 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterogeneous photocatalytic nanomaterials: prospects and challenges in selective transformations of biomass-derived compounds
Juan Carlos
Colmenares
*a and
Rafael
Luque
b
aInstitute of Physical Chemistry, Polish Academy of Sciences, ul. Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: jcarloscolmenares@ichf.edu.pl
bDepartamento de Quimica Organica, Universidad de Cordoba, Campus de Rabanales, Edificio Marie Curie, E-14014, Cordoba, Spain
First published on 11th November 2013
Abstract
Heterogeneous photocatalysis has become a comprehensively studied area of research during the past three decades due to its practical interest in applications including water–air depollution, cancer therapy, sterilization, artificial photosynthesis (CO2 photoreduction), anti-fogging surfaces, heat transfer and heat dissipation, anticorrosion, lithography, photochromism, solar chemicals production and many others. The utilization of solar irradiation to supply energy or to initiate chemical reactions is already an established idea. Excited electron–hole pairs are generated upon light irradiation of a wide-band gap semiconductor which can be applied to solar cells to generate electricity or in chemical processes to create/degrade specific compounds. While the field of heterogeneous photocatalysis for pollutant abatement and mineralisation of contaminants has been extensively investigated, a new research avenue related to the selective valorisation of residues has recently emerged as a promising alternative to utilise solar light for the production of valuable chemicals and fuels. This tutorial review will focus on the potential and applications of solid photonanocatalysts for the selective transformation of biomass-derived substrates.
 Juan Carlos Colmenares | Prof. Juan Carlos Colmenares Q. was educated in Colombia through high school, and then moved to Poland in 1991. Prof. Colmenares graduated from the Warsaw University of Technology (Chem. Eng., 1995) and obtained his MSc (1997) in Catalysis for Organic Technology and PhD (2004) in Chemical and Material Sciences from the same university. His interests range from materials science, nanotechnology and heterogeneous catalysis to biomass/CO2 valorisation, biofuels, sonication, photocatalysis and water–air purification. After his PhD he worked at the University of Cordoba, Spain (2005–2006) in Prof. Marinas group as a post-doctoral fellow and at the University of Southern California, Los Angeles (USA) (2006–2009) in Prof. G.A. Olah (Nobel Prize in Chemistry) group as a post-doctoral research associate. He is a Marie Sklodowska-Curie Fellow and has participated in many research projects as a main investigator and co-leader. |
 Rafael Luque | Prof. Rafael Luque (PhD in 2005 from the Universidad de Cordoba, Spain) has significant experience in biomass and waste valorisation practices for materials, fuels and chemicals over the past 10 years after spending a 3-year postdoctoral placement in the Green Chemistry Centre of Excellence at the University of York. Since 2009, he has been a Ramon y Cajal Fellow at UCO in Spain. Rafael was awarded the Marie Curie Prize from the Instituto Andaluz de Quimica Fina in Spain (2011), the Green Talents award from the Federal Ministry of Education and Research in Germany (2011), the TR35 Spain 2012 from MIT (USA) as one of the top 10 innovative young entrepreneurs of Spain and the recent 2013 RSC Environment, Sustainability and Energy Division Early Career Award. He has been recently appointed as a Visiting Professor and Distinguished Engineering Fellow at the Department of Chemical and Biomolecular Engineering (CBME) from HKUST in Hong Kong. |
Key learning points(1) Photocatalysis can bring several benefits from energy and environmental viewpoints.(2) Heterogeneous photocatalysts can play an important role in developing new technologies for the chemical sector at room temperature and atmospheric pressure (complex transformations in one-pot processes). (3) Photochemical reactions under milder conditions to those generally present in thermal processes can facilitate the design of short and efficient reaction sequences, minimizing side processes and making use of sunlight as a completely renewable source of energy. (4) Photocatalysis has a significant potential for the conversion of lignocellulosics to valuable products. |
1. Introduction
Developing artificial systems that can mimic natural photosynthesis to directly harvest and convert solar energy into usable or storable energy resources has been the dream of scientists for many years. The use of solar energy to drive organic syntheses is indeed not a novel concept. The idea was originally proposed by Ciamician as early as 1912.1 However, the common use of the generally accepted term photocatalysis and significant developments in this field properly started in the 1970s after the discovery of water photolysis on a TiO2 electrode by Fujishima and Honda.2The energy crisis provided a strong impulse to research alternative energy sources, hoping that mankind could mimic nature by exploiting solar energy for the generation of fuels (e.g. hydrogen via water splitting).2,3 Additionally, the pollution concerns and the increasing demand for more sustainable sources of chemicals also prompted the search for alternative solutions potentially able to clean up water and air avoiding the addition of further chemicals and to explore new “green” routes for chemicals production. A third later application in chemical synthesis was strongly related to the fact that the same photocatalysts were applied.4 Very important is the fact that all highlighted three applications can be grouped into green and sustainable processes.
Photocatalysis, in which solar photons are used to drive redox reactions to produce chemicals (e.g. fuels), is the central process to achieve this aim. Despite significant efforts to date, a practically viable photocatalyst with sufficient efficiency, stability and low cost is yet to be demonstrated. It is often difficult to simultaneously achieve these different performance metrics with a single material component. The ideal heterogeneous photocatalysts with multiple integrated functional components could combine individual advantages to overcome the drawbacks of single component photocatalysts. For further information, readers are kindly referred to recent overviews on the development of advanced photocatalysts.4,5
As a fundamental and applied field of science, heterogeneous photochemistry continues to be an important component of modern chemistry in the 21st century. Research in this field has significantly evolved during the last three decades (especially titanium oxide chemistry) with enhanced knowledge on mechanisms, development of new technologies for storage and conversion of solar energy, environmental detoxification of liquid and gaseous ecosystems, and the photochemical production of new materials. More recently, a new research avenue related to selective transformations of biomass and residues to high added value products has emerged as a potentially useful alternative to conventional heterogeneously catalyzed processes. This contribution is aimed to provide an overview of recent work conducted along the lines of selective photochemical transformations, particularly focused on photocatalysis for lignocellulose-based biomass valorization. Future prospects and work in progress in this field will also be emphasized. The effective utilization of clean, safe, and abundant solar energy is envisaged to provide energy, chemicals as well as solving environmental issues in the future and an appropriate semiconductor-photoinduced biomass/waste conversion can be the key for such transformations.
2. Fundamental principles of semiconductor photocatalysis
