 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spiropyran-based dynamic materials
Rafal Klajn
Department of Organic Chemistry, Weizmann Institute of Science, Rehovot 76100, Israel. E-mail: rafal.klajn@weizmann.ac.il
First published on 27th August 2013
Abstract
In the past few years, spiropyran has emerged as the molecule-of-choice for the construction of novel dynamic materials. This unique molecular switch undergoes structural isomerisation in response to a variety of orthogonal stimuli, e.g. light, temperature, metal ions, redox potential, and mechanical stress. Incorporation of this switch onto macromolecular supports or inorganic scaffolds allows for the creation of robust dynamic materials. This review discusses the synthesis, switching conditions, and use of dynamic materials in which spiropyran has been attached to the surfaces of polymers, biomacromolecules, inorganic nanoparticles, as well as solid surfaces. The resulting materials show fascinating properties whereby the state of the switch intimately affects a multitude of useful properties of the support. The utility of the spiropyran switch will undoubtedly endow these materials with far-reaching applications in the near future.
1. Introduction
For centuries, nature has awed scientists with its rich repertoire of materials and systems that can reversibly adjust their structure and properties in response to environmental stimuli. Wide-ranging examples include heat-shock response in bacteria,1 camouflage in caphalopods and chameleons,2 colour changes in echinoderms in response to light,3 and avian flocking in the presence of a predator.4 In sharp contrast, nearly all traditional man-made materials are static in both form and function, and only quite recently are synthetic materials chemists gradually shifting their attention to dynamic materials.5–9 Such dynamic materials have multiple advantages over their static counterparts: selected properties of interest can be reversibly “turned on” and “off” at will and the ability to reconfigure these materials imparts upon them many uses. Emerging applications include “smart” windows for the construction of energy-efficient buildings, self-erasing (reusable) paper or self-healing coatings, to name just a few.Among the different forms of external inputs that can influence the state of a material, light has numerous advantages:10–16 it can be delivered with high spatial and temporal precision, no chemical contaminants are introduced, closed systems can be actuated, and finally, light of specific wavelengths can be delivered. The last feature is of great value when photoswitchable molecules are used as light-harvesting elements; azobenzene,17,18 for example, isomerises between two forms when exposed to near-UV (∼350 nm) and blue (∼420 nm) light, respectively. Accordingly, various photoswitchable molecules – azobenzenes,17,18 stilbenes,19,20 spiropyrans,21–24 diarylethenes,25,26 fulgides,27,28 and others29–31 – have been widely investigated and employed for the construction of light-responsive systems and materials. Each of these photoswitches has its advantages and disadvantages; azobenzenes, for example, are structurally simple and readily accessed synthetically; unfortunately the yield of the trans → cis conversion is usually far from quantitative. Diarylethenes, on the other hand, are not ideal for the design of mechanically switchable architectures since their isomerisation is accompanied by a relatively small change in molecular conformation, but this class of molecules has shown superb resistance to photodegradation11,32 – a drawback which has traditionally been associated with spiropyrans (cf., however, Section 1.3.6).
What makes spiropyrans unique among this broad spectrum of photoswitches, however, is that its two isomers (see Section 1.1) have vastly different properties. As a consequence, spiropyran is far more than just a simple photoswitch; the range of stimuli able to induce its reversible isomerisation is truly impressive and includes different solvents, metal ions, acids and bases, temperature, redox potential, and mechanical force. This versatility vis-à-vis input method highlights the far-reaching capabilities of new spiropyran-based dynamic materials. It is important to emphasise, however, that in order for the dynamic materials to be robust and ultimately meet the requirements of real-world applications, it is necessary for the active components – the spiropyran units – to be covalently attached to the support33 (as elaborated in Section 1.3). For this reason, examples based on non-covalent association34–37 of small-molecule spiropyrans with macromolecules or surfaces are not included in the current review.
Materials and systems covered in this review are divided according to the type of support spiropyrans are immobilised onto/within. These supports include polymer chains (Sections 2.1–2.7), biomacromolecules (Sections 3.1–3.8), inorganic nanoparticles (NPs) (Sections 4.1 and 4.2), and solid surfaces (Sections 5.1–5.5). For consistency, the name “spiropyran” in this review applies to both the closed- and the open-ring isomers. The closed-ring isomer is abbreviated as “SP”; the open-ring isomer (or merocyanine) is abbreviated as “MC”.
1.1. Isomerisation of spiropyran
The structural formula of the parent closed-ring isomer of spiropyran is represented as 1 in Fig. 1A. The molecule comprises an indoline and a chromene moiety bound together via a spiro junction and oriented perpendicular with respect to one another. The optical spectrum of the closed-ring isomer shows two localised transitions (Fig. 1B, gray trace); the band located at ∼272–296 nm is attributed to the π–π* electronic transition in the indoline part of the molecule, and the ∼323–351 nm band corresponds to the chromene moiety.38,39 UV (λ = 365 nm) irradiation of SP gives rise to the open-ring isomer (MC; 2 in Fig. 1A) in a first-order process40 whose mechanism has been investigated extensively; the transformation begins with the cleavage of the Cspiro–O bond, resulting in cis-MC41–43 (see 5, 6 Fig. 2) – an ephemeral species detectable by using transient absorption spectroscopy immediately after the UV pulse. The rotation about the central C–C bonds44,45 in cis-MC ultimately yields trans-MC.18,19 Interestingly, the SP → MC isomerisation can also be accomplished using near-infrared (NIR) radiation (via two-photon excitation).46–50 This property is important since (i) the use of an NIR laser significantly reduces photodegradation of the switch as compared to the wavelengths (UV) used for single-photon processes, and (ii) it opens the way to perform isomerisation in biological samples as discussed in Section 2.1. The MC → SP reverse isomerisation usually occurs spontaneously, again following first-order kinetics,51 and can be accelerated by visible light.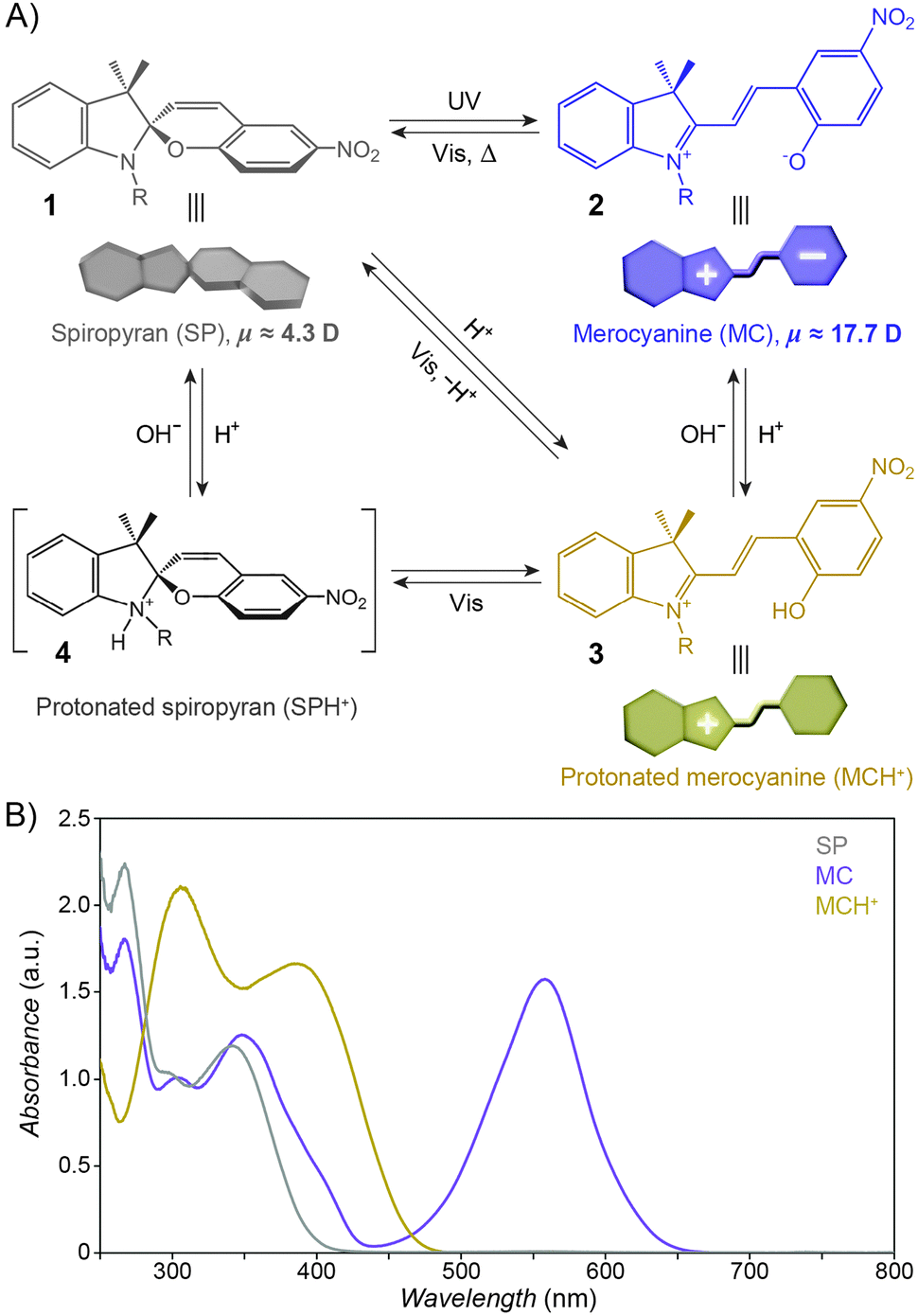 | ||
| Fig. 1 Photochromism and acidochromism of spiropyran. (A) Reversible transformations between the four states: spiropyran (SP) 1, merocyanine (MC) 2, protonated merocyanine (MCH+) 3, and protonated spiropyran (SPH+) 4 (note that although 4 is represented with the extra proton on the spiro N atom, it is also possible that the proton resides on the spiro O atom, or on the nitro group306). (B) UV-Vis spectra of the parent spiropyran (1′,3′,3′-trimethyl-6-nitrospiro[chromene-2,2′-indoline]) before (gray) and after (purple) UV irradiation (5 min; I = 0.7 mW cm−2), and after the addition of 20 eq. of HCl (yellow). Spectra were recorded on a c = 0.231 mM solution in acetonitrile; optical path length = 10 mm. | ||
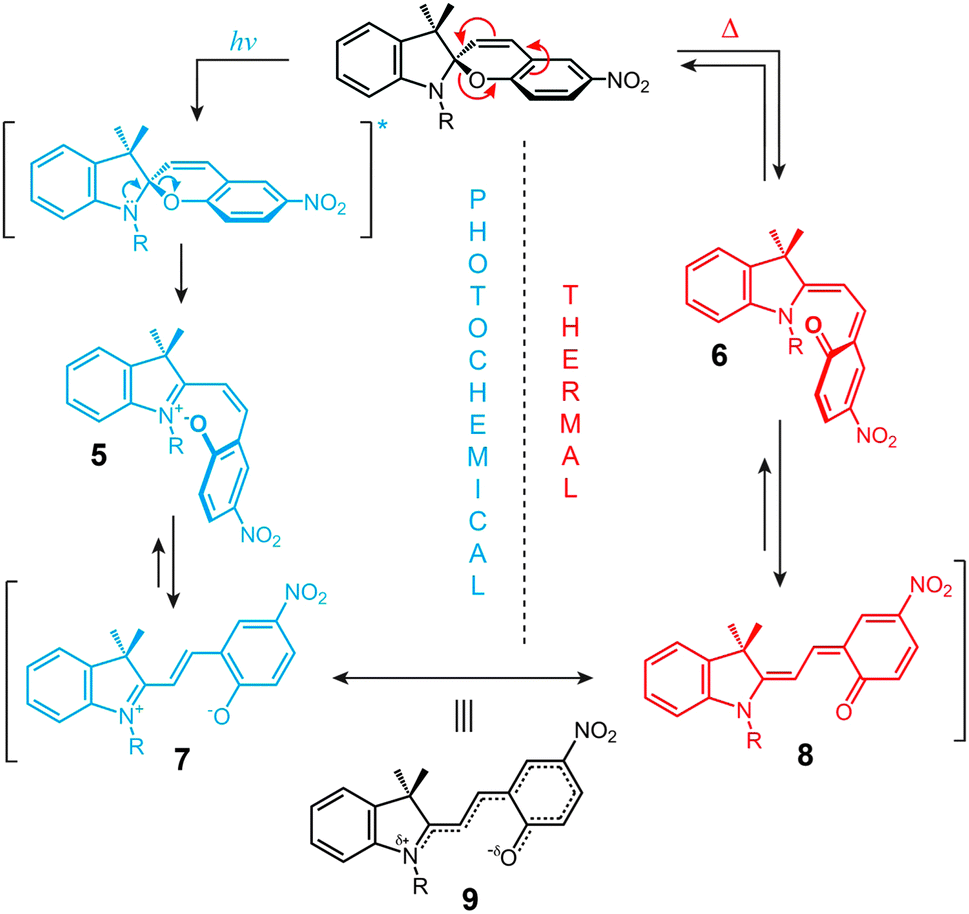 | ||
| Fig. 2 Mechanism of photochemical and thermal isomerisation of spiropyran. | ||
The ring-opening reaction can be represented either as a heterolytic C–O bond cleavage (Fig. 2, left) or as a 6π electrocyclic ring opening (Fig. 2, right), leading to the zwitterionic (7) or the quinoidal (8) resonance forms, respectively.52 The final MC product is a hybrid of these resonance forms (9 in Fig. 2). Due to its planar structure and an extended π-conjugation between the indoline and the chromene moieties, MC shows a single delocalised transition shifted to the visible region, with λmax = 550–600 nm in most non-polar solvents. The exact location of this band is dictated by the relative contributions of the two extreme resonance forms. Non-polar media, which preferentially stabilise the quinoidal form,53–55 decrease the energy gap between the ground and excited states of MC, resulting in a bathochromic shift of the MC band43,56 (“negative solvatochromism” of MC57,58). The strong dependence of MC absorption on the environment has been exploited for the construction of microcapillary-based systems capable of detecting specific solvents.59,60
The widespread utility of the spiropyran switch lies in the fact that the SP and MC isomers have vastly different physicochemical properties. First and foremost, the charge separation in MC gives rise to a large electric dipole moment, particularly in comparison with the SP isomer. Density functional theory calculations61 as well as electrical interferometry62 and electrooptical absorption measurements63 have shown that while the dipole moment of the parent (Fig. 1A) SP is in the range of ∼4–6 D, this changes drastically to ∼14–18 D for the MC form. Secondly, the SP and MC states show significant structural differences, whereby SP occupies less volume than MC. An elegant manifestation of these differences is the reversible increase, as a result of UV irradiation, in surface pressure within monolayers of a spiropyran-functionalised PMMA densely packed at the water–air interface.64–66 Thirdly, the SP isomer is optically transparent in the visible region whereas MC absorbs strongly at λmax = 550–600 nm and appears deep blue. Fourthly, SP and MC differ markedly in their emission behaviour: while the SP isomer does not exhibit strong emission, ring-opening results in the appearance of an intense emission band centered at λmax ≈ 650 nm (cf. Fig. 3A67). The resulting red emission can subsequently be “turned off” as the pyran ring re-forms and the extended π-conjugation is broken. Fifthly, the MC isomer is significantly more basic than SP, and its protonation leads to MCH+ with a characteristic band at ∼420 nm (Fig. 1B). Still, while the acidic character of MCH+ originates from the 2-hydroxy-4-nitrophenyl moiety, the pKa of MCH+70 (∼2.25) is much lower than that of the parent 4-nitrophenol (pKa = 7.1571). This dramatic stabilisation of the phenoxide anion (reduced basicity) can be attributed to the electronic conjugation within the molecule, with the quinoidal resonance form (8 in Fig. 2) favouring72 the deprotonated MC. The low pKa value is also due to the electron-withdrawing effect of the NO2 group located at the para position with respect to the phenolic OH – without it, the pKa of MCH+ was estimated to be 6–773 (compare with pKa = 10.0 for phenol). It should be noted, however, that the solution value of pKa = 2.25 often is not accurate for the immobilised spiropyrans discussed in this review; the acidity of constrained species74 is, in principle, system-dependent. Finally, the MC form has a higher affinity to different chemical species, in particular other zwitterions and metal ions.
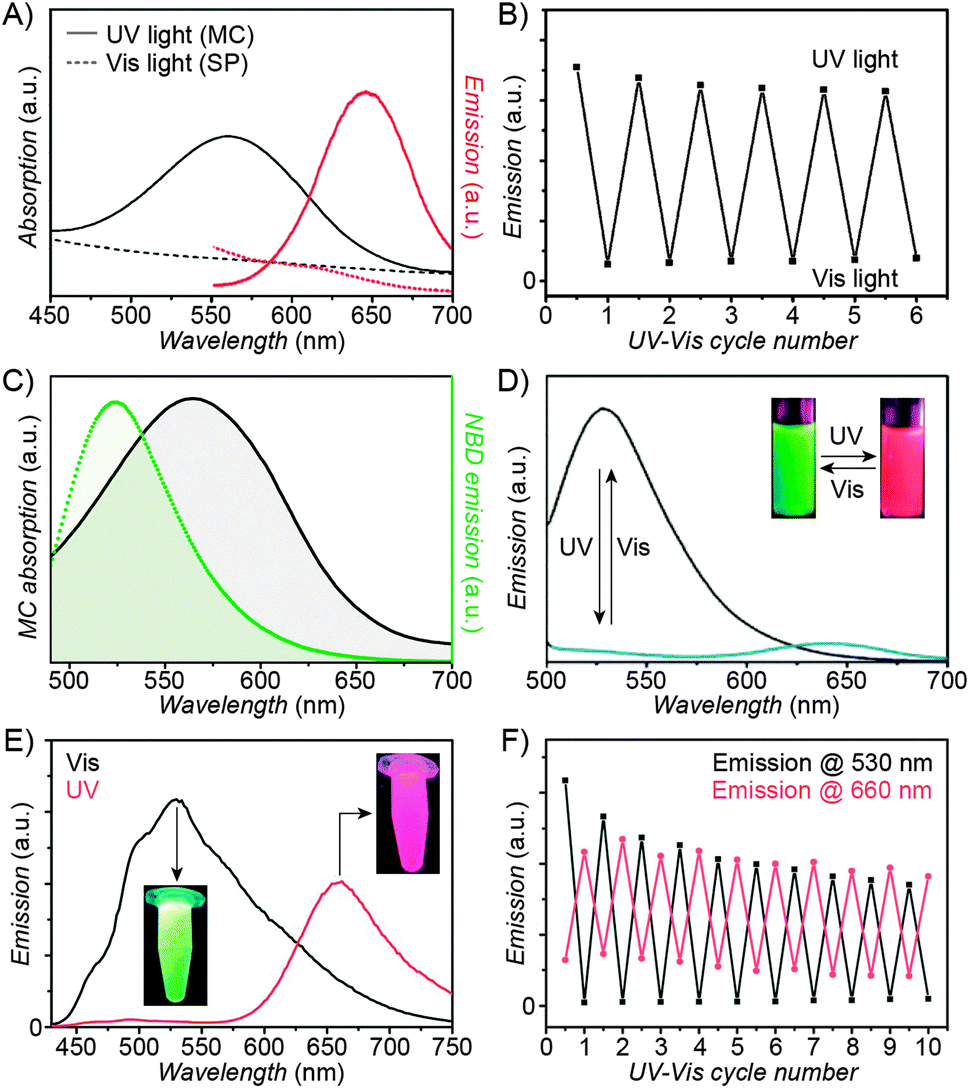 | ||
| Fig. 3 Fluorescent properties of the SP–MC system. (A) Typical absorption (black) and emission (red) profiles of SP (dashed lines) and MC (solid lines). (B) Reversible fluorescence (λem = 645 nm) switching in a spiropyran-decorated polymer upon exposure to UV and Vis light. (C) Overlap of the nitrobenzoxadiazolyl (NBD) fluorophore emission (green) and MC quencher absorption (gray) – a prerequisite for efficient FRET. (D) Photoswitchable FRET in dual-emissive polymer NPs based on spiropyran and NBD. (E) Photoswitchable FRET in dual-emissive polymer NPs based on spiropyran and a polythiophene derivative. (F) Green emission is quenched as red emission rises in dual-emissive polymer NPs shown in (E). [Adapted with permission from ref. 67 (Copyright 2010 Wiley-VCH) (A and B), ref. 159 (Copyright 2008 Royal Society of Chemistry) (C and D) and ref. 190 (Copyright 2003 Royal Society of Chemistry) (E and F).] | ||
The above differences in the characters of SP and MC are intimately linked to another unique feature of spiropyran: its responsiveness to multiple stimuli. In addition to being photochromic, its reversible isomerisation can be realised by several other independent stimuli, which include temperature21 (thermochromism), pH75–77 (acidochromism), solvent polarity78 (solvatochromism), redox potential79 (electrochromism), metal ions,45,46 and even mechanical force80 (mechanochromism). For example, treating SP with acids (Fig. 1A) or metal ions can induce ring opening even in the absence of any UV irradiation because of the high affinity of the open-ring form to H+ and metal ions (see Section 2.6). Likewise, polar environments – including solvents,81,82 silica,83–86 or reverse micelles87 – can stabilise the zwitterionic MC to the extent that the SP → MC transition occurs spontaneously in the dark. Under these conditions, SP represents the metastable state, which can exist only if the system is exposed to visible light. This property is referred to as negative photochromism58,88 (sometimes also called inverse or reverse photochromism), and is of particular relevance in the context of water-based biological environments (see Sections 3.1–3.8).
1.2. Aggregation of the open-ring isomer
An important consequence of the molecular structure of the MC isomer is its tendency to aggregate. Driven predominantly by the dipole–dipole interactions (along with the π–π stacking), the aggregation occurs readily in hydrophobic solvents.89,90 The MC units can stack in two different ways: “head-to-tail” (parallel) arrangement of the dipoles gives rise to so-called J-aggregates, whereas “side-by-side” (antiparallel; compare Fig. 7C) stacking yields H-aggregates. These two types of packing can easily be identified in the absorption spectra: while J-aggregation shifts the MC band to higher wavelengths (bathochromic/red shift), H-aggregation is manifested by the hypsochromic/blue shift (cf. Fig. 7A). Both J-aggregates91–96 and H-aggregates97–100 of MC are well known, and occasionally are found to co-exist.101,102In addition to its tendency to aggregate, MC can also form complexes with SP units.63,64 Elegant experiments suggesting the existence of such heterocomplexes were performed with ∼900 nm silica spheres and planar quartz surfaces – both functionalised with spiropyran. As expected, there were no attractive interactions between the spheres and the surfaces in the dark, but exposure of the system to UV induced adsorption of the silica onto quartz. Interestingly, such adsorption could also be induced when only one of the two components was UV-irradiated (and had its SP moieties converted to MC), therefore confirming that the SP–MC interactions were responsible for adsorption.103
MC aggregation stabilises the open-ring isomer and therefore it strongly retards101,104 or even completely blocks105 the ring-closing reaction. As such, the aggregation is counterproductive to the development of efficiently switching systems. Fortunately, immobilisation of the chromophore units can protect individual MC units from aggregation. Still, partial aggregation is often observed, with MC → SP decolouration kinetics that are best fitted by the superposition of two first-rate reactions.106,107 For example, MC immobilised on the surface of silica spheres faded with k1 = 4.2 × 10−3 s−1 and k2 = 1.3 × 10−3 s−1, which was attributed to the isomerisation of isolated and aggregated MC moieties, respectively.108 Such biexponential decay of MC was also observed within monolayers on solid SiO2, wherein transient Brewster angle reflectometry showed that while the quantum efficiency of the ring opening occurred with a well-defined quantum efficiency of ∼0.1, the MC signal decayed with quantum efficiencies of ∼0.2 and ∼0.03 assigned to isolated and stacked MC units, respectively.109
1.3. Benefits of immobilisation
Covalent attachment of spiropyran has numerous advantages over non-covalent association to the support:Fortunately, bimolecular events become largely suppressed by placing the spiropyran units on supports.123 In a study that compared photodegradation of spiropyran molecules moving around freely in solution with that of their immobilised counterparts, ten switching cycles induced degradation of ∼55% of small molecules, but only ∼21% of the immobilised version under the same irradiation conditions.124 In another example, PMMA-based spiropyran homopolymers showed significant fatigue after only several isomerisation cycles whereas a copolymer containing 20 mol% of the chromophore units was significantly more stable.125 Finally, 50 switching cycles induced degradation of ∼40% of spiropyran immobilised on ∼2 μm polystyrene beads126 – compared with ∼50% degradation of small molecules in solution after only 13 cycles.120 The decay of the switch on the beads was likely due to bimolecular events caused by the beads coming into contact with one another. Yet when spiropyran was attached to a planar surface, an impressive 370 switching cycles were realised without significant fatigue!127
Second, the support can largely affect isomerisation kinetics: while a small-molecule MC dissolved in ether isomerised within a few minutes, the colour of the same dye residing on a chitosan chain, also dissolved in ether, persisted for 24 hours.131 In another example demonstrating the buffering effect of the polymer “hosts”, addition of hydrophilic mica particles to a toluene solution of a small-molecule MC stabilised the coloured form and greatly reduced the kinetics of decolouration, whereas it had virtually no effect on the fading of polymer-immobilised MC under otherwise identical conditions.130 It is also worth noting that polymer-immobilised MC does not exhibit significant solvatochromism, in contrast to individual MC units (see Sections 1.2 and 1.3). The properties of the photoswitch can even be modulated solely by the length of the linker connecting it to the support: accordingly, ring-closing of MC attached to the surface of silica proceeded with kn=8 = 0.52 × 10−3 s−1 and kn=16 = 4.2 × 10−3 s−1 when the dye was connected through linkers comprising 8 and 16 atoms, respectively.108 In principle, longer linkers allow for more conformational flexibility and encourage solvation by the solvent molecules – which consequently leads to faster decolouration.132–134 It is important to emphasise here the need to decouple the photoswitch from the underlying surface (that is, a minimal linker length is necessary) to achieve efficient isomerisation.135 While the direct attachment of spiropyran to polystyrene beads via physisorption was possible, the isomerisation yield was only ∼20% of that of the switch which had been chemisorbed through an eight-carbon chain linker.126 Likewise, isomerisation of SP physisorbed on planar gold136 proceeded with a quantum yield of only ∼10−10.
The effect of the support can in fact be strong enough in some cases to induce a transition between positive and negative photochromism.129 For example, spiropyran on a poly(N,N-dimethylacrylamide) backbone displayed positive (“normal”) photochromism despite the polymer being surrounded by a strongly hydrophilic silica gel matrix.137 Interestingly, the SP ↔ MC equilibrium can also be affected by the surface of a nanoparticle: chromophores bound to CdSe NPs prepared with tri-n-octylphosphine as the capping agent showed positive photochromism, but in similar NPs prepared in the presence of sodium dioctylsulfosuccinate, negative photochromism was observed.138 The authors attributed the stabilisation of the MC isomer to the charged defects on the surface of NPs prepared by the latter approach.
2. Spiropyran-functionalised polymers
Various approaches to spiropyran-bound polymers have been developed. The spiropyran moiety is compatible with most polymerisation conditions; therefore routes based on both polymerisation of spiropyran-based monomers and grafting on pre-formed polymer chains have been employed. The grafting-on approach has been used to functionalise a variety of polymers, including polytetrafluoroethylene (PTFE),140 polyaniline,141 polyacrylates,39,142,143 polysulfones,82,144 polyphosphazenes,106 and Pluronic.145 Homopolymers are typically synthesised by means of ring-opening metathesis polymerisation (ROMP).146,147Random copolymers are most commonly prepared via AIBN-initiated free radical polymerisation of terminal alkenes148–154 (usually methacrylates) or by polycondensation reactions,155–158 whereas for the synthesis of block copolymers, atom transfer radical polymerisation (ATRP)129,159,160 and reversible addition–fragmentation transfer (RAFT) polymerisation128,161,162 have proven successful. These controlled polymerisation methods have also been used to derivatise solid surfaces with spiropyran polymers – in such cases, the solids (e.g. glass,163 silica colloids164) are pre-functionalised with polymerisation initiators and used as starting materials.
Examples of polymers incorporating disubstituted spiropyran units as a part of the polymer backbone are relatively rare; these polymers can be obtained by various polycondensation methods, including polyesterification,165,166 diol-diisocyanide polycondensation,167 and bis(indoline)-bis(salicylaldehyde) polycondensation.168 Polymers having precisely one photoswitchable unit as a part of the polymer chain were synthesised by controlled polymerisation methods (single-electron transfer living radical polymerisation (SET-LRP)169 and ATRP170) using disubstituted spiropyrans as initiators. Finally, end-labeled polymers were obtained by nucleophilic substitution reactions involving small-molecule spiropyrans and pre-formed polymer chains,171,172 via ATRP using a spiropyran-based tertiary bromide initiator,173 or by solid-phase synthesis.174
2.1. Photocontrol of polymer fluorescence
Over the past several years, fluorescent properties of spiropyran polymers have been studied extensively due to their potential applications in detection and imaging. Ideal for such applications are photoresponsive polymers in the form of spherical NPs, which have successfully been prepared using methods such as emulsion polymerisation68 and ATRP followed by micellisation.159 The resulting NPs are very bright (due to the compact packing of the fluorophores), often biocompatible, their surfaces can be functionalised with biomolecules,116 and, most importantly, their fluorescence can be modulated remotely in a reversible fashion (Fig. 3 and 4).In the simplest case, fluorescence of NPs is “turned on” and “off” upon exposure to UV and visible light, respectively. Such NPs, designated as “1st generation” in Fig. 4, were originally synthesised by Li and co-workers by means of emulsion polymerisation of a mixture containing N-isopropylacrylamide (NIPAM), styrene, divinylbenzene, and a spiropyran-methacrylate monomer.68 Whereas the shells of the resulting NPs were hydrophilic (PNIPAM), thus providing them with good water solubility, the SP units resided in the hydrophobic cores. This gave rise to strong fluorescence of the MC form while significantly reducing photodegradation of the chromophore, as evidenced by only an ∼5% decrease in MC fluorescence after five switching cycles.68 This fatigue resistance, however, came at the expense of slow isomerisation kinetics of the photoswitch within the compact polymer matrix.68,69 As a result, it took as long as ∼5 min to reach the photostationary state under UV irradiation (compared with ∼5 s for switching on semiconductor NPs under similar irradiation conditions175), and ∼2 min (vs. ∼90 s on the same semiconductor NPs) under visible light irradiation. The diameters of the NPs were readily controlled, in the 40–400 nm range, by varying the ratio of the monomers. This ability to control the particle size is important: ideally, NPs should be large enough to give an intense optical signal; however they should not be too large so as to minimise undesired light scattering.
