of the carbonyl groups in TCAHs with a pyridyl group, a much stronger H-bond acceptor (e.g., 11 and 12), has led to systems having an appreciable bias in favor of the E configuration (Scheme 6).26,27 The basic nature of the pyridyl group allows for the modulation of its H-bond accepting ability using acid input, which in turn makes the Z isomer the sole configuration upon protonation. This approach has provided prototypical molecular switches that can be fully, effectively, and reversibly switched between the E and Z configurations, using acid and base inputs.
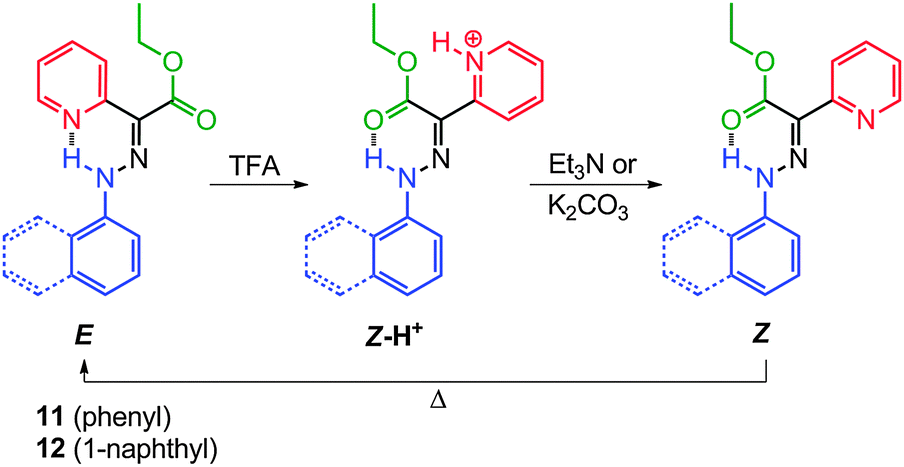 | ||
| Scheme 6 The acid–base modulated E ⇌ Z switching in 11 and 12. | ||
Compounds 11 and 12 have high E:Z isomer ratios (93:7 and 97:3, respectively), in deuterated acetonitrile (CD3CN), indicating that the E isomers are more stable than the Z ones by 1.5 and 2.0 kcal mol−1, respectively (Scheme 6). Similar to TCAHs, the energy barrier for the interconversion between the E and Z isomers of 11 and 12 falls within the timeframe of 1H NMR spectroscopy (ca. 24 kcal mol−1), leading to two sets of signals in the 1H NMR spectrum. The signals are usually very well separated, especially those for the intramolecular H-bonded N–H protons, which usually have a ca. 2 ppm upfield shift going from the E to Z isomer. Compounds 11-E and 12-E can be converted quantitatively to 11-Z-H+ and 12-Z-H+, respectively, upon treatment with trifluoroacetic acid (TFA). When 11-Z-H+ and 12-Z-H+ are passed over a plug of K2CO3, the “metastable” Z isomers (11-Z and 12-Z) are observed, which thermally equilibrate to give back 11-E and 12-E in their original isomer ratios, thus completing a full switching cycle.
Mechanistic studies have shown that 11-E and 12-E form the kinetic intermediates 11-E-H+ and 12-E-H+, respectively, upon protonation before isomerizing into 11-Z-H+ and 12-Z-H+via a single rate limiting step. The first-order rate constants of the Z → E process (Scheme 6) show dependence on solvent polarity with the associated enthalpies of activation ranging from −30 to −60 kcal mol−1. Based on calculations, these results stem from an increase in the dipole moments of the transition states as compared to the ground states. These data, in conjugation with the DFT calculations, led to the proposal of a tautomerization followed by rotation mechanism for the E Z isomerization in these systems (Scheme 5b).
The delicate balance between the two anchors (i.e., the pyridyl and ester H-bond acceptors) in the rotor part proves to be crucial for the efficient function of the switches.28 Replacing the ester group in 11 with hydrogen (13), a methyl (14) or a cyano (15) group leads to a dominant configuration in which the pyridyl ring is not involved in H-bonding (Scheme 7). On the other hand, the substitution with an acetyl group (16) leads to incomplete acid-activated switching. These results show that a moderate H-bond acceptor is necessary to ensure the correct isomer ratio and switching efficiency in these systems.
 | ||
| Scheme 7 2-Pyridylarylhydrazones 13–16 with different rotors (only the stable isomer(s) in solution and their protonation products are shown). | ||
Replacing the 1-naphthyl ring in 12 with an 8-quinolinyl one leads to the tristable switch 17 (Scheme 8).29 The introduction of an additional H-bond acceptor (quinolinyl nitrogen) in the stator moiety locks its conformation and opens the way to control its position as a function of pH (i.e., rotation around the C–N bond upon protonation).
 | ||
| Scheme 8 The acid–base induced configurational and conformational switching in 17. | ||
The initial protonation of 17-E by TFA leads to 17-Z-H+ through a configurational change (i.e., rotation around the CN bond) because pyridyl is a stronger base than quinolinyl (Scheme 8). Further protonation leads to rotation about the C–N single bond generating a new conformational isomer, 17-Z-H22+, that retains the Z configuration. After deprotonation with K2CO3 or triethylamine (Et3N), both 17-Z-H+ and 17-Z-H22+ convert into a neutral metastable species 17-Z, which eventually equilibrates to give back the thermodynamically stable 17-E isomer.
The configuration and conformation of the system can be modulated based on the sequence in which the acid and base are added, leading to a switch that can be prompted to perform precisely controlled rotations around two different axles. Therefore, a molecular choreography can be achieved by programming the quantity of acid and base used, leading to complicated sequences of rotations in 17.
2.3 Switching via coordination-coupled deprotonation (CCD)
Coordination-coupled deprotonation (CCD) is a process commonly observed in biological systems whereupon the coordination of transition metals with protein residues leads to the release of proton(s) to the environment.30 CCD provides a great potential for operating chemically activated molecular switches under mild or neutral conditions.31,32Switch 17-E can bind to Zn2+ through its quinolinyl and hydrazone N–H nitrogens, leading to the deprotonation of the latter, and subsequent transfer of the proton to the pyridyl nitrogen (Scheme 9).31 Overcrowded in a positively charged environment, the protonated pyridyl group is forced to rotate around the CN bond, leading to the coordination of Zn2+ with the ester group (Zn(17-Z)). Tetra-n-butylammonium cyanide (nBu4NCN) can then be used in removing Zn2+ from Zn(17-Z), which regenerates 17-E, and renders the switching process fully reversible.
 | ||
| Scheme 9 Proposed mechanism for the E → Z isomerization of 17 through CCD. | ||
As an orthogonal input for switches that are only responsive to protons (e.g., 11), Zn2+ can be used to turn CCD-activated switches into proton sources for subsequent switching events.32 This has been demonstrated with 18, which is similar to switch 17 but has a methylimidazolyl group in the rotor instead of a pyridyl one (Scheme 10). As in the case of 17, the binding with Zn2+ converts 18-E into Zn(18-Z-H+), and when this switching process occurs in the presence of 11-E, it yields Zn(18-Z) and 11-Z-H+. Interestingly, 11-E is protonated and switched by a proton relay from Zn(18-Z-H+), upgrading a single CCD-activated switching event into a switching cascade. Both Zn(18-Z) and 11-Z-H+ can be reinstated to 18-E and 11-E, respectively, using nBu4NCN (Scheme 10).
 | ||
| Scheme 10 The reversible switching cascade triggered by Zn2+ coordination. | ||
Although the only difference between 17 and 18 is the N-heteroaromatic group in the rotor, the switching cascade is not observed for 17. In order for the cascade to proceed, there has to be a significant decrease in the basicity of the methylimidazolium group in 18 so that it can protonate 11. The electrostatic repulsion between Zn2+ and the imidazolium group results in the required reduction in the pKa of Zn(18-Z-H+). However, this same process happens in 17 as well, and so another factor must be at play. It turns out that the highly planar skeleton in Zn(17-Z) leads to a very strong H-bonded pyridyl proton, which is not available for proton relay. On the other hand the methyl group in 18 pushes the system out of planarity, leading to a weak H-bonded proton that participates in the switching cascade.