The fundamental principles of heterogeneous photocatalysis have been extensively reported in previous reports.5 A photocatalytic transformation is initiated when a photoexcited electron is promoted from the filled valence band (VB) of a semiconductor photocatalyst (e.g., TiO2) to the empty conduction band (CB) as the absorbed photon energy, hν, equals or exceeds the band gap of the semiconductor photocatalyst. As a consequence, an electron and hole pair (e−–h+) are formed (eqns (1)–(4)).| Photon activation: TiO2 + hν → e− + h+ | (1) |
| Oxygen adsorption: (O2)ads + e− → O2˙− | (2) |
| Water ionization: H2O → OH− + H+ | (3) |
| Superoxide protonation: O2˙− + H+ → HOO˙ | (4) |
HOO˙ radicals (eqn (4)) also have scavenging properties similar to oxygen, thus prolonging the photohole lifetime (eqns (5) and (6)).
| HOO˙ + e− → HO2− | (5) |
| HOO− + H+ → H2O2 | (6) |
Redox processes can take place at the surface of the photoexcited photocatalyst. Very fast recombination between electron and hole occurs unless oxygen (or any other electron acceptor) is available to scavenge the electrons to form superoxides (O2˙−) (Fig. 1), hydroperoxyl radicals (HO2˙) and subsequently H2O2.
 | ||
| Fig. 1 Photo-activation of a semiconductor and primary reactions occurring on its surface. | ||
In contrast to conventional catalysis thermodynamics, not only spontaneous reactions (ΔG < 0) but also non-spontaneous reactions (ΔG > 0) can be promoted by photocatalysis. The input energy is used to overcome the activation barrier in spontaneous reactions so as to facilitate photocatalysis at an increased rate or under milder conditions. In comparison, for non-spontaneous processes, part of the input energy is converted into chemical energy that is accumulated in the reaction products.
The generated holes have high potential to directly oxidize organic species (the mechanism is not definitively proven) or indirectly via the combination with ˙OH abundant in water solution (eqns (7)–(9)).5a,b,6
| H2O + h+ → OH˙ + H+ | (7) |
| R–H + OH˙ → R˙ + H2O | (8) |
| R˙ + h+ → R+˙ → Mineralization products | (9) |
The corresponding mineral acid of the non-metal substituent is formed as a by-product (e.g. in organic waste maybe a different heteroatom including Cl, N, S) (eqn (10)).
| Organic waste ⇒ photocatalyst/O2/ hν ≥ Eg ⇒ Intermediate(s) ⇒ CO2 + H2O + Mineral acid | (10) |
2.1. Reactive oxygen species (ROS) formed during a photocatalytic process
Several highly reactive ROS can oxidize a large variety of organic pollutants in heterogeneous photocatalysis. These includeApart from the important role of ROS in photocatalytic degradation processes, the control and generation of ROS is essential in heterogeneous photocatalysis to be potentially able to design and predict pathways in selective organic photocatalytic oxidations.
3. Chemicals from lignocellulosic biomass
Lignocellulose is a highly complex and rather recalcitrant feedstock comprising three major polymeric units: lignin, cellulose and hemicellulose. Cellulose (a high molecular weight polymer of glucose, ca. 45 wt%) forms bundles that are additionally attached together by hemicellulose (ca. 20 wt%). Cellulose and hemicellulose are surrounded by lignin (less than 25 wt%) which provides extra rigidity and recalcitrance to the entire material.Lignocellulosic feedstocks (i.e., forestry waste, agricultural residues, municipal paper waste and certain food waste residues) can be converted into a variety of useful products in multi-step processes.7,8 However, due to the large complexity of such feedstocks, lignocellulosics have to be broken down by several processes and technologies into simpler fractions which can be converted into desired products. These include major routes such as gasification, pyrolysis and pre-treatment hydrolysis/fragmentation steps. Upon deconstruction, the obtained solid, liquid or gaseous fractions need to be upgraded via various processes which yield a plethora of chemicals.7,8
From a sustainability viewpoint, the development of low-temperature, highly selective (photo)catalytic routes for the direct transformation of lignocellulosics into valuable chemicals or platform molecules (e.g. glucose, carboxylic acids) is of great significance.7 These compounds can then be subsequently converted into useful products. However, the selective conversion of lignocellulosics under mild conditions still remains a significant challenge owing to the high recalcitrant structure of cellulose and lignin fractions. For further information on topics related to cellulose and lignin depolymerization, readers are kindly referred to recent literature overviews.9–11 Subsequent sections have been aimed to illustrate the potential of photocatalytic processes for the conversion of selected lignocellulosic feedstocks into chemicals and fuels.
4. Photocatalytic reforming of lignocellulose-based organic waste: hydrogen production
Biomass sources have been utilized for sustainable hydrogen production.12 A number of processes have been developed for this purpose which include fast pyrolysis, supercritical conversion and steam gasification and many others which however require harsh reaction conditions (e.g., high temperatures and/or pressures) and consequently imply high costs.12 Photocatalytic reforming may be a promising alternative as mild reaction conditions driven by solar light (i.e., room temperature and atmospheric pressure) can be comparatively advantageous to such energy-consuming thermochemical processes.Hydrogen production by photocatalytic reforming of lignocellulosic biomass may also be more feasible and practical as compared to photocatalytic water-splitting due to its potentially higher efficiency.13 The thermodynamics of photochemical water splitting were reported to store a maximum of only 12% of the incident light energy.13
Lignocellulosic biomass-derived compounds can also serve as sacrificial agents (electron donors) to reduce the photocatalyst recombination e−–h+ rate. A large variety of organic compounds (most of them model compounds of lignocellulose structure, e.g. alcohols, polyols, sugars, as well as organic acids) have been used as electron donors for photocatalytic hydrogen production. For further information, readers are kindly referred to recent accounts of the most pioneering studies by leading authors on biomass (photo)catalytic reforming for chemical (hydrogen included) production.14
5. Heterogeneous photocatalysis for selective chemical transformations
The utilization of heterogeneous photocatalysis for environmental purification (both in the liquid and gas phase) has been extensively investigated as photoactivated semiconductors have proven activities to unselectively mineralize various types of toxic, refractory and non-biodegradable organic pollutants under mild conditions.15 Closely related to selective synthesis, structured photocatalytic systems have also been employed for the oxofunctionalization of hydrocarbons via selective oxidations (Fig. 2).16 | ||
| Fig. 2 Production of different target compounds by semiconductor photo-induced catalytic routes. | ||
Reported organic photosynthetic reactions include oxidations, reductions, isomerizations, substitutions, polymerizations and condensations. These reactions can be carried out in inert solvents and/or their combinations with water. For more details on photocatalytic synthetic transformations based on different photo-active solid materials, readers are kindly referred to leading reviews in the field.17
The following fundamental requirements should be met by any photocatalytic system harvesting and converting solar energy into chemical energy:
(a) the photoresponse of the system should optimally match the solar spectrum;
(b) photoexcited charges must be efficiently separated to prevent recombination;