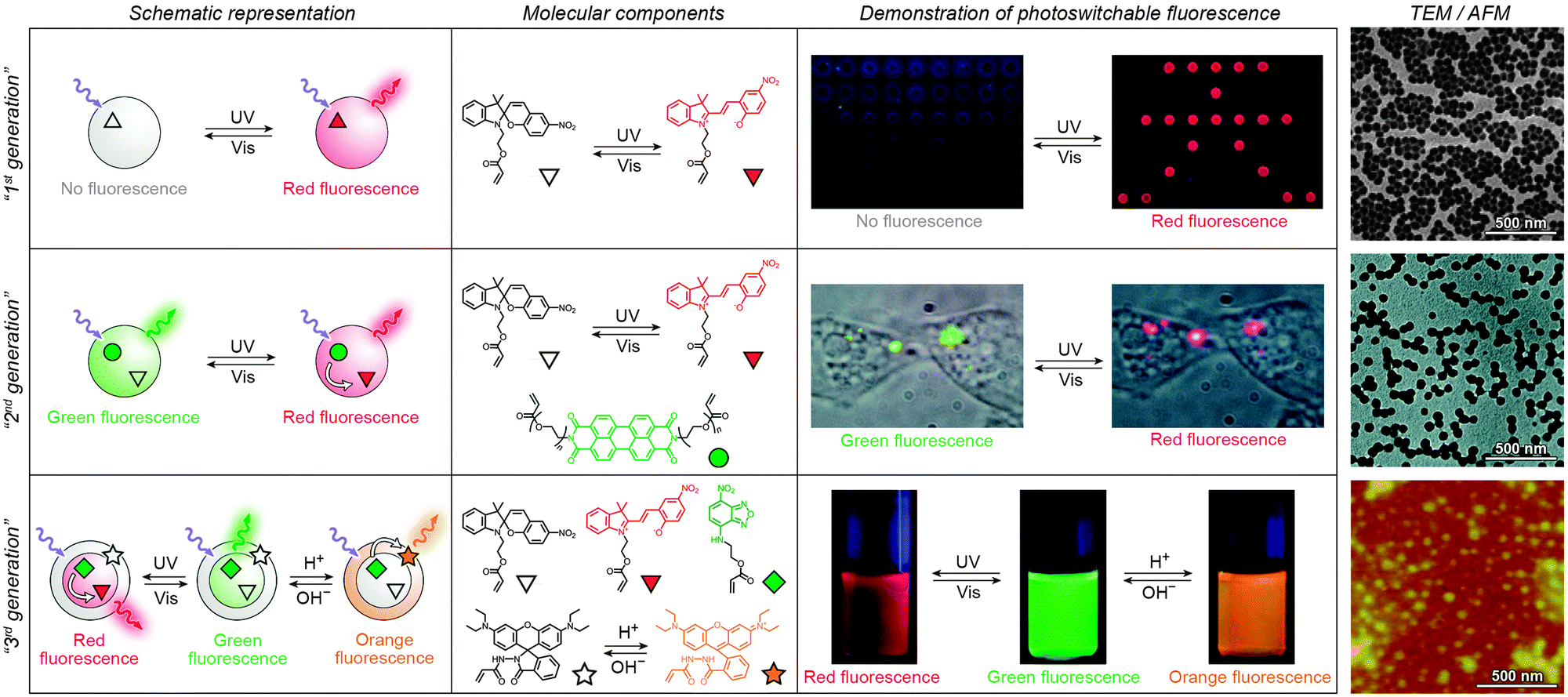 | ||
| Fig. 4 Polymer NPs exhibiting photoswitchable fluorescence. Top panel: In “1st generation” fluorescent NPs, MC fluorescence is reversibly “turned on” and “off”. Middle panel: “2nd generation” fluorescent NPs, whereby MC lights up at the expense of emission from a nearby fluorophore. Bottom panel: “3rd generation” fluorescent NPs capable of emitting light of three different wavelengths, depending on the environmental conditions. [Adapted with permission from ref. 68 (Copyright 2006 American Chemical Society) (top panel), ref. 186 (Copyright 2007 American Chemical Society) (middle panel) and ref. 161 (Copyright 2010 Wiley-VCH) (bottom panel).] | ||
Switchable fluorescence is the basis of localisation microscopy176 – a recently developed177,178 technique which enables imaging with nanometer-scale resolution – well below the diffraction limit – using standard fluorescence microscopy tools. Li et al. identified spiropyran as a switchable fluorophore which is well-suited for this application,179 and developed a variant called photoactuated unimolecular logical switching-attained reconstruction (PULSAR) microscopy.180–182 The principle of PULSAR is as follows: the sample is first irradiated with red light so as to set all the photoswitches to the dark state (closed-ring isomer), and to photobleach all adventitious (non-photoswitchable) fluorophores absorbing in that region. In each imaging cycle, a brief UV pulse (λ = 375 nm) is used to “turn on” a small fraction of MC emitters, which are then imaged (with λexcitation = 561 nm182) until photobleaching/back-isomerisation takes place. Assuming that the distances between the active emitters are greater than the Abbe diffraction limit, each of them can be localised with nanometer precision by fitting the summed intensity data to a Gaussian mask. The process is then repeated over many cycles until the entire population of the fluorophores is photobleached. An overall image is reconstructed from the positions of individual MC molecules recorded during each cycle. The resolution of PULSAR is determined by the number of photons a single MC can emit before it photobleaches. This number is as large as 1.8 × 105, giving rise to imaging resolution down to 10 nm.180
Imaging capabilities of PULSAR microscopy are demonstrated in Fig. 5. Fig. 5B shows a single (left), two (center), and four (right) 70 nm spiropyran-polymer NPs arranged in a row. PULSAR clearly resolves individual 70 nm NPs180,181 whereas conventional fluorescence microscopy (Fig. 5A) is unable to do so. Furthermore, the possibility to chemically functionalise the surfaces of these NPs115 can turn them into valuable imaging markers. For example, NPs with polyacrylic acid-rich shells exhibited affinity to CaCl2 microcrystals and could be used to acquire high-resolution images thereof: the PULSAR image shown in Fig. 5D clearly shows a monolayer of NPs decorating a round crystal of CaCl2 (as compared to the diffraction-limited image of the same crystal in Fig. 5C).182
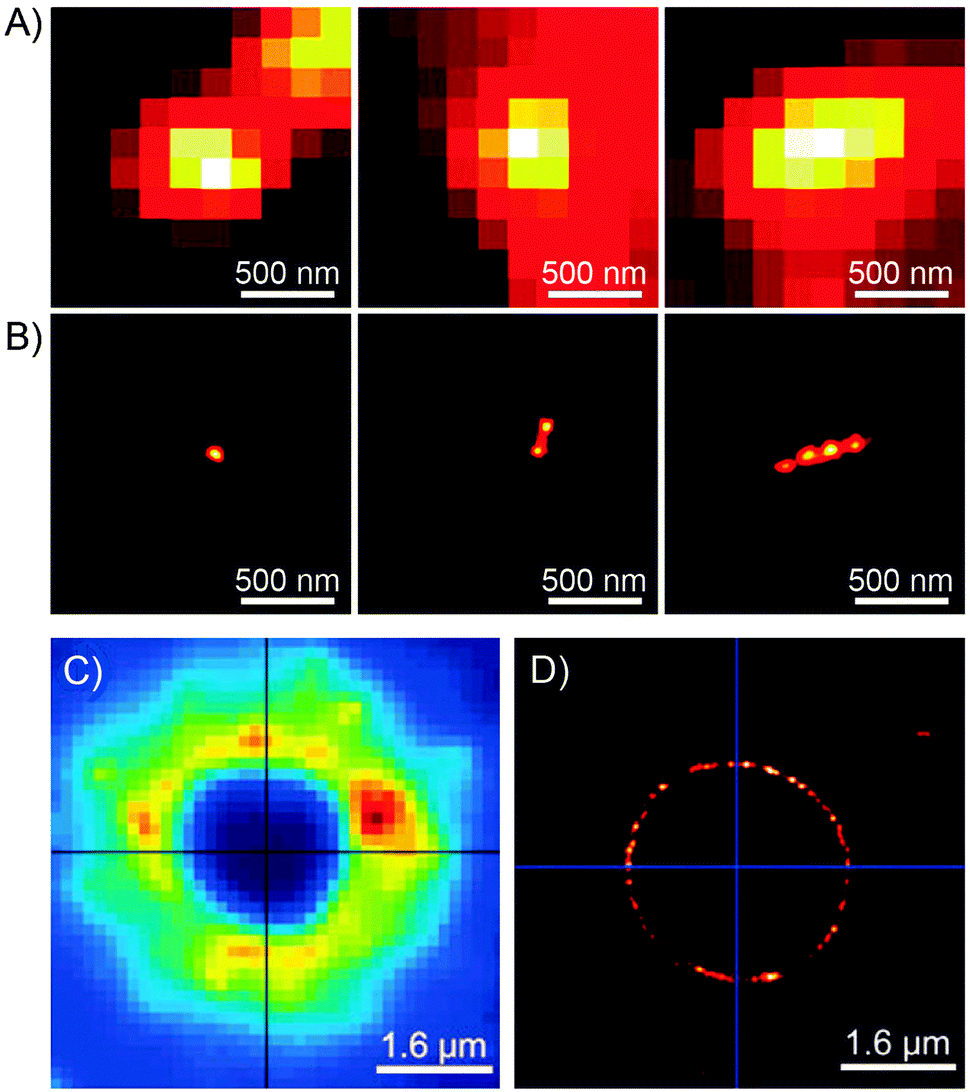 | ||
| Fig. 5 Imaging power of the PULSAR microscopy. (A) Reversibly fluorescent NPs, 70 nm in diameter, imaged using conventional fluorescence microscopy. (B) Reconstructions of the same NPs obtained using PULSAR. Resolution improves by a factor of ∼25. (C and D) Reversibly fluorescent NPs as markers for CaCl2 crystals – images obtained using conventional fluorescence microscopy (C) and PULSAR (D). [Adapted with permission from ref. 180 (Copyright 2008 American Chemical Society) (A and B) and ref. 182 (Copyright 2011 Royal Society of Chemistry) (C and D).] | ||
The combination of high resolution with the possibility to activate fluorescence “on demand” makes PULSAR microscopy of particular interest for imaging biological systems, where false positive signals due to cell autofluorescence are a ubiquitous problem. This background fluorescence, being non-photoswitchable, can easily be extracted from the signals due to the MC probe. Importantly, UV-induced cytotoxicity is not an inevitability since the SP–MC isomerisation can be induced not only by UV, but also by NIR light as discussed in Section 1.1. The excitation wavelength of ∼780 nm lies within the so-called NIR window, where both the absorption and scattering of biological tissues are minimal. Moreover, irradiation with an NIR laser enables not only the isomerisation, but also two-photon fluorescence of MC, thereby overcoming the problem of back-isomerisation of MC typically accompanying single-photon fluorescence.
A desirable feature of fluorescent probes – in the context of biological imaging – is the ability to reversibly switch between two different colours of emitted light (as opposed to a dark-bright transition). Although the parent SP (1 in Fig. 1A) is non-fluorescent, Li et al. have recently engineered dual-colour fluorescence in a series of spiropyrans by functionalising the photoswitch with electron-donating or -withdrawing substituents.183,184 For example, the MC form of a 5-cyano-substituted switch emitted red fluorescence, whereas the SP form was blue-fluorescent. The authors also prepared polymer NPs incorporating these novel spiropyrans and demonstrated the ability to unambiguously stain intracellular objects by taking advantage of the reversible, two-colour fluorescence.183
Switching between two different wavelengths of emitted light can also be realised by using fluorescence resonance energy transfer (FRET). The advantage of this approach over dual-emitting dyes (previous paragraph) is that it offers more flexibility in terms of optical output. Recall that FRET efficiency is governed by the extent of overlap between the fluorophore emission band and the MC excitation band – cf. Fig. 3C159 – as well as by the average distance between the two moieties;185 both of these parameters are readily controllable within spiropyran-polymer NPs. One such example is shown in Fig. 4, middle panel (“2nd generation” switchable NPs), whereby spiropyran has been co-polymerised with a perylene diimide dye to form spherical, ∼50 nm polymer NPs.186 In the closed form of the switch, these NPs emit green fluorescence due to the PDI units. An SP-to-MC photoisomerisation, however, activates a PDI-to-MC FRET, and the resulting NPs emit red light. A related example of emission control is shown in Fig. 3D,159 whereby a fluorescent nitrobenzoxadiazolyl (NBD)-based dye within spiropyran-polymer NPs emitted green fluorescence while SP was in its closed form.67,187,188 UV-triggered ring-opening induced FRET to the MC form resulting in red emission. Other fluorophores attached to/incorporated in spiropyran polymers include polythiophene189,190 (Fig. 3E and F), poly(fluorenyl-co-benzothiadiazole) (PFBT),116 boron-dipyrromethene (BODIPY),191 diphenylanthracene,192 naphthalimide,193,194 and even the green fluorescent protein (GFP).42,195 In all of these examples, photoswitchable dual-colour emission was achieved.
More recently, approaches have emerged for the development of spiropyran-polymer NPs whose fluorescence can be controlled by multiple orthogonal stimuli (“3rd generation” NPs in Fig. 4). Thermoresponsive polymers, such as PNIPAM, can be used to introduce temperature responsiveness. PNIPAM is readily hydrated and highly water-soluble at room temperature. Upon warming of an aqueous solution, it undergoes volume phase transition and precipitates at the temperature (T = 32 °C) which corresponds to its lower critical solution temperature (LCST) (“cloud point”),196,197 as a result of entropically driven dehydration.198 Liu and co-workers reported block copolymers comprising (i) PNIPAM copolymerised with an NBD acrylate-based fluorescence donor, and (ii) an MC methacrylate-based acceptor.164 The block copolymers were supported on silica particles, resulting in overall core@shell@shell morphology. These particles could emit light of three different colours, depending on the external conditions: (1) under visible light irradiation, the spiropyran was in the SP (“off”) form and the colour of emitted light was green (and not affected by temperature). (2) Under UV irradiation at T = 20 °C (below the LCST), the solution appeared orange due to the large average distance between the FRET donors (NBD) and acceptors (MC), causing FRET to occur with moderate efficiency. (3) Under UV irradiation at T = 35 °C, the solution appeared red as a result of PNIPAM collapsing, decreasing the average distance between the NBD and the MC groups, and enhancing FRET efficiency.164 An even more sophisticated system is shown in Fig. 4, bottom panel. In addition to being photo- and thermoresponsive, this block copolymer (poly(St-co-NBD-co-SP)-b-poly(NIPAM-co-Rh), where St = styrene and Rh = rhodamine), incorporates rhodamine, whose fluorescence can be modulated by pH. The resulting NPs can exist in as many as eight different states, which can be toggled between each other by exposure to three orthogonal stimuli.161
2.2. Photocontrol of polymer solubility
The large difference in polarity between SP and MC can be exploited to construct systems in which aggregation is induced by light. Different studies demonstrating such photoinduced changes propose three alternative explanations for the behaviour. First, preferential intramolecular “solvation” of the MC isomer by the polymer backbone was suggested to play a role, as postulated by Irie et al., who observed that UV irradiation of benzene solutions of a spiropyran-decorated PMMA resulted in decreased viscosities. This effect was attributed to intramolecular stabilisation of MC by the PMMA's ester moieties.44,199 The same rationale was used to explain the UV-induced decrease in the viscosity of SP-decorated poly(methacrylic acid) in methanol.200 Evidence supporting this scenario was provided by studies of copolymers with different contents of spiropyran: in all cases, the photoinduced effect was most pronounced for copolymers containing relatively low (e.g., ∼10%200 or ∼18%44) molar percentage of the switch.In contrast, other experiments showed a linear dependence of the viscosity change on the spiropyran content, and suggested that direct interactions between the chromophore units are involved.201 Depending on the solvent, these attractive interactions could take place between SP units, resulting in visible light-induced viscosity decrease (e.g. in DMSO)39/precipitation (in water; Fig. 6A202), or, more commonly, between the MC units,99,203–205 giving rise to UV-induced aggregation (e.g. in dioxane; Fig. 6B206,207). The contribution of direct MC–MC interactions is also corroborated by MC fluorescence quenching which accompanies polymer aggregation.208
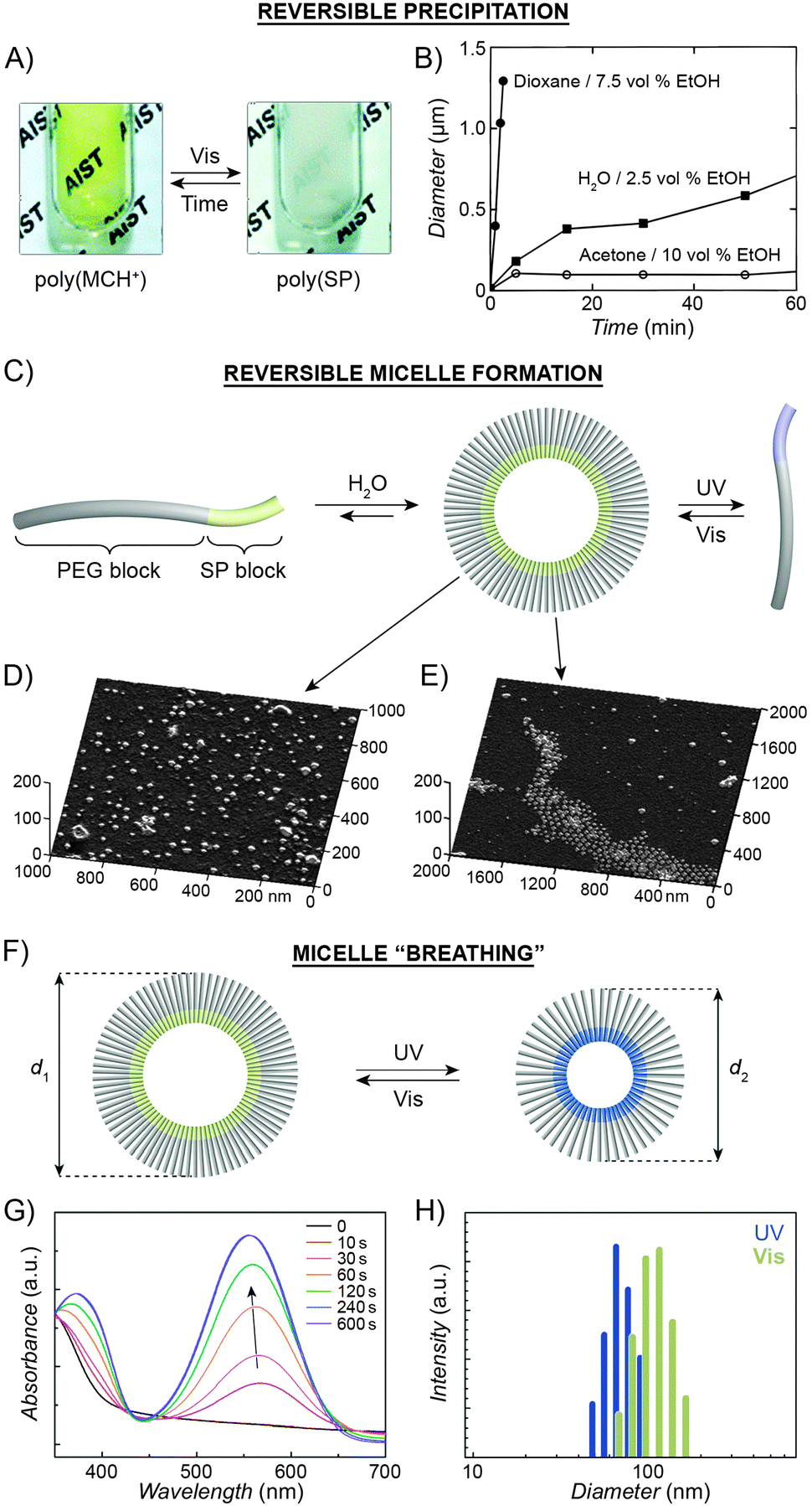 | ||
| Fig. 6 Light-controlled aggregation of spiropyran-functionalised polymers. (A and B) Depending on the solvent, aggregation can be induced by either the SP (A) or the MC (B) state. (C) Schematic representation of light-induced micelle formation. (D) AFM image of micelles formed by self-assembly of a block copolymer containing a PEG block and an SP-functionalised block. (E) AFM image of micelles formed by exposure of the sample in (D) to UV light (disassembles the micelles) followed by Vis light (micelles re-form). (F) Schematic representation of reversible size change in micelles self-assembled from a block copolymer containing a PEG block and a (PMMA-co-SP) block. (G) Changes in the UV-Vis spectra of SP-rich micelles (left in (F)) exposed to UV. (H) Micelle size distribution as a function of light wavelength. [Adapted with permission from ref. 202 (Copyright 2006 Royal Society of Chemistry) (A), ref. 206 (Copyright 1997 American Chemical Society) (B), ref. 160 (Copyright 2007 Wiley-VCH) (D and E) and ref. 218 (Copyright 2010 Chemical Society of Japan) (G and H).] | ||
Finally, the third plausible explanation is based on the photoinduced loss of the solvation layer. Spiropyran-functionalised polystyrene precipitated from a cyclohexane solution when irradiated with UV light.117 Comparison with a small-molecule analogue supported the negligible role of direct MC–MC interactions. This monomer, when exposed to UV, aggregated by means of MC–MC interactions, which stabilised the resulting aggregates and consequently inhibited redissolution by visible light. In contrast, the aggregated MC-polymer could be redissolved easily, thereby suggesting that direct MC–MC interactions play only a negligible role.117 Overall, the light-induced aggregation behaviour is highly system-dependent and can likely be explained by a combination of the above mechanisms.
UV-Vis spectroscopy provides a convenient way to monitor interactions between the MC moieties as UV-irradiated polymers aggregate. Fig. 7A shows a series of absorption spectra of a spiropyran-functionalised methacrylate in toluene upon increasing exposure to UV light. The initially observed band centered at ∼585 nm, attributed to individual MC units, develops a shoulder at ∼560 nm, which is attributed to MC stacks (H-aggregation).100 Subsequent relaxation spectra clearly show that MC within these stacks re-isomerises considerably slower than the non-stacked MCs, such that after sufficiently long relaxation times, only the peak at ∼560 nm can be observed (Fig. 7B).
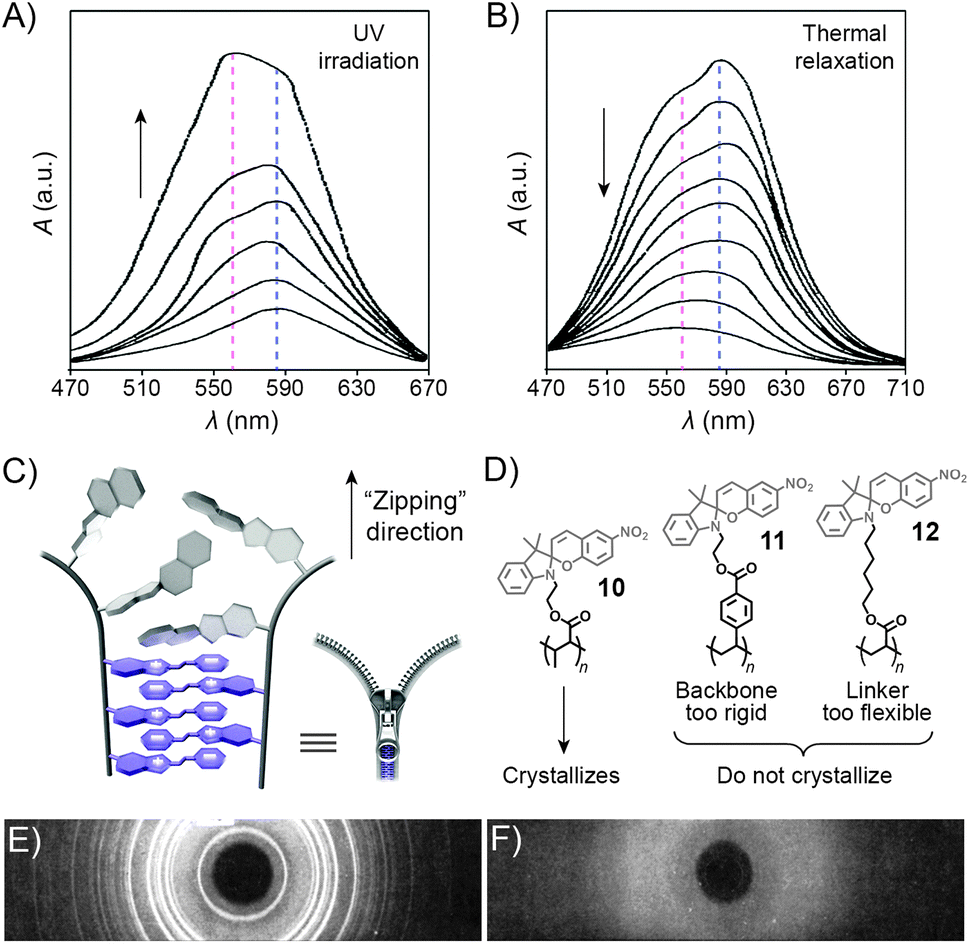 | ||
| Fig. 7 Spectroscopic and structural evidence for MC stack formation. (A) SP → MC isomerisation accompanied by MC aggregation during UV irradiation of a spiropyran-functionalised polymer 10. (B) Thermal relaxation of the sample obtained in (A). (C) Schematic representation of “zipper crystallisation”. (D) Structural formulas of spiropyran-functionalised polymers 10–12 differing in their crystallisation behaviour. (E) X-ray powder diffraction pattern of 10. (F) X-ray powder diffraction pattern of 11. [Adapted with permission from ref. 100 (Copyright 1984 American Chemical Society) (A and B) and ref. 107 (Copyright 1984 American Chemical Society) (E and F).] | ||
The controlled formation of such MC stacks governs a fascinating process first reported by Krongauz et al. which involves spontaneous ring-opening of SPs residing on polymer chains.209 Slow solvent evaporation from 2-methyltetrahydrofuran (MTHF) solutions of poly(MMA-SP) (10 in Fig. 7D) was found to result in red, crystalline (Fig. 7E) solids. In sharp contrast, fast evaporation of MTHF from the same solution yielded a white, amorphous precipitate. The red colour suggested that crystallisation entailed the SP → MC isomerisation – indeed, UV-Vis spectra of the crystals showed only one band centered at λ ≈ 560 nm, attributed to stacked MCs. Equally impressive was the stability of MC within the resulting crystals: it did not back-isomerise even upon heating to 150 °C (at which point the polymer decomposes) – in contrast to MC within amorphous aggregates obtained from the same polymers, which faded within a few seconds at T ≈ 50 °C.107 Surprisingly, such controlled crystallisation is interrupted by UV light – a stimulus usually used to induce SP → MC isomerisation – in fact, crystalline solids could only be obtained in the dark. The crystallisation process is thought to involve stepwise isomerisation along the polymer chain as the crystal forms – in other words, crystallisation and isomerisation mutually stimulate each other. The process can therefore be thought of as a molecular scale analog of closing a zipper and hence has aptly been called “zipper crystallisation”210 (Fig. 7C). It is important that the polymer chain has certain degree of flexibility – when the SP units were located on a more rigid polystyrene chain, no crystalline order in the resulting material was observed (11 in Fig. 7D and F).107 On the other hand, the spiropyran groups cannot have too much conformational freedom: no crystals were observed in the case when spiropyran was attached to the polymer backbone through a long and flexible alkyl chain linker (12 in Fig. 7D).
Advances in controlled polymerisation methods have enabled selective placement of the photoswitchable units at desired fragments of the polymer chains.211–214 The resulting block copolymers can have a tendency to spontaneously assemble into micelles or vesicles, such as in the process called “polymerisation-induced self-assembly and reorganisation”.128 Matyjaszewski and co-workers used ATRP to prepare block copolymers containing a long poly(ethylene glycol) (PEG) block appended with a short spiropyran-based block.160 The hydrophobic nature of SP induced the formation of micelles in aqueous solutions (Fig. 6C and D). When UV-irradiated, the micelles disassembled. Subsequent exposure to visible light restored the SP isomer and regenerated the original micelles, as shown in Fig. 6E. The same concept was demonstrated using block copolymers comprising (i) a PEG block and a spiropyran-decorated poly-L-glutamic acid block,215 as well as (ii) a spiropyran-appended PMMA block and a polysaccharide block.216 Interestingly, however, opposite behaviour was observed in the case of block copolymers bearing precisely one spiropyran unit at the terminal position of the polymer chain173 – in this case, UV-irradiation of well-solvated, PEG-rich polymers induced micellisation resulting from attractive MC–MC interactions.
Dual-responsiveness within spiropyran-incorporating block copolymers was encoded by substituting the PEG segment with a thermoresponsive block. Ji et al. reported a system based on a PDEGMMA-b-poly(SP) copolymer (where PDEGMMA = thermoresponsive poly(2-(2-methoxyethoxy)ethyl methacrylate)), which could exist in three different states – single molecules, micelles, and reverse micelles, depending on the environmental conditions.217
Finally, increasing the hydrophobic character of the spiropyran-incorporating block can stabilise the micellar structure such that no disassembly takes place even upon SP → MC isomerisation. The resulting MC-rich blocks, instead of being solvated by water molecules, pack more compactly due to strong MC–MC interactions, and the average micelle size decreases. This process is reversible, giving rise to oscillations of the micelle diameters between ∼110 nm and ∼90 nm (“micelle breathing”; Fig. 6F and H).218
2.3. Photocontrol of volume phase transitions in thermoresponsive polymers
An interesting case of light-induced aggregation of polymers is the behaviour of thermoresponsive polymers incorporating spiropyran, and is best exemplified by PNIPAM-based copolymers. In Section 2.1, we saw how thermally induced collapse of the PNIPAM chains decreased the distance between fluorescence acceptors (MC) and donors, thereby modulating FRET efficiency. The temperature at which this phase transition occurs – the LCST – can be increased or reduced by the incorporation of hydrophilic and hydrophobic groups, respectively, in the polymer backbone.219 Being able to transform reversibly between a hydrophobic and a hydrophilic isomer, the spiropyran switch offers the possibility to prepare PNIPAM-based copolymers with solubilities controlled not only by temperature, but also by light. For a given poly(NIPAM–X) copolymer, a range of temperatures exist where the polymers are water-soluble when X = the hydrophilic MC, but readily precipitate when X = the hydrophobic SP.220This behaviour is illustrated in Fig. 8 in an acidic (pH 4) solution of the copolymer. Under these conditions, the open form of the switch is protonated and the system exhibits negative photochromism, with the SP ring spontaneously opening in the dark. As shown in Fig. 8B, LCST of the thermally equilibrated, MCH+-rich polymer is relatively high (TMC ≈ 35 °C), whereas the closed-ring isomer reduces the LCST to TSP ≈ 30 °C. As a consequence, the initially yellow (due to MCH+), transparent solution exposed to visible light in the temperature range TMC < T < TSP turns colourless and opaque in less than a minute (Fig. 8C and D). The fact that the effect can be observed with remarkably small amounts of the chromophore units – the copolymer in Fig. 8 had only 1 mol% of SP and the solution concentration was 0.1 wt% – led the authors to hypothesise73,221 that the SP ring closing and the polymer dehydration could possibly accelerate each other. Another interesting aspect of the process is proton release accompanying the volume phase transition: 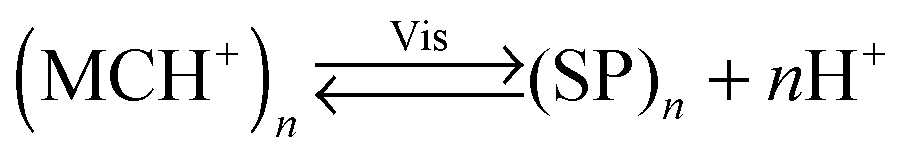 . Indeed, a ten-fold increase in the H+ concentration was observed221 upon visible light irradiation, suggesting that these polymers can be used for light-controlled proton delivery.
. Indeed, a ten-fold increase in the H+ concentration was observed221 upon visible light irradiation, suggesting that these polymers can be used for light-controlled proton delivery.
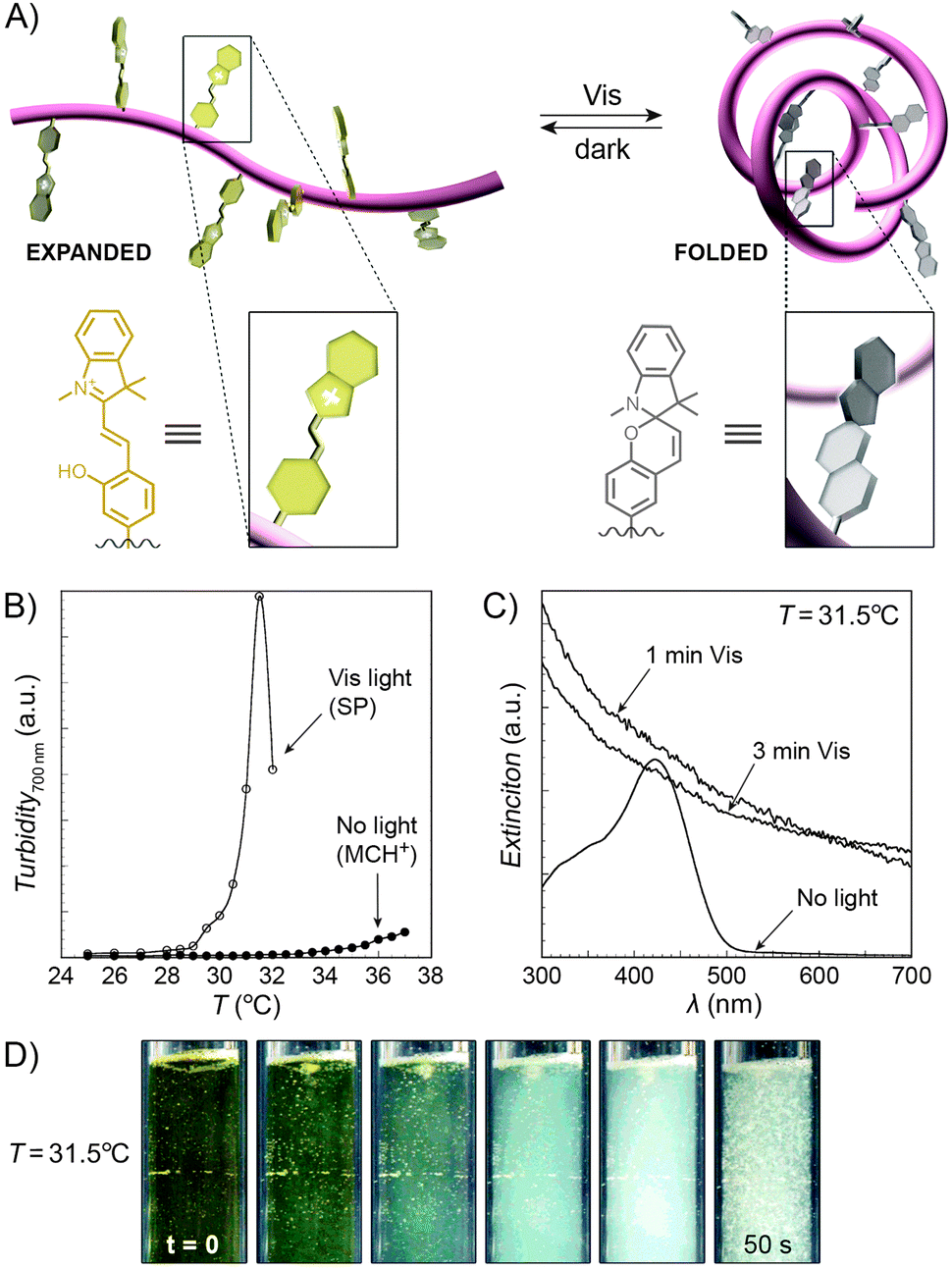 | ||
| Fig. 8 Light-controlled aggregation of thermoresponsive polymers. (A) Schematic representation of the process. Conversion of the hydrophilic MCH+ to the hydrophobic SP induces dehydration of the polymer chain, which initiates the phase transition. (B) Thermally induced precipitation of SP- and MC-rich PNIPAM. (C) Photoinduced precipitation of spiropyran-functionalised PNIPAM taking place at a temperature T such that LCSTpoly(NIPAM–MC) < T < LCSTpoly(NIPAM–SP). (D) Visible light-induced precipitation of poly(NIPAM–MC) from an aqueous solution. [Adapted with permission from ref. 73 (Copyright 2004 American Chemical Society) (B–D).] | ||
Despite the fact that the volume phase transition of the poly(NIPAM–SP) copolymers takes place abruptly at a well-defined temperature (LCST), continuous dehydration of the polymer222 backbone can be observed well below the LCST using the SP–MC pair as a probe.223,224 Fig. 9A shows the dependence of MC absorbance on temperature: absorption decreases in a roughly linear fashion starting at T ≈ 5 °C, although precipitation does not take place until T ≈ 35 °C. It is important to note that this decrease in MC absorption accompanying a gradual MC → SP ring closing is indeed due to an increasingly non-polar environment, and not a result of thermal isomerisation as evidenced by control experiments on monomeric spiropyran showing that temperature rise led to an increase in MC absorption (due to negative photochromism).223 Additionally, a slight red-shift of the MC band can also be seen with increasing temperature. This can be attributed to an increasing contribution of the quinoidal resonance form of MC at the expense of its zwitterionic form (see Section 1.1) – yet another indication of dehydration. This latter effect is more pronounced in a related system based on PNIPAM incorporating a nitrospiropyran, as shown in Fig. 9C–F.224 The dependence of λmax on T is nearly linear over a broad range of temperatures (10–34 °C) and wavelengths (519–547 nm), indicating that this system can serve as a colorimetric thermometer within this temperature window. The large difference in λmax values suggests significant contributions of the zwitterionic and quinoidal resonance forms at the low and high temperatures, respectively.