2.4 Switching beyond the molecular level
One of the major objectives of the field of molecular switches and machines is the propagation of the structural changes on the molecular level to dimensional scales beyond their own sizes, i.e., from microscopic to mesoscopic and macroscopic scales.14Liquid crystals are supramolecular assemblies whose properties have been successfully manipulated by photochemically activated switches,33 and less so by chemically activated ones. Being highly modular, the hydrazone switch 11 can be easily converted into a mesogenic liquid crystalline system (19), having a cholesteryl group in each of the rotor and stator parts (Scheme 11).
 | ||
| Scheme 11 The acid–base modulated E ⇌ Z switching in 19, accompanied by a change in the relative orientation of the cholesteryl units. | ||
Hydrazone 19 exists in an E/Z isomer ratio of 91:9 in deuterated dichloromethane (CD2Cl2) solution, and exhibits a similar switching behavior to 11 upon exposure to acid/base.34 DFT calculations using the hybrid B3LYP-D3 (6-31G*) yielded minimum energy structures for 19-E, where the cholesteryl units are oriented in a syn-fashion (Fig. 2a), and 19-Z-H+, in which the cholesteryl units are oriented in an anti-fashion (Fig. 2b). Based on the calculations, the protonation-driven switching process leads to a 10.4% increase in the solvent accessible surface area.
 | ||
| Fig. 2 DFT (B3LYP-D3/6-31G*) calculated structures of (a) 19-E and (b) 19-Z-H+; and, (c) cross-polarized view of the reflected color upon switching 19/NP5. | ||
Doping of 19 (5 wt%) into the achiral LC material Nematic Phase 5 (NP5) yields a chiral nematic phase. Upon exposure to TFA, 19/NP5 (5 wt%) in 90° twisted cells changes its reflected color from green to purple under cross-polarized view (Fig. 2c), accompanied by a decrease in the helical twisting power (HTP) from 56 to 46 μm−1 (wt%). The color change can be reversed by exposing the mixture to Et3N, which also reverts the HTP to 56 μm−1 (wt%).
The observed change in the reflected color originates from the structural change in the LC supramolecular assembly brought about by the pH activated rotary motion in the chiral switch, and the accompanied increase in the solvent accessible surface area. This process exemplifies the use of a chemically activated molecular switch in effecting mesoscopic level response in bulk materials.
3 Dynamics of hydrazone-based metallo-assemblies
Metallogrids,15 short for metallosupramolecular grid complexes, are oligonuclear metal complexes where the ligands are connected in the form of square or rectangular grids by spatially well-defined metal cation arrays. Among the variety of molecular scaffolds for building metallogrids, hydrazone-based ligands have been by far the most widely used systems, especially bis-hydrazones connected by symmetric heteroaromatic cores, including pyridine, pyrimidine and triazine. Two recent review articles3a,15b have comprehensively covered progresses in the design, synthesis, properties and applications of metallogrids, most of which are hydrazone-based systems (Scheme 12). Hence, we will direct our focus here on the dynamic processes of such metallogrids, such as shape manipulation, stimuli-induced structural interconversion and other dynamic phenomena. | ||
| Scheme 12 Structures of hydrazone ligands used in metallo-assemblies. | ||
3.1 Dynamic shape adaptation
Oligomeric hydrazone ligands usually exist as contracted helical strands in solution.35,36 In the presence of large excess of metal cations, these helical coils extend to give linear shaped strands, in order to accommodate the metals. This process can bring about significant changes in the shape of the ligands. Stoichiometry can be a determining factor in controlling the shape of the obtained metalloassemblies, especially when the ligands provide multiple binding modes. For example, ligand 20 forms a [2 × 2] metallogrid35 [Pb4204]8+ and a double helicate36 [Ag2202]2+ with 1 equiv. of lead(II) triflate (Pb(OTf)2) and silver(I) tetrafluoroborate (AgBF4), respectively (Fig. 3). When the amount of metal cations is increased, both assemblies lead to the formation of 1:2 rack-type linear complexes [Pb220]4+ and [Ag220]2+. Similarly, the [2 × 2] metallogrid [Ag4214]4+ can be converted into the 1:2 linear rack [Ag221]2+. Meanwhile, the addition of hydroxymethyl37,38 and acryloyl39 groups to the terminal pyridines in bis-hydrazone scaffolds such as in 20, can also affect the self-assembly behavior. For example, these groups inhibit the formation of [2 × 2] grids by blocking access to the metal centers. Moreover, the hydroxymethyl groups contribute to the extended supramolecular structure of the complexes through metal coordination and H-bonding.40
 | ||
| Fig. 3 Ligand 20 forms various stoichiometry-dependent metallo-assemblies. | ||
By extending the coordination units in the ligands, the motional shape adaption can be used to form larger assemblies.35 When ligand 22 is treated with large excess of Pb(OTf)2, the initial [4 × 4] metallogrid [Pb16228]32+ turns into a linear complex [Pb422]8+ (Fig. 4).
 | ||
| Fig. 4 Stoichiometry-controlled motional shape adaption in 22. | ||
Ligands 23 and 2441 on the other hand undergo a three-stage conversion from double pincer to pincer and finally to a linear rack when the corresponding ligand/Pb2+ ratios are changed from 1:2 to 1:1, and then to 2:1 (Fig. 5).
 | ||
| Fig. 5 Stoichiometry-controlled three-stage complex conversion in 23 and 24. | ||
Shape modulation can also be achieved by the isosteric modification of binding site geometry in the ligands. For example, ligand 2542 is an isosteric isomer of 21, and although both lead to linear racks upon coordination with Zn2+, 25 has to undergo a “twirling”-type motion, whereas 21 undergoes a “flapping”-type motion. Such effects become more prominent when the ligands grow longer, and have more coordination units, such as in the case of 26 with Pb2+ (Fig. 6).
 | ||
| Fig. 6 “Twirling” motion in 26 upon coordination to Pb2+. | ||
Integrating the bis-hydrazone moiety into macrocycles converts the local shape changes upon coordination into overall conformational switching of the macrocyles.43 The “S”-shaped 44-membered [2 + 2] macrocycles 27 and 28 each consist of two bis-acylhydrazone units connected by flexible linkers. Upon coordination with La3+, Eu3+ and Dy3+, 27 and 28 twist from their “S” conformation to accommodate the metal cations with their two NNNOO-binding units (Fig. 7).
 | ||
| Fig. 7 The crystal structures of 27 (top) and the complex [Eu27]3+ (bottom). | ||
When the element of two-fold symmetry is lost in basic hydrazone ligands, they can potentially be used for preparing homochiral metalloassemblies.44,45 Hydrazone 4,44 studied as a candidate for configurational switching, is also a tridentate ligand that can form a tris-chelated complex [Fe43]2+ with iron(II) perchlorate (Fe(ClO4)2). As a result of the steric hindrance originating from the C-methyl group in 4, [Fe43]2+ is obtained only as an optically pure fac-Λ isomer (Fig. 8). The unsymmetrical bis-hydrazone 29 reacts with mercury(II) triflate (Hg(CF3SO3)2) to give both parallel and antiparallel binuclear double helicates [Hg2292]4+ in solution.45 In the solid state, however, [Hg2292]4+ selectively crystallizes into the antiparallel enantiomeric pair because of the favorable H-bonds and π–π interactions in this structure (Fig. 8).
 | ||
| Fig. 8 The crystal structures of [Fe43]2+ (fac-Λ isomer) and [Hg2292]4+ (left-handed form, right). | ||
The addition of other binding features to the hydrazone ligands allows for the construction of high order metalloassemblies. Ligand 3046 possesses a terminal oxime group, in addition to the NNNO coordination site common for acylhydrazone ligands, and forms a dodecanuclear 3 × [2 × 2] grid with Ni2+. The three [2 × 2] grids are fused together through a total of six deprotonated μ-(N,O) oxime bridges from three pairs of orthogonal units of 30 (Fig. 9).
 | ||
| Fig. 9 The crystal structure of the 3 × [2 × 2] grid formed by 30 and Ni2+. | ||
3.2 Stimuli-induced structural interconversion
The structural interconversion between metallogrids and other forms of supramolecular assemblies takes advantage of the structural flexibility of the hydrazone ligands and the tunable coordination modes of the metal centers. For instance, ligands 2347 and 2441 are triazine-based bis-hydrazones that can host metal cations in linear or “U”-shaped conformations (Fig. 10). When dissolved in nitromethane (CH3NO2), 23 and 24 assemble with cobalt(II) tetrafluoroborate (Co(BF4)2) into [2 × 2] grids ([Co4234]8+ and [Co4244]8+) in which 23 and 24 extend their skeletons in a linear shape. Upon switching the solvent from CH3NO2 to CH3CN, the grids convert into mononuclear helical pincer complexes ([Co(CH3CN)223]2+ and [Co(CH3CN)224]2+) where 23 and 24 contract to adopt a “U” shape. | ||
| Fig. 10 The solvent induced dynamic interconversion between [2 × 2] grids and pincers. | ||
Ligand 31,48 on the other hand, can form a tetranuclear [2 × 2] metallogrid [Cu4314]8+ and a double helicate [Cu2312]2+ with copper(II) triflate (Cu(CF3SO3)2) and copper(I) triflate (Cu(CD3CN)4CF3SO3), respectively, as a result of the difference in the coordination modes of Cu2+ and Cu+ (Fig. 11). The dynamic conversion of [Cu4314]8+ into [Cu2312]2+ is achieved by reduction with ascorbic acid in the presence of triflic acid (CF3SO3H) followed by neutralization with Et3N. Alternatively, [Cu2312]2+ can be transformed into [Cu4314]8+ by either aerobic oxidation during crystallization or the substitution of Cu2+ with Cu+.