(c) charges should have sufficient energy to carry out the desired chemical reactions (e.g. selective oxidations);
(d) the photocatalyst must be photo-stable, chemically and biologically inert and of low cost.
For photon transfer optimization, decisive operational parameters include light intensity, nature and concentration of the substrate, nature and concentration of the photocatalyst, pH of the solution, reaction temperature, type of the reactor, presence of oxygen and the content of ions.18
5.1. Photocatalytic selective oxidations of cellulose-based chemicals
Catalytic oxidations have traditionally been carried out in environmentally harmful chlorinated organic solvents at high temperatures and pressures by employing stoichiometric amounts of various inorganic oxidants as oxygen donors (e.g. chromate and permanganate species). Such oxidants are expensive and toxic and they also produce large amounts of hazardous waste, therefore needing to be replaced by safer systems.Comparably, a photocatalytic process can bring about significant benefits in terms of milder and more environmentally sound reaction conditions and better selectivity for the desired product. Substances unstable at high temperatures may be synthesized via selective light-assisted processes.
Alcohol oxidation to their corresponding ketones, aldehydes, and/or carboxylic acids is one of the most important transformations in organic synthesis.19 Semiconductor photocatalysts have not been frequently employed in selective synthetic oxidation processes as the replacement of traditional oxidation methods has been mostly covered with heterogeneously catalyzed systems.
According to thermodynamics, an alcohol molecule with singlet electronic configuration cannot directly react with an unactivated dioxygen molecule, which has a triplet electronic configuration.20 The working mechanism in metal oxidation catalysts involves an electron transfer-mediated step by the metal which thereby induces the formation of singlet oxygen species.20 In this context, the search for an oxidation photocatalyst capable of directly activating dioxygen under solar light is an interesting as well as challenging task.
Catalytic selective photo-oxidation of biomass can provide a wide range of high added-value chemicals including some of the so-called sugar-derived platform molecules (e.g., succinic, 2,5-furandicarboxylic, 3-hydroxypropionic, gluconic, glucaric and levulinic acids as well as 3-hydroxybutyrolactone).21Table 1 summarizes recent selected studies on photocatalytic selective oxidation of lignocellulose-based model compounds to valuable chemicals.
| Entry | Model biomass-based molecule | Photocatalyst synthesis | Reaction conditions | Photocatalytic behavior (activity/selectivity) | Ref. |
|---|---|---|---|---|---|
| 1 | Methanol | Anatase-type TiO2 particles (ST-01) having a large surface area of 300 m2 g−1 | UV light (UV intensity: 1800 μW cm−2). Reaction in gas phase in the presence of air with a fixed bed of the catalyst | Highly selective (91 mol%) photooxidation of methanol to methyl formate with no catalyst deactivation. The conversion of methanol increased with the increase of the reaction temperature up to 250 °C (conversion is three times higher than that at room temperature but the selectivity decreases) | 29 |
| 2 | Ethanol, 1-propanol, 2-propanol, 2-butanol | TiO2 rutile phase (3.6 and 7.7 m2 g−1) | Catalyst suspensions irradiated with 366 nm UV light | Selective production of ethanol, propanol and propanone with 0.101, 0.104 and 0.101 quantum yields at 20 °C, respectively | 22 |
| 3 | 2-Propanol crotyl alcohol | TiO2, Me-TiO2 (Me = Pd, Pt, Zr, Fe, Zn, Ag, Au). Catalysts prepared by ultrasound- and microwave-assisted sol–gel procedures | UV light (λmax = 365 nm). Reaction in liquid phase (for crotyl alcohol) with a solid catalyst in suspension. Reaction in gas phase (for 2-propanol) with a fixed bed of the catalyst. Water used for cooling was thermostated at 10 °C for reactions with crotyl alcohol and 20 °C for 2-propanol |
For crotyl alcohol, conversions between 8% and 38% for t = 30 min or 32% and 95% for t = 300 min for the two extreme catalysts (TiO2:Fe and TiO2:Au, respectively). When the influence of the metal is considered, iron, palladium and zinc exhibit lower conversions than the corresponding bare-titania, whereas the presence of silver, zirconium and especially gold is beneficial to photoactivity
In the case of 2-propanol, platinum-containing solids showed quite high selectivity values to acetone (in the 78–80% range at 22–28% conversion) |
23 |
| 4 | Malic acid | TiO2-guanidine-(Ni,Co)-Fe2O4 | Catalysts suspended in malic acid aqueous solution and illuminated under visible light (150 W Quartz Halogen Lamp, λ > 400 nm) | Selectivity close to 80% for formic acid could be achieved in less than 2 hours of reaction. Efficient separation of the photocatalyst after reaction | 24 |
| 5 | Acetic, propionic, n-butanoic and n-pentanoic acids | Pt/TiO2 (rutile). Pt was deposited by illuminating each powdered semiconductor suspended in water–ethanol solution | 30 mL of water–organic acid (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) mixture and catalyst in suspension irradiated with a 500 W Xe lamp, pH < 2.0 :1 v/v) mixture and catalyst in suspension irradiated with a 500 W Xe lamp, pH < 2.0 |
In the absence of oxygen, aliphatic carboxylic acids (especially C4–C5 acids) are decarboxylated to the corresponding reduced hydrocarbons: acetic (156 micromol RH/10 h), propionic (1470 micromol RH/10 h), n-butanoic (996 micromol RH/10 h), n-pentanoic (1018 micromol RH/10h) acids | 25 |
| 6 | Glucose | Bare-TiO2 and supported titania on zeolite Y. Catalysts were synthesized by a modified ultrasound-assisted sol–gel method | Catalysts suspended in a glucose solution (50%H2O/50%CH3CN solvent composition) and illuminated with a 125 W mercury lamp (λmax = 365 nm) | High photoselectivity for glucaric and gluconic acid production (68.1% total selectivity, after 10 min. illumination time) especially for TiO2 supported on zeolite Y. Apart from photocatalyst properties, it was found that reaction conditions, especially solvent composition and short illumination times, also have considerable effect on the activity/selectivity of tested photocatalysts | 26 and 27 |
| 7 | Glucose | Fe-TiO2 and Cr-TiO2 supported on zeolite Y (SiO2:Al2O3 = 80) and prepared by an ultrasound-assisted wet impregnation method (rotary evaporator was coupled with ultrasonic bath) | Catalysts suspended in a glucose solution (50%H2O/50%CH3CN solvent composition) and illuminated with a 125 W mercury lamp (λmax = 365 nm) |
Fe-TiO2 zeolite-supported systems total selectivity for GUA + GA is 94.2% after 20 min of illumination
Cr-TiO2 zeolite-supported systems total selectivity for GUA + GA is 99.7% after 10 min of illumination |
28 |
| 8 | Lignocellulose | Commercial TiO2 | Solid state mixture of titania and lignocellulose UV irradiated (λmax = 360 nm) | Photocatalysis pretreatment used for an efficient biological saccharification of lignocellulosic materials. Titania did not disturb the biological reactions by cellulase and yeast | 36 |
| 9 | Kraft lignin | Ta2O5-IrO2 thin film (prepared using a thermal decomposition technique) as electrocatalyst and TiO2 nanotube (prepared using electrochemical anodization) as photocatalyst | Photochemical-electrochemical process in liquid phase. The intensity of the UV light was ca. 20 mW cm−2 (main line of emission, 365 nm) | Oxidation of lignin gave vanillin and vanillic acid | 37 |
Photo-assisted catalytic dehydrogenation reactions that take place at room temperature and ambient pressure offer an interesting route for aldehyde synthesis. C1–C4 alcohols are easily converted in the liquid and gas phase and in the presence of oxygen into their corresponding aldehydes or ketones, which may be further transformed by non-catalytic processes into acids.22,23