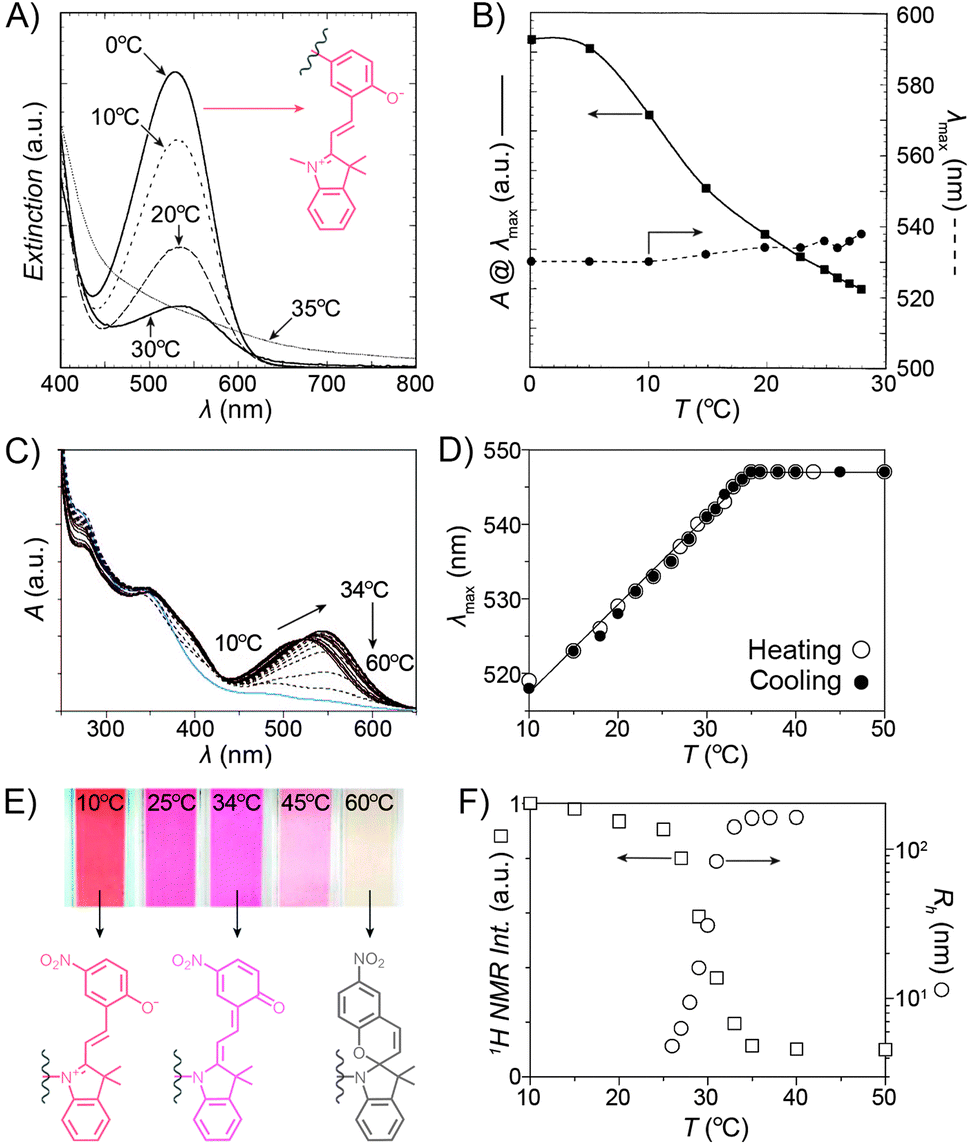 | ||
| Fig. 9 Spiropyran as a probe for microenvironment polarity. (A) Thermally induced MC → SP transition (0–30 °C) precedes precipitation of poly(NIPAM–SP) (35 °C). (B) Changes in maximum absorbance values and wavelength absorption maxima at temperatures below the volume phase transition. (C) Optical response of a poly(NIPAM–SP) polymer pre-irradiated with UV as it is gradually heated from T = 10 °C to T = 60 °C. (D) Temperature-dependent changes in wavelength absorption maxima of the same copolymer. (E) Colour changes accompanying a gradual zwitterion → quinoid conversion (observed spectrophotometrically in (C)). (F) Precipitous drop of the integrated intensity of the NIPAM's CH proton resonances (left) coincides with the rapid increase of the hydrodynamic radius (right). [Adapted with permission from ref. 223 (Copyright 2004 American Chemical Society) (A and B) and ref. 224 (Copyright 2009 American Chemical Society) (C–F).] | ||
In order to develop robust PNIPAM-based photoresponsive materials and ultimately functional devices, use of crosslinked polymers, as opposed to linear polymer chains, becomes necessary. Crosslinking is typically achieved with N,N′-methylenebisacrylamide225–227 and leads to hydrogels whose hydration (expansion) and dehydration (shrinkage) can be achieved upon exposure to UV and visible light, respectively. In an exemplary demonstration of the phenomenon, thin films of crosslinked gels exhibiting negative photochromism were exposed to blue light through a mask, causing volume phase transition and shrinkage in the irradiated regions. Irradiation times as short as 3 s were sufficient to decrease film thickness by ∼30%.227 Moreover, multilevel patterns could be created by irradiating different areas of the same film for different amounts of time. MCH+–SP isomerisation in the irradiated areas was confirmed by preferential adsorption of negatively charged latex particles onto non-irradiated (carrying more positive charges) areas. Similar reversible shrinkage–swelling behaviour was reported for poly(NIPAM–SP) gels in the form of colloidal particles. Upon consecutive cycles of visible light irradiation and thermal equilibration in the dark, these particles performed “breathing” motion, with their hydrodynamic diameters oscillating between ∼200 nm and ∼160 nm.226
Optical control of the thermal threshold for volume changes has also been reported for thermoresponsive polymers other than PNIPAM – for example, the LCST of poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) containing 1.3 mol% of spiropyran in the form of MC could be shifted from 44 °C to 34 °C upon exposure to visible light.129 Block copolymers comprising thermoresponsive poly(N,N-dimethylacrylamide) (PDMA) and poly(2-(2-methoxyethoxy)ethyl methacrylate) (PDEGMMA) units were also investigated.228 An intriguing aspect of the latter study is that two different photoswitchable groups – spiropyran and azobenzene – were incorporated into the polymer backbones and their combined effect was studied.
As demonstrated in this section, spiropyran allows for the control of properties characteristic of thermoresponsive polymers (i.e. volume phase transition) by means of light, thereby effectively rendering them photoresponsive, and paving the way towards the development of a conceptually new family of photoswitchable materials. Structure and property changes which could previously be brought about thermally229 can now be directed by light – using excellent spatial and temporal control – as well. The ability to shrink and expand crosslinked polymer gels in particular is interesting in the context of controlling transport phenomena (see Section 2.4).
2.4. Photocontrol of transport through polymeric systems
Sumaru and co-workers have pioneered the use of crosslinked poly(NIPAM–SP) hydrogels as the key components of light-actuated microvalves.230–233 As described in the previous section, these gels contain spiropyran in the MCH+ form, and can be dehydrated upon exposure to blue light, triggering ring closing and PNIPAM chain collapse. An example of the process is shown in Fig. 10A, where the surface area of a circular piece of gel irradiated with blue light decreases by a factor of two. The switchable polymers can conveniently be deposited in a region of interest – e.g., thin channels of PDMS-based microfluidic devices – by UV-induced in situ free-radical polymerisation of a mixture containing NIPAM, a spiropyran monomer, a crosslinker, and an initiator. Fig. 10B shows how three valves, separated by less than 2 mm from each other, can be opened independently by local irradiation with low-intensity (20 mW cm−2) blue light for less than 30 seconds. Although these valves could be opened by gentle heating as well, only the use of light allowed each valve to be addressed individually. Other original approaches to induce flow by means of light have been developed,234 for example one in which channels are created in a single step in thin, flat sheets of poly(NIPAM–SP) gels by exposing them to blue light through a mask in the shape on the channel.232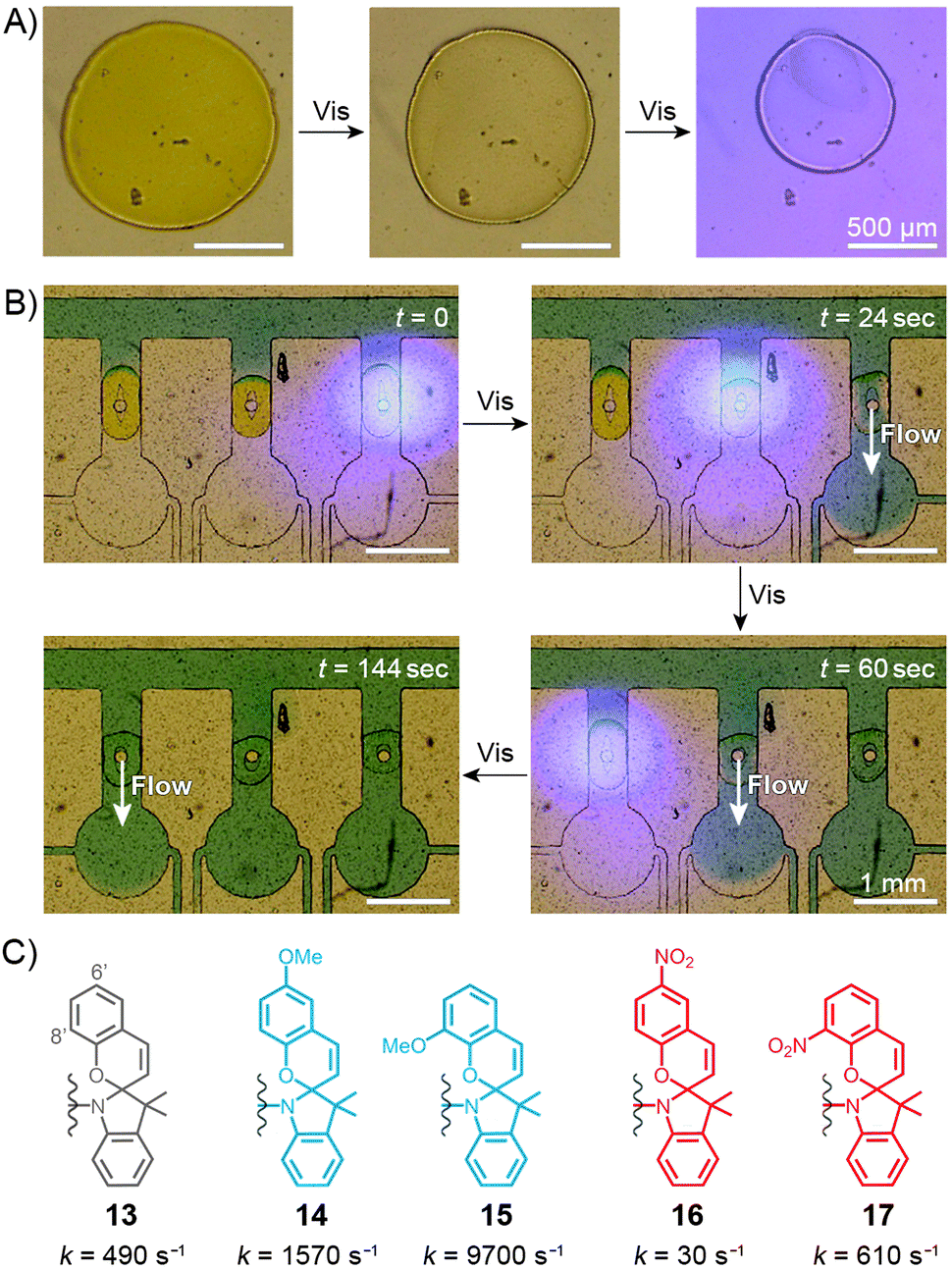 | ||
| Fig. 10 Spiropyran-based microvalves. (A) Contraction of a poly(NIPAM–SP) gel induced by blue light. (B) Remote control of liquid flow by consecutive opening of poly(NIPAM–SP)-based microvalves using blue light. (C) Controlling the rate of SP ring closure by the substitution pattern on the chromene moiety. [Adapted with permission from ref. 231 (Copyright 2007 Elsevier) (A and B).] | ||
In all of the above systems based on the unsubstituted (13 in Fig. 10C) spiropyran, reversibility is an issue: although blue light-induced ring closure (and so the valve opening) is fast, it takes more than one hour for the SP rings to reopen, for the gels to swell, and for the valves to close. To tackle this problem, Sumaru et al. investigated the effect of substituents on the kinetics of spontaneous ring opening.235 Ring opening events occurring in polar solvents involve a transition state with a partial negative charge on the pyran oxygen atom – therefore, the electron-donating methoxy substituent was placed at the 6′ position of the benzopyran moiety (14 in Fig. 10C) with the assumption that it would reduce the activation free energy of the reaction. Indeed, the ring-opening rate increased by a factor of three; conversely, installing an electron-withdrawing nitro group at the same position (16 in Fig. 10C) reduced the rate nearly 15 times. In addition, any type of substituent placed at the 8′ position led to a higher reaction rate – these two effects combined led to a 20-fold increase of the reaction rate in the case of an 8′-OMe derivative (15 in Fig. 10C), as compared to its unsubstituted counterpart.235 Therefore gels prepared from poly(NIPAM–SP–8′-OMe) exhibited excellent reversible swelling performance, with both the light-induced shrinking and spontaneous re-swelling to the original state completed within several minutes.
In the examples discussed above, spiropyran switching caused changes in hydration and the degree of swelling of a thermoresponsive polymer. Several photoinduced flow systems have also been developed which exploit different affinities of the switch to the solvent/solute molecules.236–240 In one prominent example,238 a molecularly imprinted polymer designed to bind tryptophan was prepared by polymerising a mixture of terminal alkenes containing a spiropyran derivative. The procedure was carried out under UV irradiation, which likely resulted in the formation of “binding sites” for the zwitterionic tryptophan in the proximity of the MC residues. Following polymerisation and extraction of the amino acid, the resulting materials were used as dialysis membranes. Permeability of tryptophan indeed depended on the state of the switch, with the diffusion through the MC-rich membrane faster by well over one order of magnitude.238 Control experiments revealed that the membranes were tryptophan-specific – other molecules diffused slowly and with rates independent of the conformation of spiropyran – and also confirmed the importance of the binding sites: a spiropyran polymer prepared in the absence of the template showed similar (and very low) permeability for tryptophan under both UV and visible light irradiation. This strategy was used to successfully develop a photoswitchable catalysis system, which comprised a spiropyran-functionalised polyacrylamide immobilising the enzyme α-chymotrypsin;241 while the diffusion of an amino acid-based substrate – and its access to the enzyme – was suppressed in SP-rich polymers, the system could be activated by UV light: under these conditions, the substrate readily diffused – and was converted into the product – through MC-rich copolymers. In another interesting example, commercial polyethersulfone (PES) ultrafiltration membranes were rendered photoresponsive by a grafting-from polymerisation process.242 As expected, the hydrophobic SP isomer encouraged non-specific absorption of proteins, which, in turn, translated into lower flow rate of the buffer solution.
Finally, transport properties can be controlled through physical changes to the overall polymer structure.82,144 For example, PTFE membranes coated with a poly(acrylamide-spiropyran) copolymer exhibited enhanced permeability towards water–methanol mixtures when irradiated with UV light – a property which could be correlated with UV-induced solution precipitation of this copolymer dissolved in the same mixture of solvents.140 This “surface precipitation” behaviour was also utilised to control flow through glass filters functionalised with a similar copolymer.163,243
In addition to the systems presented here, light-controlled transport has been demonstrated using various biopolymers and nanoporous inorganic materials derivatised with spiropyran – these examples are covered in Sections 3.3, 3.7, and 5.2.
2.5. Photocontrol of polymers' mechanical properties (and mechanical control of polymers' optical properties)
Copolymers discussed in this section contain spiropyran as part of their polymer backbones. When the backbone is attached to the two different (that is, indoline and chromene) rings of the switch, the isomerisation process (which is associated with large structural changes of the spiropyran moiety) is expected to affect the overall structure of the polymer chains, and therefore the macroscopic properties of the polymer. Copolymers of this type were first prepared and studied by Smets et al., who observed ∼2% contraction in thin films of rubbery (glass transition temperature, Tg ≈ −15 °C) polyesters containing up to 1 mol% of the spiropyran units.165,244 Upon storage in the dark, slow chain length recovery occurred and the process could be repeated multiple times.245 The shrinking was thought to be entropically driven, with the planar MC moieties increasing both the mobility of the polymer chains and their ability to pack more efficiently.Three decades later, a multidisciplinary team at UIUC considered the opposite – that is, the possibility to induce SP ring opening by applying mechanical force to polymer chains incorporating the chromophores in their backbones.167,169,170,246–249 A hint that this type of behaviour would be possible was provided by earlier studies which showed80 that small-molecule spiropyran underwent ring-opening upon grinding, thereby demonstrating the unique feature of the switch of being a mechanophore (undergoes chemical transformation in response to a mechanical stimulus) that is additionally mechanochromic (exhibits colour change upon the application of mechanical force). To investigate the effect of mechanical force on the isomerisation of SP, the team first prepared a linear poly(methyl acrylate) (PMA; molecular weight, Mw ≈ 170 kDa) having precisely one SP unit near the center of the chain (at the position of greatest stress under chain elongation; Fig. 11).169 The choice of the 5 and 8′ positions as the attachment points was suggested by DFT calculations which predicted246 that increasing the distance between these two points transmits the force efficiently to the C–O bond and leads to its rupture. Indeed, when an acetonitrile solution of the polymer was subjected to sonication, the colour of the solution changed from colourless to red, indicating the SP → MC reaction (Fig. 11A). The process was reversible in that the solution turned transparent upon exposure to visible light. No such effects were observed for PMA end-terminated171 with SP, indicating that the isomerisation was indeed induced by stress, and not, for example, by temperature change.
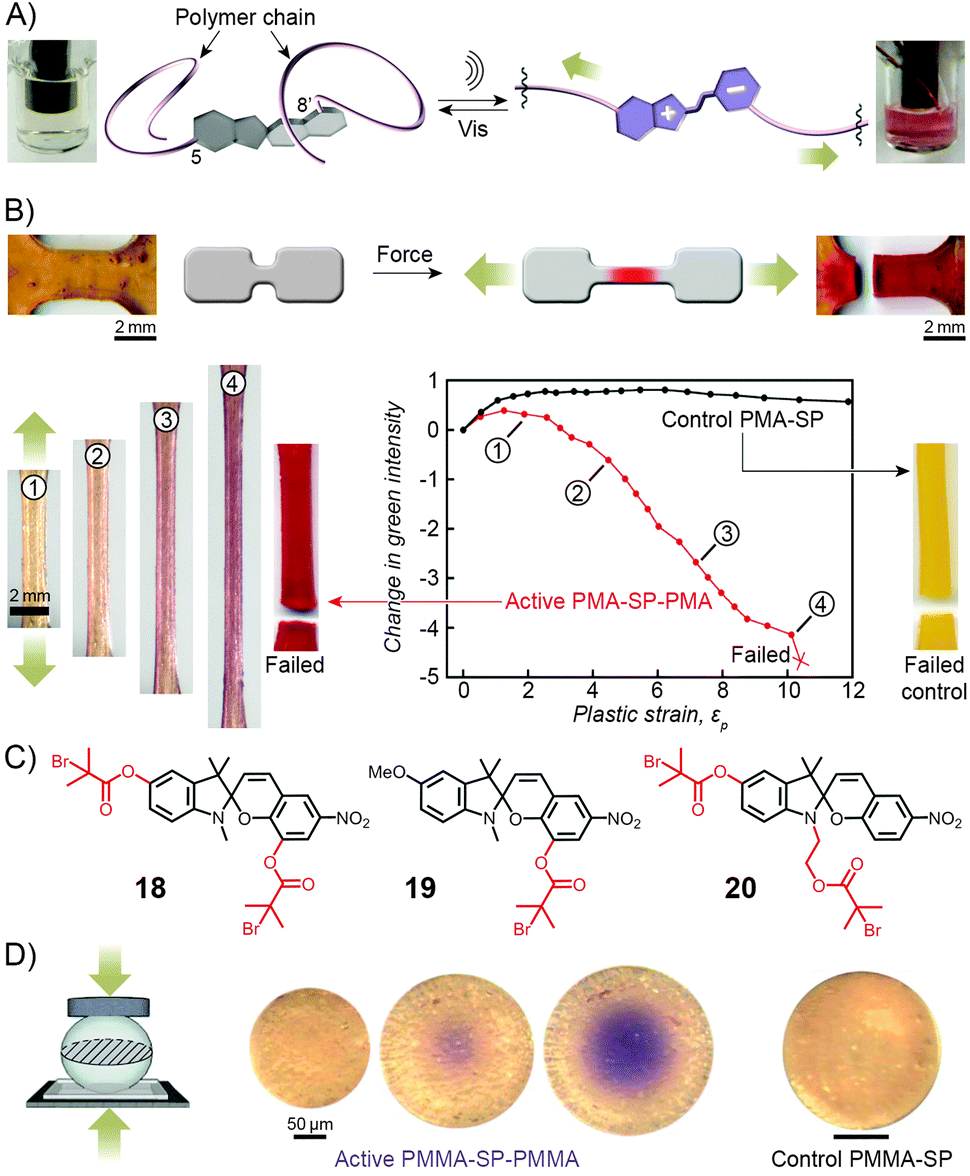 | ||
| Fig. 11 Spiropyran as a mechanophore. (A) Effect of sonication on the colour of the solution of a spiropyran whose chromene and indoline moieties are functionalised with PMA chains (PMA-SP-PMA; prepared using precursor 18 in (C)). (B) Effect of tensile loading on a dogbone-shaped specimen moulded from a PMA-SP-PMA polymer (bottom left). For comparison, a control sample moulded from a polymer synthesised from a monofunctionalised spiropyran 19 failed without a colour change (bottom right). (C) Structural formulas of precursors of various spiropyran polymers used in the study of mechanoresponsiveness. (D) Colorimetric response of a glassy bead made of PMMA-SP-PMMA to compression (centre). Control bead prepared from a monofunctionalised spiropyran does not change colour upon compression (right). [Adapted with permission from ref. 169 (Copyright 2007 American Chemical Society) (A) and ref. 246 (Copyright 2009 Nature Publishing Group) (B–D).] | ||
The spiropyran mechanophore could also be incorporated into chains of bulk polymers.246 Fig. 11B shows dogbone-shaped samples of elastomeric PMA prepared by SET-LRP using the bifunctional spiropyran 18 (Fig. 11C) as an initiator. When the samples were subjected to tensile loading, red colour – an indication of mechanochemical ring opening – emerged, and its intensity increased with increasing levels of strain. Samples moulded from control polymers – one lacking the polymer chain on the indole ring of spiropyran (prepared from 19 in Fig. 11C), the other one having both polymer chains attached to the same side of the spiro junction (prepared from 20 in Fig. 11C) – did not show any colour changes upon stretching, since neither presumably resulted in a significant force being transmitted to the sensitive C–O bond.
Similar behaviour was observed for SP-containing glassy PMMA prepared in the form of 100–500 μm spheres (Fig. 11D).246 Upon compression, an intense purple colour emerged as a result of tensile stress in the direction perpendicular to the loading direction, with the maximum tensile stress in the center of the bead. Importantly, intense colours, in all cases discussed here, appeared well before the samples failed at high strain levels, suggesting possible uses of these materials for detection and mapping of mechanical stress (in, e.g., climbing ropes or bridges) prior to catastrophic failure.166,246
2.6. Photocontrol of metal ion complexation
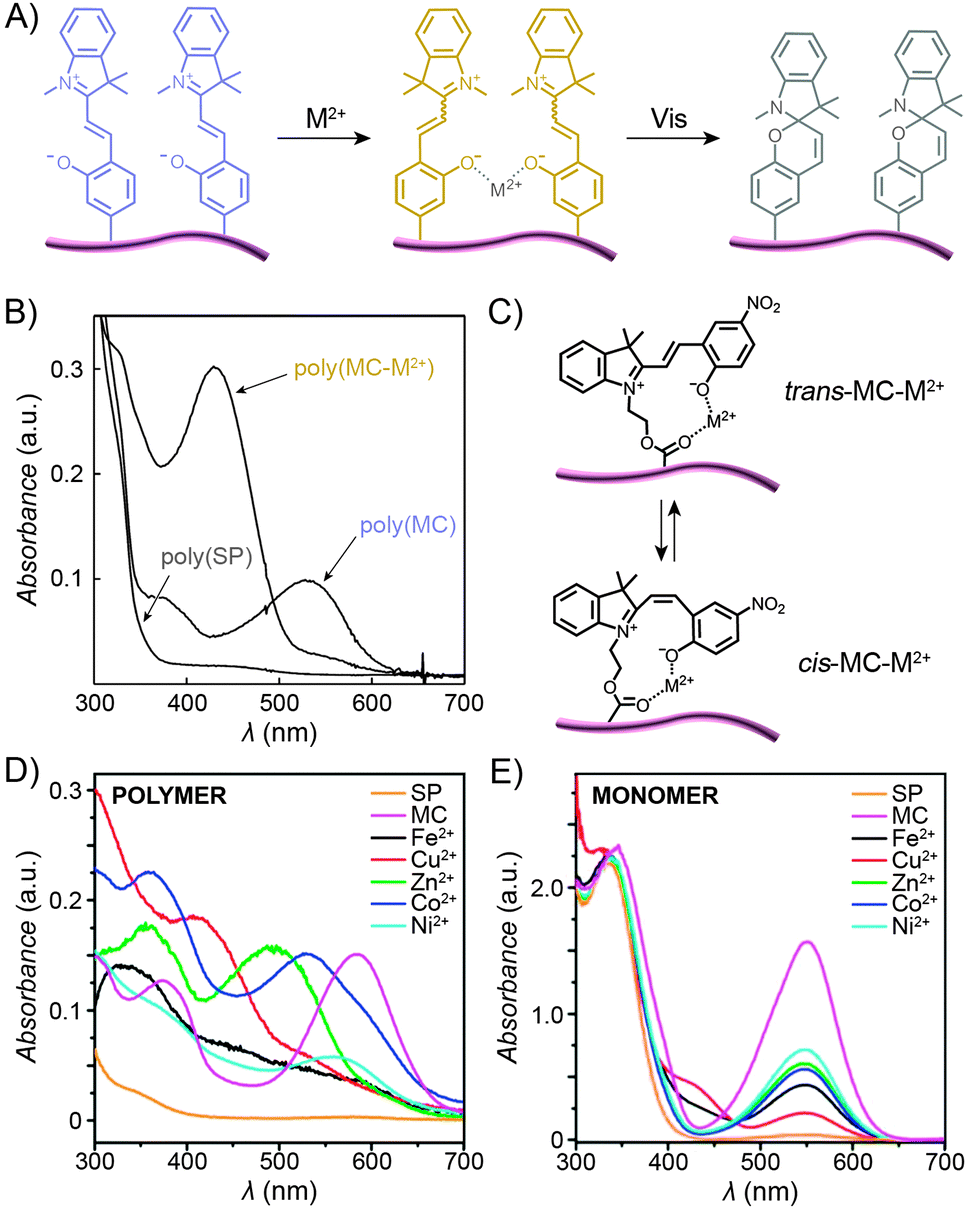
In contrast to the SP isomer, the MC form has a tendency to bind different metal ions (in an MC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mn+ 2:1 stoichiometry), with the interaction taking place through the MC's phenolate oxygen.250,251 Although the binding strength of the individual MC moieties to metal ions is usually rather weak, the stability of the complex can be increased by pre-organising two or more MC groups – for example, on a polymer backbone (Fig. 12A). Despite the higher complexation constants, MC in such complexes can still be converted to SP upon exposure to visible light, thereby resulting in the expulsion of the metal ions. This behaviour is exemplified by a spiropyran-decorated PNIPAM as it interacts with Pb2+ ions.252 As shown in Fig. 12B, the UV-Vis spectrum of this polymer shows an intense MC band at λmax ≈ 540 nm due to negative photochromism. Addition of metal ions (in this case Pb2+) leads to the appearance of a new peak at λmax ≈ 430 nm, attributed to an MC–metal ion complex.253,254 This example is of further interest since combining the metal ion binding properties of the polymer with its thermoresponsive nature allows for quantitative removal of Pb2+ from water. This goal was achieved by (i) spontaneous binding of the metal ions by MC, and (ii) subsequent gentle heating above the LCST.252 Irradiation of the resulting precipitate with visible light led to an ∼50% release of the Pb2+ ions from the solid state.252,255 It is important to emphasise that the mild binding strength of MC to metal ions is essential for the photoswitchable binding and release: if the interaction energy was too weak, neither isomer of the switch would bind metal ions; if it was too strong, the binding (and ring-opening reaction) would be irreversible.124
:Mn+ 2:1 stoichiometry), with the interaction taking place through the MC's phenolate oxygen.250,251 Although the binding strength of the individual MC moieties to metal ions is usually rather weak, the stability of the complex can be increased by pre-organising two or more MC groups – for example, on a polymer backbone (Fig. 12A). Despite the higher complexation constants, MC in such complexes can still be converted to SP upon exposure to visible light, thereby resulting in the expulsion of the metal ions. This behaviour is exemplified by a spiropyran-decorated PNIPAM as it interacts with Pb2+ ions.252 As shown in Fig. 12B, the UV-Vis spectrum of this polymer shows an intense MC band at λmax ≈ 540 nm due to negative photochromism. Addition of metal ions (in this case Pb2+) leads to the appearance of a new peak at λmax ≈ 430 nm, attributed to an MC–metal ion complex.253,254 This example is of further interest since combining the metal ion binding properties of the polymer with its thermoresponsive nature allows for quantitative removal of Pb2+ from water. This goal was achieved by (i) spontaneous binding of the metal ions by MC, and (ii) subsequent gentle heating above the LCST.252 Irradiation of the resulting precipitate with visible light led to an ∼50% release of the Pb2+ ions from the solid state.252,255 It is important to emphasise that the mild binding strength of MC to metal ions is essential for the photoswitchable binding and release: if the interaction energy was too weak, neither isomer of the switch would bind metal ions; if it was too strong, the binding (and ring-opening reaction) would be irreversible.124
 | ||
| Fig. 12 Light-controlled complexation of metal cations by spiropyran polymers. (A) Metal ions are bound by MC-polymers, and released following Vis-irradiation. (B) Optical spectra corresponding to the three states in (A). (C) Metal ions can be complexed by either trans- or cis-MC. (D) Optical properties of complexes formed between MC-polymers and metal ions are strongly dependent on the metal ion. (E) In contrast, no such diversity is seen when the same metal ions are complexed by a monomeric MC. [Adapted with permission from ref. 252 (Copyright 2004 Royal Society of Chemistry) (B) and ref. 258 (Copyright 2010 Royal Society of Chemistry) (D and E).] | ||
The polymer backbone itself can also be modulated to enable selective detection of a specific metal ion – for example copolymers rich in sulfobetaine moieties give rise to an environment in which the pendant MC groups bind Cu2+ with high selectivity (over, e.g., Zn2+, Ni2+, and Co2+).256 End-functionalised PMMAs have also been prepared:172 addition of metal ions to these polymers induced the formation of 2:1 complexes thereby enabling selective and reversible dimerisation of polymer chains.225,257
If the MC unit is equipped with an additional metal ion-binding group, the resulting bidentate ligand can be used to form complexes of 1:1 stoichiometry. Locklin and co-workers prepared a polymer with a unique capability to bind and distinguish multiple divalent metal ions from each other.258 In this design, MC was connected to a PMMA chain via an ester group – the latter serving as an additional coordinative group (Fig. 12C). Fig. 12D shows a collection of UV-Vis spectra of the polymer pre-irradiated with UV light and exposed to different metal ions, including Fe2+, Cu2+, Zn2+, Co2+ and Ni2+. The spectra feature (i) bands centered at λmax ≈ 500 nm, which can be assigned to trans-MC–M2+; the different hypsochromic shifts are thought to be due to varying degrees of deplanarisation of MC; and (ii) bands centered at λmax ≈ 400 nm attributed to cis-MC–M2+ complexes. This variety of optical responses can be contrasted to the behaviour of an analogous small-molecule MC, which lacked similar selectivity – for example, UV-Vis spectra of MC-bound Zn2+, Co2+, and Ni2+ were all indistinguishable from each other (Fig. 12E) – thus confirming the critical role of MC immobilisation for inducing selectivity of binding. In a subsequent report, the same group demonstrated the possibility to selectively detect two metal cations simultaneously.259 Meanwhile, Chan et al. reported a creative method to quantitatively detect Cu2+, with sensitivities covering essentially the whole spectrum of physiological Cu ion levels.260 The method is based on the combination of two types of polymer NPs, ∼10–30 nm in diameter, both decorated with spiropyrans. Upon excitation, the first of these polymers, poly(2,5-dialkylphenylene-1,4-ethynylene) (PPE), emits blue light, and the other, PFBT, emits green while also quenching the PPE fluorescence (provided the two are in close proximity). As a result, aggregation of the NPs, driven by copper-induced MC dimerisation, can initiate the energy transfer, with an effectiveness – measured as the ratio of the green to the blue emission – proportional to the concentration of Cu2+.260
In addition to sensing capabilities, metal cation binding to spiropyran polymers has other interesting implications. For instance, light-controlled complexation of Zn2+ ions resulted in reversible modulation of ionic conductivity.261 While conductivity changes were small (<10%), a related polymer incorporating both spiropyran and crown ether moieties262,263 (capable of reversibly binding Li+) provided light-controlled two-fold modulation of ionic conductivity.264 The bound metal ions can also be chemically reduced, as shown for Pd2+ ions complexed by spiropyran polymers in the form of so-called honeycomb films.265 Exposure of these Pd-rich films to a borohydride solution resulted in the formation of metallic palladium with a morphology reflecting that of the underlying honeycomb films.142 The complexed Pd2+ could also serve as a catalyst for electroless plating of silver.266 Notably, the latter two studies represent new approaches to creating nanostructured metallic surfaces.