 | ||
| Fig. 11 The reversible transformation between the [2 × 2] metallogrid [Cu4314]8+ and the double helicate [Cu2312]2+. | ||
3.3 Polymers and gels
The versatility of the hydrazone ligands enables their use in advanced applications as well, such as metallosupramolecular polymers and gels. A series of ditopic acylhydrazone ligands (32–34) featuring oligo-dimethylsilanoxyl-based spacers were used as monomers for preparing linear coordination polymers with Ni2+ and Zn2+.49 A simultaneous deprotonation occurs when two hydrazone NNO sites bind the metal cations, leading to the formation of neutral polymers (35–38, Scheme 13), with average molecular weights ranging from 15000 to 45000 g mol−1. While 36 is obtained as a polymeric gum, 35, 37 and 38 give transparent free-standing films. The Zn2+-based polymers (35–37) are fluorescent whereas the Ni2+-based 38 is nonfluorescent, possibly as a result of quenching by Ni2+.
 | ||
| Scheme 13 The structures of neutral metallo-polymers 35–38. | ||
Given the reversibility of the coordination connectivity, homopolymers can be used for preparing polymer blends by dynamic exchange. For example, when stacks of 36 and 37 in the form of neat films are heated to 50 °C for 24 h, the constitutional metal–ligand exchange between 36 and 37 yields the heteropolymer 39. This dynamic process opens up a new approach towards the construction of metallodynamers with tunable mechanical and optical properties,50 as compared to their covalently built counterparts.51
The introduction of long urea side chains to the 4,6-disubstituted pyrimidyl-based bis-acylhydraone skeleton leads to precursor 40,52 which can form a [2 × 2] metallogrid gelator [Zn4404]8+ upon coordination with Zn2+. With a minimum concentration of gelation (MCG) of 18 mg ml−1 in toluene at room temperature, [Zn4404]8+ forms organogels from the fibrillar network of metallogrid-based polymers (Fig. 12). This approach results in neutral organometallogels that are not affected by counterion effects, and can lead to [2 × 2] metallogrids with potentially interesting optical, electronic and magnetic properties.
 | ||
| Fig. 12 (a) Photograph of the metallo-supramolecular polymer organogel formed by [Zn4404]8+ at 20 mg ml−1 in toluene, and (b) the SEM image of a dried sample of the organogel (scale bar represents 1000 nm). Reproduced from ref. 52 with permission from the Centre National de la Recherche Scientifique (CNRS) and The Royal Society of Chemistry. | ||
4 Sensing applications
The broad range of the chemical and supramolecular reactivities of the hydrazone functional groups (Fig. 1) enables their use in the detection of cations, anions and other species. Here we summarize several recent examples of the use of hydrazones in sensing applications.4.1 Cation sensing
In 2006, Tong et al.53 reported an “off–on” fluorescent chemosensor for Cu2+ based on a rhodamine B-based hydrazone (41). As a spirolactam, 41 is weakly fluorescent, and upon coordination with Cu2+, the lactam C–N bond breaks and the complex [Cu41]+ is formed via coordination with the ONO binding site (Scheme 14). The ring opening reaction enhances the fluorescence emission of 41, which enables the fluorescence sensing of Cu2+ at the submicromolar level in buffered aqueous solutions. The binding constant between 41 and Cu2+ is larger than 106 M−1 in Tris-HCl (10 mM, pH = 7.0) and water–CH3CN (v/v = 1:1) buffer, which leads to a detection range between 25 nM and 3.3 μM, and high selectivity against Fe3+, Fe2+, Zn2+, Pb2+ and Hg2+.
 | ||
| Scheme 14 The ring-opening reaction of 41 upon binding to Cu2+. | ||
By replacing the rhodamine B group in 41 with a rhodamine 6G group (42), Tong et al.54 enhanced the Cu2+ sensing performance in both absorptiometric and fluorometric methods (Scheme 15). In sodium acetate–acetic acid (10 mM, pH = 7.0) water–ethanol (v/v = 1:1) buffer, 42 offers a limit of detection (LoD) of 10 nM with a linear range of 0.05–5.00 μM in the absorptiometric method, whereas in the fluorometric one it has a LoD of 25 nM with a linear range of 0.1–3.6 μM.
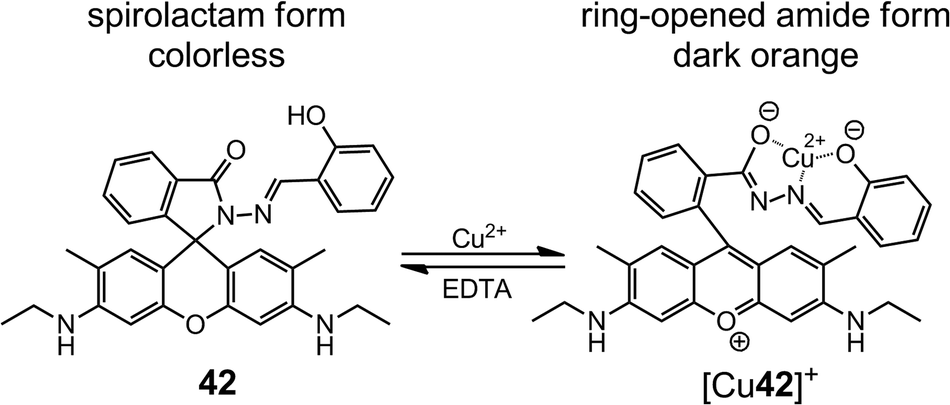 | ||
| Scheme 15 The ring-opening reaction of 42 upon binding to Cu2+. | ||
When the chromic group is changed to fluorescein (43),55 the absorption profile of 43 becomes pH-dependent because of the two phenolic groups in the fluorescein unit (Scheme 16). Therefore, 43 can behave as a colorimetric YES and INHIBIT logic gate that can be modulated by both the concentration of Cu2+ and pH.
 | ||
| Scheme 16 The ring-opening reaction of 43 upon binding to Cu2+, and the different products obtained at various pH values. | ||
The lactam ring-opening reaction in rhodamine-derived hydrazones has since gained enormous popularity in the development of optical sensors for metal cations. The coordination strength of the hydrazones can be tuned by changing the aldehydes or ketones used in the condensation with the rhodamine hydrazides. For instance, 4456 is prepared by the condensation between rhodamine B hydrazide and 4-N,N-diethylaminosalicylaldehyde, instead of salicylaldehyde (41), which switches the selectivity of the metal from Cu2+ to Zn2+. By forming a 1:1 complex with Zn2+, 44 is able to detect Zn2+ in the concentration range of 0–10.0 μM with an LoD of 0.05 μM in HEPES (10 mM, pH = 7.0) water–ethanol (v/v = 1:9) buffer. The selectivity towards Zn2+ and sensing capability can be further enhanced by combining fluorescein hydrazide and 8-formyl-7-hydroxyl-4-methylcoumarin (45),57 which leads to a LoD of 15 nM (Scheme 17).
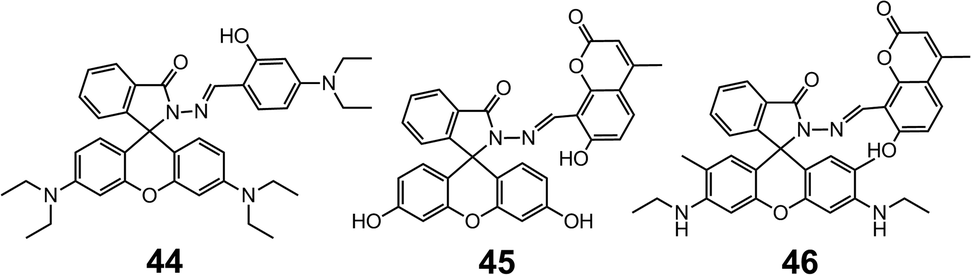 | ||
| Scheme 17 The structures of hydrazones 44–46. | ||
Interestingly, when the fluorescein group in 45 is replaced by a rhodamine 6G group (46),58 the system becomes highly selective for Ca2+ over other alkali-, alkaline earth- and transition metal cations. The binding with Ca2+ is accompanied by a 64-fold increase in fluorescence, leading to a LoD of 0.75 μM, and a linear working concentration range of 20–60 μM (Scheme 17).