A range of different titania-based systems synthesized through sol–gel processes by varying the precursor and/or the ageing conditions (magnetic stirring, ultrasound, microwave or reflux) were recently reported for the liquid-phase selective photo-oxidation of crotyl alcohol to crotonaldehyde. The gas-phase selective photooxidation of 2-propanol to acetone was also chosen as a model reaction (Table 1, entry 3).23 Both reactions showed relatively similar results in terms of influence of precursor and metal, despite having very different reactant/catalyst ratios and contact times. Titanium isopropoxide provided better results as compared to those achieved with titanium tetrachloride. The presence of iron, palladium or zinc in the systems was found to be detrimental for the activity. Zirconium and particularly gold improved the results as compared to pure titania (Table 1, entry 3).
A higher reactivity has been generally reported for primary alcohols22 (Table 1, entry 2), which opens up a promising selective synthesis method for the production of hydroxycarboxylic acids. The selective dehydrogenation of various secondary alcohols was also possible. If the alcohol is unsaturated, isomerization may occur, yielding the corresponding saturated aldehyde.
The photocatalytic oxidation of alcohols is highly dependent on the type of alcohol. Generally, the conversion per pass of primary alcohols is low (with a slightly higher value for secondary alcohols), but high selectivities are generally achieved (>95%). The initial step of the proposed mechanism is the interaction of a surface hole with the hydroxyl group of the alcohol, forming metal-oxo species with proton removal. This proton removal step becomes easier with increasing carbon chain length and branching. The higher the number of adjacent hydrogen atoms present, the simpler the removal and thus improved conversions can be achieved.
Aliphatic carboxylic acids can be transformed to shorter chain acids (e.g. malic to formic acid)24 or decarboxylated to the corresponding reduced hydrocarbons or hydrocarbon dimers in the absence of oxygen and in pure aqueous or mixed aqueous/organic solutions by means of photo-Kolbe-type processes (Table 1, entry 5).25 As an example, the selective aqueous conversion of malic acid into formic acid has been conducted under visible light irradiation using a magnetically separable TiO2-guanidine-(Ni,Co)-Fe2O4 nanocomposite (Table 1, entry 4). The photocatalyst featured a simple magnetic separation and offered the possibility to work under visible light and sunlight irradiation due to titania modification with guanidine, which remarkably decreased the band gap of the metal oxide semiconductor.24 Comparably, acetic, propionic, butanoic and n-pentanoic acids could be decarboxylated to hydrocarbons in the absence of oxygen. For the case of acetic acid (a common product of biological processes), the aforementioned photo-Kolbe reaction could be combined with biological waste treatments to generate combustible fuels.
The efficiency of heterogeneous nano-TiO2 catalysts in the selective photocatalytic oxidation of glucose into high-valued organic compounds has also been recently reported (Fig. 3).26 This reaction was found to be highly selective (>70%) towards two organic carboxylic acids, namely glucaric (GUA) and gluconic acids (GA). These carboxylic acids are important building blocks for pharmaceutical, food, perfume or fuel industries.18 CO2 and traces of light hydrocarbons were also detected in the gas phase. Among all photocatalytic systems tested, the best product selectivity was achieved with titania synthesized by an ultrasound-modified sol–gel methodology (TiO2(US)).26 Solvent composition and short illumination times were proved to have a considerable effect on photocatalysts activity/selectivity. Total organic compound selectivity was found to be 39% and 71% for liquid phase reactions using 10%Water/90%Acetonitrile and 50%Water/50%Acetonitrile, respectively. These values were obtained using the optimum TiO2(US) photocatalyst.
 | ||
| Fig. 3 Acid catalytic hydrolysis of cellulose followed by catalytic partial oxidation of glucose to gluconic acid. | ||
Such results suggested that synthesized nano-TiO2 material could in principle be used in the decomposition of waste from the food industry with the simultaneous production of high-value chemicals when residues (here glucose) act as electron donors.18
In an attempt to improve selectivities to glucaric (GUA) and gluconic (GA) acids, different nano-titania systems supported on a zeolite type Y (SiO2:Al2O3 = 80) have been prepared.27 These photocatalysts were synthesized using the proposed ultrasound-assisted sol–gel protocol fully detailed in Fig. 4.
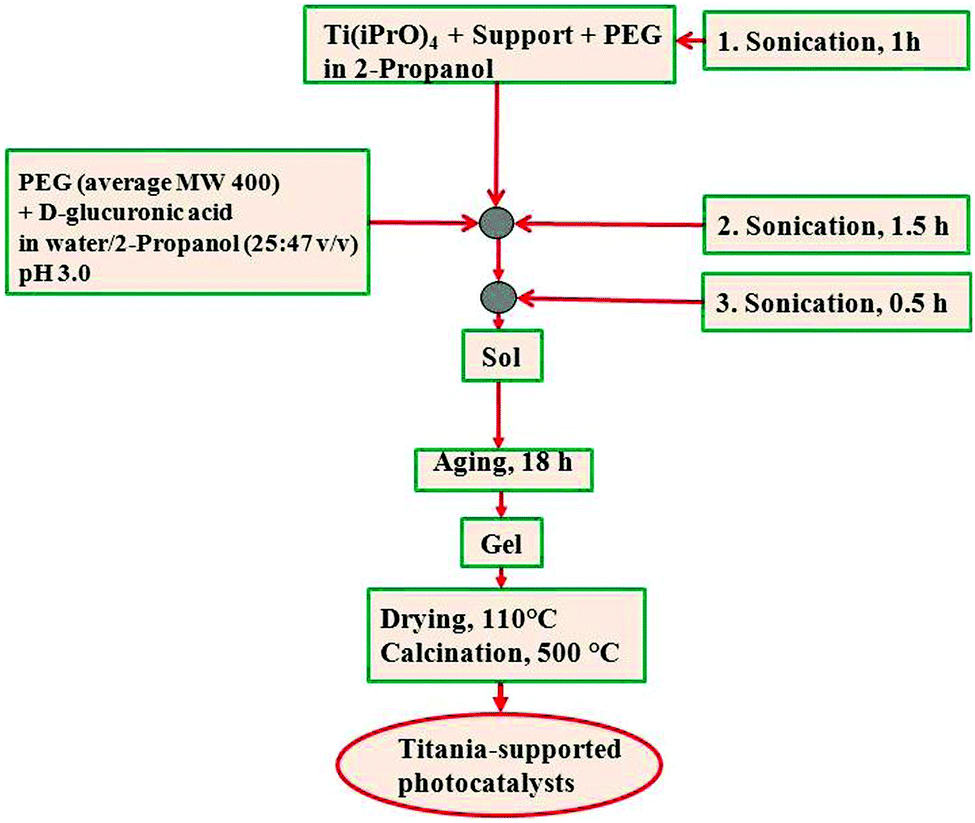 | ||
| Fig. 4 Schematic chart of TiO2 supported on different materials (e.g., zeolite, silica) prepared by an ultrasound-assisted sol–gel methodology.27 | ||
Homogeneously distributed TiO2 on zeolite Y provided an improved selectivity for glucose oxidation towards glucaric acid (GUA) and gluconic acid (GA). Total selectivity was ca. 68% after 10 min illumination time using a 1:1 H2O–acetonitrile solvent composition. Results were comparably superior to those of unsupported TiO2 and commercially available Evonik P-25 photocatalyst (Table 1, entry 6). Importantly, the more acidic character of the zeolite in comparison to silica-supported, bulk TiO2(US) and Evonik P-25 may relate to the higher selectivity to carboxylic acids. Moreover, Y type zeolites are negatively charged materials which adsorb cationic substrates and repulse anionic analogues by electrostatic attraction. This behavior can facilitate the selective photocatalytic oxidation of glucose, as carboxylic acids formed under the reaction conditions (pH lowered from 6–7, depending on glucose solvent composition, to about 4–5 after 4 h of photocatalytic reaction) are repulsed, preventing at the same time their complete photo-oxidation/mineralization.
Further photocatalyst optimization via development of transition-metal containing supported nanotitania materials (Fig. 5)28 provided advanced systems incorporating Fe or Cr able to achieve improved selectivities to carboxylic acids. No metal leaching (Fe, Cr, Ti) was detected after photoreaction, with Fe–TiO2 systems being most selective (94% after 20 min of illumination under similar conditions previously reported, Table 1, entry 7).
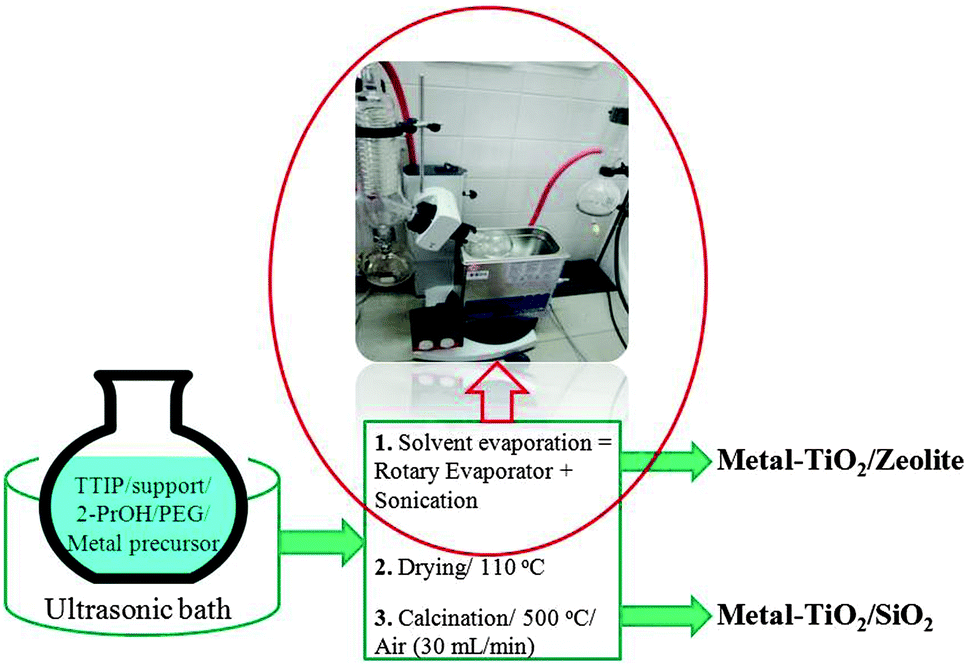 | ||
| Fig. 5 General procedure to synthesize metal-containing TiO2 supported on different materials (e.g., zeolite, silica) by a sonication-assisted impregnation method (titanium tetraisopropoxide, TTIP; polyethylene glycol 400 molecular weight, PEG).28 | ||
The gas-phase selective photo-oxidation of methanol to methyl formate is another interesting process.29 The reaction was carried out in a flow-type reactor to avoid deep oxidation of methanol in the presence of TiO2 particles and under UV light irradiation. A high selectivity to methyl formate was observed (91%) with no catalyst deactivation observed under the investigated conditions (Table 1, entry 1).
The oxidation mechanism, efficiency and selectivity of the photocatalytic oxidation of alcohols using TiO2 Evonik P25 were reported to strongly depend on the nature of the dispersing medium. Shiraishi et al.16 and Morishita et al.30 demonstrated the positive influence of acetonitrile on epoxide formation in the selective photo-oxidation of alkenes. The addition of small amounts of water to CH3CN strongly inhibited alcohol adsorption and its subsequent oxidation as evidenced by ESR-spin trapping investigations.31 The reactivity of alcohols on the surface of photoexcited TiO2 was also found to be affected by the nature of their hydrophobic aliphatic chain.31 Molecules including geraniol and citronellol were observed to be more susceptible to water content as compared to shorter chain analogues such as trans-2-penten-1-ol and 1-pentanol. Optimum reaction conditions were achieved in the photocatalytic oxidation of geraniol, citronellol, trans-2-penten-1-ol, and 1-pentanol to the corresponding aldehydes with good selectivity (>70%).