2.7. Photocontrol of other polymer properties
In addition to binding metal ions, the MC isomer can also interact strongly with amino acids and cyanide anions, and the binding abilities of various spiropyran polymers could indeed be reversibly activated with UV light,267 with the detection limit of CN− down to the impressive 500 nM.268 When the photoisomerisation process occurs at relatively low pH values, it is accompanied by proton capture/release, according to the equation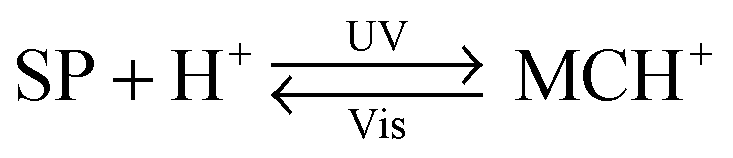 . Accordingly an ∼10-fold increase in the solution concentration of the H+ ions was observed as the protonated merocyanine units were exposed to visible light.221 Angiolini et al. took advantage of this light-regulated proton binding to construct a family of chiroptical switches based on polymers functionalised with both spiropyran and an optically active pyrrolidinyl moiety linked to an azopyridine group. With the increasing basicity of the building blocks in the order SP < azopyridine < MC, proton shuffling between azopyridine and the SP–MC pair was enabled and observed both within copolymers incorporating spiropyran and azopyridine as parts of the same polymer chain,269 and between poly(spiropyran) and poly(azopyridine) homopolymers.270,271 The proton transfer occurred reversibly and resulted in pronounced changes in the circular dichroism (CD) spectra.
. Accordingly an ∼10-fold increase in the solution concentration of the H+ ions was observed as the protonated merocyanine units were exposed to visible light.221 Angiolini et al. took advantage of this light-regulated proton binding to construct a family of chiroptical switches based on polymers functionalised with both spiropyran and an optically active pyrrolidinyl moiety linked to an azopyridine group. With the increasing basicity of the building blocks in the order SP < azopyridine < MC, proton shuffling between azopyridine and the SP–MC pair was enabled and observed both within copolymers incorporating spiropyran and azopyridine as parts of the same polymer chain,269 and between poly(spiropyran) and poly(azopyridine) homopolymers.270,271 The proton transfer occurred reversibly and resulted in pronounced changes in the circular dichroism (CD) spectra.
A variety of polyacrylates272–275 and polysiloxanes276,277 incorporating both SP and mesogenic units have been synthesised. Although the primary motivation was the development of reversible optical data storage,278–281 other fascinating properties, such as transient birefringence98,282 and second harmonic generation,283–285 have also been observed in these systems. In another study, spiropyran-decorated polyacrylamide hydrogel was used to fill the empty spaces between regularly arranged polystyrene colloidal spheres, giving rise to so-called polymerised crystalline colloidal arrays (PCCAs).286 The regular packing of the colloids resulted in the diffraction of incident light of a wavelength which was defined by the particle spacing. Photoisomerisation of spiropyran resulted in reversible contraction and expansion of the gel matrix – during which the crystalline arrangement of the colloidal spheres was retained. As a result, a reversible shift in the diffraction wavelength of as much as ∼11 nm was observed.286 An as of yet unrealised challenge is the ability to control the electronic properties of conductive polymers using light. Several polymers have been synthesised for this purpose,141,287 including ones with the spiropyran units incorporated into the polymer chain.288–290 Finally, in an interesting application demonstrated recently,291 a thin layer of a poly-spiropyran lying between solid surfaces and multilayer films grown by the layer-by-layer technique enabled an easy, light-induced detachment of these films from the underlying substrate – a task which could be difficult to realise otherwise.
3. Spiropyran-functionalised biopolymers
Functionalisation of Nature's macromolecules with artificial molecular photoswitches opens up the attractive possibility of influencing various biological processes using light. In the highly polar environments of biological systems, spiropyran exists in the MC form, and the isomerisation to SP can be accomplished with visible light; SP → MC reisomerisation takes place spontaneously. The negative photochromism represents a major advantage by enabling the reversible switching to be accomplished without the use of UV – a stimulus which is (i) potentially harmful as far as the often sensitive biological systems are concerned; and (ii) induces fatigue to a much greater extent than visible light (consequently, many systems covered in this section have been reported to be “fully reversible”).Functionalisation of various biomacromolecules with spiropyran is straightforward and can be accomplished in a single step using reagents 21 and 22 (Fig. 13A) – a spiropyran anhydride and an active ester, respectively. These molecules readily react with free amino groups on the surfaces of biopolymers via amide bond formation, and have emerged as universal reagents for rendering biopolymers photoresponsive.
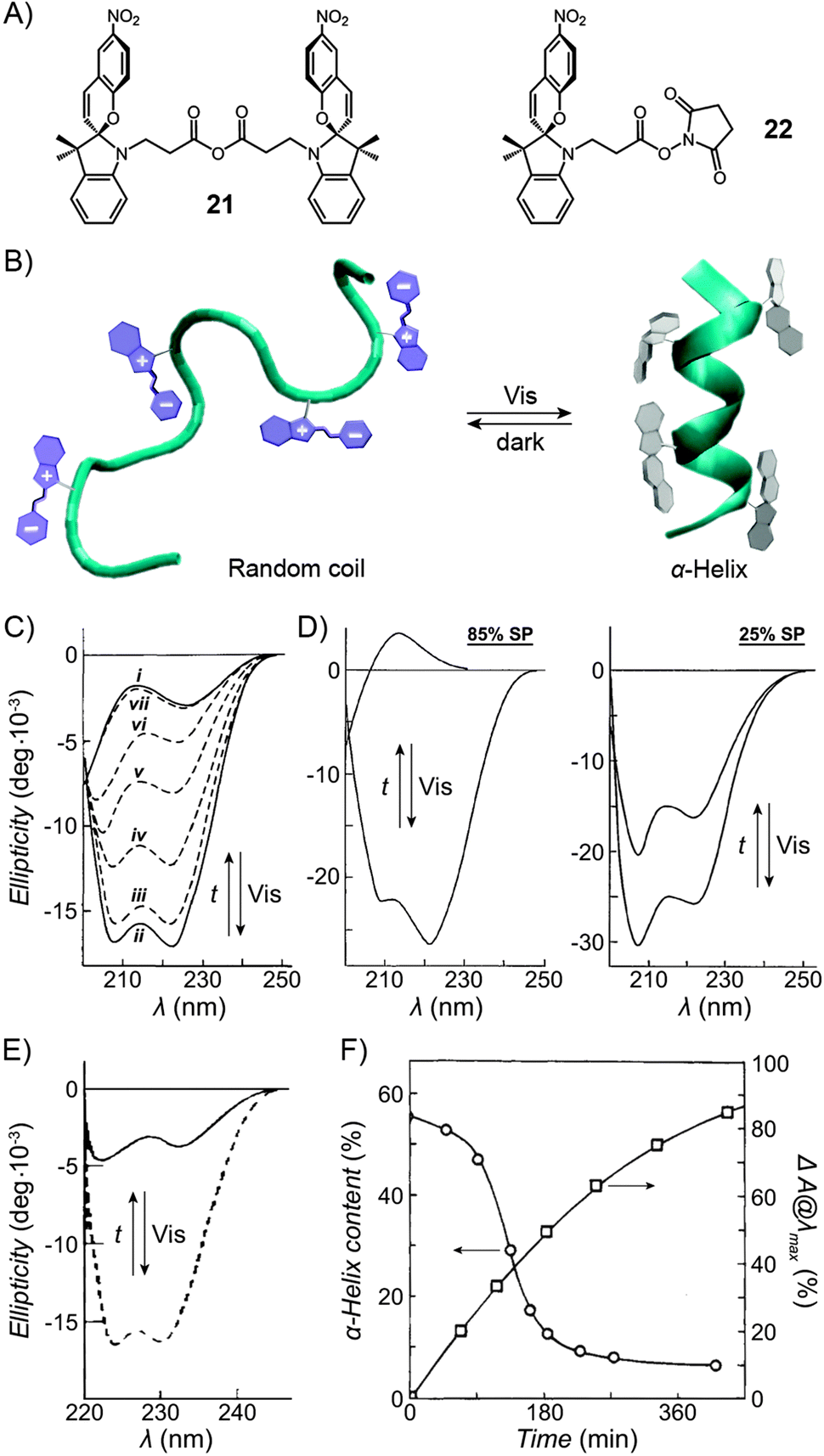 | ||
| Fig. 13 Light-controlled folding of polypeptide chains. (A) Structural formulas of reactive spiropyran derivatives used for functionalisation of various biopolymers. (B) Schematic representation of light-controlled folding. (C) Changes in the CD spectra of MC-functionalised poly-L-glutamic acid as it is exposed to visible light (i → ii), and during thermal equilibration (ii → vii). (D) Effect of spiropyran content in poly-L-glutamic acid on the helix ↔ random coil transformation. (E) Photocontrol of poly-L-lysine conformation. (F) Dependence of the α-helix content on the fraction of MC in a thermally equilibrating modified poly-L-lysine. Time = 0 represents a Vis-irradiated sample; ΔA indicates the percentage of the MC state. While the SP decay follows first order kinetics, the helix content drops precipitously once enough MC has accumulated. [Adapted with permission from ref. 433 (Copyright 2009 American Institute of Physics) (B), ref. 295 (Copyright 1989 American Chemical Society) (C), ref. 301 (Copyright 1998 Elsevier) (D), ref. 304 (Copyright 1992 American Chemical Society) (E) and ref. 434 (Copyright 1995 American Chemical Society) (F).] | ||
3.1. Photocontrol of polypeptide conformation
Inspired by the elegance with which biological systems use photons to initiate biochemical processes, several groups have investigated the structural properties of polypeptides decorated with spiropyran units.292 First such photochromic polypeptides – spiropyran-functionalised poly-L-tyrosine293 and poly-L-lysine294 – were synthesised by Smets and co-workers, but no effects of photoswitching on conformational changes (Fig. 13B) were reported until two decades later when Ciardelli et al. studied the behaviour of spiropyran-decorated poly-L-glutamic acid.295 Fig. 13C shows representative CD spectra of hexafluoropropanolic solutions of poly-L-glutamic acid having 41 mol% of the side chain –COOH groups functionalised with spiropyran. In this figure, i corresponds to a dark-adapted, MC-rich polypeptide, and the spectrum is typical of that of a random coil. By contrast, exposure to visible light generated a colourless (SP) solution with spectrum ii, whereby the two minima at 208 and 222 nm are characteristic of the α-helix (the fraction of the chain adopting the helical conformation was estimated at 45%). The change was remarkably fast and took as little as 5 s of irradiation with λ = 525–575 nm.295 Thermal relaxation to the original disordered conformation (ii → vii) was significantly longer (e.g. 24 hours), but could be accelerated by placing different substituents on the spiropyran moiety.296 The reversible helix ↔ random coil transformation could be repeated over many cycles, and was shown297 to occur in a stepwise manner via an intermediate thought to be a solvated helix.In addition to changes in the CD spectra, the folding process could be followed by viscosity measurements. A significant decrease in viscosity accompanying the MC → SP isomerisation could not be rationalised by the isomerisation reaction alone; recall from Section 2.2 that the viscosities of spiropyran-decorated polymethacrylate solutions decreased by up to 50% upon visible light irradiation, whereas those of functionalised poly-L-Glu dropped by 250–300%.298 In the present example, this enhanced effect was attributed to the conformational change, the α-helix being much more compact than the random coil. Interestingly, such photoinduced conformational changes were also observed at the water–air interface.299,300
It is important to emphasise that the SP isomer does not, strictly speaking, stabilise the helical conformation – in fact, the native poly-L-Glu lacking any SP readily folds into an α-helix under similar conditions – rather, the helical structure is disrupted by the MC isomer. This can be appreciated from Fig. 13D, where poly-L-glutamic acids decorated with 25 and 85 mol% SP both have ∼85% helical content. Upon thermal relaxation, however, the 85 mol% MC polypeptide adopted a completely disordered structure, whereas the 25 mol% MC one retained 55% of the helical content.301 In other words, the fraction of the photoinduced α-helix could be regulated by the degree of functionalisation of the side chain –COOH groups with spiropyran.
How exactly MC destabilises the α-helical conformation has been a matter of some debate; however, strong MC–MC interactions involving the open form of the switch seem to play a key role. These interactions can be either attractive298 or repulsive,302 depending on whether the open form is in the zwitterionic (MC) or in the protonated (MCH+) state, respectively. In one elegant study, the unfolding process was shown, by means of UV-Vis spectroscopy, to be accompanied by the formation MC dimers.301 On the other hand, molecular dynamics simulations suggest the existence of hydrogen bonding interactions between the MC's phenolates and the unfunctionalised Glu's carboxylic acid groups.303
Photoinduced conformational changes are not limited to poly-L-Glu: for example, poly-L-lysine – another polypeptide with a tendency to adopt an α-helical conformation – showed analogous behaviour upon the attachment of spiropyran moieties to the side chain –NH2 groups (Fig. 13E).304,305 However, it is important to note that structural properties of both poly-L-Glu and poly-L-Lys (and other polypeptides in general) are strongly environment-dependent306,307 and that the reversible, light-induced conformational changes described above were observed only in selected solvents. Recently, a more general approach to controlling polypeptide conformations was introduced, which is based on a spiropyran-based crosslinker within a rationally designed peptide sequence.308 This crosslinker acted as a reversible “hinge”, inducing different degrees of helicity (48% for the dark-adapted MC vs. 62% for the Vis-exposed SP) in an otherwise disordered polypeptide chain (12% helicity in the native peptide). Although the light-induced conformational changes were only moderate, the advantage of this approach is that it is expected to operate in a range of different environmental conditions.
3.2. Photocontrol of enzymatic activity
Enzymes whose catalytic activity can be modulated using light represent a highly attractive target as far as the control of the properties of biomacromolecules with external stimuli is concerned. Research in this field has been pioneered by Suzuki et al., who demonstrated three different modes of photoregulating enzymatic activity – all based on immobilised spiropyrans (Fig. 14).309 Initial studies based on this approach date back to as early as 1975 and deal with the enzyme α-amylase310,311 – an example which is here discussed as a case study. α-Amylase, the enzyme hydrolysing α-linked polysaccharides, was functionalised with excess of 21, and a solution of the resulting protein had a red colour indicating that the spiropyran was in the MC form. As could be expected, this modification significantly (by 64%) decreased the enzymatic activity as compared to the native α-amylase; importantly, however, exposure to visible light (amylose·MC → amylose·SP) reduced the activity by a further 18–36%.310,311 While being rather small, this change can be appreciated given that only 2 out of ∼900 amino acid residues were modified with spiropyran. When left in the dark, the protein regained its original activity due to thermal SP → MC re-isomerisation. It has been speculated that the hydrophobic nature of SP prevented the enzyme from interacting efficiently with its substrate – the hydrophilic amylose – resulting in decreased catalytic activity. A related enzyme β-amylase responded similarly, and the changes were much more pronounced: exposure to visible light reduced the activity by as much as 87%.312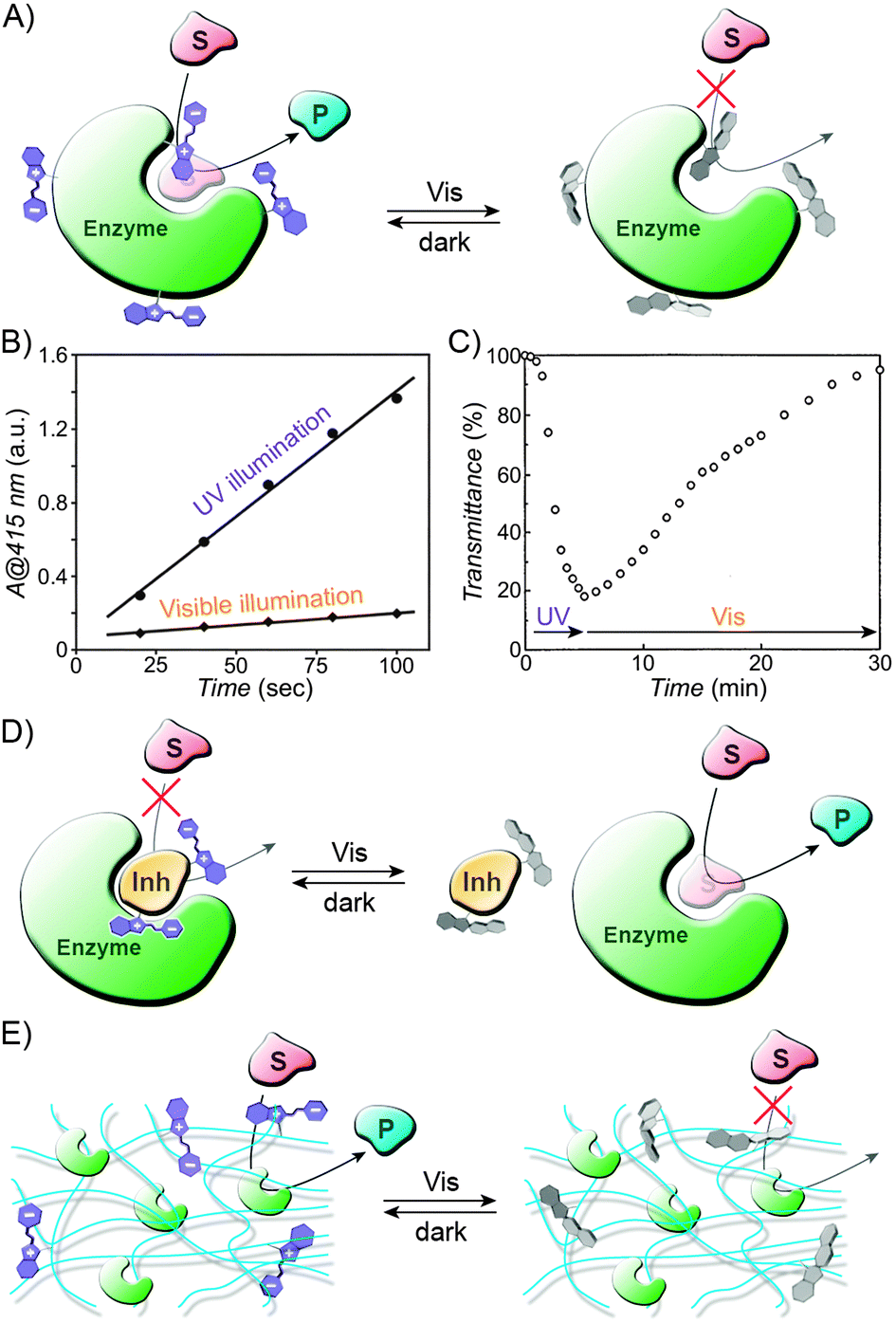 | ||
| Fig. 14 Light-controlled catalysis using spiropyran-functionalised biopolymers. (A) Schematic representation of light-controlled catalysis using spiropyran-modified enzymes. Note that the MC → SP isomerisation can either activate or deactivate the enzyme. (B) Effect of light on the catalytic activity of spiropyran-derivatised horseradish peroxidase. In this case, Vis-irradiation deactivated the enzyme. (C) The opposite trend was achieved with spiropyran-functionalised subtilisin. (D) Another approach to photoswitchable catalysis based on spiropyran-functionalised macromolecular inhibitors. (E) Light-controlled catalysis using enzymes bound to spiropyran-functionalised porous media. [Adapted with permission from ref. 320 (Copyright 1999 Elsevier) (B) and ref. 321 (Copyright 1999 Nature Publishing Group) (C).] | ||
In principle, however, the effect of switching on catalytic activity is difficult to predict a priori: modified β-glucosidase (another enzyme which hydrolyses polysaccharides), for example, was more active when its spiropyran residues were in the closed form.312 A mechanism by which the MC groups deactivate their host enzymes might be based on intramolecular attractive MC–MC (or repulsive MCH+–MCH+) interactions perturbing the active conformation of the protein – in fact, it has been shown by dynamic light scattering (DLS) that modified concanavalin A undergoes shrinkage upon SP → MC isomerisation,313,314 resulting in decreased binding315 of the protein's monosaccharide substrates. Similarly, α-chymotrypsin could be partially deactivated by converting the SP moieties on its surface to MC.312,316 Other enzymes which were modified by the same approach and subsequently showed photoregulated catalytic properties include urease,317 glucose oxidase (GOX),318,319 and horseradish peroxidase (HRP).320 This last example is worth highlighting because of the large changes in activity exhibited by the enzyme upon photoirradiation. HRP equipped with 8–9 spiropyran groups per protein molecule readily catalysed the oxidation of a model compound when the switch was in the MC form, whereas exposure to visible light reduced the reaction rate by as much as 92% (Fig. 14B).320 An even more impressive change was facilitated by an approach based on the light-induced precipitation of the enzyme from the solution. To achieve this goal, Ito and co-workers prepared a construct comprising the enzyme subtilisin and a photoswitchable random copolymer pre-synthesised from (i) methacrylic acid, (ii) methyl methacrylate, and (iii) a spiropyran-appended methacrylate.321 The resulting hybrid subtilisin carried as many as ∼30–80 spiropyran moieties per protein molecule, which provided a unique hybrid system with dramatic photoresponsiveness. For example, the hybrid formed well-defined solutions in toluene, from which it could be quantitatively precipitated upon exposure to UV light (Fig. 14C), thus effectively shutting down its catalytic activity in a transesterification reaction. Notably, the catalytic efficiency of the hybrid enzyme did not change after three precipitation–solubilisation cycles.321
The second strategy to manipulate enzymatic activity with light is based on photoswitchable inhibitors – that is, inhibitors whose affinities to native enzymes depend on the light to which they are exposed (Fig. 14D).322 For example, ovomucoid, a protein also known as trypsin inhibitor, was functionalised with spiropyran using the generic reagent 21.323 The photoinhibition effect of the modified ovomucoid towards native trypsin – defined as the ratio of the amount of trypsin inhibited by unit mass of the inhibitor under visible light vs. in the dark – depended on the degree of functionalisation with SP; it was established that the highest effect was exhibited by ovomucoid having 2 of its 10 amino groups modified with spiropyran. Specifically, this modified inhibitor showed a photoinhibition effect of 0.72, indicating that the inhibitor functionalised with MC interacted with trypsin more strongly than when appended with SP.323 This difference has been attributed to the electrostatic stabilisation of the enzyme–(inhibitor·MC) binding (the activity of the inhibitor decreased with increasing ionic strength of the solution).
Finally, photoswitchable catalysis can be achieved using native enzymes which have been non-covalently associated with photoswitchable membranes (Fig. 14E). Such photoresponsive environments can be easily prepared by modifying inexpensive biomacromolecules such as collagen fibrils and agarose gels with 21. Suzuki et al. were the first to show that various enzymes embedded in such membranes display light-dependent activities, despite not being chemically attached to the SP species. Generally, these different systems all showed that the rate of the catalytic reaction (e.g. urease-catalysed urea hydrolysis; lactate dehydrogenase-catalysed lactate oxidation) was decelerated upon exposure of the MC-gels (negative photochromism) to visible light. The changes, however, were small: the activity of trypsin,324 lactate dehydrogenase,325 and urease326 – all embedded in spiropyran-functionalised collagen – dropped by 22%, 37%, and ∼50%, respectively. Similarly, the rate of hydrolysis catalysed by trypsin associated with a spiropyran-agarose gel decreased by 33%.327 The differences in reaction rates could be attributed to environment-dependent diffusion coefficients of the reaction substrates: MC-rich media can be hydrated more readily than their SP counterparts, thus enabling efficient transport of solutes and their delivery to the enzymes (it has been shown, for example, that urea hydrolysis catalysed by a spiropyran–collagen gel is a diffusion-limited process328). In certain cases, however, the visible light-decreased reactivities could not be accounted for by the diffusion effects: e.g. both lactate and NAD – substrates for lactate dehydrogenase – diffused faster through SP- than through MC-collagen membranes (presumably due to their attractive interactions with the MC groups). It was speculated that the hydrophobic microenvironment around the lactate dehydrogenase imposed by the SP moieties may directly destabilise the enzyme.325
Whether an enzyme-catalysed reaction will be accelerated or suppressed by visible light depends also on the nature of the reaction. A chymotrypsin-catalysed plastein synthesis – a process during which peptide bonds form as opposed to being hydrolysed – proceeded more efficiently in SP-rich agarose gels.329 The authors hypothesised that the hydrophobic microenvironment promoted a reaction in which water molecules are consumed.329 In a remarkable example supporting this reasoning, the direction of a reaction – esterification vs. ester hydrolysis – could be controlled by the state of spiropyran.330 Specifically, a Vis-irradiated spiropyran-agarose gel promoted esterification of N-acetyl-L-tyrosine with ethanol, whereas in the dark, the respective ester was hydrolysed. In an elegant demonstration of this behaviour, this ester could reversibly be formed and subsequently quantitatively hydrolysed upon exposure to visible light and dark adaptation, respectively, over several cycles.330
3.3. Photocontrol of transport through biopolymers
In the last mode of light-controlled catalysis discussed above,325 the rate of diffusion through spiropyran-functionalised gels depended on the state of the switch. Another example of such behaviour was found during studies on cytochrome c (Cyt c), which interacted more strongly with the SP isomer of spiropyran immobilised on an agarose gel, such that ∼46% of the protein bound under visible light could subsequently be released in the dark.331 While the interactions with SP/MC alone are not selective enough as far as mixtures of proteins are concerned, co-functionalisation of the stationary media with the photoswitchable groups and specific enzyme inhibitors at the same time could greatly increase binding selectivities of selected proteins, which, in combination with the light responsiveness, has become the basis of a technique called light-enhanced affinity chromatography. The principle of this technique is based on the empirical observation332,333 that the strength of an enzyme's interaction with immobilised inhibitors depends on whether the nearby photoswitch molecules are in the SP vs. the MC form. For example, trypsin interacted strongly with spiropyran-functionalised agarose gel appended with the soybean trypsin inhibitor in the dark (whereby spiropyran existed in the MC state due to negative photochromism), but could be released upon exposure to visible light. The potential of this technique was demonstrated by the purification of crude trypsin from bovine pancreas: light-induced elution from a column packed with the modified agarose gel led to 21-fold purification of the enzyme.333 Notably, the opposite binding pattern was observed when the spiropyran-agarose gel – now co-functionalised with aspartate residues – interacted with the enzyme asparaginase. While the enzyme bound strongly to the visible light-irradiated, SP-rich gel, it was readily released in the dark, leading to 90-fold purification of the crude enzyme.332 It is worth emphasising that asparaginase interacted with the aspartate residues, and not with the photoswitch – in fact, no binding was observed between the enzyme and a spiropyran gel lacking any aspartates. Instead the mode of action of the switch was thought to involve reversible modulation of the ionic strength in the proximity of the enzyme–ligand interaction site.3.4. Photocontrol of DNA hybridisation
The ability to reversibly unwind the DNA double helix using light could be used to phototrigger a variety of biological processes. A rationale behind using spiropyran as a building block for photoswitchable DNA is that its MC isomer bears much more structural resemblance to the DNA bases than the SP form. Consequently, one might expect a preferential ability of MC to participate in π–π stacking interactions with the DNA bases, and consequently to increase the stability of the double helix. This hypothesis was verified in a study based on a spiropyran-terminated homothymidine (T8). Indeed, the melting temperature of this oligonucleotide increased from 18.0 °C to 21.6 °C upon SP → MC isomerisation,334 suggesting that within this narrow temperature window, reversible, light-induced duplex formation/melting was possible. Functionalisation of long-chain DNA was also reported, whereby control of the extent of functionalisation with SP was demonstrated – again, efficient isomerisation of the switch was observed.335 Recently, more sophisticated spiropyran-functionalised DNA molecules were synthesised, incorporating the photoswitch at the center of the chains. Within this context, spiropyran was found to be resistant to UV irradiation.336 The authors hypothesised that this lack of responsiveness was due to quenching of the triplet state of spiropyran by neighbouring DNA bases.3.5. Photocontrol of volume phase transitions in thermoresponsive polypeptides
Elastin-like polypeptides (e.g. (VPGVG)n polymers) show behaviour analogous to that of thermoresponsive polymers (cf. Sections 2.1 and 2.3) in that they precipitate from aqueous solutions concurrent to a temperature increase. The temperature at which the transition occurs is referred to as the inverse transition temperature (Tt; compare with LCST).337 To investigate the possibility of modulating phase transitions in elastin-like polymers using light, Rodríguez-Cabello and co-workers prepared a random copolymer [(VPGVG)0.74(VPGEG)0.26]n, and functionalised the side chain –COOH groups with spiropyran moieties via esterification.338 The dark-adapted photostationary state of the resulting polymer contained approximately equal fractions of SP and MC, and the polypeptide had a Tt ≈ 13.5 °C, a value which could be decreased to ∼10 °C and increased to ∼20 °C upon exposure to visible and UV light, respectively. Subjecting a solution of this polymer to alternate cycles of visible and UV light irradiation at T = 14 °C increased the population of SP and MC, respectively, to the extent that the polymer reversibly precipitated and redissolved. Remarkably, these drastic solubility changes could be achieved with a polymer equipped with as little as 2.3 spiropyran groups per 100 amino acid residues.3.6. Photocontrol of protein folding
A fascinating idea of employing the reversible SP ↔ MC isomerisation for triggering conformational changes of nearby protein molecules was investigated by Akiyoshi et al.339 These researchers prepared nanoparticles of gels (“nanogels”) made of the polysaccharide pullulan functionalised with spiropyran. An intentionally misfolded protein – citrate synthase (CS) – was then interfaced with the nanogels, and the rate of its refolding to the native conformation, estimated by the rate of recovery of the enzyme activity, was probed under different types of irradiation. Specifically, denatured CS exhibited 32% of the native enzyme activity when no nanogels were added. Upon the addition of a nanogel containing both the SP and the MC forms of the switch, CS activity increased to 61%. Interestingly, however, when the protein was first incubated with the same SP + MC gel for a short time, and then exposed to visible light which gave rise to an all-SP nanogel, 81% of the native CS activity was regained. Clearly, the MC → SP isomerisation induced extra refolding in a process perhaps reminiscent of the mode of action of the natural chaperones, which undergo ATP-driven conformational changes thereby refolding misfolded proteins bound in their cavity. Unfortunately, only one switching cycle was reported, and the question of if and how the amount of refolded protein would increase by repeated UV-Vis cycling remains. Nevertheless, this study represents an important step toward a rational development of synthetic machinery with chaperone-like activity.3.7. Photocontrol of transport through natural nanopores
The large change in polarity accompanying the SP ↔ MC isomerisation is critical in the design of light-actuated nanovalves.340,341 Such “smart” nanovalves are based on a natural channel protein found in Escherichia coli known as the mechanosensitive channel of large conductance (MscL). In the bacteria, the valves are embedded within the membranes and, under normal conditions (that is, no significant difference in osmotic pressure on the two sides of the membrane), the native MscL is “closed” – as a consequence of hydrophobic interactions between non-polar amino acid residues located inside the channel. It has been found, however, that substituting a single, strategically positioned hydrophobic glycine residue with polar amino acids leads to increased hydration of the pore, and consequently increased channel mechanosensitivity.342 Based on this knowledge, Feringa, Meijberg, and co-workers prepared an MscL mutant wherein the Gly residue was substituted with cysteine, and then post-synthetically modified the resulting protein using a cysteine-selective alkylating agent comprising an iodoacetate group and a spiropyran moiety.340 As predicted, the reversible hydrophobic ↔ hydrophilic isomerisation of spiropyran which was thus installed within the channel enabled opening and resealing of the channel, respectively. The utility of these nanovalves was demonstrated by performing light-controlled transport of a fluorescent dye across a membrane incorporating the modified nanopores.3.8. Spiropyran as a probe for the analysis of biological microenvironments
We saw in Section 2.3 how the open form of spiropyran could act as a colorimetric sensor of the polarity of its microenvironment in the context of phase transitions in thermoresponsive polymers (cf. Fig. 9E). Importantly, environment-dependent properties of the switch can also be employed in order to tackle meaningful biological questions. Imanishi et al. synthesised several spiropyran-functionalised oligopeptides, all with the sequence corresponding to the shortest active fragment of melittin, and equipped them with the photoswitch at different positions of the sequence (melittin is the active component of honey bee venom, and it is responsible for cell membrane lysis). These different peptides were exposed to UV light, and the rate of thermal MC → SP re-isomerisation in the presence of lamellar vesicles (mimicking cell membranes) was followed spectroscopically.343 The isomerisation rate was shown to be dependent on the location of the photoswitch, ranging from fast decay of the MC signal when the probe was at the C-terminus of the peptide, to an indefinite stabilisation of the coloured form for the N-terminus-functionalised peptide (negative photochromism). Placing the MC probe at the center of the peptide sequence resulted in an intermediate decay rate. These results indicate that the C-terminus of the peptide was located in the hydrophobic part of the vesicles, whereas the N-terminus was exposed to the aqueous environment. In an interesting extension of this study, light-stimulated lysis of membranes incorporating spiropyran-containing melittin fragments was demonstrated.344Similarly, Mihara et al. developed a technique called photochromism-based assay, which is based on spiropyran-functionalised polypeptides having specific protein-binding sequences. Again, the peptides were pre-exposed to UV light, and, the rate of MC → SP re-isomerisation was followed by measuring the decay of fluorescence due to the MC isomer.345 Attractive interactions with the target protein molecules were evidenced by an emission decay rate different from that of the same peptide in a protein-free solution. These differences could stem either from the different dielectric constant of the new microenvironment around the switch, or from the steric hindrance imposed by the target protein molecule. Based on how a given protein affected the emission behaviour of eight different spiropyran-containing polypeptides, it was assigned a specific “barcode”, by which it could then be recognised. Using this strategy, six different proteins could unambiguously be identified. The same authors also described a closely related method: the chromism-based assay (CHROBA), which has been useful in detecting the phosphorylation activity of kinases.346 The assay is similarly based on spiropyran-functionalised polypeptides – in this case substrates for specific kinases. For example, one such peptide contained the so-called Kemptide sequence (LRRASLG-NH2) – a kinase A-specific substrate – having spiropyran covalently attached to its C-terminus (S indicates the serine residue which can be phosphorylated). Following phosphorylation (or lack thereof), this peptide was incubated with polyelectrolytes – poly(L-lysine), poly(L-arginine), and/or poly(L-aspartic acid) (Fig. 15). Since each phosphorylation event entails the introduction of two negative charges (at near-physiological pH), phosphorylated peptides are expected to interact differently with the polyelectrolytes: specifically, their binding with poly(lysine) and poly(arginine) should be stronger, whereas with poly(aspartic acid) – weaker upon phosphorylation. Association with a polyelectrolyte is, in turn, expected to suppress the rate of spontaneous SP → MC isomerisation (solutions are pre-exposed to visible light), now taking place in a viscous microenvironment of the polyelectrolyte.347 This isomerisation could be followed fluorometrically or even colorimetrically348 – that is, visual inspection of the samples allows one to discriminate the original, non-phosphorylated substrates from the modified ones, or, in other words, to detect the presence of a given kinase (e.g. it was established that the lower limit for the detection of kinase A is ∼0.1–1.0 μg mL−1). A significant advantage of CHROBA over traditional phosphorylation assays is that it avoids the need to use immobilised protein substrates or laborious isolation steps.