The substitution of the phenol group in 42 with electron-donating (methyl, 47) and electron-withdrawing (dicyano, 48) allylic groups59,60 leads to sensors having great affinity towards Pd2+ cation. The coordination proceeds with 2:1 stoichiometry through a similar spirolactam ring opening process. As fluorometric sensors, 47 and 48 have LoDs of 1.80 × 10−7 and 1.70 × 10−6 M, repsectively, and excellent selectivity for Pd2+ over a variety of metal cations, especially other platinum-group elements (PGEs), such as Pt2+, Ru3+ and Rh3+.
By introducing weak field ligands such as thiol and carboxylate groups, spirolactam-based hydrazones can effectively bind with the soft metal cation Hg2+. Yang and Xu et al.61 reported the sensing of Hg2+ with 49, which was obtained from 2-furaldehyde and thiooxorhodamine B hydrazide. The fluorescence of 49 shows a 120-fold enhancement upon binding to Hg2+, which proceeds in 2:1 stoichiometry in sodium acetate–acetic acid (pH = 7.0) water–DMF (v/v = 1:1) buffer with a detection range of 0.1–1 μM. Sensor 49 was also used for the in vitro Hg2+ fluorescence imaging in Rat Schwann cells (Fig. 13), showing the efficacy of these systems in biological applications (Scheme 18).
 | ||
| Fig. 13 Bright-field transmission images (a–c) and fluorescence images (d–e) of Rat Schwann cells incubated with 0 μM, 10 μM and 50 μM of Hg2+ ions for 30 min, respectively, in the presence of 49 (10 μM) upon excitation with green light. Reproduced from ref. 61. | ||
 | ||
| Scheme 18 The structures of hydrazones 47–49. | ||
Park and Yoon et al.62 prepared hydrazones 50 and 51 through the condensation of rhodamine B hydrazide with 2-mercaptobenzaldehyde and 2-carboxybenzaldehyde, respectively (Scheme 19). Compounds 50 and 51 form 2:1 and 1:1 complexes with Hg2+ in a water–CH3CN (v/v = 99:1) mixture, respectively, with their corresponding LoDs being 1 and 4.2 nM, respectively. Moreover, both 50 and 51 were used in the in vivo imaging of nanomolar quantities of Hg2+ in the nematode Caenorhabditis elegans using fluorescence microscopy.
 | ||
| Scheme 19 The binding modes of hydrazones 50 and 51 with Hg2+, respectively. | ||
Simple hydrazones can also bind to metals through their NNN or NNO binding sites. For example, 2-pyridylhydrazone 5263 forms a stable 1:1 complex with Pd2+ (Fig. 14) with an association constant of 5.5 × 104 M−1 in methanol. Upon binding with Pd2+, a drastic color change from yellow to red can be observed by the naked eye, originating from the π–π* transitions in the complex [Pd52]2+.
 | ||
| Fig. 14 The crystal structure of the complex [Pd52Cl]. | ||
Hydrazone 53 is obtained by replacing the pyridine-2-carboxaldehyde in 52 with quinoline-2-carboxaldehyde, which acts as a fluorescence emitter.64 In alkaline methanol, the fluorescence of 53 can be quenched by Pd2+ in a ratiometric manner, making it a fluorescence “on–off” sensor for Pd2+. The coupling between 2-imidazolyl hydrazine and pyridine-2-carboxaldehyde leads to 54,65 which selectively binds Cu2+ through the imine, pyridyl and imidazolyl nitrogens. Sensor 54 can be used in detecting 2.0–20 μM of Cu2+ in a water–methanol (v/v = 99:1) mixture (Scheme 20).
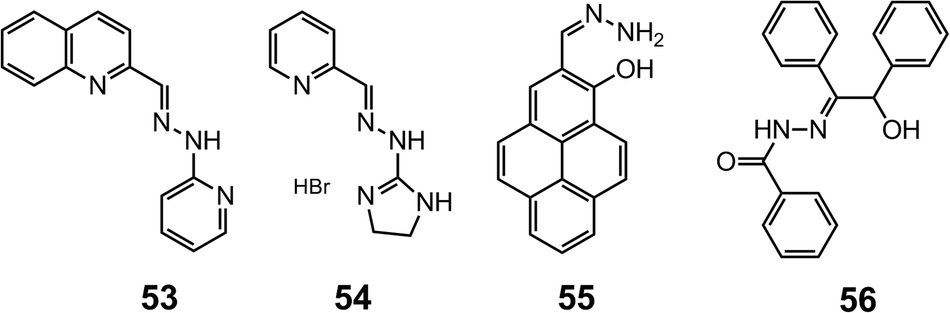 | ||
| Scheme 20 The structures of hydrazones 53–56. | ||
The simple non-substituted hydrazone 5566 is built on a pyrene fluorophore and can bind Zn2+ with its NNO binding site. Upon the formation of [Zn55]2+, the fluorescence is drastically enhanced because the photo-induced electron transfer (PET) from the hydrazone nitrogen to pyrene is blocked. Compound 55 is able to detect intracellular Zn2+ at the micromolar level in HaCaT and pancreatic β cells in vitro.
Acylhydrazones offer another NNO or ONO binding motif for metals. Hydrazone 5667 can be fabricated into microelectrodes with polyvinyl chloride (PVC) support for Er3+ sensing. This ion-selective electrode can achieve Er3+ detection in the range of 1.0 mM–0.3 nM with a LoD of 0.2 nM within the pH range of 3.0–9.0 using potentiometic methods.
Yang et al.69 reported the ratiometric detection of Cu2+ with 57, which has an ONO binding site (Fig. 15). Fluorescence quenching measurements show that 57 is able to selectively sense Cu2+ at 1 μM level in CH3CN through photo-induced metal-to-ligand electron/energy transfer. The sensitivity of acylhydrazone-based Cu2+ sensors can be improved by integrating the mono ligand into a triphenylamine scaffold (58).68 Compound 58 can form a 1:3 complex with Cu2+ through each of the three NOO binding sites. The fluorescence of 58 suffers drastic quenching when bound to Cu2+ because of the reverse PET process from the triphenylamine to the electron-deficient hydrazone nitrogens and phenolic oxygens. With an LoD for Cu2+ of 9.4 nM in a water–CH3CN (v/v = 7:3) mixture, 58 can be used for the fluorescence imaging of micromolar amounts of Cu2+ in HeLa cells with low cytotoxicity (Fig. 16) (Scheme 21).
 | ||
| Fig. 15 The crystal structure of the complex [Cu57(DMF)ClO4]. | ||
 | ||
| Fig. 16 (a) Bright-field transmission image of HeLa cells; (b) fluorescence images of Cu2+ in HeLa cells incubated with 58 (1 μM), and (c) when stained with Hoechst 33342. Reproduced from ref. 68. | ||
 | ||
| Scheme 21 The structure of hydrazone 58. | ||
The introduction of pyrazinyl (59, Scheme 22)70 and quinoxalinyl (60, Scheme 22)71 groups into the hydrazones has led to the development of highly selective and sensitive sensors for Al3+ and Ni2+, respectively. While compound 59 is able to detect Al3+ at the submicromolar level, compound 60 has a LoD of 1.47 μM for Ni2+.
 | ||
| Scheme 22 The binding modes of hydrazones 59 and 60 with Al3+ and Ni2+, respectively. | ||
Tweezer-type72 hydrazones 6173 and 6274 (Scheme 23) have also been developed as fluorescence “turn-on” cation sensors, by taking advantage of the restriction of the CN bond rotation, which otherwise contributes to non-radiative decay. For instance, in the presence of Zn2+, 6173 experiences a 49-fold increase in quantum yield, and this occurs with great selectivity especially over Cd2+. Similarly, 6274 exhibits a 250-fold increase in quantum yield upon binding to Al3+, providing a LoD of 0.77 μM in a water–methanol (v/v = 1:1) solution.