Along these lines, a partial photooxidation of diols including 1,3-butanediol, 1,4-pentanediol and vicinal-diols (e.g. 1,2 propanediol) could also be effectively conducted using TiO2 in dichloromethane.32 CO2 was not observed as a reaction product but the observed main reaction products included two hydroxy-carbonyl compounds for each 1,n-diol. 1,2-propanediol mainly gave rise to hydroxyacetone (90% selectivity), with only traces of pyruvic acid. Comparatively, 1,3-butanediol was converted into 3-hydroxybutyraldehyde and 4-hydroxy-2-butanone while 1,4-pentanediol gave 4-hydroxypentanal (75% selectivity) and certain quantities of 3-acetyl 1-propanol.
Similarly, glycerol is a relevant polyol currently produced in large quantities as a by-product of the biodiesel industry. The mechanism of the selective photocatalytic oxidation of glycerol was also recently reported in the presence of TiO2 Evonik P25 and Merck TiO2 (Fig. 6).33 The product distribution observed at low glycerol concentration (glyceraldehyde and dihydroxyacetone) changes after a sharp maximum giving formaldehyde and glycolaldehyde as main products for P25 Evonik (mechanism derived from a direct electron transfer). Interestingly, mainly glyceraldehyde and dihydroxyacetone were observed on Merck TiO2 (˙OH-based mechanism), a material characterized by a lower density and more uniform population of hydroxyl groups at surface sites. These findings suggested that photocatalyst structures can significantly influence product distribution in photo-assisted processes. This example is particularly relevant to engineering of valuable products from a biofuel-derived by-product.
 | ||
| Fig. 6 Pathways for glycerol photo-oxidation to valuable chemicals. | ||
A comprehensive investigation on advanced mechanistic aspects of polyol photooxidation on photoactivated metal oxides was subsequently reported aiming to better understand the interaction of sugars with the surface of some semiconductor materials.34 The authors demonstrated that carbohydrates are oxidized at sites involved in the formation of oxo bridges between the chemisorbed carbohydrate molecule and metal ions at the oxide surface (e.g. TiO2, α-FeOOH, and α-Fe2O3). Bridging was claimed to inhibit the loss of water, promoting a rearrangement that leads to elimination of formyl radicals. For natural carbohydrates, the latter reaction mainly involves carbon-1, whereas the main radical products of the oxidation were observed to be radicals arising from H atom loss centered on carbon-1, -2, and -3 sites. Photoexcited TiO2 oxidizes all carbohydrates and polyols, whereas α-FeOOH oxidizes some of the carbohydrates, α-Fe2O3 being unreactive.34
Detailed mechanistic studies on photocatalytic oxidation of related organic compounds (e.g., methane to methanol, partial oxidation of geraniol to produce only citral, glycerol oxidation and propene epoxidation among others) are obviously out of the scope of the present contribution, but readers are kindly referred to a recently reported comprehensive overview on this subject.35
5.2. Lignocellulosics photo-assisted pretreatments and selective photo-oxidations of aromatic alcohols as lignin model compounds
Lignocellulosics pretreatment using (bio)chemical methodologies has attracted a great deal of attention in recent years. Enzymatic saccharification (SA) and subsequent fermentation (FE) are conventionally utilized processes for bio-ethanol production from cellulose materials. Importantly, an efficient biological saccharification of lignocellulosic material requires certain pre-treatment steps. Several reported pre-treatments include the use of dilute H2SO4, alkali or pressured hot water and even the utilisation of ionic liquids. Photocatalytic pre-treatments of lignocellulosics have however rarely been reported in the literature. Interestingly, Yasuda et al.36 have recently devised a 2 step process for biological bioethanol production from lignocellulose via feedstock pre-treatment with titania photocatalyst under UV-illumination. Selected lignocellulosic feedstocks were napiergrass (Pennisetum purpureum Schumach) and silver grass (Miscanthus sinensis Andersson) as summarized in Table 1, entry 8. The photocatalytic pre-treatment did not significantly affect the final product distribution, demonstrating that TiO2 was not interfering with biological reactions promoted by cellulases and yeast. Most importantly, the photocatalytic pre-treatment was remarkably effective in reducing the time in SA and FE reactions with respect to untreated and NaOH pre-treated feedstocks. The photocatalytic pretreatment was also proved to be an environmentally sound replacement for acid- and alkali-based processes. This report constitutes the first finding on photocatalytic pre-treatments featuring important insights into bioethanol production from cellulose.Another example of the combination of a photocatalytic process with biomass oxidation comprised an integration of photochemical and electrochemical oxidation processes for the modification and degradation of kraft lignin applied (Table 1, entry 9).37 Ta2O5–IrO2 thin films were used as electrocatalysts with TiO2 nanotube arrays as photocatalyst. Lignin deconstruction provided vanillin and vanillic acid, relevant compounds with important applications in the food and perfume industries.
In spite of these recent reports that exemplify the potential of photocatalysis for lignocellulosics conversion, selective photocatalytic transformations of lignocellulosic feedstocks are rather challenging due to the complex structure of lignocellulose, particularly related to the highly recalcitrant lignin fraction. Comparatively, relevant research has been conducted on model compounds which have been selected on the basis of representing structural motifs present in lignocellulose.
Aromatic alcohols have been used as model lignin compounds to be converted into various compounds via photocatalytic processing (Table 2). Among selective oxidation processes of alcohols, the conversion of benzyl alcohol to benzaldehyde is particularly attractive. Benzaldehyde is in fact the second most important aromatic molecule (after vanillin) used in the cosmetics, flavor and perfumery industries. Synthetic benzaldehyde is industrially produced via benzylchloride hydrolysis derived from toluene chlorination or through toluene oxidation. In this regard, alternative protocols able to selectively produce benzaldehyde from benzyl alcohol are in demand. While most research on selective production of benzaldehyde has been reported via heterogeneously catalyzed protocols, there are some relevant examples of such reactions under photo-assisted conditions. Aerobic aqueous suspensions of Au supported on cerium(IV) oxide (Au/CeO2) were reported to be able to quantitatively oxidize benzyl alcohol to benzaldehydes under green light irradiation for a reaction time of 36 h (Table 2, entries 4–7).38 Comparatively, the oxidation of benzyl alcohol was performed using differently prepared TiO2 catalysts, reaching selectivities three- to seven fold superior to those of commercial TiO2 catalysts.39
| Entry | Reactants | Products | Conversion [%] | Selectivity [%] |
|---|---|---|---|---|
| Immobilized TiO2 on silica cloth. Reaction conditions: alcohol, 1.43 mmol min−1; O2/alcohol = 22; 2.5-L annular photoreactor (average contact time 32 s); temperature = 190 °C; conversion after 2-h illumination (UV light) time.49 | ||||
| 1 |