 | ||
| Fig. 15 “Chromism-based assay” for the detection of phosphorylation activity. See text for explanation. [Adapted with permission from ref. 348 (Copyright 2006 Royal Society of Chemistry).] | ||
4. Spiropyran-functionalised inorganic nanoparticles
Decades of research in the modification of planar inorganic surfaces with organic ligands provide important lessons as far as modification of nanostructured particles is concerned. In principle, the choice of the ligand monolayer depends on the chemistry, but not the curvature of the solid substrate – that is, the same chemical functionalities can be used for the modification of planar and nanostructured surfaces. Accordingly, gold NPs have been functionalised with spiropyran-terminated thiols,349,350 silica shells of upconverting nanocrystals (UNCs) were modified with a spiropyran-terminated silane, and so on. An attractive alternative to the above place-exchange methods is provided by “post-synthetic” modification of SAMs – a strategy particularly useful in the case of chemically sensitive NPs. For example, alkyne-terminated CdSe quantum dots underwent further modification via the copper-catalysed [3+2] cycloaddition – e.g. with an azide-appended small-molecule spiropyran.351,3524.1. Photocontrol of nanoparticle fluorescence
The ability of spiropyran to switch between an optically transparent SP and a strongly absorbing, fluorescent MC form enables the reversible control of FRET; several examples of modulating the fluorescence of organic fluorophores353 have been covered in Section 2.1. This concept can be extended to inorganic fluorescent NPs by functionalising their surfaces with spiropyran molecules,9 and has to date been demonstrated for four different classes of such materials: CdSe–ZnS core–shell quantum dots (QDs), gold clusters, carbon nanodots, and rare-earth nanophosphors. In all of these cases, the inherent fluorescence of the NP cores can effectively be “turned off” and “on” as a consequence of the reversible isomerisation of the photoswitch. It is worth recalling, however, that the excitation wavelength should be adjusted such that it does not induce the SP → MC or MC → SP photochemical isomerisation.Fortunately, quantum dots can be excited with a wide spectrum of wavelengths, such that those fulfilling the above condition can easily be identified. Medintz and co-workers were the first to interface CdSe–ZnS core–shell QDs with spiropyran, employing a rather complex immobilisation approach based on decorating the ZnS shells with a pentahistidine sequence appended to the C-terminus of the maltose binding protein, which had been modified with spiropyran using an active ester similar to 22 in Fig. 13.354 The number of switch units per protein molecule could be varied between 1 and 5, thereby allowing placement of various numbers of spiropyran moieties on each QD, and ultimately control of FRET efficiency (e.g. ∼25% and ∼60% for proteins carrying 1 and 5 spiropyran units, respectively). The same NPs were subsequently functionalised with spiropyran using a simpler approach based on thiolate self-assembled monolayers (SAMs).175 When covered with the SP isomer and excited with λ = 420 nm, these 5 nm NPs emitted green light, as shown in Fig. 16A, left. Exposure to only 5 seconds of UV, however, activated FRET and the excited solution assumed a red colour typical of MC emission (Fig. 16A, right). The process is reversible and NPs' emission can be restored using visible light (Fig. 16B).
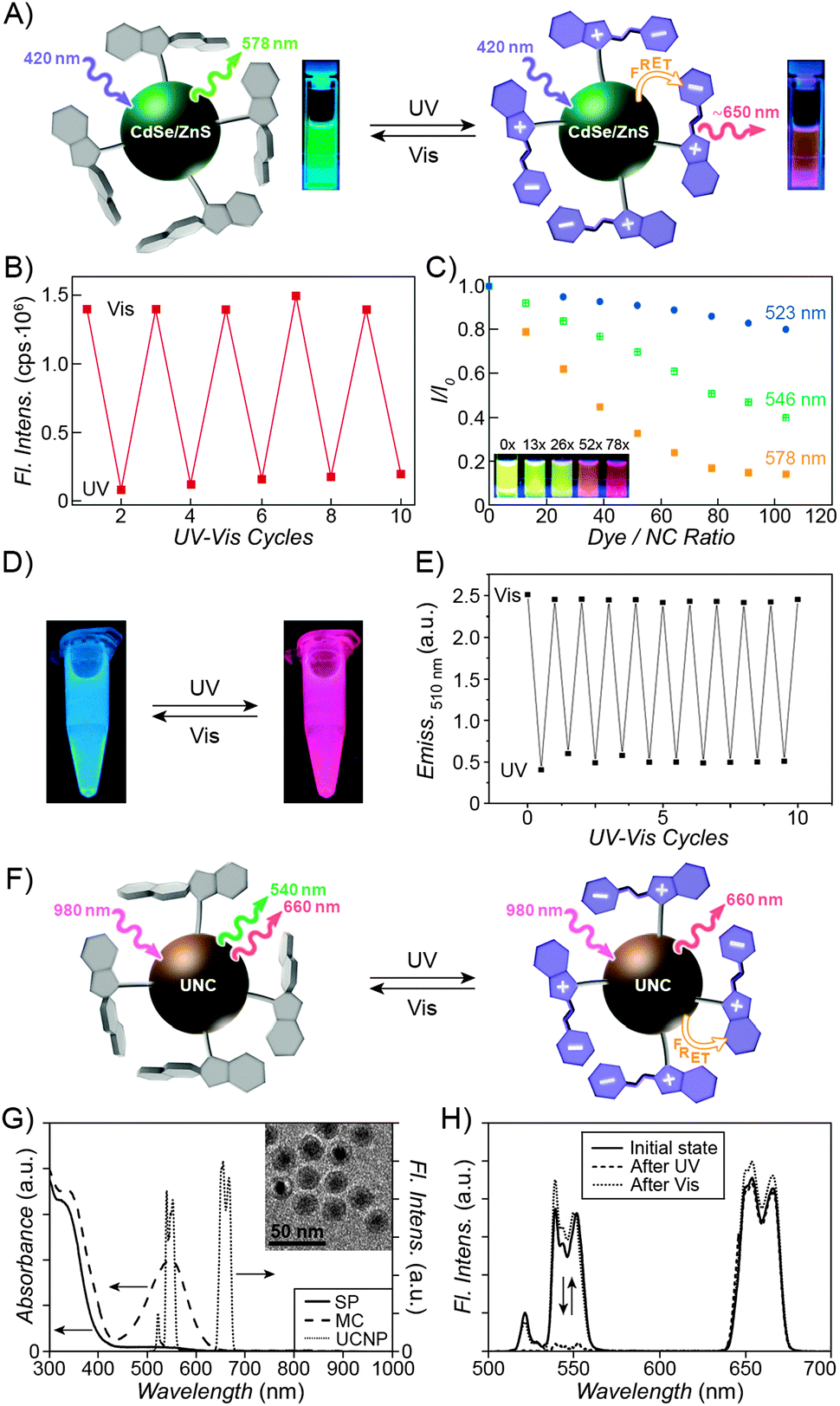 | ||
| Fig. 16 Light-controlled modulation of fluorescence of inorganic nanoparticles. (A) Reversible FRET between a green-emitting core and an MC shell in spiropyran-coated quantum dots. (B) Reversible quenching of the quantum dots' emission at 546 nm. (C) Dependence of fluorescence quenching efficiency on the overlap of quantum dots' emission and MC absorption. Inset: fluorescence emission as a function of spiropyran loading. (D) Dual-emissive spiropyran-modified carbon nanodots. (E) Reversible quenching of carbon nanodots' emission at 510 nm. (F) Selective quenching of only one emission wavelength of dual-emissive upconverting nanocrystals. (G) Absorption spectra of SP and MC along with the emission spectrum of UNCs. (H) Selective, reversible quenching of the green emission of the UNCs. [Adapted with permission from ref. 175 (Copyright 2005 American Chemical Society) (A–C), ref. 356 (Copyright 2013 Royal Society of Chemistry) (D and E) and ref. 357 (Copyright 2012 Wiley-VCH) (G and H).] | ||
Fig. 16C provides an elegant manifestation of the importance of the overlap355 of MC's absorption and the nanocrystals' emission for effective FRET. One can see that for any given number of MC per NP, FRET is most efficient for the largest, 6 nm QDs. This seemingly counter-intuitive result can be understood by taking into account the maximum emission wavelengths of the differently sized QDs: λem ≈ 523 nm for 4 nm NPs, λem ≈ 546 nm for 5 nm NPs, and λem ≈ 578 nm for 6 nm NPs (compare with MC absorption at ∼580 nm). Hence, MC absorption coincides with the emission of the QDs that are 6 nm in diameter (it is worth emphasising that as little as 10% surface coverage of the 6 nm NP with the MC groups can reduce as much as ∼50% of the original emission intensity). On the other hand, the poor overlap of MC absorption with the emission of the 4 nm QDs is reflected by only mediocre FRET values of these small NPs.
Analogous results were reported for spiropyran-functionalised fluorescent gold nanoclusters.349 The emission of these dots, comprising on average only eight gold atoms each (λmax ≈ 480 nm), partially overlapped with the absorption spectrum of the open form of spiropyran. As a result, fluorescence of Au8 decorated with a spiropyran thiol could efficiently (by ∼90%) and reversibly be quenched upon irradiation with UV light. The same methodology has been applied to spiropyran-modified 3 nm carbon nanodots,356 with a blue emission at λmax ≈ 510 nm (Fig. 16D, left). Following UV exposure, efficient FRET occurred, and the dots emitted red light (Fig. 16D, right). As Fig. 16E shows, blue emission intensity decreased by ∼80% even after ten cycles of switching. Finally, Yan et al. reported a very interesting example of selective silencing of only one emission channel of dual-emissive NPs (Fig. 16F).357 These researchers synthesised a trimethoxysilyl-terminated spiropyran and used it to functionalise silica-coated NaYF4:Yb,Er@CaF2 UNCs (“rare-earth nanophosphors”) (Fig. 16G, inset). The fluorescence spectrum of as-prepared UNCs is shown in Fig. 16G: when excited with a near-infrared (980 nm) laser, they show two sharp bands centered at ∼540 nm and ∼660 nm, corresponding to green and red emission, respectively. Upon exposure to UV, SP → MC isomerisation takes place, and the resulting MC absorption strongly overlaps with the green emission channel of the UNCs; consequently, green emission is quenched by as much as 94% (Fig. 16H; note that, interestingly, MC's own emission coincides with the red emission channel of the UNCs). The system was remarkably reversible, with barely any degradation observed after ten switching cycles.357
4.2. Photocontrol of nanoparticle solubility
Control of the stability of colloidal suspensions using light can be achieved by decorating the particle surfaces with monolayers of spiropyran. Typically, the non-polar character of the SP isomer ensures good solubility of SP-coated particles in apolar solvents; these suspensions can then be destabilised – and the particles flocculated – upon generation of the polar MC isomer by exposure to UV light. In addition, aggregation can also take place due to attractive MC–MC and MC–SP interactions between moieties located at different particles. Seminal reports on the photocontrol of colloidal stability were published in the mid-nineties by Ueda and co-workers, who worked with spiropyran-functionalised 150 nm silica particles.108,358,359 They found, for example, that such (transparent) suspensions in CCl4 were relatively stable under ambient light, but when exposed to UV, red solids rapidly precipitated. These initial studies highlighted the importance of the solvent on the particles' behaviour: when the medium was too hydrophobic (e.g. cyclohexane), NPs quickly sedimented irrespective of irradiation, whereas other solvents (e.g. CHCl3) stabilised NPs decorated with both isomers of the switch equally well (compare with Fig. 17D).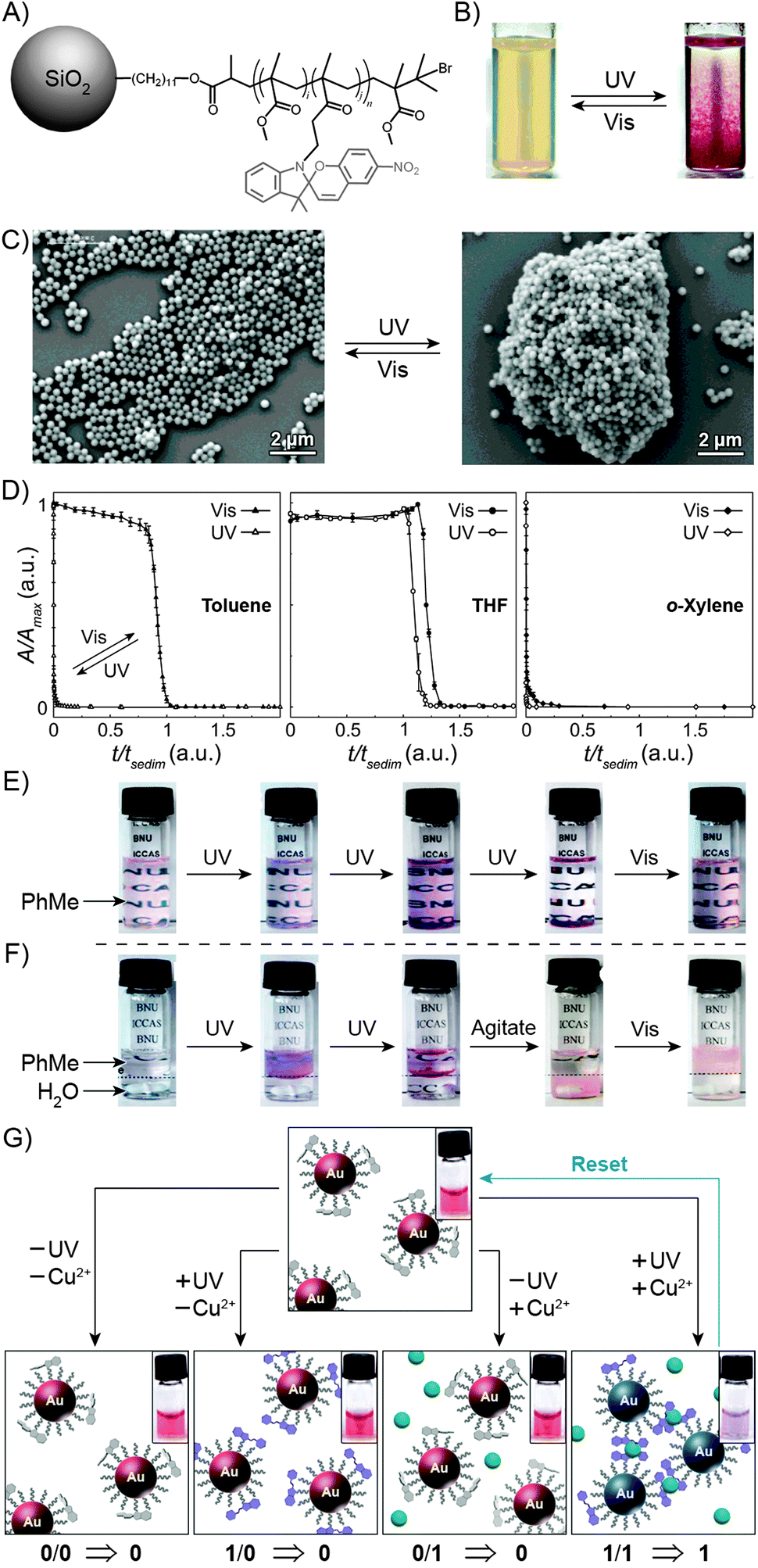 | ||
| Fig. 17 Spiropyran enables light-controlled aggregation of nanoparticles. (A) Structural formula of a photoresponsive copolymer used to functionalise silica colloids. (B) Light-controlled aggregation of spiropyran-coated silica. (C) SEM images of photoresponsive silica colloids under visible (left) and UV (right) light irradiation. (D) Solvent-dependent behaviour of spiropyran-functionalised silica particles. (E) Light-controlled precipitation of silica colloids from toluene. (F) Light-controlled phase transfer of silica colloids between toluene and water. (G) Dual-controlled aggregation of spiropyran-functionalised gold NPs. [Adapted with permission from ref. 125 (Copyright 2006 American Chemical Society) (B–D), ref. 102 (Copyright 2010 American Chemical Society) (E and F) and ref. 350 (Copyright 2011 Wiley-VCH) (G).] | ||
More recently, Bell et al. worked with similar systems comprising ∼300 and ∼900 nm silica spheres functionalised with spiropyran polymers synthesised via surface-initiated ATRP (Fig. 17A).49,360 Exposing the relatively stable toluene suspension of these particles to UV resulted in rapid precipitation of red solids, and SEM confirmed the presence of aggregates (Fig. 17B and C).125 Again, the behaviour was strongly solvent-dependent, as evidenced by rates of sedimentation under UV or Vis light – for example, while the strong effect of light on solubility was observed in toluene (specifically, the sedimentation rate was enhanced ∼335 times in the presence of UV), particles decorated with either SP or MC sedimented rapidly in o-xylene, and could efficiently be stabilised in THF irrespective of the isomer (Fig. 17D). These light-controlled dispersibility changes imply that in a biphasic system comprising a polar and an apolar solvent, the particles might be able to undergo a reversible phase transfer. A realisation of this remarkable concept has recently been reported using ∼400 nm diameter SiO2 spheres which have been chemically functionalised with a spiropyran polymer.102 The behaviour of this system is shown in Fig. 17E and F; in pure toluene, UV irradiation caused an almost colourless suspension of the particles to turn purple and quickly sediment (Fig. 17E). The presence of the additional aqueous phase allowed this precipitate to be effectively resuspended (Fig. 17F). In both cases, exposure to visible light regenerated the original toluene suspension via simple resuspension or phase transfer for the one- and two-phase systems, respectively. Notably, at intermediate stages of irradiation the silica spheres behaved as “colloidal surfactants” able to stabilise water–toluene emulsions.102
What are the implications of reversible, light-controlled self-assembly of nanoparticles? In one interesting example, Louie et al. showed that light-induced aggregation of spiropyran-coated magnetic iron oxide NPs could modulate the spin–spin (T2) relaxation time of adjacent water protons.361 Surface chemistry of these NPs was tailored so as to induce negative photochromism and ensure good water solubility of the particles in the dark. Indeed, DLS showed that aggregation commenced upon exposure to visible light, and it entailed reduction in the T2 relaxation time from ∼37 to ∼25 milliseconds – results which are of potential interest for the development of new types of “smart” MRI contrast agents.
As first demonstrated by Ueda's studies on silica colloids, aggregation properties of SP/MC-coated particles depend strongly on the polarity of the medium. In solvents stabilising both isomers the NPs display good solubility, yet aggregation can still be induced upon the addition of metal ions – provided the switch is present in the MC form (compare Section 2.6). This property inspired Wang, Jiang et al. to develop a series of logic gates whose key components were spiropyran-functionalised gold NPs in combination with metal ions.350 One such logic gate capable of performing an AND operation is shown in Fig. 17G. Neither UV light nor Cu2+ alone affects the state of the NPs dissolved in ethanol; however, when both of these “inputs” are present, aggregation commences, resulting in the colour change (Fig. 17G, bottom right). Importantly, the NP aggregates could be disassembled (and the gate allowed to “reset”) upon exposure to visible light, which re-isomerised MC back to SP. Similarly, OR and INHIBIT logic gates were realised by using other metal ions and EDTA, respectively, as additional components.350 In addition to metal cations, MC-coated NPs can also interact with amino acids, which suppress the MC → SP re-isomerisation rate.362
5. Spiropyran-functionalised solid surfaces
Having discussed spiropyran-functionalised polymers, biopolymers and inorganic NPs, we conclude with inorganic solid-state materials having their surfaces covalently modified by spiropyrans. Similar to inorganic NPs, the surface modification strategy is dictated by the chemical composition of the solid substrate. However, UV-Vis spectroscopy, which is routinely used to monitor the SP–MC isomerisation in solution, is hardly applicable48,363,364 to single layers of molecules on solid substrates because of the very short optical pathways. This forces one to resort to other tools as discussed below in Section 5.1. Conversely, immobilisation on solid surfaces offers one the unique opportunity to study the isomerisation process using force microscopy techniques, and several meaningful conclusions from such studies follow. Picraux et al. employed interfacial force microscopy to probe isomerisation within diluted monolayers of spiropyrans on glass.365 Studies performed in air showed an increase in the dipole moment upon UV irradiation, with the dipoles pointing towards the surface (consistent with the mode of attachment of the switch to the substrate). Reversible changes in the dipole moment were observed upon alternating UV and Vis irradiation, and the gradual decay of the difference could be attributed to the fatigue effects. Measurements carried out on the same monolayers in a polar liquid, on the other hand, provided evidence for a partial positive charge of the open-ring isomer (due to MCH+). Depending on whether the tip was negatively (bare silica) or positively (amine-coated silica) charged, the long-range force was attractive or repulsive, respectively.365 In another elegant study, Kimura and co-workers went a step further and immobilised spiropyran on both the AFM tip and the substrate.366 Adhesion forces between the tip and the surface increased substantially (by ∼50%) under in situ UV irradiation in ethanol, which could be attributed to direct MC–MC interactions. In toluene, however, no differences were seen under UV vs. Vis radiation – as expected from the low stability and fast re-isomerisation of the MC isomer in the non-polar medium.3665.1. Photocontrol of surface wettability
Perhaps the most obvious manifestation of the surface polarity switching is the change in wetting properties.367 The MC isomer, being more polar than SP, would be expected to increase the wettability of spiropyran-functionalised surfaces by polar solvents upon exposure to UV light. Fig. 18A shows this phenomenon for water droplets lying on glass substrates decorated with mixed monolayers comprising spiropyran-terminated silanes diluted with shorter, “dummy” silanes.78 The dilution with background ligands, while decreasing the surface concentration of spiropyran, enhanced wettability change by virtue of promoting efficient isomerisation, increasing contact angle variation from ∼5° to ∼13°. Still, the change seen in Fig. 18A is much smaller than what one might expect from a hydrophobic-to-zwitterionic change in the character of the photoswitch. Some clues of why this may be come from the employment of diverse experimental techniques to study on-surface isomerisation processes. For example, Brewster angle reflectometry,109 X-ray photoelectron spectroscopy,48 and atomic force microscopy365,368 all suggest that the yield of ring opening is far from quantitative (∼10–20%), even within diluted monolayers very similar to those from Fig. 18A.109 On the other hand, sum frequency generation (SFG) vibrational spectroscopy proved invaluable in probing the orientation of the photoswitch with respect to the underlying surface.369 When p-polarised beams are used for the SFG measurements, the method is sensitive to vibrational modes perpendicular with respect to the underlying surface. The authors identified symmetric and antisymmetric stretching modes of the spiropyran's nitro group as diagnostic of the isomerisation process, and concluded that in the open form, the NO2 group points away from the surface. This orientation of the molecule hinders the negatively charged oxygen atom away from the environment, and as a consequence, MC-functionalised surfaces retain much of the hydrophobic character of the SP-decorated ones369 – in agreement with the high values of the water contact angles before and after UV irradiation.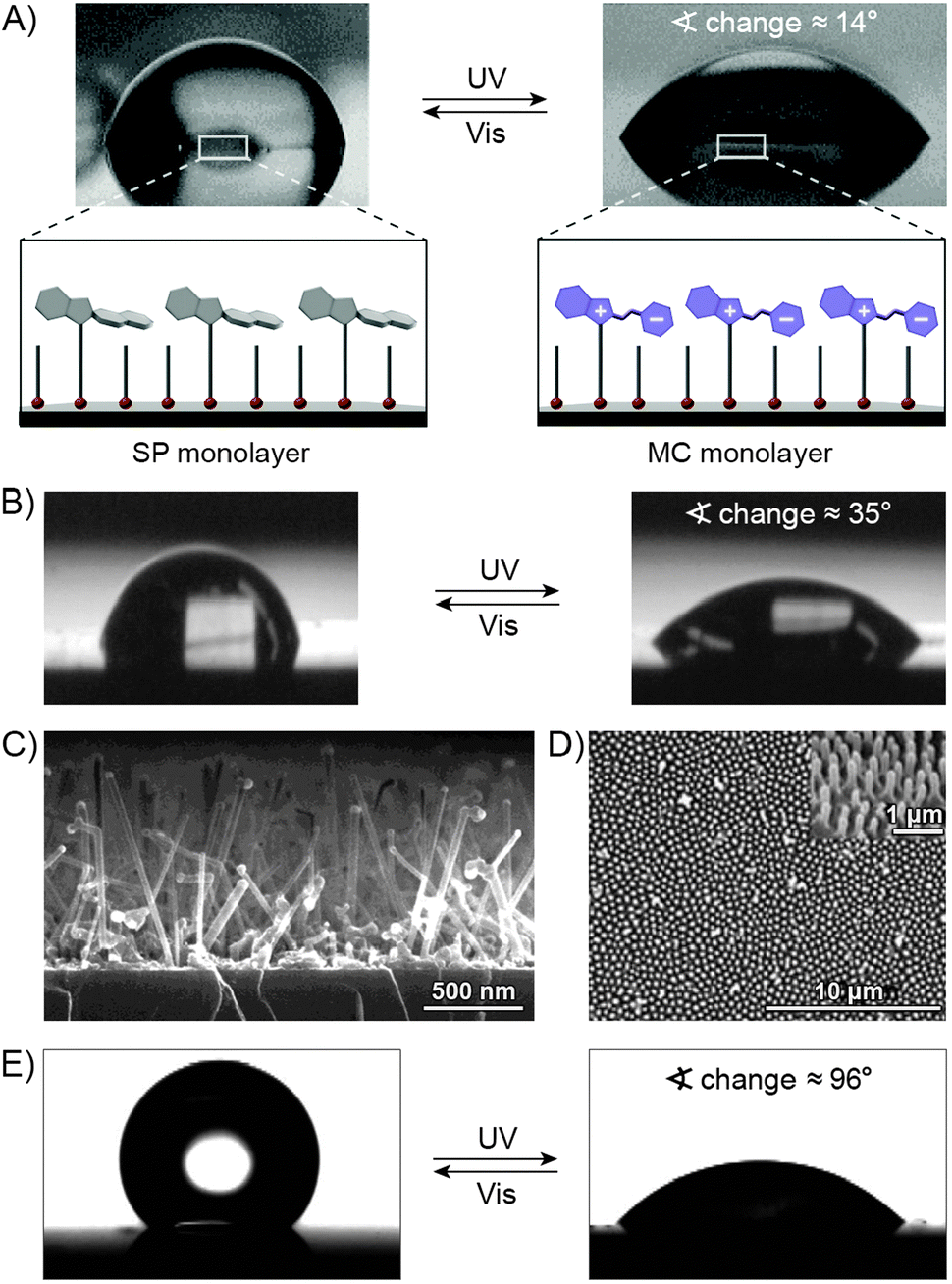 | ||
| Fig. 18 Photocontrolled wetting of spiropyran-functionalised surfaces. (A) Wettability change of spiropyran-functionalised glass. (B) Photoinduced change in the contact angle of an aqueous solution of Co2+ on spiropyran-functionalised silicon. (C) SEM image of a spiropyran-functionalised rough Si surface. (D) SEM image of a spiropyran-functionalised array of polymer nanorods. (E) Wettability change of a spiropyran-functionalised nanostructured surface. [Adapted with permission from ref. 78 (Copyright 2002 American Chemical Society) (A), ref. 90 (Copyright 2008 Royal Society of Chemistry) (B), ref. 370 (Copyright 2004 American Chemical Society) (C), ref. 373 (Copyright 2010 Royal Society of Chemistry) (D) and ref. 372 (Copyright 2012 Wiley-VCH) (E)]. | ||
Research in the field of photoswitchable surfaces is motivated in part by the quest for new, efficient ways to direct the motion of liquids in thin channels of the ever-shrinking microfluidic devices. The photoswitchable wettability change shown in Fig. 18A was sufficient to raise the water level in 500 μm-thick glass capillaries functionalised with the same monolayer by 2.8 mm – an interesting demonstration dubbed “photocapillarity”78 – but was not large enough to induce a reverse effect upon MC → SP re-isomerisation. A general condition for a droplet to move33 along a spiropyran-functionalised surface is that the advancing contact angle on an MC-rich surface be smaller than the receding contact angle on an SP-rich one – which, in principle, requires a significant difference in the advancing contact angles on surfaces coated with the two isomers. Accordingly, different approaches have been undertaken in an effort to increase contact angle variation. Locklin et al. reported that simply the addition of metal salts to a water droplet lying on a spiropyran-functionalised surface can increase contact angle difference from ∼15° to ∼35°, as shown in Fig. 18B for a Co2+ salt.90 The reason is that under these conditions, the isomerisation takes place between SP and the MC·Co2+ complex as opposed to a free MC – the complex having a more hydrophilic character than the uncomplexed MC. A disadvantage of this approach is that it requires the presence of a dissolved metal salt in the droplet.