 | ||
| Scheme 23 The binding modes of hydrazones 61 and 62 with Zn2+ and Al3+, respectively. | ||
4.2 Anion sensing
The acidic nature of the hydrazone N–H group allows for the detection of anionic species through H-bond interactions that alter the photophysical properties of the system. These spectroscopic changes make hydrazones ideal for anion sensing applications, especially for the fluoride anion.75–77 Fluoride usually forms H-bonded adducts [R–N–H⋯F−] with hydrazones, and in certain cases, the interaction between the anion and the hydrazone N–H proton is so strong that deprotonation occurs, leading to the formation of HF2− species, and further enhancement of the photophysical changes. For example, the indole-derived hydrazone 6378 can form a 1:1 complex 63⋯F− in the presence of less than 2 equiv. of F−, above which the hydrazone N–H is deprotonated (Scheme 24). The association with F− causes a red-shift and an increase in the UV-vis absorption and fluorescence emission of 63, respectively. This property makes 63 a dual-channel fluoride receptor. Similarly, hydrazone 6479 has a reduced HOMO–LUMO gap in its fluoride complex 64⋯F−, which gives rise to a large red-shift to the near IR region in its UV-vis absorption spectrum (Scheme 25). This hydrazone is a sensitive sensor for F−, and allows for the “naked-eye” detection of the anion at levels as low as 50 ppm in toothpaste solution samples.
 | ||
| Scheme 24 The reaction between 63 and F−. | ||
 | ||
| Scheme 25 The structure of the 64⋯F− adduct. | ||
The imine (CN) bond in hydrazones can be susceptible to nucleophilic attack, especially by cyanide (CN−), making these systems good candidates for reaction-based sensing.16a,c For instance,80 the addition of CN− to the imine group of 65 converts it into a disubstituted hydrazine species 65′ through a proton transfer (Scheme 26). This transformation gives rise to a new absorption peak at 525 nm, which is responsible for the drastic color change of the solution from faint yellow to dark red. Qualified as a “naked-eye” CN− sensor, 65 is capable of detecting submillimolar CN− with a LoD of 1.5 μM in a water–DMSO (v/v = 1:1) solution.
 | ||
| Scheme 26 The nucleophilic addition of CN− to 65. | ||
When a fluorophore is present in conjugation with the hydrazone system, such as in 66 and 67,81 the fluorescence is usually quenched as a result of excited state charge transfer (ESCT). The nucleophilic addition of CN− can effectively disrupt the ESCT process in 66 and 67, leading to remarkable fluorescence recovery. In this way, compounds 66 and 67 are capable of detecting submicromolar quantities of CN− with LoDs at the 10 nM level (Scheme 27).
 | ||
| Scheme 27 The structures of hydrazones 66–68. | ||
The addition of CN− can also be used in turning on the fluorescence resonance energy transfer (FRET) in hydrazones containing energy donor–acceptor pairs. The energy donor 4-(N,N-dimethylamino)benzamide and the energy acceptor fluorescein are connected by a hydrazone linkage in 68,82 where FRET is suppressed since fluorescein exists in the closed, non-fluorescent spirolactam form. Upon reaction with CN−, fluorescein undergoes ring opening and becomes fluorescent, which turns on the FRET process. As a two-channel fluorescence sensor for CN−, 68 works in the CN− concentration range of 0–90 μM, with a LoD of 0.44 μM in a water–DMF (v/v = 1:9) solution.
Metal–hydrazone complexes have been developed as anion sensors as well, by taking advantage of anion–metal interactions. The fluorescent Al3+ complex of 62 reported by Goswami et al.74 shows affinity towards pyrophosphate (PPi, P2O74−) via Al3+–PPi interactions. The complex [Al62]3+ exhibits strong blue fluorescence that is quenched upon the addition of PPi (Fig. 17). This process provides a LoD of 1.7 μM for PPi with great discrimination over common anions, especially adenosine diphosphate (ADP) and triphosphate (ATP).
 | ||
| Fig. 17 Changes in fluorescence intensity (monitored at 508 nm) of [Al62]3+ after the addition of 2.0 equiv. of each anion in water–methanol (v/v = 1:1, pH = 7.5). Inset shows the fluoresence intensity vs. the concentration of PPi. Reproduced from ref. 74. | ||
The Zn2+ complex of the naphthol-based symmetric acylhydrazone 6983 is weakly emissive (Scheme 28); however, upon coordination with PPi, the fluorescence intensity increases and is accompanied by a color change from colorless to yellow in HEPES (20 mM, pH = 7.4) buffer. With a LoD of 0.16 μM, the complex [Zn69]2+ is used as a PPi-specific fluorescent probe for DNA pyrosequencing, and is able to detect trace amounts of PPi from DNA polymerase chain reactions even in the presence of large excess of ATP (Fig. 18).
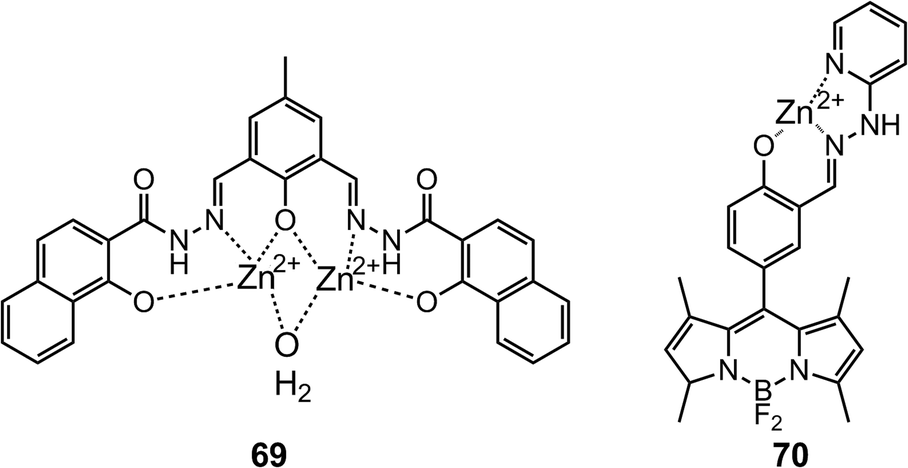 | ||
| Scheme 28 The structures of Zn2+–hydrazone complexes [Zn69]2+ and [Zn70]2+. | ||
 | ||
| Fig. 18 (a) Mechanism of sensing PPi released from PCR by [Zn69]2+; (b) gel electrophoresis of finished PCR mixtures (M = DNA marker). (c) Fluorescence intensity of the sensor [Zn69]2+ at 525 nm upon addition of the finished PCR product mixture. (i) 10 μM of the finished PCR product mixture performed without template DNA; 10 μM of the finished PCR product mixture performed with template DNA after (ii) 29 cycles, (iii) 30 cycles, (iv) 31 cycles, (v) 32 cycles, and (vi) 35 cycles. Reprinted with permission from S. Anbu, S. Kamalraj, C. Jayabaskaran and P. S. Mukherjee, Inorg. Chem., 2013, 52, 8294–8296. Copyright 2013 American Chemical Society. | ||
The fluorescence quantum yield (ΦF) of acylhydrazone 70,84 which is functionalized with a BODIPY fluorophore, drops from 0.27 to 0.038 when forming a 1:1 complex with Zn2+ ([Zn70]2+). Nonetheless, this complex can be used in detecting PPi and ATP with micromolar LoDs of 1.0 and 2.4 μM, respectively, which is below their normal biological concentrations (Scheme 28).
4.3 Other sensing applications
Trishydrazones 71–7385 are C3-symmetric compounds with planar cores stabilized by peripheral intramolecular H-bonds. The structural distortion caused by the steric hindrance between the peripheral substituents can lower the degree of overall π-conjugation, and hence a blue shift is observed for these systems as crowdedness increases from 71 to 73. Similar changes can occur when the intramolecular H-bonding is disrupted, hence compounds 71–73 are spectroscopically responsive towards H-bond acceptors. For instance, when a thin film of 71 is exposed to vapors of volatile primary and secondary amines, it undergoes a fast color change from pink to dark-orange as a result of conformational switching. Using this sensing mechanism Lee et al. have demonstrated the “naked-eye” sensing of volatile amines (Scheme 29). | ||
| Scheme 29 The structures of trishydrazones 71–73. | ||
The intrinsic characteristics of the hydrazone group has allowed its use in bifunctional sensing applications as well. For example, the 4,6-naphthalimide-based hydrazone 7486 emits green fluorescence in CH3CN, which is completely quenched upon complexation with Cu2+. The same effect is observed with F−, which can effectively deprotonate the hydrazone N–H group, leading to quenching. Therefore, 74 can be used as a bifunctional “turn-off” chemosensor for sensing either Cu2+ or F− at micromolar levels (Scheme 30).
 | ||
| Scheme 30 The bifunctional sensing of Cu2+ and F− with 74. | ||
Acetate (AcO−) effectively binds with hydrazones mainly through H-bonding, the breaking of which can be used to quantify the disruptive additive. Chang et al.87 reported the detection of water in organic solvents using the hydrazone–acetate complexes [75AcO]− and [76AcO]−. Both complexes undergo color changes from red to yellow, indicating the breaking of the hydrazone–acetate interaction, as a result of the formation of the more favored aqueous acetate species (Scheme 31). While [75AcO]− provides a LoD as low as 0.071% for water in THF, [76AcO]− has a LoD of 0.12% for water in CH3CN.
 | ||
| Scheme 31 The sensing of water with [75AcO]− and [76AcO]−. | ||
Similarly, hydrazone–cation complexes can be used for the sensing of metallophilic species. Complex [Cu41]+ (Scheme 14),88 formed during the detection of Cu2+, is a weakly fluorescent species because of the fluorescence inner filter effect (IFE). Upon the addition of Cys, IFE is halted as a result of the dissociation of [Cu41]+, leading to emission turn-on that can be used in the detection of micromolar Cys with a LoD of 0.17 μM. Although [Cu41]+ shows great selectivity for Cys over a variety of amino acids, it cannot differentiate Cys from other thiol-based molecules, such as homocysteine (Hcy) and glutathione.