|

|
35 | >95 |
| 2 |

|

|
97 | 7 (83% styrene) |
| 3 |

|
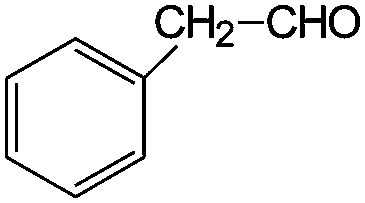
|
53 | 26 |
| Oxidation over 1 wt% Au/CeO2 under green light. Reaction conditions: benzyl alcohols 33 μmol; Au/CeO2 50 mg; 5 mL water; 1 bar O2; 25 °C; LED 530 nm; conversion after 20-h illumination time.38 | ||||
| 4 |

|

|
>99 | >99 |
| 5 |

|
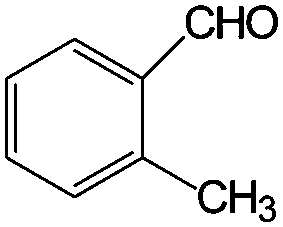
|
>99 | >99 |
| 6 |

|

|
>99 | >99 |
| 7 |

|

|
>99 | >99 |
| Carbon Quantum Dots (CQDs) under NIR light irradiation. Reaction conditions: 10 mmol alcohol; H2O2 (10 mmol, 30 wt% in water); 8 mg of CQD catalyst; 60 °C; NIR irradiation for 12 h.46 | ||||
| 8 |

|

|
92 | 100 |
| 9 |

|
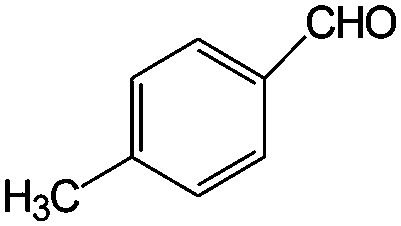
|
88 | >99 |
| 10 |

|

|
86 | >99 |
| 11 |

|

|
90 | >96 |
| 12 |

|

|
88 | >99 |
| Au2(DP673)/P25 catalyst. Reaction conditions: 5 mL toluene (solvent); 10 μmol alcohol; 20 mg catalyst; 1 bar O2; 30 °C; 4 h irradiation time (98% of light involves the visible range).47 | ||||
| 13 |

|

|
79 | 100 |
| 14 |

|

|
85 | 93 |
| 15 |

|

|
>99 | >100 |
| 16 |

|

|
>99 | 91 |
| 17 |

|

|
83 | 99 |
| 18 |

|

|
>99 | >100 |
| 19 |

|

|
81 | 99 |
| Multi-Pd core–CeO2 shell nanocomposite. Reaction conditions: 20 h of visible light (λ > 420 nm) irradiation time.48 | ||||
| 20 |