An alternative strategy to amplify the change in contact angle is based on the immobilisation of spiropyran onto rough surfaces,370–373 and takes advantage of the well-known lotus effect.374 SEM images of some such surfaces are shown in Fig. 18C and D, whereby the roughness originates from arrays of silicon nanowires and polymeric nanorods, respectively. It is generally assumed375 that while the pronounced water-repellent properties of nanostructured hydrophobic surfaces originate from the air trapped in the nanopores, the enhanced wettability of the hydrophilic ones can be attributed to water penetrating the pores by the capillary action. Fig. 18E demonstrates such enhanced wettability change on porous silicon substrates, with the water contact angle decreasing – reversibly372 – by as much as 96°. A record value of 118° was reported in a conceptually different system based on a poly(NIPAM–SP) (cf. Sections 2.1 and 2.3) copolymer.373 In the dark, the polymer was rich in the MCH+ units and readily hydrated; exposure to visible light, however, generated SP, triggering dehydration of the polymer. The authors then systematically investigated the effect of substrate morphology, and showed that the transition from flat to microstructured to nanostructured surface increased contact angle difference from ∼24° to ∼79° to ∼118°.373
5.2. Photocontrol of transport through porous media
The photoswitchable systems discussed in this section are based on inorganic porous materials functionalised with monolayers of spiropyrans, and are complementary to the systems based on organic polymers covered in Section 2.4. Perhaps the simplest of such porous materials is commercial silica gel, which can be readily functionalised with desired monolayers. For example, Kimura et al. attached their “crowned spiropyrans” (see Section 2.6) to silica and used the resulting particles as the stationary phase for the separation of alkali metal ions.376 The binding ability of these modified crown ethers towards metal ions is enhanced by the UV-induced generation of the phenolate group – with the enhancement being metal-dependent. For example, Li+ and K+ could readily be separated when the chromatography column was exposed to UV light, but virtually no separation was achieved under visible light.376 A similar setup was employed by Zharov and co-workers, who prepared pads made from densely packed monodisperse, ∼170 nm silica spheres. These pads were ∼5 μm-thick (Fig. 19A) and the colloidal spheres comprising them had been derivatised with spiropyran monolayers.377 The authors monitored the flux of a positively charged redox probe – [Ru(NH3)6]3+ – by measuring the voltammetric limiting current, with the assumptions that the current is proportional to the flux under steady-state conditions, and that the SP → MCH+ isomerisation would reduce the flux as a result of the electrostatic repulsion between the positively charged probe and the like-charged porous medium. At a low (1.8) pH, however, the switch existed in the MCH+ form irrespective of irradiation, whereas photoisomerisation at pH 6.6 took place between two neutral states (SP ↔ MC) – as a consequence, only a minor (albeit reversible; cf. Fig. 19B) effect of light on the flux was observed.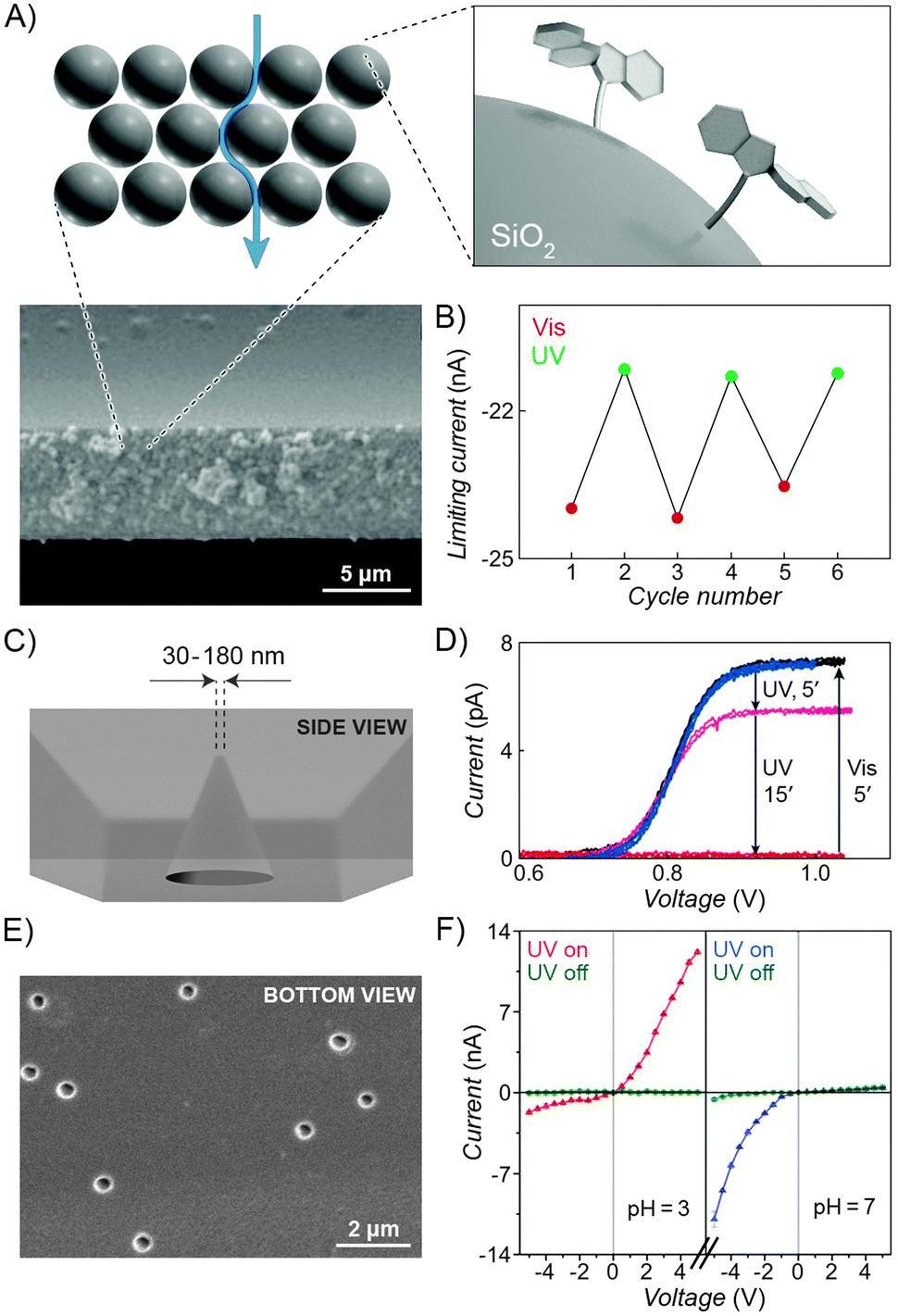 | ||
| Fig. 19 Light-controlled transport through porous materials. (A and B) Effect of irradiation on the flux of a charged redox probe through densely packed, spiropyran-functionalised colloidal spheres. (C) Schematic representation of a conical nanopore with spiropyran-functionalised interior. (D) Light-controlled transport through a conical nanopore. Formation of the positively charged MCH+ inhibits the flux of a positively charged probe. (E) SEM image showing multiple spiropyran-functionalised nanopores. (F) Ionic current rectification modulated by pH and light. [Adapted with permission from ref. 377 (Copyright 2010 Springer) (A and B), ref. 378 (Copyright 2006 Royal Society of Chemistry) (D) and ref. 379 (Copyright 2012 Wiley-VCH) (E and F)]. | ||
Another study from the same group has demonstrated that efficient switching between the neutral SP and the positively charged MCH+ states can enable high selectivities of transport of a positively charged probe [Fe(bpy)3]2+. In this case, the setup was based on the glass nanopore electrode, which comprised a Pt disk electrode embedded at the bottom of a conical pore (Fig. 19C). The electrode was immersed in an acidic acetonitrile solution. In order to reach the electrode, the solution molecules had to diffuse through the nanopore orifice whose diameter was in the range of 30–180 nm.378 The voltammetric response of the channel with the narrowest (30 nm) orifice is shown as a black trace in Fig. 19D and corresponds to diffusion-limited current. This current decreased with UV irradiation (pink trace after 5 minutes), until it was reduced to background levels (red trace after 20 minutes of UV) – under these conditions, the local concentration of the MCH+ groups immobilised at the pore orifice was sufficiently high so as prevent any of the like-charged [Fe(bpy)3]2+ from crossing the orifice and reaching the electrode. At the same time, visible light (5 min) induced regeneration of the neutral SP, with the faradaic current reaching the original values (blue trace), and the sequence could be repeated over at least several cycles. This mechanism of transport control based on repulsive electrostatic interactions is supported by three experimental observations, where no light-induced current modulation was observed (i) using small nanopores lacking the spiropyran coating, (ii) using large-diameter (4 μm) pores functionalised with spiropyran, and (iii) using small nanopores functionalised with spiropyran in the presence of high concentrations of a strong electrolyte, which screened the surface charge originating from MCH+.378 In another system, similarly shaped nanochannels functionalised with spiropyran monolayers were shown to act as light- and pH-operated nanofluidic diodes.379 Depending on whether the surface of nanochannels was positively or negatively charged, they were anion- or cation-selective, respectively. In addition, the conical shape of such charged nanochannels enabled preferential flow of ions in one direction (from the tip to the base) – that is, absolute values of ionic current at the applied voltage = +n vs. −n V were different, resulting in current rectification. Zhai et al. functionalised the interiors of conical (∼350 nm base – Fig. 19E – and ∼12 nm tip) nanochannels with a spiropyran monolayer and showed that ionic current under visible light (neutral SP) was, in fact, negligible and independent of pH (Fig. 19F, green traces). When the system was exposed to UV light at pH 3, however, a high current rectification ratio of ∼7 was observed (Fig. 19F, red trace), which could be attributed to the positively charged MCH+ moieties on the channel walls. Interestingly, UV irradiation at pH 7 resulted in an even higher value of rectification (∼30), with the current flowing in the opposite direction (indicating that cations are the majority carriers). This behaviour could be rationalised by the fact that although the zwitterionic MC groups carry zero net charge, the negatively charged oxygen atoms are more exposed to the solution compared with the positively charged nitrogen atoms (which act as a point of attachment to the nanopore surface).
A conceptually different approach to control transport properties has enabled Smirnov et al. to construct light-activated “burst” valves.380,381 These researchers used mixed monolayers comprising spiropyran and hydrophobic molecules to functionalise nanoporous alumina membranes. In the “off” (SP) state, no transport through the membranes was observed simply because water did not wet the strongly hydrophobic, narrow (∼20 nm) nanopores. Exposure to UV light, however, generated MC and triggered the admission of water to the nanopores and the consequent flow. However, capillary forces prevented the dewetting of the nanopores upon subsequent MC → SP re-isomerisation – therefore, these “burst” valves could be opened, but not resealed using light.380 Other porous materials which have been functionalised with spiropyran include zeolite L382 and mesoporous silica.383–385 The latter material has enabled “on-demand” release of encapsulated dyes upon exposure to UV light.385
5.3. Photocontrol of binding to surfaces
As discussed in Section 2.6, MC can bind different metal ions with varying strengths, and the distinctive optical properties of the resulting complexes are key to the development of sensing devices. The spiropyran-based systems are unique in their need to be pre-activated with UV light in order to bind metal cations,386,387 and particularly attractive in that once they have performed their sensing function, they can be “reset” using visible light, to be reused again later. These characteristics have led Diamond et al. to propose the concept of remotely activatable colorimetric sensors,388 which are normally in their “passive” state, but can be “turned on” at will – when the measurement is required.389 This ability to activate and deactivate sensors “on demand” can significantly increase their lifetime. In order to develop practical sensing devices, however, it is necessary to immobilise spiropyrans onto solid supports. Fig. 20A, for example, shows spiropyran-functionalised glass slides that, having been pre-activated with UV, were exposed to a series of divalent metal cations, giving rise to differently coloured complexes. Metal cations can then be released from the surfaces and recaptured over multiple cycles using visible and UV light, respectively.390 Interestingly, capturing of metal ions on the surfaces of MC-decorated electrodes could be followed by electrochemical reduction – giving rise to the corresponding metallic NPs, as was shown for silver391,392 and cobalt.393 Although the resulting metallic layers could not be released upon exposure to visible light, they readily redissolved upon electrooxidation. This method has enabled high-resolution patterning of electrode surfaces, with the smallest feature sizes on the order of only a few micrometers.392,393 | ||
| Fig. 20 Light-controlled adsorption to spiropyran-functionalised surfaces. (A) Glass slides functionalised with a spiropyran polymer brush before (SP) and after (MC) irradiation with UV light, and after exposure of the latter to a series of metal ions. (B) MCH+ → SP transformation cancels out the electrostatic interactions between the positively charged surface of a mesoporous material and negatively charged dendrimers, triggering dye release. (C) Light-controlled adsorption of the anti-DNP antibody onto a spiropyran-functionalised surface. (D) Following reversible adsorption of the anti-DNP Ab using quartz crystal microbalance analysis. (E) Following reversible adsorption of the same antibody onto an SP-functionalised surface by means of surface plasmon resonance. Dotted lines = addition of the anti-DNP Ab. Dashed lines = exposure to UV followed by rinsing. (F) Site-selective cell release from areas not exposed to UV. [Adapted with permission from ref. 435 (Copyright 2008 Royal Society of Chemistry) (A), ref. 394 (Copyright 2007 Wiley-VCH) (B), ref. 397 (Copyright 1997 American Chemical Society) (D), ref. 398 (Copyright 1999 American Chemical Society) (E) and ref. 403 (Copyright 2005 American Chemical Society) (F).] | ||
On the other hand, immobilised MCH+ groups can interact electrostatically and bind various negatively charged molecules. These interactions were explored for the construction of a light-activated release system shown in Fig. 20B.394 In this design, mesoporous silica MCM-41 was surface-functionalised with MCH+, which bound negatively charged generation 1.5 poly(amidoamine) (PAMAM) dendrimers from solution. With their fairly large molecular sizes (∼2.8 nm), the dendrimers acted as “stoppers” for MCM-41's nanopores (diameter ∼2.1 nm) pre-loaded with an optically active [Ru(bpy)3]2+ “cargo” (Fig. 20B, left). The release of the probe could be triggered upon exposing the system to visible light, which generated the SP isomer and cancelled the attractive interactions between the surfaces and the dendritic “plugs” (Fig. 20B, right). To increase the specificity of binding beyond electrostatic interactions, Willner et al. performed electropolymerisation of 4-thioaniline anchored to the surfaces of gold NPs co-functionalised with spiropyran, in the presence of a small-molecule template N,N′-bis(3-sulfatopropyl)-4,4′-bipyridinium. The affinity of the resulting NP “sponges” to the template molecule could be controlled using both optical and electrical stimuli.395 Larger molecules can also be reversibly adsorbed onto surfaces – the same authors found that spiropyran bearing two NO2 groups (dinitrospiropyran) can act as an antigen for the anti-dinitrophenol (anti-DNP) antibody (AB) when it exists in the closed – but not in the open – form (Fig. 20C). Accordingly, gold surfaces functionalised with dinitrospiropyran monolayers could capture and release this specific AB,396 and the reversibility of the process was confirmed by quartz crystal microbalance analysis (Fig. 20D)397 as well as by surface plasmon resonance (Fig. 20E)398 and impedance399 spectroscopies.
The complexity of the species whose reversible adsorption onto spiropyran-decorated surfaces has been demonstrated ranges from metal ions through small molecules through proteins, all the way to living cells. Higuchi and co-workers investigated interactions between mesenchymal stem cells and glass plates coated with a PMMA-based copolymer comprising spiropyran as a function of UV light.400 Irradiation with low-intensity UV light released ∼90% of the cells originally adhering to the surface, almost all of which remained alive. Control experiments with analogous surfaces lacking spiropyran confirmed that the release was indeed due to the generation of MC as opposed to the direct interaction of the cells with UV light. It is interesting to point out that the same trend was observed in experiments aimed at investigating interactions at a single molecule level; in a recent study,401 AFM tips functionalised with fibronectin – a protein involved in mediating cell adhesion – were interfaced with spiropyran-decorated polymers, and the adhesion forces were measured under UV and visible irradiation. As expected, the adhesion force of the protein to the SP-rich surfaces was higher (by ∼50%) than to the MC-rich ones.
Interestingly, surfaces exhibiting the opposite effect – that is, adhesion of cells to the UV-irradiated locations – have also been developed. This design is based on previous studies402 which found that the adhesion of cells to the thermoresponsive PNIPAM is dramatically reduced upon dehydration of the polymer (that is, above the LCST). With the possibility to modulate the LCST of poly(NIPAM–SP) using light (compare Sections 2.1 and 2.3), Sumaru et al. employed this photoresponsive copolymer as the key component of “photoresponsive cell culture surfaces” (PRCS).403–405 The behaviour of a PRCS is demonstrated in Fig. 20F, whereby the upper half of the sample was exposed to UV light. Subsequent washing at a lower temperature T, such that LCSTpoly(NIPAM–MC) < T < LCSTpoly(NIPAM–SP), removed CHO-K1 cells selectively from the non-irradiated area. An important advantage of using light to promote/inhibit cell adhesion is that it enables patterning of surfaces with cells with high spatial precision.403,405
5.4. Photocontrol of electrochemical properties
Light-induced adsorption of various species onto surfaces as discussed in the previous section results in decreased contact of solution molecules with the surface. This has some interesting implications: consider, for example, the possibility of employing light to affect the communication of solubilised redox probes with the surfaces of electrodes – such control could be used as a way to modulate the electrode potential using light. A variety of such “smart electrodes” based on spiropyran-functionalised surfaces have been developed by Willner and co-workers, and are the focus of this section.The binding of the anti-DNP antibody to SP-functionalised electrodes insulates the electrochemically active surface from the molecules in the solution. An example is shown in Fig. 21B, where the originally high amperometric signal, corresponding to reversible [Fe(CN)6]3−/[Fe(CN)6]4− conversion on an MC-decorated electrode, gradually decreased upon exposure to visible light in the presence of the anti-DNP AB. A near-complete loss of electrochemical activity was observed after 16 min of irradiation. The activity of thus-generated AB-insulated SP-electrode was partially restored upon subsequent irradiation with UV – a process which regenerated a surface coated with the MC isomer, to which the anti-DNP AB did not bind.406 Similarly, reversible passivation of electrodes with the same AB controlled the rate of oxidation of catechols to quinones.407
 | ||
| Fig. 21 Modulation of electrochemical properties of electrodes using light. (A) Reversible electrode insulation from redox-active molecules in the solution. (B) Gradual changes (see the arrow) in cyclic voltammograms of an SP-functionalised electrode in the presence of [Fe(CN)6]3− following the addition of the anti-DNP Ab. (C) Electrocatalytic currents for glucose oxidation in the presence of an SP/MCH+-coated electrode in the presence and in the absence of the anti-DNP Ab. (D) Photocontrol of the Frumkin effect: negatively charged species are attracted to the MCH+-functionalised electrode, facilitating the redox reactions (left). No such acceleration takes place on a neutral electrode (right). (E) Cyclic voltammograms of a spiropyran-functionalised electrode in the presence of PQQ before (i) and after (ii) exposure to visible light, and after thermal treatment (iii). (F) Electrocatalytic current for H2O2 reduction in the presence of (i) SP- and (ii) MCH+-coated electrodes, and after exposure of the latter electrode to visible light (iii). [Adapted with permission from ref. 406 (Copyright 1994 Royal Society of Chemistry) (B), ref. 397 (Copyright 1997 American Chemical Society) (C), ref. 411 (Copyright 1995 Wiley-VCH) (E), ref. 416 (Copyright 2007 American Chemical Society) (F).] | ||
Light-controlled electrode insulation could also be transduced into amplified amperometric responses. Here, the electrodes were reversibly protected from molecules acting as electrocatalysts for a model reaction. An example of this concept is provided by the same SP-functionalised electrodes interfaced with ferrocene-modified GOX.397 Trace (i) in Fig. 21C indicates high electrocatalytic current resulting from the efficient GOX-catalysed oxidation of glucose enabled by an effective communication between the enzyme and the SP-coated electrode. Addition of the anti-DNP antibody blocked the access to the electrode and suppressed the catalytic process (Fig. 21C, trace (ii)). When the AB was introduced to the same system having the electrode coated with the MC isomer, minute changes in the electrocatalytic current were seen (trace (iii) vs. (iv) in Fig. 21C). Moreover, the process showed good reversibility – the current at 0.4 V oscillated between ∼0.5 μA and 5 μA upon treatment with UV and Vis light. Unfortunately, the UV-released AB remained adsorbed on the electrode surface following the SP → MC isomerisation, and had to be washed off in order for high electrocatalytic activity to be regained.408
The second mode of regulating the properties of electrodes using light explores the so-called Frumkin effect409 – that is, the dependence of electrochemical reaction kinetics on the surface charge of the electrodes. With their ability to switch surface charge upon exposure to light, spiropyran-decorated electrodes provide an ideal platform for studying the Frumkin effect. Pyrroloquinoline quinone (PQQ3−), for example, is a widely used redox probe bearing three negative charges at pH 7; as such, it is expected to exhibit attractive electrostatic interactions with MCH+-decorated electrodes, resulting in efficient interfacial electron transfer,410 as manifested, for example, by the dashed and dotted lines in Fig. 21E. Electrochemical reduction of PQQ3− is blocked, however, after exposing the electrode to visible light, as the resulting SP-coated electrode loses its ability to electrostatically interact with the probe (solid line in Fig. 21E).411 The electrochemical activity could be restored by thermal regeneration of the open-ring isomer. Interestingly, the positive surface charge of the MCH+-coated electrodes allowed for the selection of substrates from the solution based on their net charge. For example, electrooxidation was accomplished selectively on the negatively charged (3,4-dihydroxyphenyl)acetate in the presence of the positively charged protonated dopamine.407 When functionalised with the SP isomer, however, the electrode did not discriminate between these two catechols.
Apart from charged redox-active substrates, the light-controlled Frumkin effect has also been demonstrated for charged (i) electron transfer mediators, (ii) cofactors, (iii) enzymes, and (iv) catalytic NPs. First, glucose oxidase (GOX) reconstituted on the electrode surface was used to catalyse oxidation of glucose to gluconic acid in the presence of a positively charged electron relay.412 When the electrode was co-functionalised with MCH+ moieties, the reaction was significantly slowed down on account of electrostatic repulsions between the electron transfer mediator and the like-charged surface. Isomerisation to SP increased the electrocatalytic current by over ten fold.412 Second, PQQ covalently attached to the electrode surface acted as an electrocatalyst for the oxidation of nicotinamide adenine dinucleotide phosphate (NADPH). This reaction required the presence of the Ca2+ “cofactor”, whose complexation by PQQ could be regulated by the state of spiropyran co-adsorbed with PQQ as a mixed SAM.319,410 While the SP isomer did not affect the reaction kinetics, Ca2+ was repelled from the MCH+-decorated monolayer and, as a consequence, the electrocatalysed oxidation was slowed down.
Third, the same principle was used to control the interaction of the positively charged enzyme Cyt c with spiropyran-functionalised electrodes. Under visible light, efficient electron transfer between the SP-coated electrode and Cyt c took place, enabling inter-protein electron transfer to cytochrome c oxidase (COX), which catalysed the oxygen reduction reaction (ORR).413 The ORR was nearly completely shut down, however, upon isomerisation to the MCH+-electrode. The reversible reduction of Cyt c was also coupled to a bioelectrocatalysed reduction: here, the reduced Cyt c was used to activate lactate dehydrogenase towards the reduction of lactate to pyruvate.414 The opposite effect – that is, attraction of a negatively charged protein to an MCH+-electrode – has also been described and is exemplified by UV light-accelerated glucose oxidation catalysed by the negatively charged GOX appended with a ferrocene relay unit.415 Fourth, 20–30 nm platinum NPs have been employed as electrocatalysts for the reduction of H2O2 (Fig. 21F). Stabilised with the citrate anions, these NPs carried substantial negative charge, favouring their interaction with the positively charged MCH+-coated electrodes. Consequently, enhanced electrocatalytic current was observed under UV irradiation (Fig. 21F).416
5.5. Photocontrol of other surface properties
Other creative implications of isomerisation of immobilised spiropyrans are being reported; Thomas et al., for example, investigated contact electrification between steel and spiropyran polymers.417 In these experiments, steel spheres were rolled on spiropyran-functionalised surfaces, and the charges developing on the spheres were monitored. The most interesting result was found for a copolymer of spiropyran methacrylate and 4-fluorostyrene: while the spheres charged positively on poly(SP), the direction of charge transfer on poly(MC) was reversed. In an elegant experiment, the authors UV-irradiated a positively charged (+160 pC) sphere rolling on poly(SP) and observed a gradual decrease of charge down to −160 pC within 100 s of irradiation. Finally, some efforts have been devoted to the concept of aligning liquid crystal molecules using the underlying spiropyran-functionalised surfaces;418–420 however, experimental realisation of the concept has yet to be demonstrated.4216. Conclusions and outlook
Reversible isomerisation of spiropyrans was first reported as early as 1952.422 Six decades later, a resurgence of interest in this molecular switch has been prompted by advances in controlled polymerisation reactions and other synthetic methods, the ever-improving capabilities of high-resolution imaging and spectroscopy techniques, and – most of all – an emerging focus on dynamic materials. This review has discussed a variety of dynamic materials resulting from covalent immobilisation of spiropyran onto the following types of organic and inorganic supports: polymer chains, biomacromolecules, inorganic nanoparticles, and solid surfaces. The resulting materials often have fascinating properties whereby the support affects the properties of the switch, and – more importantly – vice versa: spiropyran isomerisation can control the emission of quantum dots, folding of polypeptides, and the dimensions of polymeric micelles, among many other properties. Potential applications are well within reach and include reusable sensors, high-resolution imaging in biological samples and the detection and mapping of mechanical stresses.What more can we expect in the decade to come? The superb fatigue resistance of properly immobilised spiropyrans127 can enable the realisation of information storage devices and holographic optical materials.423,424 In this context, the strong dependence of the isomerisation kinetics on the microenvironment is particularly attractive, as it can allow for tuning of the information storage time.425 An ambitious challenge that has yet to be addressed is the reversible control of the remarkable physical properties of graphene by means of isomerisation of surface-immobilised spiropyrans. The development of such hybrid materials could take advantage of earlier studies426–428 in which spiropyran was covalently attached to the surfaces of carbon nanotubes. Another direction is the development of materials which are responsive to multiple external stimuli in an orthogonal fashion. Here, the state-of-the-art is represented by a triblock copolymer designed to self-assemble in response to (i) light, (ii) pH, (iii) metal ions, and (iv) thermal treatment (“quadrupole responsiveness”).162 But, arguably, the most room for creativity lies in the derivatisation of biological systems. Here, studies aimed at structural modification of the parent spiropyran to increase its stability in aqueous media are ongoing.429 Of particular importance is the ability to place the spiropyran units at precise locations within the protein structures. In one elegant example, the chaperonin GroEL was equipped with spiropyran groups selectively at the portal regions; this functionalisation enabled self-assembly of individual GroEL units into one-dimensional supramolecular polymers.430 Spiropyran has several important advantages over the structurally simpler azobenzene (whose applications in biological systems are also emerging431,432) – most notably, isomerisation can be accomplished using NIR light and its yield can be near-quantitative in both directions. The ultimate goal would be to use light in order to control various biological processes within living organisms in a spatiotemporal fashion.
Abbreviations
| AB | Antibody |
| AIBN | Azobisisobutyronitrile |
| ATRP | Atom transfer radical polymerisation |
| bpy | Bipyridyl |
| CD | Circular dichroism |
| CHROBA | Chromism-based assay |
| COX | Cytochrome c oxidase |
| CS | Citrate synthase |
| Cyt c | Cytochrome c |
| DLS | Dynamic light scattering |
| DNP | 2,4-Dinitrophenyl |
| FRET | Fluorescence resonance energy transfer |
| GOX | Glucose oxidase |
| HRP | Horseradish peroxidase |
| LCST | Lower critical solution temperature |
| MC | Mero (“open”) form of spiropyran |
| MMA | Methyl methacrylate |
| MscL | Mechanosensitive channel of large conductance |
| MTHF | 2-Methyltetrahydrofuran |
| NAD | Nicotinamide adenine dinucleotide |
| NBD | Nitrobenzoxadiazolyl |
| NIPAM | N-Isopropylacrylamide |
| NIR | Near-infrared |
| NP | Nanoparticle |
| ORR | Oxygen reduction reaction |
| PAMAM | Poly(amidoamine) |
| PDEGMMA | Poly(2-(2-methoxyethoxy)ethyl methacrylate) |
| PDMA | Poly(N,N-dimethylacrylamide) |
| PDMAEMA | Poly(2-(dimethylamino)ethyl methacrylate) |
| PEG | Poly(ethylene glycol) |
| PES | Polyethersulfone |
| PFBT | Poly(fluorenyl-co-benzothiadiazole) |
| PMA | Poly(methyl acrylate) |
| PMMA | Poly(methyl methacrylate) |
| PNIPAM | Poly(N-isopropylacrylamide) |
| PPE | Poly(2,5-dialkylphenylene-1,4-ethynylene) |
| PQQ | Pyrroloquinoline quinone |
| PRCS | Photoresponsive cell culture surface |
| PTFE | Polytetrafluoroethylene |
| PULSAR | Photoactuated unimolecular logical switching-attained reconstruction |
| QD | Quantum dot |
| SAM | Self-assembled monolayer |
| SEM | Scanning electron microscopy |
| SET-LRP | Single-electron transfer living radical polymerisation |
| SFG | Sum frequency generation |
| SP | Spiro (“closed”) form of spiropyran |
| THF | Tetrahydrofuran |
| UNC | Upconverting nanocrystal |
| UV | Ultraviolet light |
| Vis | Visible light |
Acknowledgements
This work was supported by the Israel Science Foundation (grant no. 1463/11). The author acknowledges Dr S. Das and Mr K. Livanov for performing the initial literature search, and Dr E. M. Schuster for critical reading of the manuscript.References
- Stress Response in Microbiology, ed. J. M. Requena, Calster Academic Press, Norfolk, UK, 2012 Search PubMed
.