In analogy to some fluorescence “turn-on” hydrazone-based metal sensors (Scheme 22), fluorescence enhancement by restricting CN bond rotation can also be used for Cys or Hcy detection.89 The fluorescence from the naphthalimide group in 77 is quenched by the E Z isomerization of the CN bond. Upon treatment with Cys and Hcy, thiazolidine and thiazinane derivatives are formed, respectively, through the cyclization reaction with the aldehyde group in 75 (Scheme 32). Therefore, the ΦF of 77 increases from 0.012 to 0.625, as the CN isomerization in 77 is restricted by intramolecular H-bonding between the positively charged thiazolidine or thiazinane N–H proton and the imine nitrogen. Compound 77 has been used for the in vitro detection of submillimolar Cys in both buffer solutions and Tetrahymena thermophila cells.
 | ||
| Scheme 32 The sensing of Cys and Hcy with 77 by restricting the CN bond rotation. | ||
Machado et al.90 reported the hydrolysis of acetylthiochiline (ATCh) by the unsubstituted hydrazone 78. This process provides a ratiometric electrochemical response to the concentration of ATCh, making it an acetylcholinesterase (AChE) mimic. Compound 78 first forms a supramolecular complex with ATCh through cation–π interaction, followed by a nucleophilic attack on the carbonyl group by the hydrazone nitrogen, forming the acylhydrazone 78′ that is prone to hydrolysis (Scheme 33). Differential pulse voltammetry reveals a linear response of the current to the concentration of ATCh in the presence of 78, which allows for its detection in the millimolar to submillimolar range with a LoD of 20 μM.
 | ||
| Scheme 33 Mechanism for the hydrolysis of ATCh by the AChE mimic 78. | ||
5 Concluding remarks
For some time now hydrazones have been bundled under the imine banner,91 because of the similarities between both functional groups. This point is made evident by the scarcity of focused reviews and/or monographs dealing with only hydrazone chemistry, in spite of its vast richness and popularity. In this review we aimed at showcasing the uniqueness of hydrazones in the context of supramolecular chemistry, albeit it is clear to us that this functional group is important and pervasive in other fields as well. There are a number of factors that distinguish the hydrazone functional group from its imine counterpart, namely: (i) the stability of the CN double bond to hydrolysis under neutral conditions because of a mesomeric effect; (ii) the existence of an extra amino-type nitrogen in the system that enhances this group's coordination capability; and (iii) the acidic N–H proton that can be utilized in intramolecular H-bonding, anion sensing and even coordination with metals. These qualities not only set this functional group apart from imines, but they also open up complementary research directions that are not easily accessible by the latter. We are hopeful that this review will lead to a divergence in thinking about hydrazones as only a subgroup of imines, and facilitate the exploration of its rich chemistry92 as it relates to supramolecular chemistry and beyond.
Acknowledgements
We would like to thank Dartmouth College and the National Science Foundation CAREER program (CHE-1253385) for their generous support. We are also thankful to Prof. David S. Glueck for his input.References
- (a) M. Hidai and Y. Mizobe, Chem. Rev., 1995, 95, 1115–1133 CrossRef CAS; (b) D. Enders, L. Wortmann and R. Peters, Acc. Chem. Res., 2000, 33, 157–169 CrossRef CAS PubMed; (c) R. Lazny and A. Nodzewska, Chem. Rev., 2010, 110, 1386–1434 CrossRef CAS PubMed; (d) S. Kobayashi, Y. Mori, J. S. Fossey and M. M. Salter, Chem. Rev., 2011, 111, 2626–2704 CrossRef CAS PubMed.
- (a) P. Vicini, F. Zani, P. Cozzini and I. Doytchinova, Eur. J. Med. Chem., 2002, 37, 553–564 CrossRef CAS; (b) C. Loncle, J. Brunel, N. Vidal, M. Dherbomez and Y. Letourneux, Eur. J. Med. Chem., 2004, 39, 1067–1071 CrossRef CAS PubMed; (c) L. Savini, L. Chiasserini, V. Travagli, C. Pellerano, E. Novellino, S. Cosentino and M. Pisano, Eur. J. Med. Chem., 2004, 39, 113–122 CrossRef CAS PubMed; (d) M. Cocco, C. Congiu, V. Lilliu and V. Onnis, Bioorg. Med. Chem., 2006, 14, 366–372 CrossRef CAS PubMed; (e) A. Masunari and L. C. Tavares, Bioorg. Med. Chem., 2007, 15, 4229–4236 CrossRef CAS PubMed; (f) P. Vicini, M. Incerti, P. La Colla and R. Loddo, Eur. J. Med. Chem., 2009, 44, 1801–1807 CrossRef CAS PubMed.
- (a) J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC; (b) J.-M. Lehn, in Topics in Current Chemistry, ed. M. Barboiu, Springer, Berlin, 2012, vol. 322, book section Constitutional Dynamic Chemistry: Bridge from Supramolecular Chemistry to Adaptive Chemistry, pp. 1–32 Search PubMed; (c) J.-M. Lehn, Angew. Chem., Int. Ed., 2013, 52, 2836–2850 CrossRef CAS PubMed.
- (a) F. J. Uribe-Romo, C. J. Doonan, H. Furukawa, K. Oisaki and O. M. Yaghi, J. Am. Chem. Soc., 2011, 133, 11478–11481 CrossRef CAS PubMed; (b) D. N. Bunck and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 14952–14955 CrossRef CAS PubMed; (c) X.-P. Zhou, Y. Wu and D. Li, J. Am. Chem. Soc., 2013, 135, 16062–16065 CrossRef CAS PubMed.
- (a) S. Rowan, S. Cantrill, G. Cousins, J. Sanders and J. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef; (b) P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed; (c) Dynamic Combinatorial Chemistry, Drug Discovery, Bioorganic Chemistry, and Materials Science, ed. B. L. Miller, John Wiley and Sons, Hoboken, New Jersey, 2010 Search PubMed; (d) Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- R. Raue, A. Brack and K. Lange, Angew. Chem., Int. Ed. Engl., 1991, 30, 1643–1644 CrossRef.
- R. Lygaitis, V. Getautis and J. V. Grazulevicius, Chem. Soc. Rev., 2008, 37, 770–788 RSC.
- C. Serbutoviez, C. Bosshard, G. Knopfle, P. Wyss, P. Pretre, P. Gunter, K. Schenk, E. Solari and G. Chapuis, Chem. Mater., 1995, 7, 1198–1206 CrossRef CAS.
- (a) F. R. Japp and F. Klingemann, Ber. Dtsch. Chem. Ges., 1887, 20, 2942–2944 CrossRef; (b) F. R. Japp and F. Klingemann, Ber. Dtsch. Chem. Ges., 1887, 20, 3284–3286 CrossRef; (c) F. R. Japp and F. Klingemann, Ber. Dtsch. Chem. Ges., 1887, 20, 3398–3401 CrossRef; (d) F. R. Japp and F. Klingemann, Justus Liebigs Ann. Chem., 1888, 247, 190–225 CrossRef.
- A. C. Day and M. C. Whiting, Org. Synth., 1988, 6, 10–12 Search PubMed.
- G. Stork and J. Benaim, Org. Synth., 1988, 6, 242–244 Search PubMed.
- S. Wagaw, B. H. Yang and S. L. Buchwald, J. Am. Chem. Soc., 1998, 120, 6621–6622 CrossRef CAS.
- V. Lefebvre, T. Cailly, F. Fabis and S. Rault, J. Org. Chem., 2010, 75, 2730–2732 CrossRef CAS PubMed.