|

|
28 | 100 |
| 21 |

|
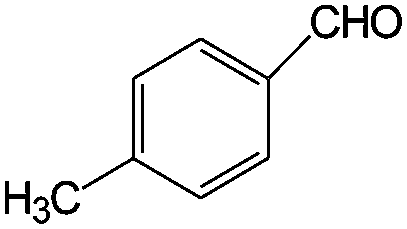
|
10 | 100 |
| 22 |

|
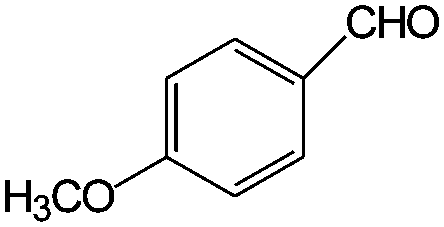
|
12 | 100 |
In terms of benzyl alcohol derivatives, the aqueous phase photocatalytic oxidation of 4-methoxybenzyl alcohol was achieved by using uncalcined brookite TiO2. A maximum selectivity of ca. 56% towards aldehydes (i.e. ca. 3 times higher than that obtained with commercial TiO2) was reported.
Heteropolyoxometalate (POM) catalysts of the type [S2M18O62]4+ (M = W, Mo)40 have also been developed to carry out the photo-oxidation of aromatic alcohols under sunlight and UV/Vis light in acetonitrile. Mechanistic investigations of photocatalysis by these nontoxic compounds indicate that near-UV/Vis irradiation of a POM solution results in an oxygen-to-metal charge-transfer excited state that has strong oxidation ability, responsible for the oxidation of organic substrates. Photoexcited POMs were observed to be reduced by the transfer of one or two electron(s) from the organic substrate. Other types of POMs including water tolerant materials by combining homogeneous POMs with photoactive and inactive supporting materials (e.g., mesoporous molecular sieves, titania in amorphous phase) and silica-encapsulated H3PW12O40 have also been reported in similar chemistries.
The acceleration of the aerobic photo-oxidation of similar alcohols has also been described to take place on TiO2 or SiO2/TiO2 samples without any loss of selectivity via surface loading of Bronsted acids (reaction time 2–7 h, conversion of the starting molecules ca. 42–100 mol%, selectivity 73–100 mol%).41 The effect of Bronsted acids was confirmed and enhanced when a small quantity of SiO2 was incorporated into TiO2 after the acid pretreatment, due to the presence of more acid sites on the TiO2 surface. The acceleration effect was attributed to the protons of Bronsted acids that effectively promote the decomposition of the formed surface peroxide-Ti species. This was demonstrated by means of in situ spectroscopy titration measurements which indicated that this phenomenon leads to surface site regeneration, contributing to an enhancement of the cycle efficiency without any loss of selectivity.
A similar selective photocatalytic oxidation of aromatic alcohols to aldehydes was systematically studied under visible-light irradiation utilizing anatase TiO2 nanoparticles.42 The unique features of the protocol rely on the possibility to use visible light irradiation apart from UV due to the formation of surface complex species by the adsorbed aromatic alcohol. Studies using fluorinated TiO2 pointed to a dramatic decrease of photocatalytic activity under visible light irradiation which suggested a significant role played by the adsorption of the aromatic alcohol species on the surface of the solid involving the surface OH species.42Fig. 7 summarizes the reaction pathway proposed by Higashimoto et al.42a for the selective photocatalytic oxidation of benzyl alcohol in the presence of oxygen and titania. The surface complexes formed by the close interaction of the alcohol group or the aromatic ring with the hydroxyl surface group are able to absorb in the visible region, and hence the photoexcited complex undergoes hydrogen abstraction in the CH2OH group by the photoproduced holes forming a water molecule. The organic radical also releases one electron to form benzaldehyde. At the same time the photoinduced electrons are trapped by oxygen in the reacting medium.
 | ||
| Fig. 7 Proposed reaction mechanism for the photocatalytic selective oxidation of benzyl alcohol under visible light irradiation.42a | ||
Benzyl alcohol and some of its derivatives (para-structures) could be converted into their corresponding aldehydes at high conversions and selectivities (ca. 99%) under both UV and visible light irradiation. The only exception to this behavior was 4-hydroxybenzyl alcohol which was oxidized to 4-hydroxy benzaldehyde (selectivity of ca. 23% at ca. 85% conversion) along with some unidentified products. The authors claimed that OH groups from the TiO2 surface reacted with the aromatic compound to form Ti–O–Ph species, exhibiting strong absorption in the visible region by ligand to metal charge transfer. These findings confirmed the results reported by Kim et al.43 which proposed a direct electron transfer from the surface complexes to the conduction band of the TiO2 upon absorbing visible light. Importantly, results evidenced that visible light can induce reactions by substrate–surface complexation enabling visible light absorption. Similar observations were recently reported by Li and co-workers44 with nanorods of rutile titania phase synthesized by a hydrothermal reaction using rutile TiO2 nanofibers obtained from calcination of composite electrospun nanofibers. The authors proposed a tentative mechanism where selective photocatalytic activities were proposed to be due to the visible-light absorption ability of the benzyl alcohol–TiO2 nanorod complex and the unique properties of rutile TiO2 nanorods. Excellent properties of these materials including a high surface-to-volume ratio, unidirectional 1D channels and superior survivability of electrons may contribute to more efficient electron transport, further required for benzaldehyde formation (>99% selectivity to benzaldehyde).
Visible light illumination of nitrogen-doped TiO2 prepared by a sol–gel method brings about the selective oxidation of benzyl and cinnamyl alcohols to the corresponding aldehydes. Upon UV light irradiation, the alcohol conversion rate is faster but the reaction is unselective with respect to aldehyde formation.45 The reaction takes place in oxygenated dry nitrile solvents (CH3CN as the optimum solvent) and is totally inhibited in the presence of water. Alcohols were proved to be weakly adsorbed (do not readily capture the photogenerated holes) and the interaction of acetonitrile with the surface was demonstrated by its quenching of luminescence from band gap energy levels introduced by nitrogen. The formation of active oxygen species, particularly superoxide radicals, was proposed to be a key channel leading to aldehyde formation. Nevertheless, the possibility of dry acetonitrile playing an active role in the reaction mechanism cannot be ruled out as acetonitrile has been previously reported to promote the formation of active radical species.45
A new class of carbon nanomaterials known as Carbon Quantum Dots (CQDs) with sizes below 10 nm can also function as an effective near infrared (NIR) light driven photocatalyst for the selective oxidation of benzyl alcohol to benzaldehyde.46 Based on the NIR light driven photo-induced electron transfer property and its photocatalytic activity for H2O2 decomposition, this metal-free catalyst could efficiently provide high yields of benzaldehyde under NIR light irradiation (Table 2, entries 8–12). Hydroxyl radicals were the main active oxygen species in benzyl alcohol selective oxidative reaction as confirmed by terephthalic acid photoluminescence probing assay, selecting toluene as a substrate. Such metal-free photocatalytic system also selectively converts other alcohol substrates to their corresponding aldehydes with high conversion, demonstrating a potential application of accessing traditional alcohol oxidation chemistry.
Plasmonic photocatalysts (noble metal – semiconductor hybrids) have been reported for selective formation of organic molecules.47 Nanosized (<5 nm) gold nanoparticles supported on anatase–rutile interphase (Evonik P-25 photocatalyst) by a deposition–precipitation technique could take advantage of plasmonic effects to achieve the photocatalytic selective production of several aromatic aldehydes from their corresponding aromatic alcohols (Table 2, entries 13–19). This photocatalysis can be promoted via plasmon activation of the Au particles by visible light followed by consecutive electron transfer in the Au/rutile/anatase interphase contact. Activated Au particles transfer their conduction electrons to rutile and then to adjacent anatase which catalyzes the oxidation of substrates by the positively charged Au particles along with reduction of oxygen by the conduction band electrons on the surface of anatase titania. Oxygen peroxide species have been found to be responsible for alcohol oxidation. Au2(DP673)/P25 produced 17 μmol of acetophenone in the dark as a result of the high activity of the small Au particles. Light irradiation further enhanced the reaction to produce 74 μmol of acetophenone (over 4 times that obtained in the absence of light). Visible light illumination of Au/P25 catalyst clearly enhances the oxidation process.
Recent progress in metal core-semiconductor shell nanohybrids has also demonstrated that these systems can be potentially utilized as photocatalysts for selective organic oxidations.48 A (Pt)–CeO2 nanocomposite has been prepared in an aqueous phase with tunable core–shell and yolk–shell structure via a facile and green template-free hydrothermal approach, as illustrated in Fig. 8A.48 The authors found that the core–shell nanocomposite could serve as an efficient visible-light-driven photocatalyst for the selective oxidation of benzyl alcohol to benzaldehyde using molecular oxygen as a green oxidant. The same authors also prepared various hybrids based on other metals (e.g. Pd) and carried out photocatalytic selective oxidation of benzylic alcohols over the multi-Pd core@CeO2 shell nanocomposite (Table 2, entries 20–22). Electron–hole pairs are produced under visible light irradiation. Electrons are then trapped by the Pd cores and adsorbed benzyl alcohol interacts with holes to form the corresponding radical cation. Further reaction with dioxygen or superoxide radical species will lead to the formation of the corresponding aldehydes, with the plausible mechanism illustrated in Fig. 8B.
 | ||
| Fig. 8 (A) TEM image of as-synthesized multi-Pd-core–CeO2-shell nanoparticles; (B) a plausible mechanism for the selective oxidation of aromatic alcohols to aldehydes in the presence of multi-Pd-core–CeO2-shell photocatalyst under visible light irradiation (adapted from ref. 48, Royal Society of Chemistry). | ||