- M. Stevens and S. Merilaita, Philos. Trans. R. Soc. London, Ser. B, 2009, 364, 423–427 CrossRef PubMed
- J. Aizenberg, A. Tkachenko, S. Weiner, L. Addadi and G. Hendler, Nature, 2001, 412, 819–822 CrossRef CAS PubMed
- M. Ballerini, N. Calbibbo, R. Candeleir, A. Cavagna, E. Cisbani, I. Giardina, V. Lecomte, A. Orlandi, G. Parisi, A. Procaccini, M. Viale and V. Zdravkovic, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1232–1237 CrossRef CAS PubMed
- M. Fialkowski, K. J. M. Bishop, R. Klajn, S. K. Smoukov, C. J. Campbell and B. A. Grzybowski, J. Phys. Chem. B, 2006, 110, 2482–2496 CrossRef CAS PubMed
- A. Grinthal and J. Aizenberg, Chem. Soc. Rev., 2013, 42, 7072–7085, 10.1039/c3cs60045a
- M.-M. Russew and S. Hecht, Adv. Mater., 2010, 22, 3348–3360 CrossRef CAS PubMed
- C. Roche and V. Percec, Isr. J. Chem., 2013, 53, 30–44 CrossRef CAS
- R. Klajn, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2010, 39, 2203–2237 RSC
- S. Das, P. Ranjan, P. S. Maiti, G. Singh, G. Leitus and R. Klajn, Adv. Mater., 2013, 25, 422–426 CrossRef CAS PubMed
- M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed
- R. Klajn, K. J. M. Bishop and B. A. Grzybowski, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10305–10309 CrossRef CAS PubMed
- M. Yamada, M. Kondo, J.-i. Mamiya, Y. Yu, M. Kinoshita, C. J. Barrett and T. Ikeda, Angew. Chem., Int. Ed., 2008, 47, 4986–4988 CrossRef CAS PubMed
- O. Chovnik, R. Balgley, J. R. Goldman and R. Klajn, J. Am. Chem. Soc., 2012, 134, 19564–19567 CrossRef CAS PubMed
- R. Klajn, K. J. M. Bishop, M. Fialkowski, M. Paszewski, C. J. Campbell, T. P. Gray and B. A. Grzybowski, Science, 2007, 316, 261–264 CrossRef CAS PubMed
- R. Klajn, K. P. Browne, S. Soh and B. A. Grzybowski, Small, 2010, 6, 1385–1387 CrossRef CAS PubMed
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC
- J. Henzl, M. Mehlhorn, H. Gawronski, K. H. Rieder and K. Morgenstern, Angew. Chem., Int. Ed., 2006, 45, 603–606 CrossRef CAS PubMed
- W. Fuss, C. Kosmidis, W. E. Schmid and S. A. Trushin, Angew. Chem., Int. Ed., 2004, 43, 4178–4182 CrossRef CAS PubMed
- J. Zhang, J. K. Whitesell and M. A. Fox, Chem. Mater., 2001, 13, 2323–2331 CrossRef CAS
- V. I. Minkin, Chem. Rev., 2004, 104, 2751–2776 CrossRef CAS PubMed
- G. Berkovic, V. Krongauz and V. Weiss, Chem. Rev., 2000, 100, 1741–1753 CrossRef CAS PubMed
- R. Guglielmetti, in Photochromism: Molecules and Systems, ed. H. Durr and H. Bouas-Laurent, Elsevier, Amsterdam, 2003, pp. 314–466 Search PubMed
- R. C. Bertelson, in Organic Photochromic and Thermochromic Compounds, vol. 1, Main Photochromic Families, ed. J. C. Crano and R. J. Gugliemetti, Kluwer Academic Publishers, 2002, pp. 11–83 Search PubMed
- K. Matsuda and M. Irie, J. Photochem. Photobiol., C, 2004, 5, 169–182 CAS
- T. Kudernac, S. J. van der Molen, B. J. van Wees and B. L. Feringa, Chem. Commun., 2006, 3597–3599 RSC
- Y. Chen, C. M. Wang, M. G. Fan, B. L. Yao and N. Menke, Opt. Mater., 2004, 26, 75–77 CrossRef CAS PubMed
- B. Heinz, S. Malkmus, S. Laimgruber, S. Dietrich, C. Schulz, K. Rueck-Braun, M. Braun, W. Zinth and P. Gilch, J. Am. Chem. Soc., 2007, 129, 8577–8584 CrossRef CAS PubMed
- N. Koumura, R. W. J. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152–155 CrossRef CAS PubMed
- P. Zhao, C. F. Fang, C. J. Xia, Y. M. Wang, D. S. Liu and S. J. Xie, Appl. Phys. Lett., 2008, 93, 013113 CrossRef
- M. R. di Nunzio, P. L. Gentili, A. Romani and G. Favaro, ChemPhysChem, 2008, 9, 768–775 CrossRef CAS PubMed
- Y.-C. Jeong, D. G. Park, I. S. Lee, S. I. Yang and K.-H. Ahn, J. Mater. Chem., 2009, 19, 97–103 RSC
- R. Klajn, Pure Appl. Chem., 2010, 82, 2247–2279 CrossRef
- A. Setaro, P. Bluemmel, C. Maity, S. Hecht and S. Reich, Adv. Funct. Mater., 2012, 22, 2425–2431 CrossRef CAS
- A. R. Jang, E. K. Jeon, D. Kang, G. Kim, B. S. Kim, D. J. Kang and H. S. Shin, ACS Nano, 2012, 6, 9207–9213 CrossRef CAS PubMed
- P. Joo, B. J. Kim, E. K. Jeon, J. H. Cho and B.-S. Kim, Chem. Commun., 2012, 48, 10978–10980 RSC
- A. Fontcuberta i Morral and F. Stellacci, Nat. Mater., 2012, 11, 272–273 CrossRef CAS PubMed
- N. W. Tyer and R. S. Becker, J. Am. Chem. Soc., 1970, 92, 1289–1294 CrossRef CAS
- M. Moniruzzaman, C. J. Sabey and G. F. Fernando, Polymer, 2007, 48, 255–263 CrossRef CAS PubMed
- H. Gorner, Phys. Chem. Chem. Phys., 2001, 3, 416–423 RSC
- S. A. Krysanov and M. V. Alfimov, Chem. Phys. Lett., 1982, 91, 77–80 CrossRef CAS
- Z. Y. Tian, W. W. Wu, W. Wan and A. D. Q. Li, J. Am. Chem. Soc., 2011, 133, 16092–16100 CrossRef CAS PubMed
- J. T. C. Wojtyk, A. Wasey, P. M. Kazmaier, S. Hoz and E. Buncel, J. Phys. Chem. A, 2000, 104, 9046–9055 CrossRef CAS
- M. Irie, A. Menju and K. Hayashi, Macromolecules, 1979, 12, 1176–1180 CrossRef CAS
- C. Lenoble and R. S. Becker, J. Phys. Chem., 1986, 90, 62–65 CrossRef CAS
- D. A. Parthenopoulos and P. M. Rentzepis, Science, 1989, 245, 843–845 CAS
- S. O. Konorov and A. M. Zheltikov, Opt. Express, 2003, 11, 2440–2445 CrossRef CAS
- O. Ivashenko, J. T. van Herpt, B. L. Feringa, P. Rudolf and W. R. Browne, Langmuir, 2013, 29, 4290–4297 CrossRef CAS PubMed
- M. C. George, A. Mohraz, M. Piech, N. S. Bell, J. A. Lewis and P. V. Braun, Adv. Mater., 2009, 21, 66–70 CrossRef CAS
- A. S. Dvornikov, J. Malkin and P. M. Rentzepis, J. Phys. Chem., 1994, 98, 6746–6752 CrossRef CAS
- J. Whelan, D. Abdallah, J. Wojtyk and E. Buncel, J. Mater. Chem., 2010, 20, 5727–5735 RSC
- S. Swansburg, E. Buncel and R. P. Lemieux, J. Am. Chem. Soc., 2000, 122, 6594–6600 CrossRef CAS
- X. Q. Song, J. W. Zhou, Y. T. Li and Y. W. Tang, J. Photochem. Photobiol., A, 1995, 92, 99–103 CrossRef CAS
- R. Rosario, D. Gust, M. Hayes, J. Springer and A. A. Garcia, Langmuir, 2003, 19, 8801–8806 CrossRef CAS
- S. R. Keum, S. J. Roh, S. M. Ahn, S. S. Lim, S. H. Kim and K. Koh, Dyes Pigm., 2007, 74, 343–347 CrossRef CAS PubMed
- K. Patel, A. Castillo-Muzquiz and M. C. Biewer, Tetrahedron Lett., 2002, 43, 5933–5935 CrossRef CAS
- S. R. Keum, M. S. Hur, P. M. Kazmaier and E. Buncel, Can. J. Chem., 1991, 69, 1940–1947 CrossRef CAS
- J. W. Zhou, Y. T. Li, Y. W. Tang, F. Q. Zhao, X. Q. Song and E. C. Li, J. Photochem. Photobiol., A, 1995, 90, 117–123 CrossRef CAS
- L. Florea, A. Hennart, D. Diamond and F. Benito-Lopez, Sens. Actuators, B, 2012, 175, 92–99 CrossRef CAS PubMed
- L. Florea, A. McKeon, D. Diamond and F. Benito-Lopez, Langmuir, 2013, 29, 2790–2797 CrossRef CAS PubMed
- Q. Shen, Y. Cao, S. Liu, M. L. Steigerwald and X. Guo, J. Phys. Chem. C, 2009, 113, 10807–10812 CAS
- M. Levitus, G. Glasser, D. Neher and P. F. Aramendia, Chem. Phys. Lett., 1997, 277, 118–124 CrossRef CAS
- M. Bletz, U. Pfeifer-Fukumura, U. Kolb and W. Baumann, J. Phys. Chem. A, 2002, 106, 2232–2236 CrossRef CAS
- H. Gruler, R. Vilanove and F. Rondelez, Phys. Rev. Lett., 1980, 44, 590–592 CrossRef CAS
- R. Vilanove, H. Hervet, H. Gruler and F. Rondelez, Macromolecules, 1983, 16, 825–831 CrossRef CAS
- I. Panaiotov, S. Taneva, A. Bois and F. Rondelez, Macromolecules, 1991, 24, 4250–4254 CrossRef CAS
- J. Chen, F. Zeng and S. Z. Wu, ChemPhysChem, 2010, 11, 1036–1043 CrossRef CAS PubMed
- M. Q. Zhu, L. Y. Zhu, J. J. Han, W. W. Wu, J. K. Hurst and A. D. Q. Li, J. Am. Chem. Soc., 2006, 128, 4303–4309 CrossRef CAS PubMed
- Y. Wu, X. Z. Qu, L. Y. Huang, D. Qiu, C. L. Zhang, Z. P. Liu, Z. Z. Yang and L. Feng, J. Colloid Interface Sci., 2010, 343, 155–161 CrossRef CAS PubMed
- A. A. Garcia, S. Cherian, J. Park, D. Gust, F. Jahnke and R. Rosario, J. Phys. Chem. A, 2000, 104, 6103–6107 CrossRef CAS
- D. D. Perrin, B. Dempsey and E. P. Serjeant, Prediction for Organic Acids and Bases, Chapman and Hall, London, 1981 Search PubMed
- R. A. Hall, P. J. Thistlethwaite and F. Grieser, Langmuir, 1994, 10, 3743–3748 CrossRef CAS
- K. Sumaru, M. Kameda, T. Kanamori and T. Shinbo, Macromolecules, 2004, 37, 4949–4955 CrossRef CAS
- C. D. Bain and G. M. Whitesides, Langmuir, 1989, 5, 1370–1378 CrossRef CAS
- F. M. Raymo and S. Giordani, J. Am. Chem. Soc., 2001, 123, 4651–4652 CrossRef CAS
- J. T. C. Wojtyk, A. Wasey, N. N. Xiao, P. M. Kazmaier, S. Hoz, C. Yu, R. P. Lemieux and E. Buncel, J. Phys. Chem. A, 2007, 111, 2511–2516 CrossRef CAS PubMed
- S. R. Keum, K. B. Lee, P. M. Kazmaier and E. Buncel, Tetrahedron Lett., 1994, 35, 1015–1018 CrossRef CAS
- R. Rosario, D. Gust, M. Hayes, F. Jahnke, J. Springer and A. A. Garcia, Langmuir, 2002, 18, 8062–8069 CrossRef CAS
- K. Wagner, R. Byrne, M. Zanoni, S. Gambhir, L. Dennany, R. Breukers, M. Higgins, P. Wagner, D. Diamond, G. G. Wallace and D. L. Officer, J. Am. Chem. Soc., 2011, 133, 5453–5462 CrossRef CAS PubMed
- D. S. Tipikin, Russ. J. Phys. Chem., 2001, 75, 1720–1722 Search PubMed
- Y. Shiraishi, M. Itoh and T. Hirai, Phys. Chem. Chem. Phys., 2010, 12, 13737–13745 RSC
- J. Ratner, N. Kahana, A. Warshawsky and V. Krongauz, Ind. Eng. Chem. Res., 1996, 35, 1307–1315 CrossRef CAS
- K. Kinashi, S. Nakamura, Y. Ono, K. Ishida and Y. Ueda, J. Photochem. Photobiol., A, 2010, 213, 136–140 CrossRef CAS PubMed
- K. Kinashi, S. Nakamura, M. Imamura, K. Ishida and Y. Ueda, J. Phys. Org. Chem., 2012, 25, 462–466 CrossRef CAS
- P. Calero, E. Aznar, J. M. Lloris, M. D. Marcos, R. Martinez-Manez, J. V. Ros-Lis, J. Soto and F. Sancenon, Chem. Commun., 2008, 1668–1670 RSC
- J. Allouche, A. Le Beulze, J.-C. Dupin, J.-B. Ledeuil, S. Blanc and D. Gonbeau, J. Mater. Chem., 2010, 20, 9370–9378 RSC
- J. Sunamoto, K. Iwamoto, M. Akutagawa, M. Nagase and H. Kondo, J. Am. Chem. Soc., 1982, 104, 4904–4907 CrossRef CAS
- I. Shimizu, H. Kokado and E. Inoue, Bull. Chem. Soc. Jpn., 1969, 42, 1730–1734 CrossRef CAS
- C. P. McCoy, L. Donnelly, D. S. Jones and S. P. Gorman, Tetrahedron Lett., 2007, 48, 657–661 CrossRef CAS PubMed
- S. Samanta and J. Locklin, Langmuir, 2008, 24, 9558–9565 CrossRef CAS PubMed
- V. A. Krongauz and E. S. Goldburt, Nature, 1978, 271, 43–45 CrossRef CAS
- T. Seki, K. Ichimura and E. Ando, Langmuir, 1988, 4, 1068–1069 CrossRef CAS
- H. Tachibana, Y. Yamanaka, H. Sakai, M. Abe and M. Matsumoto, J. Lumin., 2000, 87–89, 800–802 CrossRef CAS
- P. Uznanski, Synth. Met., 2000, 109, 281–285 CrossRef CAS
- Y. Unuma and A. Miyata, Thin Solid Films, 1989, 179, 497–502 CrossRef CAS
- E. Ando, J. Miyazaki, K. Morimoto, H. Nakahara and K. Fukuda, Thin Solid Films, 1985, 133, 21–28 CrossRef CAS
- I. Cabrera, F. Shvartsman, O. Veinberg and V. Krongauz, Science, 1984, 226, 341–343 CAS
- I. Cabrera and V. Krongauz, Macromolecules, 1987, 20, 2713–2717 CrossRef CAS
- E. Goldburt and V. Krongauz, Macromolecules, 1986, 19, 246–247 CrossRef CAS
- E. Goldburt, F. Shvartsman, S. Fishman and V. Krongauz, Macromolecules, 1984, 17, 1225–1230 CrossRef CAS
- H. Tomioka and T. Itoh, J. Chem. Soc., Chem. Commun., 1991, 532–533 RSC
- Y. Wu, C. L. Zhang, X. Z. Qu, Z. P. Liu and Z. Z. Yang, Langmuir, 2010, 26, 9442–9448 CrossRef CAS PubMed
- M. Piech, M. C. George, N. S. Bell and P. V. Braun, Langmuir, 2006, 22, 1379–1382 CrossRef CAS PubMed
- T. Seki and K. Ichimura, J. Phys. Chem., 1990, 94, 3769–3775 CrossRef CAS
- H. Eckhardt, A. Bose and V. A. Krongauz, Polymer, 1987, 28, 1959–1964 CrossRef CAS
- H. R. Allcock and C. Kim, Macromolecules, 1991, 24, 2846–2851 CrossRef CAS
- E. Goldburt, F. Shvartsman and V. Krongauz, Macromolecules, 1984, 17, 1876–1878 CrossRef CAS
- M. Ueda, K. Kudo and K. Ichimura, J. Mater. Chem., 1995, 5, 1007–1011 RSC
- S. Gorelik, S. Hongyan, M. J. Lear and J. Hobley, Photochem. Photobiol. Sci., 2010, 9, 141–151 CAS
- M. Plaschke, R. Czolk and H. J. Ache, Anal. Chim. Acta, 1995, 304, 107–113 CrossRef CAS
- I. Oehme, S. Prattes, O. S. Wolfbeis and G. J. Mohr, Talanta, 1998, 47, 595–604 CrossRef CAS
- M. H. Lee, X. D. Li and E. Kim, Mol. Cryst. Liq. Cryst., 2000, 349, 51–54 CrossRef CAS
- X. D. Li, E. Kim and M. H. Lee, Polymer, 2005, 29, 25–31 CAS
- K. Arai, Y. Shitara and T. Ohyama, J. Mater. Chem., 1996, 6, 11–14 RSC
- M. Q. Zhu, G. F. Zhang, C. Li, M. P. Aldred, E. Chang, R. A. Drezek and A. D. Q. Li, J. Am. Chem. Soc., 2011, 133, 365–372 CrossRef CAS PubMed
- Y.-H. Chan, M. E. Gallina, X. Zhang, I. C. Wu, Y. Jin, W. Sun and D. T. Chiu, Anal. Chem., 2012, 84, 9431–9438 CAS
- M. Irie, T. Iwayanagi and Y. Taniguchi, Macromolecules, 1985, 18, 2418–2422 CrossRef CAS
- G. Baillet, G. Giusti and R. Guglielmetti, J. Photochem. Photobiol., A, 1993, 70, 157–161 CrossRef CAS
- G. Baillet, M. Campredon, R. Guglielmetti, G. Giusti and C. Aubert, J. Photochem. Photobiol., A, 1994, 83, 147–151 CrossRef CAS
- X. L. Li, J. L. Li, Y. M. Wang, T. Matsuura and J. B. Meng, J. Photochem. Photobiol., A, 2004, 161, 201–213 CrossRef CAS
- R. Matsushima, M. Nishiyama and M. Doi, J. Photochem. Photobiol., A, 2001, 139, 63–69 CrossRef CAS
- M. Sakuragi, K. Aoki, T. Tamaki and K. Ichimura, Bull. Chem. Soc. Jpn., 1990, 63, 74–79 CrossRef CAS
- J. Whelan, J. T. C. Wojtyk and E. Buncel, Chem. Mater., 2008, 20, 3797–3799 CrossRef CAS
- A. Radu, R. Byrne, N. Alhashimy, M. Fusaro, S. Scarmagnani and D. Diamond, J. Photochem. Photobiol., A, 2009, 206, 109–115 CrossRef CAS PubMed
- N. S. Bell and M. Piech, Langmuir, 2006, 22, 1420–1427 CrossRef CAS PubMed
- S. Scarmagnani, Z. Walsh, C. Slater, N. Alhashimy, B. Paull, M. Macka and D. Diamond, J. Mater. Chem., 2008, 18, 5063–5071 RSC
- A. Radu, S. Scarmagnani, R. Byrne, C. Slater, K. T. Lau and D. Diamond, J. Phys. D: Appl. Phys., 2007, 40, 7238–7244 CrossRef CAS
- C. Q. Huang, Y. Wang, C. Y. Hong and C. Y. Pan, Macromol. Rapid Commun., 2011, 32, 1174–1179 CrossRef CAS PubMed
- D. S. Achilleos and M. Vamvakaki, Macromolecules, 2010, 43, 7073–7081 CrossRef CAS
- R. Adelmann, P. Mela, M. O. Gallyamov, H. Keul and M. Moller, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1274–1283 CrossRef CAS
- M. Bertoldo, S. Nazzi, G. Zampano and F. Ciardelli, Carbohydr. Polym., 2011, 85, 401–407 CrossRef CAS PubMed
- J. Labsky, I. Koropecky, S. Nespurek and J. Kalal, Eur. Polym. J., 1981, 17, 309–313 CrossRef CAS
- J. Labsky, J. Kalal and F. Mikes, Polym. Photochem., 1982, 2, 289–295 CrossRef CAS
- J. Labsky, F. Mikes and J. Kalal, Polym. Bull., 1980, 2, 785–789 CrossRef CAS
- R. J. Byrne, S. E. Stitzel and D. Diamond, J. Mater. Chem., 2006, 16, 1332–1337 RSC
- C. Bronner, G. Schulze, K. J. Franke, J. I. Pascual and P. Tegeder, J. Phys.: Condens. Matter, 2011, 23, 484005 CrossRef PubMed
- Y. Imai, K. Adachi, K. Naka and Y. Chujo, Polym. Bull., 2000, 44, 9–15 CrossRef CAS
- M. Tomasulo, I. Yildiz and F. M. Raymo, Inorg. Chim. Acta, 2007, 360, 938–944 CrossRef CAS PubMed
- Y. Kalisky and D. J. Williams, Macromolecules, 1984, 17, 292–296 CrossRef CAS
- D. J. Chung, Y. Ito and Y. Imanishi, J. Appl. Polym. Sci., 1994, 51, 2027–2033 CrossRef
- Y. Bardavid, I. Goykhman, D. Nozaki, G. Cuniberti and S. Yitzchaik, J. Phys. Chem. C, 2011, 115, 3123–3128 CAS
- L. A. Connal, G. V. Franks and G. G. Qiao, Langmuir, 2010, 26, 10397–10400 CrossRef CAS PubMed
- S. Z. Wu, J. R. Lu, F. Zeng, Y. N. Chen and Z. Tong, Macromolecules, 2007, 40, 5060–5066 CrossRef CAS
- A. Warshawsky, N. Kahana, F. Buchholtz, A. Zelichonok, J. Ratner and V. Krongauz, Ind. Eng. Chem. Res., 1995, 34, 2825–2832 CrossRef CAS
- Y. J. Oh, J. A. Nam, A. Al-Nahain, S. Lee, I. In and S. Y. Park, Macromol. Rapid Commun., 2012, 33, 1958–1963 CrossRef CAS PubMed
- L. Hauser, A. C. Knall, M. Roth, G. Trimmel, M. Edler, T. Griesser and W. Kern, Monatsh. Chem., 2012, 143, 1551–1558 CrossRef CAS
- S. R. Keum, S. M. Ahn, S. J. Roh and S. Y. Ma, Dyes Pigm., 2010, 86, 74–80 CrossRef CAS PubMed
- B. B. Mistry, R. G. Patel and V. S. Patel, J. Appl. Polym. Sci., 1997, 64, 841–848 CrossRef CAS
- J. Verborgt and G. Smets, J. Polym. Sci., Polym. Chem. Ed., 1974, 12, 2511–2523 CrossRef CAS
- J. H. Su, J. Chen, F. Zeng, Q. M. Chen, S. Z. Wu and Z. Tong, Polym. Bull., 2008, 61, 425–434 CrossRef CAS
- M. Moniruzzaman, G. F. Fernando and A. J. Bellamy, Eur. Polym. J., 2006, 42, 1455–1466 CrossRef CAS PubMed
- Y. K. Choi, E. Y. Kim and S. R. Keum, Tetrahedron Lett., 1998, 39, 8861–8864 CrossRef CAS
- H. Schenderlein, A. Voss, R. W. Stark and M. Biesalski, Langmuir, 2013, 29, 4525–4534 CrossRef CAS PubMed
- N. Negishi, K. Tsunemitsu, K. Ishihara, I. Shinohara, T. Okano, K. Kataoka, T. Akaike and Y. Sakurai, Kobunshi Ronbunshu, 1980, 37, 287–291 CrossRef CAS
- R. Nakao, N. Ueda, Y. Abe, T. Horii and E. Inoue, Polym. Adv. Technol., 1994, 5, 240–241 CrossRef CAS
- R. Nakao, N. Ueda, Y. Abe, T. Horii and E. Inoue, Polym. Adv. Technol., 1995, 6, 243–247 CrossRef CAS
- R. Nakao, N. Ueda, Y. Abe, T. Horii and H. Inoue, Polym. Adv. Technol., 1996, 7, 863–866 CrossRef CAS
- J. H. Kim, S. Y. Ban, Q. Zhang, G. W. Kim, M. J. Cho and D. H. Choi, Mol. Cryst. Liq. Cryst., 2006, 445, 307–314 CAS
- J. Chen, F. Zeng, S. Z. Wu, J. Q. Zhao, Q. M. Chen and Z. Tong, Chem. Commun., 2008, 5580–5582 RSC
- H. I. Lee, W. Wu, J. K. Oh, L. Mueller, G. Sherwood, L. Peteanu, T. Kowalewski and K. Matyjaszewski, Angew. Chem., Int. Ed., 2007, 46, 2453–2457 CrossRef CAS PubMed
- C. H. Li, Y. X. Zhang, J. M. Hu, J. J. Cheng and S. Y. Liu, Angew. Chem., Int. Ed., 2010, 49, 5120–5124 CrossRef CAS PubMed
- S. Guragain, B. P. Bastakoti, M. Ito, S.-i. Yusa and K. Nakashima, Soft Matter, 2012, 8, 9628–9634 RSC
- Y. S. Park, Y. Ito and Y. Imanishi, Macromolecules, 1998, 31, 2606–2610 CrossRef CAS
- T. Wu, G. Zou, J. M. Hu and S. Y. Liu, Chem. Mater., 2009, 21, 3788–3798 CrossRef CAS
- G. Smets and F. De Blauwe, Pure Appl. Chem., 1974, 39, 225–238 CrossRef CAS
- G. O'Bryan, B. M. Wong and J. R. McElhanon, ACS Appl. Mater. Interfaces, 2010, 2, 1594–1600 CAS
- C. K. Lee, D. A. Davis, S. R. White, J. S. Moore, N. R. Sottos and P. V. Braun, J. Am. Chem. Soc., 2010, 132, 16107–16111 CrossRef CAS PubMed
- J. Kadokawa, Y. Tanaka, Y. Yamashita and K. Yamamoto, Eur. Polym. J., 2012, 48, 549–559 CrossRef CAS PubMed
- S. L. Potisek, D. A. Davis, N. R. Sottos, S. R. White and J. S. Moore, J. Am. Chem. Soc., 2007, 129, 13808–13809 CrossRef CAS PubMed
- B. A. Beiermann, D. A. Davis, S. L. B. Kramer, J. S. Moore, N. R. Sottos and S. R. White, J. Mater. Chem., 2011, 21, 8443–8447 RSC
- T. H. Mourey, I. Noh and H. Yu, J. Chromatogr., 1984, 303, 361–369 CrossRef CAS
- K. Fukushima, A. J. Vandenbos and T. Fujiwara, Chem. Mater., 2007, 19, 644–646 CrossRef CAS
- Y. Nakahara, J. Nakamura, N. Shirotani and K. Kimura, Chem. Lett., 2012, 1142–1144 CrossRef CAS
- W. L. Zhao and E. M. Carreira, Org. Lett., 2005, 7, 1609–1612 CrossRef CAS PubMed
- L. Y. Zhu, M. Q. Zhu, J. K. Hurst and A. D. Q. Li, J. Am. Chem. Soc., 2005, 127, 8968–8970 CrossRef CAS PubMed
- S. Manley, J. M. Gillette and J. Lippincott-Schwartz, in Methods in Enzymology, ed. N. G. Walter, Elsevier, San Diego, 2010, vol. 475, pp. 109–120 Search PubMed
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642–1645 CrossRef CAS PubMed
- M. J. Rust, M. Bates and X. W. Zhuang, Nat. Methods, 2006, 3, 793–795 CrossRef CAS PubMed
- Z. Y. Tian and A. D. Q. Li, Acc. Chem. Res., 2013, 46, 269–279 CrossRef CAS PubMed
- D. H. Hu, Z. Y. Tian, W. W. Wu, W. Wan and A. D. Q. Li, J. Am. Chem. Soc., 2008, 130, 15279–15281 CrossRef CAS PubMed
- D. H. Hu, Z. Y. Tian, W. W. Wu, W. Wan and A. D. Q. Li, Microsc. Microanal., 2009, 15, 840–841 CrossRef
- Z. Y. Tian, A. D. Q. Li and D. H. Hu, Chem. Commun., 2011, 47, 1258–1260 RSC
- Z. Y. Tian, W. W. Wu, W. Wan and A. D. Q. Li, J. Am. Chem. Soc., 2009, 131, 4245–4252 CrossRef CAS PubMed
- Y. Lv, H. Liu, B. Zhao, Z. Tian and A. D. Q. Li, Isr. J. Chem., 2013, 53, 294–302 CrossRef CAS
- J. Chen, P. S. Zhang, G. Fang, C. Weng, J. Hu, P. G. Yi, X. Y. Yu and X. F. Li, Polym. Chem., 2012, 3, 685–693 RSC
- L. Y. Zhu, W. W. Wu, M. Q. Zhu, J. J. Han, J. K. Hurst and A. D. Q. Li, J. Am. Chem. Soc., 2007, 129, 3524–3526 CrossRef CAS PubMed
- J. Chen, F. Zeng, S. Z. Wu, J. Su and Z. Tong, Small, 2009, 5, 970–978 CrossRef CAS PubMed
- J. Chen, F. Zeng, S. Z. Wu, Q. M. Chen and Z. Tong, Chem.–Eur. J., 2008, 14, 4851–4860 CrossRef CAS PubMed
- I. S. Park, Y. S. Jung, K. J. Lee and J. M. Kim, Chem. Commun., 2010, 46, 2859–2861 RSC
- J. Chen, D. Wang, A. Turshatov, R. Munoz-Espi, U. Ziener, K. Koynov and K. Landfester, Polym. Chem., 2013, 4, 773–781 RSC
- M. Tomasulo, E. Deniz, R. J. Alvarado and F. M. Raymo, J. Phys. Chem. C, 2008, 112, 8038–8045 CAS
- J. Chen, P. S. Zhang, X. Y. Yu, X. F. Li, H. W. Tao and P. G. Yi, J. Macromol. Sci., Part A: Pure Appl. Chem., 2011, 48, 637–643 CrossRef CAS
- J. A. Chen, P. S. Zhang, G. Fang, P. G. Yi, X. Y. Yu, X. F. Li, F. Zeng and S. Z. Wu, J. Phys. Chem. B, 2011, 115, 3354–3362 CrossRef CAS PubMed
- J. Chen, P. S. Zhang, G. Fang, P. G. Yi, F. Zeng and S. Z. Wu, J. Phys. Chem. B, 2012, 116, 4354–4362 CrossRef CAS PubMed
- S. Mao, R. K. P. Benninger, Y. Yan, C. Petchprayoon, D. Jackson, C. J. Easley, D. W. Piston and G. Marriott, Biophys. J., 2008, 94, 4515–4524 CrossRef CAS PubMed
- Y. Hirokawa and T. Tanaka, J. Chem. Phys., 1984, 81, 6379–6380 CrossRef
- R. Yoshida, K. Uchida, Y. Kaneko, K. Sakai, A. Kikuchi, Y. Sakurai and T. Okano, Nature, 1995, 374, 240–242 CrossRef CAS
- I. Dimitrov, B. Trzebicka, A. H. E. Muller, A. Dworak and C. B. Tsvetanov, Prog. Polym. Sci., 2007, 32, 1275–1343 CrossRef CAS PubMed
- M. Irie, A. Menju, K. Hayashi and G. Smets, J. Polym. Sci., Polym. Chem. Ed., 1979, 17, 29–31 CrossRef CAS
- A. Menju, K. Hayashi and M. Irie, Macromolecules, 1981, 14, 755–758 CrossRef CAS
- M. Irie, K. Hayashi and A. Menju, Polym. Photochem., 1981, 1, 233–242 CrossRef CAS
- J. Edahiro, K. Sumaru, T. Takagi, T. Shinbo and T. Kanamori, Langmuir, 2006, 22, 5224–5226 CrossRef CAS PubMed
- V. A. Krongauz and A. Golinelli, Polym. Bull., 1982, 6, 259–262 CAS
- J. H. Kim, S. Y. Ban, S. Kaihua and D. H. Choi, Dyes Pigm., 2003, 58, 105–112 CrossRef CAS
- D. H. Choi, S. Y. Ban and J. H. Kim, Bull. Korean Chem. Soc., 2003, 24, 441–445 CrossRef CAS
- C. Konak, R. C. Rathi, P. Kopeckova and J. Kopecek, Macromolecules, 1997, 30, 5553–5556 CrossRef CAS
- C. Konak, R. C. Rathi, P. Kopeckova and J. Kopecek, Polym. Adv. Technol., 1998, 9, 641–648 CrossRef CAS
- L. Ma, J. Li, D. Han, H. Geng, G. Chen and Q. Li, Macromol. Chem. Phys., 2013, 214, 716–725 CrossRef CAS
- V. A. Krongauz and E. S. Goldburt, Macromolecules, 1981, 14, 1382–1386 CrossRef CAS
- T. Wismontski-Knittel and V. Krongauz, Macromolecules, 1985, 18, 2124–2126 CrossRef CAS
- B. Yan, J. He, P. Ayotte and Y. Zhao, Macromol. Rapid Commun., 2011, 32, 972–976 CrossRef CAS PubMed
- D. S. Achilleos, T. A. Hatton and M. Vamvakaki, J. Am. Chem. Soc., 2012, 134, 5726–5729 CrossRef CAS PubMed
- Y. Nakahara, Y. Okazaki and K. Kimura, Soft Matter, 2012, 8, 3192–3199 RSC
- S. Kado, K. Uehara, Y. Nakahara and K. Kimura, Bull. Chem. Soc. Jpn., 2011, 84, 422–426 CrossRef CAS
- V. K. Kotharangannagari, A. Sanchez-Ferrer, J. Ruokolainen and R. Mezzenga, Macromolecules, 2011, 44, 4569–4573 CrossRef CAS
- S. Menon, R. M. Ongungal and S. Das, Polym. Chem., 2013, 4, 623–628 RSC
- Q. A. Jin, G. Y. Liu and J. A. Ji, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2855–2861 CrossRef CAS