- (a) Molecular Machines and Motors, ed. J.-P. Sauvage, Springer, New York, 2001 Search PubMed; (b) E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS PubMed; (c) V. Balzani, A. Credi and M. Venturi, Molecular Decices and Machines - Concepts and Perspectives for the Nanoworld, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed; (d) M.-M. Russew and S. Hecht, Adv. Mater., 2010, 22, 3348–3360 CrossRef CAS PubMed; (e) Molecular Switches, ed. B. L. Feringa, Wiley-VCH, Weinheim, Germany, 2nd edn, 2011 Search PubMed; (f) A. Coskun, M. Banaszak, R. D. Astumian, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2012, 41, 19–30 RSC; (g) A. C. Fahrenbach, S. C. Warren, J. T. Incorvati, A.-J. Avestro, J. C. Barnes, J. F. Stoddart and B. A. Grzybowski, Adv. Mater., 2013, 25, 331–348 CrossRef CAS PubMed.
- (a) M. Ruben, J.-M. Lehn and P. Müller, Chem. Soc. Rev., 2006, 35, 1056–1067 RSC; (b) J. G. Hardy, Chem. Soc. Rev., 2013, 42, 7881–7899 RSC.
- (a) H. Miyaji and J. Sessler, Angew. Chem., Int. Ed., 2001, 40, 154–157 CrossRef CAS; (b) E. V. Anslyn, J. Org. Chem., 2007, 72, 687–699 CrossRef CAS PubMed; (c) D.-G. Cho and J. L. Sessler, Chem. Soc. Rev., 2009, 38, 1647–1662 RSC; (d) P. A. Gale, Chem. Commun., 2011, 47, 82–86 RSC; (e) M. T. Albelda, J. C. Frias, E. Garcia-Espana and H. J. Schneider, Chem. Soc. Rev., 2012, 41, 3859–3877 RSC; (f) M. Wenzel, J. R. Hiscock and P. A. Gale, Chem. Soc. Rev., 2012, 41, 480–520 RSC; (g) A. P. F. Turner, Chem. Soc. Rev., 2013, 42, 3148–3196 RSC; (h) P. A. Gale, N. Busschaert, C. J. E. Haynes, L. E. Karagiannidis and I. L. Kirby, Chem. Soc. Rev., 2014, 43, 205–241 RSC.
- (a) H. C. Yao, J. Org. Chem., 1964, 29, 2959–2963 CrossRef CAS; (b) J. C. Tobin, A. F. Hegarty and F. L. Scott, J. Chem. Soc. B, 1971, 2198–2202 RSC; (c) J. L. Wong and M. F. Zady, J. Org. Chem., 1975, 40, 2512–2516 CrossRef CAS.
- A. C. Pratt, Chem. Soc. Rev., 1977, 6, 63–81 RSC.
- A. Padwa, Chem. Rev., 1977, 77, 37–68 CrossRef CAS.
- (a) P. Courtot, J. Le Saint and R. Pichon, Bull. Soc. Chim. Fr., 1975, 2538–2542 CAS; (b) P. Courtot, R. Pichon and J. Le Saint, Tetrahedron Lett., 1976, 1181–1184 CrossRef CAS; (c) R. Pichon, J. Le Saint and P. Courtot, Tetrahedron, 1981, 37, 1517–1524 CrossRef CAS.
- S. Yamamura, T. Tamaki, T. Seki, M. Sakuragi, Y. Kawanishi and K. Ichimura, Chem. Lett., 1992, 543–546 CrossRef CAS.
- D. G. Belov, B. G. Rogachev, L. I. Tkachenko, V. A. Smirnov and S. M. Aldoshin, Russ. Chem. Bull., 2000, 49, 666–668 CrossRef CAS.
- Y. E. Semenov, V. A. Smirnov, S. M. Aldoshin and B. G. Rogachev, Russ. Chem. Bull., 2001, 50, 2471–2472 CrossRef CAS.
- M. N. Chaur, D. Collado and J.-M. Lehn, Chem.–Eur. J., 2011, 17, 248–258 CrossRef CAS PubMed.
- G. Vantomme and J.-M. Lehn, Angew. Chem., Int. Ed., 2013, 52, 3940–3943 CrossRef CAS PubMed.
- S. M. Landge and I. Aprahamian, J. Am. Chem. Soc., 2009, 131, 18269–18271 CrossRef CAS PubMed.
- S. M. Landge, E. Tkatchouk, D. Benítez, D. A. Lanfranchi, M. Elhabiri, W. A. Goddard III and I. Aprahamian, J. Am. Chem. Soc., 2011, 133, 9812–9823 CrossRef CAS PubMed.
- X. Su, T. Lessing and I. Aprahamian, Beilstein J. Org. Chem., 2012, 8, 872–876 CrossRef CAS PubMed.
- X. Su and I. Aprahamian, Org. Lett., 2011, 13, 30–33 CrossRef CAS PubMed.
- (a) T. Dudev and C. Lim, Chem. Rev., 2003, 103, 773–788 CrossRef CAS PubMed; (b) D. E. Wilcox, Inorg. Chim. Acta, 2008, 361, 857–867 CrossRef CAS PubMed.
- X. Su, T. F. Robbins and I. Aprahamian, Angew. Chem., Int. Ed., 2011, 50, 1841–1844 CrossRef CAS PubMed.
- D. Ray, J. T. Foy, R. P. Hughes and I. Aprahamian, Nat. Chem., 2012, 4, 757–762 CrossRef CAS PubMed.
- (a) K. Ichimura, Chem. Rev., 2000, 100, 1847–1873 CrossRef CAS PubMed; (b) R. Eelkema and B. L. Feringa, Org. Biomol. Chem., 2006, 4, 3729–3745 RSC; (c) I. M. Saez and J. W. Goodby, in Liquid Crystalline Functional Assemblies and Their Supramolecular Structures, ed. T. Kato, Springer-Verlag Berlin, Heidelberger Platz 3, D-14197 Berlin, Germany, 2008, vol. 128, pp. 1–62 Search PubMed; (d) Liquid Crystal Beyond Displays: Chemistry, Physics, and Applications, ed. Q. Li, Wiley-VCH, Weinheim, Germany, 2012 Search PubMed; (e) Y. Wang and Q. Li, Adv. Mater., 2012, 24, 1926–1945 CrossRef CAS PubMed.
- X. Su, S. Voskian, R. P. Hughes and I. Aprahamian, Angew. Chem., Int. Ed., 2013, 52, 10734–10739 CrossRef CAS PubMed.
- A.-M. Stadler, N. Kyritsakas, R. Graff and J.-M. Lehn, Chem.–Eur. J., 2006, 12, 4503–4522 CrossRef CAS PubMed.
- A.-M. Stadler, N. Kyritsakas, G. Vanghan and J.-M. Lehn, Chem.–Eur. J., 2007, 13, 59–68 CrossRef CAS PubMed.
- D. J. Hutchinson, L. R. Hanton and S. C. Moratti, Inorg. Chem., 2010, 49, 5923–5934 CrossRef CAS PubMed.
- D. J. Hutchinson, L. R. Hanton and S. C. Moratti, Inorg. Chem., 2011, 50, 7637–7649 CrossRef CAS PubMed.
- D. J. Hutchinson, L. R. Hanton and S. C. Moratti, Inorg. Chem., 2013, 52, 2716–2728 CrossRef CAS PubMed.
- D. J. Hutchinson, S. A. Cameron, L. R. Hanton and S. C. Moratti, Inorg. Chem., 2012, 51, 5070–5081 CrossRef CAS PubMed.
- J. Ramírez, A.-M. Stadler, L. Brelot and J.-M. Lehn, Tetrahedron, 2008, 64, 8402–8410 CrossRef PubMed.
- A.-M. Stadler, J. Ramírez and J.-M. Lehn, Chem.–Eur. J., 2010, 16, 5369–5378 CrossRef CAS PubMed.
- J. M. Klein, J. K. Clegg, V. Saggiomo, L. Reck, U. Luning and J. K. M. Sanders, Dalton Trans., 2012, 41, 3780–3786 RSC.
- C. P. Sebli, S. E. Howson, G. J. Clarkson and P. Scott, Dalton Trans., 2010, 39, 4447–4454 RSC.
- G. Schaeffer, J. M. Harrowfield, J.-M. Lehn and A. K. H. Hirsch, Polyhedron, 2012, 41, 40–43 CrossRef CAS PubMed.
- Y. S. Moroz, S. Demeshko, M. Haukka, A. Mokhir, U. Mitra, M. Stocker, P. Müller, F. Meyer and I. O. Fritsky, Inorg. Chem., 2012, 51, 7445–7447 CrossRef CAS PubMed.