These photocatalytic protocols account for lab scale systems with relatively restricted applications. However, the possibility of scaling up photocatalytic processes to selective large scale processes has also been attempted in recent years. With regard to photocatalytic selective oxidation processes, various aromatic alcohols could be converted in a gas-phase 2.5-liter annular photoreactor using immobilized TiO2 catalyst and under UV light (Table 2, entries 1–3).49 The system was found to be specifically suited for the selective oxidation of primary and secondary aliphatic alcohols to their corresponding carbonyl compounds. Benzylic alcohols gave higher conversions, however, with more secondary reaction products. The presence of oxygen was found to be critical for the photooxidation. One disadvantage of the system relates to catalyst deactivation, which is attributed to the surface accumulation of reaction products. However, the catalyst could be regenerated by calcination in air for 3 h at 450 °C, providing similar activities to those of fresh catalysts.49
Another similar scale-up protocol was developed in a solar pilot plant reactor using TiO2/Cu(II) photocatalyst.50 A maximum yield of 53.3% for benzaldehyde could be obtained with respect to initial benzyl alcohol concentration (approx. 63% selectivity, reaction time 385 min), operating with an average temperature of 38 °C. It is well known that the substitution of oxygen with potential reducing species (in this case metal ions dissolved in the solution) still enables the oxidation of the organic species present in the solution. The authors claimed that in the absence of oxygen, one of the photochemical pathways is inhibited, leading to unselective OH radical production. In fact, the formation of superoxide radicals (O2˙−) or hydrogen peroxide (H2O2) is not possible in the absence of oxygen, whose photolysis can generate OH radicals. In this case, OH radicals should form just as a result of the reaction of water with positive holes.
6. Future challenges and prospects
The concept to utilize solar light energy at room temperature and atmospheric pressure via photocatalysis to selectively convert lignocellulosic biomass to important chemicals and valuable products is a highly innovative approach that can bring several benefits from energy and environmental viewpoints. As such, photocatalytic selective processes are able to keep selectivities to products at reasonable levels at increasing conversions in the systems, as opposed to conventional thermally activated heterogeneous catalysis.Photochemical reactions also require milder conditions to those generally present in thermal processes which may allow the conception of short and efficient reaction sequences, minimizing side processes making use of sunlight as a completely renewable source of energy (leaving no residue). In the near future, integrated systems of simultaneous photocatalyst-mediated chemical evolution and water purification may be possible. These processes could then be efficiently coupled to scale-up facilities (e.g. solar plants) able to provide continuous flow equipment and large reactors for important advances in the field.
Ideal photocatalysts for commercial applications in the valorization of such organic residues should be designed to be able to maximize conversion with a compromised selectivity for the desired product under solar light irradiation in aqueous solution (or a solventless environment), avoiding complete mineralization. However, even though most reported syntheses can be virtually carried out under sunlight, there are only very few reports on sunlight utilization for such transformations due to reproducibility issues in measurements.
This contribution has been aimed to illustrate that novel nanotechnology and nanomaterials design are currently bringing innovative concepts, ideas and protocols to the design and preparation of well-defined heterogeneous photocatalysts with highly controllable and desirable properties. By combining these new synthetic routes with surface-science, fundamentals of heterogeneous photocatalysis and theoretical mechanistic studies, we should possibly develop a new generation of highly stable and selective photocatalysts for oxidative organic transformations including those of lignocellulosic derivatives from which we hope to actively participate in future years to come.
Acknowledgements
Dr Colmenares would like to thank the Marie Curie International Reintegration Grant within the 7th European Community Framework Programme and also the 2012–2014 science financial resources, granted for the international co-financed project implementation (Project Nr. 473/7.PR/2012, Ministry of Science and Higher Education of Poland). Rafael Luque gratefully acknowledges support from the Spanish MICINN via the concession of a RyC contract (ref. RYC 2009-04199) and funding under projects P10-FQM-6711 (Consejeria de Ciencia e Innovacion, Junta de Andalucia) and CTQ201-28954-C02-02 (MICINN).Notes and references
- G. Ciamician, Science, 1912, 36, 385 CAS
.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS
- J. A. Turner, Science, 2004, 305, 972 CrossRef CAS PubMed
- S. Rawalekar and T. Mokari, Adv. Energy Mater., 2013, 3, 12 CrossRef CAS
-
(a) M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725 CrossRef CAS PubMed
- A. Fujishima, T. N. Rao and D. A. Tryk, J. Photochem. Photobiol., C, 2000, 1, 1 CrossRef CAS
- M. Stoecker, Angew. Chem., Int. Ed., 2008, 47, 9200 CrossRef PubMed
- C. S. K. Lin, L. A. Pfaltzgraff, L. Herrero-Davila, E. B. Mubofu, S. Abderrahim, J. H. Clark, A. Koutinas, N. Kopsahelis, K. Stamatelatou, F. Dickson, S. Thankappan, Z. Mohamed, R. Brocklesby and R. Luque, Energy Environ. Sci., 2013, 6, 426 CAS
- A. Stark, Energy Environ. Sci., 2011, 4, 19 CAS
-
(a) S. Van de Vyver, J. Geboers, P. A. Jacobs and B. F. Sels, ChemCatChem, 2011, 3, 82 CrossRef CAS
-
(a) Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994 RSC
- M. Ni, D. Y. C. Leung, M. K. H. Leung and K. Sumathy, Fuel Process. Technol., 2006, 87, 461 CrossRef CAS PubMed
- J. R. Bolton, S. J. Strickler and J. S. Connolly, Nature, 1985, 316, 495 CrossRef CAS
-
Producing Fuels and Fine Chemicals from Biomass Using Nanomaterials, ed. R. Luque and A. M. Balu, Taylor and Francis Book Inc., New Jersey, 2013 Search PubMed
- A. Di Paola, E. García-López, G. Marcì and L. Palmisano, J. Hazard. Mater., 2012, 211–212, 3 CrossRef CAS PubMed
- Y. Shiraishi, M. Morishita and T. Hirai, Chem. Commun., 2005, 5977 RSC
-
(a) M. D. Tzirakis, I. N. Lykakis and M. Orfanopoulos, Chem. Soc. Rev., 2009, 38, 2609 RSC
- J. C. Colmenares, R. Luque, J. M. Campelo, F. Colmenares, Z. Karpinski and A. A. Romero, Materials, 2009, 2, 2228 CrossRef CAS
- S. E. Davis, M. S. Ide and R. J. Davis, Green Chem., 2013, 15, 17 RSC
-
R. Ho, J. Liebman and J. Valentine, in Active Oxygen in Chemistry, ed. C. Foote, J. Valentine, A. Greenberg and J. Liebman, Springer, Berlin, 1995, pp. 1–23 Search PubMed
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539 RSC
- P. R. Harwey, R. Rudham and S. Ward, J. Chem. Soc., Faraday Trans. 1, 1983, 79, 2975 RSC
- F. J. Lopez-Tenllado, A. Marinas, F. J. Urbano, J. C. Colmenares, M. C. Hidalgo, J. M. Marinas and J. M. Moreno, Appl. Catal., B, 2012, 128, 150 CrossRef CAS PubMed
- A. M. Balu, B. Baruwati, E. Serrano, J. Cot, J. Garcia-Martinez, R. S. Varma and R. Luque, Green Chem., 2011, 13, 2750 RSC
- T. Sakata, T. Kawai and K. Hashimoto, J. Phys. Chem., 1984, 88, 2344 CrossRef CAS
- J. C. Colmenares, A. Magdziarz and A. Bielejewska, Bioresour. Technol., 2011, 102, 11254 CrossRef CAS PubMed
- J. C. Colmenares and A. Magdziarz, J. Mol. Catal. A: Chem., 2013, 366, 156 CrossRef CAS PubMed
-
(a) J. C. Colmenares, A. Magdziarz, K. Kurzydlowski, J. Grzonka, O. Chernyayeva and D. Lisovytskiy, Appl. Catal., B, 2013, 134–135, 136 CrossRef CAS PubMed
- H. Kominami, H. Sugahara and K. Hashimoto, Catal. Commun., 2010, 11, 426 CrossRef CAS PubMed
- M. Morishita, Y. Shiraishi and T. Hirai, J. Phys. Chem. B, 2006, 110, 17898 CrossRef CAS PubMed
- A. Molinari, M. Montoncello, R. Houria and A. Maldotti, Photochem. Photobiol. Sci., 2009, 8, 613 CAS
- A. Molinari, M. Bruni and A. Maldotti, J. Adv. Oxid. Technol., 2008, 11, 143 CAS
- C. Minero, A. Bedini and V. Maurino, Appl. Catal., B, 2012, 128, 135 CrossRef CAS PubMed
- I. A. Shkrob, T. W. Marin, S. D. Chemerisov and M. D. Sevilla, J. Phys. Chem. C, 2011, 115, 4642 CAS
- V. Augugliaro, M. Bellardita, V. Loddo, G. Palmisano, L. Palmisano and S. Yurdakal, J. Photochem. Photobiol., C, 2012, 13, 224 CrossRef CAS PubMed
- M. Yasuda, A. Miura, R. Yuki, Y. Nakamura, T. Shiragami, Y. Ishii and H. Yokoi, J. Photochem. Photobiol., A, 2011, 220, 195 CrossRef CAS PubMed
- M. Tian, J. Wen, D. MacDonald, R. M. Asmussen and A. Chen, Electrochem. Commun., 2010, 12, 527 CrossRef CAS PubMed
- A. Tanaka, K. Hashimoto and H. Kominami, J. Am. Chem. Soc., 2012, 134, 14526 CrossRef CAS PubMed
- G. Palmisano, E. Garcia-Lopez, G. Marcì, V. Loddo, S. Yurdakal, V. Augugliaro and L. Palmisano, Chem. Commun., 2010, 46, 7074 RSC
- T. Ruether, A. M. Bond and W. R. Jackson, Green Chem., 2003, 5, 364 RSC
- Q. Wang, M. Zhang, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2010, 49, 7976 CrossRef CAS PubMed
-
(a) S. Higashimoto, N. Kitao, N. Yoshida, T. Sakura, M. Azuma, H. Ohue and Y. Sakata, J. Catal., 2009, 266, 279 CrossRef CAS PubMed
- S. Kim and W. Choi, J. Phys. Chem. B, 2005, 109, 5143 CrossRef CAS PubMed
- C.-J. Li, G.-R. Xu, B. Zhang and J. R. Gong, Appl. Catal., B, 2012, 115–116, 201 CrossRef CAS PubMed
- L. Samiolo, M. Valigi, D. Gazzoli and R. Amadelli, Electrochim. Acta, 2010, 55, 7788 CrossRef CAS PubMed
- H. Li, R. Liu, S. Lian, Y. Liu, H. Huang and Z. Kang, Nanoscale, 2013, 5, 3289 RSC
- D. Tsukamoto, Y. Shiraishi, Y. Sugano, S. Ichikawa, S. Tanaka and T. Hirai, J. Am. Chem. Soc., 2012, 134, 6309 CrossRef CAS PubMed
- N. Zhang, S. Liu and Y.-J. Xu, Nanoscale, 2012, 4, 2227 RSC
- U. R. Pillai and E. Sahle-Demessie, J. Catal., 2002, 211, 434 CAS
- D. Spasiano, L. P. Prieto Rodriguez, J. Carbajo Olleros, S. Malato, R. Marotta and R. Andreozzi, Appl. Catal., B, 2013, 136–137, 56 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2014 |