- Y. Arisaka, A. Tamura, K. Uchida and H. Yajima, Chem. Lett., 2010, 58–59 CrossRef CAS
- H. Feil, Y. H. Bae, J. Feijen and S. W. Kim, Macromolecules, 1993, 26, 2496–2500 CrossRef CAS
- A. E. Ivanov, N. L. Eremeev, P. O. Wahlund, I. Y. Galaev and B. Mattiasson, Polymer, 2002, 43, 3819–3823 CrossRef CAS
- K. Sumaru, M. Kameda, T. Kanamori and T. Shinbo, Macromolecules, 2004, 37, 7854–7856 CrossRef CAS
- J. I. Edahiro, K. Sumaru, T. Takagi, T. Shinbo, T. Kanamori and M. Sudoh, Eur. Polym. J., 2008, 44, 300–307 CrossRef CAS PubMed
- M. Kameda, K. Sumaru, T. Kanamori and T. Shinbo, Langmuir, 2004, 20, 9315–9319 CrossRef CAS PubMed
- Y. Shiraishi, R. Miyamoto and T. Hirai, Org. Lett., 2009, 11, 1571–1574 CrossRef CAS PubMed
- R. Byrne, C. Ventura, F. B. Lopez, A. Walther, A. Heise and D. Diamond, Biosens. Bioelectron., 2010, 26, 1392–1398 CrossRef CAS PubMed
- A. Garcia, M. Marquez, T. Cai, R. Rosario, Z. B. Hu, D. Gust, M. Hayes, S. A. Vail and C. D. Park, Langmuir, 2007, 23, 224–229 CrossRef CAS PubMed
- A. Szilagyi, K. Sumaru, S. Sugiura, T. Takagi, T. Shinbo, M. Zrinyi and T. Kanamori, Chem. Mater., 2007, 19, 2730–2732 CrossRef CAS
- Y. Zhao and L. Tremblay, J. Polym. Sci.,
Part A: Polym. Chem., 2010, 48, 4055–4066 CrossRef CAS
- X. M. He, M. Aizenberg, O. Kuksenok, L. D. Zarzar, A. Shastri, A. C. Balazs and J. Aizenberg, Nature, 2012, 487, 214–218 CrossRef CAS PubMed
- T. Satoh, K. Sumaru, T. Takagi and T. Kanamori, Soft Matter, 2011, 7, 8030–8034 RSC
- S. Sugiura, K. Sumaru, K. Ohi, K. Hiroki, T. Takagi and T. Kanamori, Sens. Actuators, A, 2007, 140, 176–184 CrossRef CAS PubMed
- S. Sugiura, A. Szilagyi, K. Sumaru, K. Hattori, T. Takagi, G. Filipcsei, M. Zrinyi and T. Kanamori, Lab Chip, 2009, 9, 196–198 RSC
- K. Sumaru, K. Ohi, T. Takagi, T. Kanamori and T. Shinbo, Langmuir, 2006, 22, 4353–4356 CrossRef CAS PubMed
- F. Benito-Lopez, R. Byrne, A. M. Raduta, N. E. Vrana, G. McGuinness and D. Diamond, Lab Chip, 2010, 10, 195–201 RSC
- T. Satoh, K. Sumaru, T. Takagi, K. Takai and T. Kanamori, Phys. Chem. Chem. Phys., 2011, 13, 7322–7329 RSC
- K. Kimura, H. Sakamoto and T. Nakamura, J. Nanosci. Nanotechnol., 2006, 6, 1741–1749 CrossRef CAS PubMed
- S. Kurihara, M. Higuchi, T. Ogata and T. Nonaka, J. Membr. Sci., 1994, 93, 69–78 CrossRef CAS
- S. Marx-Tibbon and I. Willner, Chem. Commun., 1994, 1261–1262 RSC
- Z. Walsh, P. A. Levkin, S. Abele, S. Scarmagnani, D. Heger, P. Klan, D. Diamond, B. Paull, F. Svec and M. Macka, J. Chromatogr., A, 2011, 1218, 2954–2962 CrossRef CAS PubMed
- Z. Walsh, S. Scarmagnani, F. Benito-Lopez, S. Abele, F. Q. Nie, C. Slater, R. Byrne, D. Diamond, B. Paull and M. Macka, Sens. Actuators, B, 2010, 148, 569–576 CrossRef CAS PubMed
- I. Willner, S. Rubin, R. Shatzmiller and T. Zor, J. Am. Chem. Soc., 1993, 115, 8690–8694 CrossRef CAS
- A. Nayak, H. Liu and G. Belfort, Angew. Chem., Int. Ed., 2006, 45, 4094–4098 CrossRef CAS PubMed
- Y. Ito and Y. S. Park, Polym. Adv. Technol., 2000, 11, 136–144 CrossRef CAS
- G. Smets, Pure Appl. Chem., 1975, 42, 509–526 CrossRef CAS
- G. Smets, J. Braeken and M. Irie, Pure Appl. Chem., 1978, 50, 845–856 CrossRef CAS
- D. A. Davis, A. Hamilton, J. L. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martinez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS PubMed
- B. A. Beiermann, S. L. B. Kramer, J. S. Moore, S. R. White and N. R. Sottos, ACS Macro Lett., 2012, 1, 163–166 CrossRef CAS
- C. M. Kingsbury, P. A. May, D. A. Davis, S. R. White, J. S. Moore and N. R. Sottos, J. Mater. Chem., 2011, 21, 8381–8388 RSC
- C. K. Lee, B. A. Beiermann, M. N. Silberstein, J. Wang, J. S. Moore, N. R. Sottos and P. V. Braun, Macromolecules, 2013, 46, 3746–3752 CrossRef CAS
- J. D. Winkler, K. Deshayes and B. Shao, J. Am. Chem. Soc., 1989, 111, 769–770 CrossRef CAS
- J. W. Zhou, Y. T. Li and X. Q. Song, J. Photochem. Photobiol., A, 1995, 87, 37–42 CrossRef CAS
- T. Suzuki, T. Kato and H. Shinozaki, Chem. Commun., 2004, 2036–2037 RSC
- J. T. C. Wojtyk, P. M. Kazmaier and E. Buncel, Chem. Commun., 1998, 1703–1704 RSC
- J. T. C. Wojtyk, P. M. Kazmaier and E. Buncel, Chem. Mater., 2001, 13, 2547–2551 CrossRef CAS
- T. Suzuki, Y. Kawata, S. Kahata and T. Kato, Chem. Commun., 2003, 2004–2005 RSC
- T. Suzuki, Y. Hirahara, K. Bunya and H. Shinozaki, J. Mater. Chem., 2010, 20, 2773–2779 RSC
- C. Ventura, R. Byrne, F. Audouin and A. Heise, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3455–3463 CrossRef CAS
- K. H. Fries, J. D. Driskell, S. Samanta and J. Locklin, Anal. Chem., 2010, 82, 3306–3314 CrossRef CAS PubMed
- K. H. Fries, J. D. Driskell, G. R. Sheppard and J. Locklin, Langmuir, 2011, 27, 12253–12260 CrossRef CAS PubMed
- P.-J. Wu, J.-L. Chen, C.-P. Chen and Y.-H. Chan, Chem. Commun., 2013, 49, 898–900 RSC
- N. Kobayashi, S. Sato, K. Takazawa, K. Ikeda and R. Hirohashi, Electrochim. Acta, 1995, 40, 2309–2311 CrossRef CAS
- K. Kimura, M. Sumida and M. Yokoyama, Chem. Commun., 1997, 1417–1418 RSC
- K. Kimura, M. Nakamura, H. Sakamoto, R. M. Uda, M. Sumida and M. Yokoyama, Bull. Chem. Soc. Jpn., 2003, 76, 209–215 CrossRef CAS
- K. Kimura, H. Sakamoto and R. M. Uda, Macromolecules, 2004, 37, 1871–1876 CrossRef CAS
- M. Kojima, T. Nakanishi, Y. Hirai, H. Yabu and M. Shimomura, Chem. Commun., 2010, 41, 3970–3972 RSC
- T. Nakanishi, Y. Hirai, M. Kojima, H. Yabu and M. Shimomura, J. Mater. Chem., 2010, 20, 6741–6745 RSC
- N. Negishi, T. Iida, K. Ishihara and I. Shinohara, Makromol. Chem., Rapid Commun., 1981, 2, 617–620 CrossRef CAS
- Y. Shiraishi, S. Sumiya, K. Manabe and T. Hirai, ACS Appl. Mater. Interfaces, 2011, 3, 4649–4656 CAS
- L. Angiolini, T. Benelli, L. Giorgini and F. M. Raymo, Polymer, 2009, 50, 5638–5646 CrossRef CAS PubMed
- L. Angiolini, T. Benelli, L. Giorgini and F. M. Raymo, Macromol. Chem. Phys., 2008, 209, 2049–2060 CrossRef CAS
- L. Angiolini, T. Benelli, E. Bicciocchi, L. Giorgini and F. M. Raymo, React. Funct. Polym., 2012, 72, 469–477 CrossRef CAS PubMed
- I. Cabrera, V. Krongauz and H. Ringsdorf, Mol. Cryst. Liq. Cryst., 1988, 155, 221–230 CrossRef CAS
- S. Yitzchaik, G. Berkovic and V. Krongauz, Macromolecules, 1990, 23, 3539–3541 CrossRef CAS
- S. Yitzchaik, I. Cabrera, F. Buchholtz and V. Krongauz, Macromolecules, 1990, 23, 707–713 CrossRef CAS
- E. J. C. Kellar, G. Williams, V. Krongauz and S. Yitzchaik, J. Mater. Chem., 1991, 1, 331–337 RSC
- I. Cabrera, V. Krongauz and H. Ringsdorf, Angew. Chem., Int. Ed. Engl., 1987, 26, 1178–1180 CrossRef
- L. V. Natarajan, T. J. Bunning and S. Y. Kim, Macromolecules, 1994, 27, 7248–7253 CrossRef CAS
- L. V. Natarajan, V. Tondiglia, T. J. Bunning, R. L. Crane and W. W. Adams, Adv. Mater. Opt. Electron., 1992, 1, 293–297 CrossRef CAS
- A. Y. Bobrovsky, N. I. Boiko and V. P. Shibaev, Adv. Mater., 1999, 11, 1025–1028 CrossRef CAS
- A. Y. Bobrovsky, N. I. Boiko and V. P. Shibaev, Liq. Cryst., 2000, 27, 57–62 CrossRef CAS
- A. K. Bobrovsky, N. I. Boiko and V. P. Shibaev, Liq. Cryst., 2000, 27, 219–223 CrossRef CAS
- I. Cabrera and V. Krongauz, Nature, 1987, 326, 582–585 CrossRef
- S. Yitzchaik, G. Berkovic and V. Krongauz, Adv. Mater., 1990, 2, 33–36 CrossRef CAS
- S. Yitzchaik, G. Berkovic and V. Krongauz, Chem. Mater., 1990, 2, 162–168 CrossRef CAS
- S. Yitzchaik, G. Berkovic and V. Krongauz, Opt. Lett., 1990, 15, 1120–1122 CrossRef CAS
- M. Kamenjicki Maurer, I. K. Lednev and S. A. Asher, Adv. Funct. Mater., 2005, 15, 1401–1406 CrossRef
- Q. S. Liu, K. J. Jiang, Y. Q. Wen, J. X. Wang, J. Luo and Y. L. Song, Appl. Phys. Lett., 2010, 97, 253304 CrossRef
- M. Sommer and H. Komber, Macromol. Rapid Commun., 2013, 34, 57–62 CrossRef CAS PubMed
- J. H. Yang and M. K. Ng, Synthesis, 2006, 3075–3079 CAS
- K. Wagner, M. Zanoni, A. B. S. Elliott, P. Wagner, R. Byrne, L. Florea, D. Diamond, K. C. Gordon, G. G. Wallace and D. L. Officer, J. Phys. Chem. C, 2013, 1, 3913–3916 CAS
- J. Pennakalathil and J. D. Hong, ACS Nano, 2011, 5, 9232–9237 CrossRef CAS PubMed
- G. Montagnoli, O. Pieroni and S. Suzuki, Polym. Photochem., 1983, 3, 279–294 CrossRef CAS
- P. H. Vandewyer and G. Smets, J. Polym. Sci., Part A-1: Polym. Chem., 1970, 8, 2361–2374 CrossRef CAS PubMed
- G. Smets, Pure Appl. Chem., 1972, 30, 1–24 CrossRef CAS
- F. Ciardelli, D. Fabbri, O. Pieroni and A. Fissi, J. Am. Chem. Soc., 1989, 111, 3470–3472 CrossRef CAS
- T. M. Cooper, L. V. Natarajan and C. G. Miller, Photochem. Photobiol., 1999, 69, 173–176 CrossRef CAS
- T. M. Cooper, K. A. Obermeier, L. V. Natarajan and R. L. Crane, Photochem. Photobiol., 1992, 55, 1–7 CrossRef CAS
- A. Fissi, O. Pieroni, F. Ciardelli, D. Fabbri, G. Ruggeri and K. Umezawa, Biopolymers, 1993, 33, 1505–1517 CrossRef CAS
- N. Higashi, K. Shimizu and M. Niwa, J. Colloid Interface Sci., 1997, 185, 44–48 CrossRef CAS
- B. Mecheri, P. Baglioni, O. Pieroni and G. Caminati, Mater. Sci. Eng., C, 2003, 23, 893–896 CrossRef PubMed
- N. Angelini, B. Corrias, A. Fissi, O. Pieroni and F. Lenci, Biophys. J., 1998, 74, 2601–2610 CrossRef CAS
- O. Pieroni, A. Fissi, F. Ciardelli and D. Fabbri, Mol. Cryst. Liq. Cryst., 1994, 246, 191–194 CrossRef CAS
- R. Pachter, T. M. Cooper, L. V. Natarajan, K. A. Obermeier, R. L. Crane and W. W. Adams, Biopolymers, 1992, 32, 1129–1140 CrossRef CAS
- O. Pieroni, A. Fissi, A. Viegi, D. Fabbri and F. Ciardelli, J. Am. Chem. Soc., 1992, 114, 2734–2736 CrossRef CAS
- T. M. Cooper, M. O. Stone, L. V. Natarajan and R. L. Crane, Photochem. Photobiol., 1995, 62, 258–262 CrossRef CAS
- A. Fissi, O. Pieroni, N. Angelini and F. Lenci, Macromolecules, 1999, 32, 7116–7121 CrossRef CAS
- O. Pieroni, A. Fissi and F. Ciardelli, React. Funct. Polym., 1995, 26, 185–199 CrossRef CAS
- K. Fujimoto, M. Amano, Y. Horibe and M. Inouye, Org. Lett., 2006, 8, 285–287 CrossRef CAS PubMed
- T. Sakata, Y. L. Yan and G. Marriott, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4759–4764 CrossRef CAS PubMed
- K. Namba and S. Suzuki, Chem. Lett., 1975, 947–950 CrossRef CAS
- M. Aizawa, K. Namba and S. Suzuki, Arch. Biochem. Biophys., 1977, 180, 41–48 CrossRef CAS
- M. Aizawa, K. Namba and S. Suzuki, Arch. Biochem. Biophys., 1977, 182, 305–310 CrossRef CAS
- E. Zahavy, S. Rubin and I. Willner, J. Chem. Soc., Chem. Commun., 1993, 1753–1755 RSC
- E. Zahavy, S. Rubin and I. Willner, Mol. Cryst. Liq. Cryst., 1994, 246, 195–199 CrossRef CAS
- I. Willner, S. Rubin and Y. Cohen, J. Am. Chem. Soc., 1993, 115, 4937–4938 CrossRef CAS
- M. Lion-Dagan and I. Willner, J. Photochem. Photobiol., A, 1997, 108, 247–252 CrossRef CAS
- I. Karube, Y. Nakamoto, K. Namba and S. Suzuki, Biochim. Biophys. Acta, 1976, 429, 975–981 CrossRef CAS
- I. Willner, M. Lion-Dagan and E. Katz, Chem. Commun., 1996, 623–624 RSC
- M. Lion-Dagan, E. Katz and I. Willner, J. Am. Chem. Soc., 1994, 116, 7913–7914 CrossRef CAS
- D. G. Weston, J. Kirkham and D. C. Cullen, Biochim. Biophys. Acta, 1999, 1428, 463–467 CrossRef CAS
- Y. Ito, N. Sugimura, O. H. Kwon and Y. Imanishi, Nat. Biotechnol., 1999, 17, 73–75 CrossRef CAS PubMed
- I. Karube, S. Suzuki, Y. Nakamoto and M. Nishida, Biotechnol. Bioeng., 1977, 19, 1549–1552 CrossRef CAS PubMed
- I. Karube, S. Suzuki, Y. Nakamoto and M. Nishida, J. Mol. Catal., 1979, 6, 51–56 CrossRef CAS
- Y. Nakamoto, I. Karube, S. Terawaki and S. Suzuki, J. Ferment. Technol., 1977, 55, 409–413 CAS
- Y. Nakamoto, I. Karube, S. Terawaki and S. Suzuki, J. Solid-Phase Biochem., 1976, 1, 143–149 CAS
- I. Karube, Y. Nakamoto and S. Suzuki, Biochim. Biophys. Acta, 1976, 445, 774–779 CrossRef CAS
- Y. Nakamoto, M. Nishida, I. Karube and S. Suzuki, Biotechnol. Bioeng., 1977, 19, 1115–1123 CrossRef CAS
- Y. Nakamoto, I. Karube and S. Suzuki, J. Ferment. Technol., 1975, 53, 595–598 CAS
- I. Karube, Y. Yugeta, H. Matsuoka and S. Suzuki, J. Mol. Catal., 1983, 18, 135–140 CrossRef CAS
- Y. Nakamoto, I. Karube, I. Kobayashi, M. Nishida and S. Suzuki, Arch. Biochem. Biophys., 1979, 193, 117–121 CrossRef CAS
- I. Karube, Y. Ishimori and S. Suzuki, J. Solid-Phase Biochem., 1977, 2, 9–17 CrossRef CAS
- I. Karube, Y. Ishimori, S. Suzuki and T. Sato, Biotechnol. Bioeng., 1978, 20, 1775–1783 CrossRef CAS
- I. Karube, Y. Ishimori and S. Suzuki, Anal. Biochem., 1978, 86, 100–106 CrossRef CAS
- H. Asanuma, K. Shirasuka, T. Yoshida, T. Takarada, X. G. Liang and M. Komiyama, Chem. Lett., 2001, 108–109 CrossRef CAS
- P. Zhang, J. B. Meng, T. Matsuura and Y. M. Wang, Chin. Chem. Lett., 2002, 13, 299–302 CAS
- C. Beyer and H. A. Wagenknecht, Synlett, 2010, 1371–1376 CAS
- D. W. Urry, Angew. Chem., Int. Ed. Engl., 1993, 32, 819–841 CrossRef
- M. Alonso, V. Reboto, L. Guiscardo, A. San Martin and J. C. Rodríguez-Cabello, Macromolecules, 2000, 33, 9480–9482 CrossRef CAS
- T. Hirakura, Y. Nomura, Y. Aoyama and K. Akiyoshi, Biomacromolecules, 2004, 5, 1804–1809 CrossRef CAS PubMed
- A. Kocer, M. Walko, W. Meijberg and B. L. Feringa, Science, 2005, 309, 755–758 CrossRef CAS PubMed
- W. Szymanski, D. Yilmaz, A. Kocer and B. L. Feringa, Acc. Chem. Res., 2013 DOI:10.1021/ar4000357
- K. Yoshimura, A. Batiza, M. Schroeder, P. Blount and C. Kung, Biophys. J., 1999, 77, 1960–1972 CrossRef CAS
- E. Kato, T. Ueda, S. Kimura and Y. Imanishi, Biophys. Chem., 1994, 49, 215–222 CrossRef CAS
- T. Ueda, K. Nagamine, S. Kimura and Y. Imanishi, J. Chem. Soc., Perkin Trans. 2, 1995, 365–368 RSC
- K. Y. Tomizaki and H. Mihara, J. Mater. Chem., 2005, 15, 2732–2740 RSC
- K. Y. Tomizaki, X. He and H. Mihara, Bioorg. Med. Chem. Lett., 2005, 15, 1731–1735 CrossRef CAS PubMed
- K. Y. Tomizaki and H. Mihara, J. Am. Chem. Soc., 2007, 129, 8345–8352 CrossRef CAS PubMed
- K. Y. Tomizaki and H. Mihara, Mol. Biosyst., 2006, 2, 580–589 RSC
- B. Liao, J. A. Chen, H. W. Huang, X. F. Li and B. Q. He, J. Mater. Chem., 2011, 21, 5867–5869 RSC
- D. B. Liu, W. W. Chen, K. Sun, K. Deng, W. Zhang, Z. Wang and X. Y. Jiang, Angew. Chem., Int. Ed., 2011, 50, 4103–4107 CrossRef CAS PubMed
- W. H. Binder, R. Sachsenhofer, C. J. Straif and R. Zirbs, J. Mater. Chem., 2007, 17, 2125–2132 RSC
- M. James, S. Ciampi, T. A. Darwish, T. L. Hanley, S. O. Sylvester and J. J. Gooding, Langmuir, 2011, 27, 10753–10762 CrossRef CAS PubMed
- F. May, M. Peter, A. Hutten, L. Prodi and J. Mattay, Chem.–Eur. J., 2012, 18, 814–821 CrossRef CAS PubMed
- I. L. Medintz, S. A. Trammell, H. Mattoussi and J. M. Mauro, J. Am. Chem. Soc., 2004, 126, 30–31 CrossRef CAS PubMed
- M. Tomasulo, I. Yildiz and F. M. Raymo, Aust. J. Chem., 2006, 59, 175–178 CrossRef CAS
- B. Liao, P. Long, B. Q. He, S. J. Yi, B. L. Ou, S. H. Shen and J. Chen, J. Mater. Chem. C, 2013, 1, 3716–3721 RSC
- C. Zhang, C. H. Xu, L. D. Sun and C. H. Yan, Chem.–Asian J., 2012, 7, 2225–2229 CrossRef CAS PubMed
- M. Ueda, H. B. Kim and K. Ichimura, J. Mater. Chem., 1994, 4, 883–889 RSC
- M. Ueda, H. B. Kim and K. Ichimura, Mater. Lett., 1994, 20, 245–249 CrossRef CAS
- M. Piech and N. S. Bell, Macromolecules, 2006, 39, 915–922 CrossRef CAS
- E. A. Osborne, B. R. Jarrett, C. Q. Tu and A. Y. Louie, J. Am. Chem. Soc., 2010, 132, 5934–5935 CrossRef CAS PubMed
- B. I. Ipe, S. Mahima and K. G. Thomas, J. Am. Chem. Soc., 2003, 125, 7174–7175 CrossRef CAS PubMed
- M. H. Yang and M. C. Biewer, Tetrahedron Lett., 2005, 46, 349–351 CrossRef CAS PubMed
- L. de Leon and M. C. Biewer, Tetrahedron Lett., 2000, 41, 3527–3530 CrossRef CAS
- B. C. Bunker, B. I. Kim, J. E. Houston, R. Rosario, A. A. Garcia, M. Hayes, D. Gust and S. T. Picraux, Nano Lett., 2003, 3, 1723–1727 CrossRef CAS
- S. Kado, K. Yamada, T. Murakami and K. Kimura, J. Am. Chem. Soc., 2005, 127, 3026–3030 CrossRef CAS PubMed
- D. Kessler, F. D. Jochum, J. Choi, K. Char and P. Theato, ACS Appl. Mater. Interfaces, 2011, 3, 124–128 CAS
- Y. Aburaya, H. Nomura, M. Kageshima, Y. Naitoh, Y. J. Li and Y. Sugawara, J. Appl. Phys., 2011, 109, 064308 CrossRef
- T. A. Darwish, Y. Tong, M. James, T. L. Hanley, Q. Peng and S. Ye, Langmuir, 2012, 28, 13852–13860 CrossRef CAS PubMed
- R. Rosario, D. Gust, A. A. Garcia, M. Hayes, J. L. Taraci, T. Clement, J. W. Dailey and S. T. Picraux, J. Phys. Chem. B, 2004, 108, 12640–12642 CrossRef CAS
- D. Dattilo, L. Armelao, G. Fois, G. Mistura and M. Maggini, Langmuir, 2007, 23, 12945–12950 CrossRef CAS PubMed
- D. Wang, P. Jiao, J. Wang, Q. Zhang, L. Feng and Z. Yang, J. Appl. Polym. Sci., 2012, 125, 870–875 CrossRef CAS
- G. Joseph, J. Pichardo and G. F. Chen, Analyst, 2010, 135, 2303–2308 RSC
- W. Barthlott and C. Neinhuis, Planta, 1997, 202, 1–8 CrossRef CAS
- H. S. Lim, W. H. Lee, S. G. Lee, D. Lee, S. Jeon and K. Cho, Chem. Commun., 2010, 46, 4336–4338 RSC
- Y. Nakahara, Y. Yamaguchi, H. Iwamoto, H. Sakamato and K. Kimura, Anal. Methods, 2012, 4, 4025–4029 RSC
- A. K. Bohaty, M. R. Newton and I. Zharov, J. Porous Mater., 2010, 17, 465–473 CrossRef CAS
- G. Wang, A. K. Bohaty, I. Zharov and H. S. White, J. Am. Chem. Soc., 2006, 128, 13553–13558 CrossRef CAS PubMed
- M. Zhang, X. Hou, J. Wang, Y. Tian, X. Fan, J. Zhai and L. Jiang, Adv. Mater., 2012, 24, 2424–2428 CrossRef CAS PubMed
- I. Vlassiouk, C. D. Park, S. A. Vail, D. Gust and S. Smirnov, Nano Lett., 2006, 6, 1013–1017 CrossRef CAS PubMed
- I. Vlassiouk, F. Rios, S. A. Vail, D. Gust and S. Smirnov, Langmuir, 2007, 23, 7784–7792 CrossRef CAS PubMed
- R. Q. Albuquerque, J. Kuehni, P. Belser and L. De Cola, ChemPhysChem, 2010, 11, 575–578 CrossRef CAS PubMed
- C. T. Burns, S. Y. Choi, M. L. Dietz and M. A. Firestone, Sep. Sci. Technol., 2008, 43, 2503–2519 CrossRef CAS
- L. Malfatti, S. Costacurta, T. Kidchob, P. Innocenzi, M. Casula, H. Amenitsch, D. Dattilo and M. Maggini, Microporous Mesoporous Mater., 2009, 120, 375–380 CrossRef CAS PubMed
- Z. Zhang, D. Balogh, F. Wang, R. Tel-Vered, N. Levy, S. Y. Sung, R. Nechushtai and I. Willner, J. Mater. Chem. B, 2013, 1, 3159–3166 RSC
- M. Natali and S. Giordani, Chem. Soc. Rev., 2012, 41, 4010–4029 RSC
- E. Del Canto, M. Natali, D. Movia and S. Giordani, Phys. Chem. Chem. Phys., 2012, 14, 6034–6043 RSC
- R. Byrne and D. Diamond, Nat. Mater., 2006, 5, 421–424 CrossRef CAS PubMed
- S. Heng, M.-C. Nguyen, R. Kostecki, T. M. Monro and A. D. Abell, RSC Adv., 2013, 3, 8308–8317 RSC
- G. Wen, J. Yan, Y. Zhou, D. Zhang, L. Mao and D. Zhu, Chem. Commun., 2006, 3016–3018 RSC
- M. Riskin, E. Katz, V. Gutkin and I. Willner, Langmuir, 2006, 22, 10483–10489 CrossRef CAS PubMed
- M. Riskin and I. Willner, Langmuir, 2009, 25, 13900–13905 CrossRef CAS PubMed
- M. Riskin, V. Gutkin, I. Felner and I. Willner, Angew. Chem., Int. Ed., 2008, 47, 4416–4420 CrossRef CAS PubMed
- E. Aznar, R. Casasus, B. Garcia-Acosta, M. D. Marcos and R. Martinez-Manez, Adv. Mater., 2007, 19, 2228–2231 CrossRef CAS
- D. Balogh, R. Tel-Vered, R. Freeman and I. Willner, J. Am. Chem. Soc., 2011, 133, 6533–6536 CrossRef CAS PubMed
- I. Willner and R. Blonder, Thin Solid Films, 1995, 266, 254–257 CrossRef CAS
- R. Blonder, S. Levi, G. L. Tao, I. BenDov and I. Willner, J. Am. Chem. Soc., 1997, 119, 10467–10478 CrossRef CAS
- E. Kaganer, R. Pogreb, D. Davidov and I. Willner, Langmuir, 1999, 15, 3920–3923 CrossRef CAS
- F. Patolsky, B. Filanovsky, E. Katz and I. Willner, J. Phys. Chem. B, 1998, 102, 10359–10367 CrossRef CAS
- A. Higuchi, A. Hamamura, Y. Shindo, H. Kitamura, B. O. Yoon, T. Mori, T. Uyama and A. Umezawa, Biomacromolecules, 2004, 5, 1770–1774 CrossRef CAS PubMed
- A. Gelmi, M. Zanoni, M. J. Higgins, S. Gambhir, D. L. Officer, D. Diamond and G. G. Wallace, J. Mater. Chem. B, 2013, 1, 2162–2168 RSC
- T. Okano, N. Yamada, H. Sakai and Y. Sakurai, J. Biomed. Mater. Res., 1993, 27, 1243–1251 CrossRef CAS PubMed
- J. Edahiro, K. Sumaru, Y. Tada, K. Ohi, T. Takagi, M. Kameda, T. Shinbo, T. Kanamori and Y. Yoshimi, Biomacromolecules, 2005, 6, 970–974 CrossRef CAS PubMed
- Y. Tada, K. Sumaru, M. Kameda, K. Ohi, T. Takagi, T. Kanamori and Y. Yoshimi, J. Appl. Polym. Sci., 2006, 100, 495–499 CrossRef CAS
- K. Kikuchi, K. Sumaru, J.-I. Edahiro, Y. Ooshima, S. Sugiura, T. Takagi and T. Kanamori, Biotechnol. Bioeng., 2009, 103, 552–561 CrossRef CAS PubMed
- I. Willner, R. Blonder and A. Dagan, J. Am. Chem. Soc., 1994, 116, 9365–9366 CrossRef CAS
- A. Doron, E. Katz, G. L. Tao and I. Willner, Langmuir, 1997, 13, 1783–1790 CrossRef CAS
- R. Blonder, I. BenDov, A. Dagan, I. Willner and E. Zisman, Biosens. Bioelectron., 1997, 12, 627–644 CrossRef CAS
- D. J. Gavadhan and S. W. Feldberg, J. Electroanal. Chem., 2000, 491, 103–110 CrossRef
- E. Katz, M. Lion-Dagan and I. Willner, J. Electroanal. Chem., 1995, 382, 25–31 CrossRef
- E. Katz and I. Willner, Electroanalysis, 1995, 7, 417–419 CrossRef CAS
- R. Blonder, I. Willner and A. F. Buckmann, J. Am. Chem. Soc., 1998, 120, 9335–9341 CrossRef CAS
- M. Lion-Dagan, E. Katz and I. Willner, J. Chem. Soc., Chem. Commun., 1994, 2741–2742 RSC
- I. Willner, M. Lion-Dagan, S. Marxtibbon and E. Katz, J. Am. Chem. Soc., 1995, 117, 6581–6592 CrossRef CAS
- I. Willner, A. Doron, E. Katz, S. Levi and A. J. Frank, Langmuir, 1996, 12, 946–954 CrossRef CAS
- T. Niazov, B. Shlyahovsky and I. Willner, J. Am. Chem. Soc., 2007, 129, 6374–6375 CrossRef CAS PubMed
- S. Friedle and S. W. Thomas, III, Angew. Chem., Int. Ed., 2010, 49, 7968–7971 CrossRef CAS PubMed
- K. Aoki, K. Ichimura, T. Tamaki, T. Seki and Y. Kawanishi, Kobunshi Ronbunshu, 1990, 47, 771–777 CrossRef CAS
- K. Ichimura, Y. Hayashi and N. Ishizuki, Chem. Lett., 1992, 1063–1066 CrossRef CAS
- K. Ichimura, Y. Hayashi, K. Goto and N. Ishizuki, Thin Solid Films, 1993, 235, 101–107 CrossRef CAS
- J. M. Galvin and G. B. Schuster, Supramol. Sci., 1998, 5, 89–100 CrossRef CAS
- E. Fischer and Y. Hirschberg, J. Chem. Soc., 1952, 4522–4524 CAS
- T. M. Cooper, V. Tondiglia, L. V. Natarajan, M. Shapiro, K. Obermeier and R. L. Crane, Appl. Opt., 1993, 32, 674–677 CrossRef CAS PubMed
- Z. Tokarski, L. V. Natarajan, B. L. Epling, T. M. Cooper, K. L. Hussong, T. M. Grinstead and W. W. Adams, Chem. Mater., 1994, 6, 2063–2069 CrossRef CAS
- R. Klajn, P. J. Wesson, K. J. M. Bishop and B. A. Grzybowski, Angew. Chem., Int. Ed., 2009, 48, 7035–7039 CrossRef CAS PubMed
- R. F. Khairutdinov, M. E. Itkis and R. C. Haddon, Nano Lett., 2004, 4, 1529–1533 CrossRef CAS
- E. Del Canto, K. Flavin, M. Natali, T. Perova and S. Giordani, Carbon, 2010, 48, 2815–2824 CrossRef CAS PubMed
- Y. J. Song, C. Xu, W. L. Wei, J. S. Ren and X. G. Qu, Chem. Commun., 2011, 47, 9083–9085 RSC
- T. Stafforst and D. Hilvert, Chem. Commun., 2009, 287–288 RSC
- S. Biswas, K. Kinbara, N. Oya, N. Ishii, H. Taguchi and T. Aida, J. Am. Chem. Soc., 2009, 131, 7556–7557 CrossRef CAS PubMed
- R. H. Kramer, D. L. Fortin and D. Trauner, Curr. Opin. Neurobiol., 2009, 19, 544–552 CrossRef CAS PubMed
- N. Caporale, K. D. Kolstad, T. Lee, I. Tochitsky, D. Dalkara, D. Trauner, R. Kramer, Y. Dan, E. Y. Isacoff and J. G. Flannery, Mol. Ther., 2011, 19, 1212–1219 CrossRef CAS PubMed
- A. S. Reddy, A. Izmitli and J. J. de Pablo, J. Chem. Phys., 2009, 131, 085101 CrossRef PubMed
- A. Fissi, O. Pieroni, G. Ruggeri and F. Ciardelli, Macromolecules, 1995, 28, 302–309 CrossRef CAS
- K. Fries, S. Samanta, S. Orski and J. Locklin, Chem. Commun., 2008, 6288–6290 RSC
| This journal is © The Royal Society of Chemistry 2014 |