- J. Ramírez, A.-M. Stadler, N. Kyritsakas and J.-M. Lehn, Chem. Commun., 2007, 237–239 RSC.
- A.-M. Stadler, C. Burg, J. Ramírez and J.-M. Lehn, Chem. Commun., 2013, 49, 5733–5735 RSC.
- C.-F. Chow, S. Fujii and J.-M. Lehn, Angew. Chem., Int. Ed., 2007, 46, 5007–5010 CrossRef CAS PubMed.
- C. F. Chow, S. Fujii and J.-M. Lehn, Chem.–Asian J., 2008, 3, 1324–1335 CrossRef CAS PubMed.
- T. Ono, S. Fujii, T. Nobori and J.-M. Lehn, Chem. Commun., 2007, 4360–4362 RSC.
- J. G. Hardy, X.-Y. Cao, J. Harrowfield and J.-M. Lehn, New J. Chem., 2012, 36, 668–673 RSC.
- Y. Xiang, A. J. Tong, P. Y. Jin and Y. Ju, Org. Lett., 2006, 8, 2863–2866 CrossRef CAS PubMed.
- Y. Xiang, M. Li, X. T. Chen and A. J. Tong, Talanta, 2008, 74, 1148–1153 CrossRef CAS PubMed.
- X. T. Chen, Z. F. Li, Y. Xiang and A. J. Tong, Tetrahedron Lett., 2008, 49, 4697–4700 CrossRef CAS PubMed.
- N. Li, W. X. Tang, Y. Xiang, A. J. Tong, P. Y. Jin and Y. Ju, Luminescence, 2010, 25, 445–451 CrossRef CAS PubMed.
- J. M. An, M. H. Yan, Z. Y. Yang, T. R. Li and Q. X. Zhou, Dyes Pigm., 2013, 99, 1–5 CrossRef CAS PubMed.
- J. M. An, Z. Y. Yang, M. H. Yan and T. R. Li, J. Lumin., 2013, 139, 79–83 CrossRef CAS PubMed.
- H. L. Li, J. L. Fan, F. L. Song, H. Zhu, J. J. Du, S. G. Sun and X. J. Peng, Chem.–Eur. J., 2010, 16, 12349–12356 CrossRef CAS PubMed.
- H. Li, J. Cao, J. Fan and X. Peng, Tetrahedron Lett., 2013, 54, 4357–4361 CrossRef CAS PubMed.
- H. G. Wang, Y. P. Li, S. F. Xu, Y. C. Li, C. Zhou, X. L. Fei, L. Sun, C. Q. Zhang, Y. X. Li, Q. B. Yang and X. Y. Xu, Org. Biomol. Chem., 2011, 9, 2850–2855 CAS.
- H. N. Kim, S. W. Nam, K. M. K. Swamy, Y. Jin, X. Q. Chen, Y. Kim, S. J. Kim, S. Park and J. Yoon, Analyst, 2011, 136, 1339–1343 RSC.
- S. Mukherjee, S. Chowdhury, A. P. Chattopadhyay and H. Stoeckli-Evans, Polyhedron, 2010, 29, 1182–1188 CrossRef CAS PubMed.
- S. Mukherjee, S. Chowdhury, A. K. Paul and R. Banerjee, J. Lumin., 2011, 131, 2342–2346 CrossRef CAS PubMed.
- S. Mukherjee, P. Mal and H. Stoeckli-Evans, Polyhedron, 2013, 50, 495–501 CrossRef CAS PubMed.
- Y. Zhou, H. N. Kim and J. Yoon, Bioorg. Med. Chem. Lett., 2010, 20, 125–128 CrossRef CAS PubMed.
- (a) M. R. Ganjali, F. Faridbod, P. Norouzi and M. Adib, Sens. Actuators, B, 2006, 120, 119–124 CrossRef CAS PubMed; (b) F. Faridbod, M. R. Ganjali, B. Larijani, P. Norouzi, S. Riahi and F. F. S. Mirnaghi, Sensors, 2007, 7, 3119–3135 CrossRef CAS.
- S. Anbu, S. Shanmugaraju, R. Ravishankaran, A. A. Karande and P. S. Mukherjee, Dalton Trans., 2012, 41, 13330–13337 RSC.
- H.-G. Li, Z.-Y. Yang and D.-D. Qin, Inorg. Chem. Commun., 2009, 12, 494–497 CrossRef CAS PubMed.
- Z. C. Liao, Z. Y. Yang, Y. Li, B. D. Wang and Q. X. Zhou, Dyes Pigm., 2013, 97, 124–128 CrossRef CAS PubMed.
- S. Goswami, S. Chakraborty, S. Paul, S. Halder and A. C. Maity, Tetrahedron Lett., 2013, 54, 5075–5077 CrossRef CAS PubMed.
- S. Ulrich, A. Petitjean and J.-M. Lehn, Eur. J. Inorg. Chem., 2010, 1913–1928 CrossRef CAS.
- L. N. Wang, W. W. Qin and W. S. Liu, Inorg. Chem. Commun., 2010, 13, 1122–1125 CrossRef CAS.
- S. Goswami, A. Manna, S. Paul, K. Aich, A. K. Das and S. Chakraborty, Dalton Trans., 2013, 42, 8078–8085 RSC.
- L. L. Zhou, X. H. Zhang and S. K. Wu, Chem. Lett., 2004, 850–851 CrossRef CAS.
- J. Shao, H. Lin, M. Yu, Z. S. Cai and H. K. Lin, Talanta, 2008, 74, 1122–1125 CrossRef PubMed.
- S. Z. Hu, Y. Guo, J. Xu and S. J. Shao, Org. Biomol. Chem., 2008, 6, 2071–2075 CAS.
- Q. Li, Y. Guo, J. Xu and S. J. Shao, J. Photochem. Photobiol., B, 2011, 103, 140–144 CrossRef CAS PubMed.
- R. K. Mishra, V. Kumar, U. Diwan, K. K. Upadhyay and P. K. R. Chowdhury, J. Mol. Struct., 2012, 1027, 167–174 CrossRef CAS PubMed.
- Y. Sun, Y. Liu and W. Guo, Sens. Actuators, B, 2009, 143, 171–176 CrossRef PubMed.
- Y. Sun, Y. L. Liu, M. L. Chen and W. Guo, Talanta, 2009, 80, 996–1000 CrossRef CAS PubMed.
- X. Lv, J. Liu, Y. L. Liu, Y. Zhao, M. L. Chen, P. Wang and W. Guo, Org. Biomol. Chem., 2011, 9, 4954–4958 CAS.
- S. Anbu, S. Kamalraj, C. Jayabaskaran and P. S. Mukherjee, Inorg. Chem., 2013, 52, 8294–8296 CrossRef CAS PubMed.
- O. G. Tsay, S. T. Manjare, H. Kim, K. M. Lee, Y. S. Lee and D. G. Churchill, Inorg. Chem., 2013, 52, 10052–10061 CrossRef CAS.
- H. Y. Lee, X. L. Song, H. Park, M. H. Baik and D. Lee, J. Am. Chem. Soc., 2010, 132, 12133–12144 CrossRef CAS PubMed.
- H. Yang, H. S. Song, Y. C. Zhu and S. P. Yang, Tetrahedron Lett., 2012, 53, 2026–2029 CrossRef CAS PubMed.
- Y. H. Kim, M. G. Choi, H. G. Im, S. Ahn, I. W. Shim and S.-K. Chang, Dyes Pigm., 2012, 92, 1199–1203 CrossRef CAS PubMed.
- X. F. Yang, P. Liu, L. P. Wang and M. L. Zhao, J. Fluoresc., 2008, 18, 453–459 CrossRef CAS PubMed.
- P. Wang, J. Liu, X. Lv, Y. L. Liu, Y. Zhao and W. Guo, Org. Lett., 2012, 14, 520–523 CrossRef CAS PubMed.
- L. F. Sgobbi, V. D. Pinho, M. F. Cabral, A. C. B. Burtoloso and S. A. S. Machado, Sens. Actuators, B, 2013, 182, 211–216 CrossRef CAS PubMed.
- M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC.
- (a) Y. Yang, R. P. Hughes and I. Aprahamian, J. Am. Chem. Soc., 2012, 134, 15221–15224 CrossRef CAS PubMed; (b) X. Su, M. Liptak and I. Aprahamian, Chem. Commun., 2013, 49, 4160–4162 RSC; (c) X. Su and I. Aprahamian, Org. Lett., 2013, 15, 5952–5955 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |