
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ion formation pathways of crown ether–fullerene conjugates in the gas phase†
Ina D.
Kellner
a,
Leanne C.
Nye‡
a,
Marc S.
von Gernler
a,
Jing
Li
a,
Manolis D.
Tzirakis§
b,
Michael
Orfanopoulos
b and
Thomas
Drewello
*a
aPhysical Chemistry I, Department of Chemistry and Pharmacy, University of Erlangen-Nürnberg, Egerlandstraße 3, 91058 Erlangen, Germany. E-mail: thomas.drewello@fau.de
bOrganic Chemistry, Department of Chemistry, University of Crete, 71003 Voutes, Heraklion, Greece
First published on 29th July 2014
Abstract
The ion formation of crown ether–[60]fullerene conjugates of the type (crown − H)–C60–H with crown = 12cr4, 15cr5 and 18cr6 has been studied with matrix-assisted laser desorption/ionisation (MALDI) and electrospray ionisation mass spectrometry (ESI MS). In total five different ways of ion formation are presented, including metalation (MALDI, ESI), protonation and oxidation (both in MALDI) in the positive-ion mode and deprotonation (MALDI, ESI) and reduction (MALDI) in the negative-ion mode. In line with thermochemistry, the deprotonation and electron transfer processes involve the C60 moiety as the charge-carrying entity, while protonation and metalation occur at the crown ether. Particular emphasis has been placed on the study of metal cation attachment in MALDI varying the crown ether size in the conjugate and using different alkali metal chlorides in the target preparation. Dissociation reactions of the metalated conjugates are influenced by the interaction strength of the metal cation to the crown ether fullerene conjugate. The data confirm an increase in bond strength with smaller metal cations, supporting the notion of charge density-driven interactions.
Introduction
The crown ether–fullerene conjugate is a classic in the development of making mass spectrometry (MS) useful for analysis of fullerenes and their derivatives. In 1993, Wilson and Wu synthesised the first crown ether–fullerene conjugate with the aim to provide a better way of investigating fullerenes by electrospray ionisation (ESI).1 Due to their non-polar nature, fullerenes would not necessarily be easily ionised and in fact are ionised only by difficult-to-control oxidation and reduction processes.2,3 While the Wilson-conjugate was readily metalated,1 the need to perform a synthesis prior to the ESI analysis was not attractive. However, as a proof of concept study the paper was well accepted. In the Wilson-conjugate the crown ether was linked to C60via a methylene (spiro) bridge, occupying a former double bond of the fullerene. The Cretan partner in this collaboration has recently reported a new type of crown ether–fullerene conjugate in which the crown is – without the use of a linker – directly attached via a C–C single bond to the fullerene, which also attains a hydrogen atom as the second substituent to the former fullerene double bond.4–6 The present work is motivated by the desire to study different ion formation mechanisms for these new compounds and to evaluate the gas phase behaviour of the resulting ions. The two most widely-used ionisation methods to date, electrospray ionisation7 and matrix-assisted laser desorption/ionisation (MALDI)8 have been employed. ESI cannot only be applied to these in the well-established way of metalation in the positive ion mode, but the acidic nature of the fullerene hydrogen9–13 allows through deprotonation also the use of the negative ion mode by the formation of quasi-molecular ions [M − H]−. The application of MALDI allows the formation of true molecular ions in both ion modes. In total five different ways of ion formation are presented, including metalation, protonation, oxidation, deprotonation and reduction.The localisation of the charge within the ions is important to evaluate the involvement of either crown or fullerene moiety in the ion formation mechanism. The charge carrying site has been identified by dissociation and fragment ion analysis. This concept has been used successfully before to localise the charge carrying moiety with derivatised fullerenes.14–16 Aside from the investigation of the ion formation process, the study puts particular emphasis on the analysis of three differently sized crown ether–fullerene conjugates with the five alkali metal cations. The interactions of alkali metal cations with free crown ethers have been studied comprehensively in a variety of mass spectrometry-centred experiments. Pioneering approaches included the study of solution equilibria by mass spectrometry17 and the use of tandem mass spectrometry (MS2) for dissociation studies,18–25 ion-molecule reactions23 and ion mobility.26 More recent work extends to ion spectroscopy-based methods.27–30 Pioneering work has been reviewed in the context of supramolecular chemistry in the gas phase.17,21,22,31 One key outcome is the finding that the affinity of a particular pair of crown ether and alkali metal ion is not necessarily exclusively determined by their bond strength, but can be strongly influenced by their molecular environment.
Along these lines, the present study also intends to evaluate the interaction of different combinations of alkali metal ions with differently sized crown ether–fullerene dyads and to enable comparison with the wealth of knowledge gathered for the free crown ether molecules. The post-source decay (PSD) behaviour of the various metal-cationised crown ether adducts was recorded as a function of the activating laser fluence which modulates the internal energy of the decaying ions. The fragment ion distributions allow the evaluation of the relative strength of interaction of metal cation and crown ether–fullerene conjugate.
Experimental
All solvents used were of HPLC grade purity.ESI experiments
Most electrospray measurements were conducted with a quadrupole ion trap (esquire6000, Bruker Daltonics). For spectra with higher resolution a quadrupole time-of-flight (QTOF) mass spectrometer with MS/MS capability (micrOTOF-Q II, Bruker Daltonics) was used. As the crown ether–fullerene conjugates are prone to oxidation while in solution,32 only very small amounts (0.05–0.2 mg) of the compounds were dissolved in dichloromethane (DCM) for each experiment. As the weight measurements on the available laboratory balance were not reliable below 0.1 mg, at least a twofold molar excess of alkali metal chloride was added as methanolic solution (1 g l−1). To achieve stable spraying conditions, the sample was then further diluted with acetonitrile (ACN) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume-to-volume ratio. The concentration of the crown ether–fullerene conjugate in the final solution was about 2 × 10−5 mol l−1.
:1 volume-to-volume ratio. The concentration of the crown ether–fullerene conjugate in the final solution was about 2 × 10−5 mol l−1.
MALDI experiments
Except for the negative-ion mode spectra, all MALDI spectra were acquired using a reflectron time-of-flight instrument (reflex IV, Bruker Daltonics). As matrix 2-[(2E)-3-(4-tert-butylphenyl)-2-methylprop-2-enylidene]malononitrile (DCTB) was used for all experiments. The target was prepared as follows. The crown ether–fullerene conjugates were dissolved in DCM and mixed with appropriate volumes of alkali metal chloride salt solutions (1 g l−1 in MeOH). These mixtures were diluted with DCTB (5 g l−1 in DCM) to give molar sample-to-salt-to-matrix ratios of 1:2:100. The solutions were deposited on a stainless steel target and dried in air.
The instrument parameter laser power relates to the attenuation of the laser light and is used here in arbitrary units (%), as the actual laser fluence is unknown. For all MALDI spectra a laser power of 0–2% was used, resulting in sufficiently abundant ions without inducing fragmentation.
The selection of a particular precursor ion for the post-source decay (PSD) spectra33 is achieved by “ion gating”, i.e. after delayed extraction and a short drift region, the ions are deflected by an electric field. The applied high voltage is only switched off for a short period of time to allow ions of a selected mass range to pass unhindered. The fragments travel basically with the same velocity as the precursor ions, but due to their different masses they possess different kinetic energies. As the lengths of the product ions' flight path in the reflectron depends on their kinetic energy, they can be separated and mass analysed using the reflectron. Focusing the product ions on the detector, however, only works for a certain range of kinetic energies. Therefore, the reflectron voltage has to be decreased incrementally and for each step a spectrum for the resulting mass range is recorded. These segment spectra are finally stitched together to form a complete post-source decay spectrum.33 For the fragmentation study, the PSD spectra of the three crown ether C60 adducts with Na+, K+, Rb+ and Cs+ were each measured at least three times for the laser powers 5, 10, 15 and 20%. For each segment spectrum 200 single shot spectra were averaged. As the calibration of the measurement depends on a sufficiently abundant and resolved precursor ion signal, the laser power in the first segment had to be adjusted for some experiments. Most notably for the spectra at high laser fluence, the parent ion signal of the more unstable complexes was nearly extinguished and the laser power had to be lowered to allow calibration. The laser power in all other segments was kept constant at the values mentioned above.
The MALDI spectra in negative-ion mode were acquired using a TOF/TOF instrument (ultraflex II, Bruker Daltonics), which is capable of measuring PSD in the negative ion mode. It also incorporates an argon-filled collision cell to enhance fragmentation (placed between the ion source and the precursor ion selector) and a “lift” device to allow the acquisition of a full PSD spectrum in a single scan without incrementally decreasing the reflectron voltage.34 This is referred to hereafter as the LIFT method.
Discussion
In Fig. 1 the crown ether–[60]fullerene adducts are depicted together with the five different molecular and quasi-molecular ions that could be generated in this study. The adducts are composed of a 1,2-addition of a hydrogen atom and a covalently linked crown ether, the synthesis of which was recently published.4 The crown ether moiety was varied from 12-crown-4 (12cr4) to 15-crown-5 (15cr5) and 18-crown-6 (18cr6).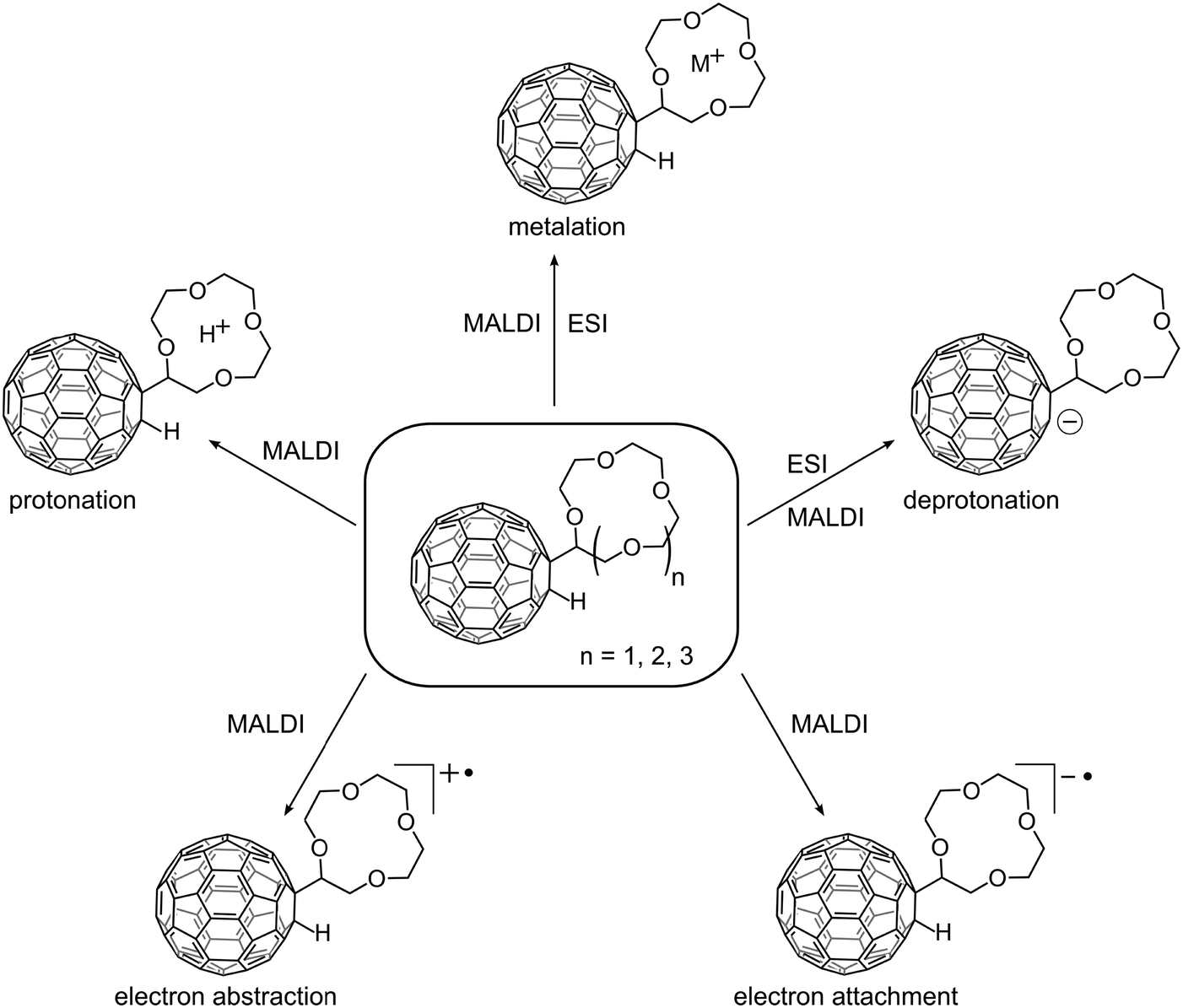 | ||
| Fig. 1 Crown ether–C60 adducts investigated in this study (centre) and their observed ions, shown only for C60H(12cr4 − H) for better legibility. | ||
As a brief summary of the discussion that will follow we emphasise that both the cation and anion radical are readily produced by electron transfer MALDI in both ion modes. ESI was used as the other ionisation method. In the negative ion mode the deprotonated molecule [M − H]− is abundantly formed. Cationisation can easily be achieved by both approaches, while the protonated molecule was only occasionally observed as a minor by-product of the MALDI process. Efforts to protonate the crown in ESI remained unsuccessful, cationisation was the sole reaction.
Negative-ion mode
The negative-ion formation is displayed in Fig. 2. Though the deprotonated molecule [M − H]− is still observable in MALDI using the electron transfer matrix DCTB, the most abundant ion is the molecular anion radical (Fig. 2a). DCTB is used throughout this study as matrix material. It was discovered as a MALDI matrix by Luftmann and co-workers.35 and functions in MALDI as an electron transfer matrix. The thermochemistry of this process has been detailed36,37 and several striking examples of its superior performance with a variety of fullerene derivatives have been published.14,32,38–42 | ||
| Fig. 2 Negative-ion mode mass spectra of C60H(18cr6 − H) and enlargements of the relevant mass ranges on the right side. (a) In MALDI both C60(18cr6 − H)− (m/z 983) and C60H(18cr6 − H)−˙ (m/z 984) are observed. (b) The MS2 (LIFT) spectrum shows both C60−˙ (m/z 720) and C60H− (m/z 721). (c) In the ESI mass spectrum only C60(18cr6 − H)− is detected, the corresponding MS2 spectrum is shown in (d). | ||
The dissociation behaviour of the radical anion was probed by LIFT which is a collision-induced/laser activation experiment on a TOF/TOF instrument.34 The spectrum reveals that the electron resides on the C60 moiety while the crown ether acts as “ligand” and is released in the dissociation (Fig. 2b). The LIFT experiment does not allow the monoisotopic selection of the precursor ion, so that both C60(18cr6 − H)− at m/z 983 and C60H(18cr6 − H)−˙ at m/z 984 may contribute to the two fragment ions C60−˙ and C60H−. The present experiments cannot clarify if C60−˙ is formed exclusively from C60(18cr6 − H)− or via C60H− from C60H(18cr6 − H)−˙. The latter precursor may also lose the crown ether as intact moiety as recently observed in the dissociation of a porphyrin–azacrown ether conjugate.43 In ESI the deprotonated molecules [M − H]− are abundantly formed as the only ions derived from the compounds (Fig. 2c). The formation of these species is perfectly in line with the fact that C60HR compounds of this type are acidic.9–13 Evidently deprotonation is more attractive than electrochemical reduction as alternative ion formation mechanism which is occasionally operative for other, non-acidic derivatised fullerenes.3 Upon activation, the crown ether radical is released which identifies the fullerene as expected as the charge carrier (Fig. 2d). In the following, the formation of positive ions applying MALDI will be discussed.
Positive-ion mode
The positive-ion mode MALDI mass spectra of C60H(15cr5 − H) are shown in Fig. 3. The 15cr5 adduct is taken as the representative of the three crown ether sizes, as some of the features lie indeed in between the two other sizes. The corresponding spectra of C60H(12cr4 − H) and C60H(18cr6 − H) are displayed for comparison in the ESI† (Fig. S1 and Fig. S2, respectively). Full MALDI mass spectra are displayed on the left side of Fig. 3, with enlargements of the molecular ion region shown on the right half. The MALDI experiments commence with the activation of a target that only contained the adduct and the DCTB matrix (ratio 1:100, Fig. 3a) without addition of any alkali salt. This is followed by experiments where alkali chlorides were added successively with increasing size of the alkali metal. The spectra show several prominent signals in the low and the high mass region, with a gap in between that roughly corresponds to the mass of C60. This clearly indicates that the C60 moiety does not play a dominant role as a charge-carrying species under the applied conditions. A noticeable exception is the radical cation, i.e. the molecular ion, of the crown ether adduct, which is seen as a relatively low abundant ion in the high mass region. Sodiation, however, although no sodium was added in this experiment is much more pronounced than the formation of the radical cation. Sodium is a well-known, omnipresent contaminant of, for instance, glass ware. Dissolving the sample, as done in the present case, could be a possible cause of the sodium contamination. The true molecular ion is most likely formed in an electron transfer reaction of matrix-derived cations with the neutral, desorbed adduct. The DCTB+˙ radical cation of the matrix is detected in all the spectra of this adduct and may therefore serve as a likely candidate to ionise the adduct by electron transfer. The ionisation energy (IE) of DCTB is with 8.5 eV37 higher than of the fullerene core (IEC60 = 7.6 eV)44,45 of the adduct, which means that the fullerene core is most likely the carrier of the positive charge. The ionisation energy of crown ethers (as discussed below) is larger than the one of DCTB so that electron removal from the crown ether moiety is highly unlikely. The dissociation behaviour of this ion (vide infra) confirms this assignment. The isotope pattern of the molecular ion reveals a small contribution of the protonated molecule C60H(crown − H)H+. Interestingly, the matrix is unlikely to be the source of the proton, since it does not contain acidic hydrogens and is so far only established as efficient electron transfer matrix.37,42 The analyte, on the other hand, may well show a considerable degree of acidity as observed for related compounds.9–13 The behaviour of the crown ether adducts in ESI confirm the assumed acidity by the abundant formation of their deprotonated anions (see above).
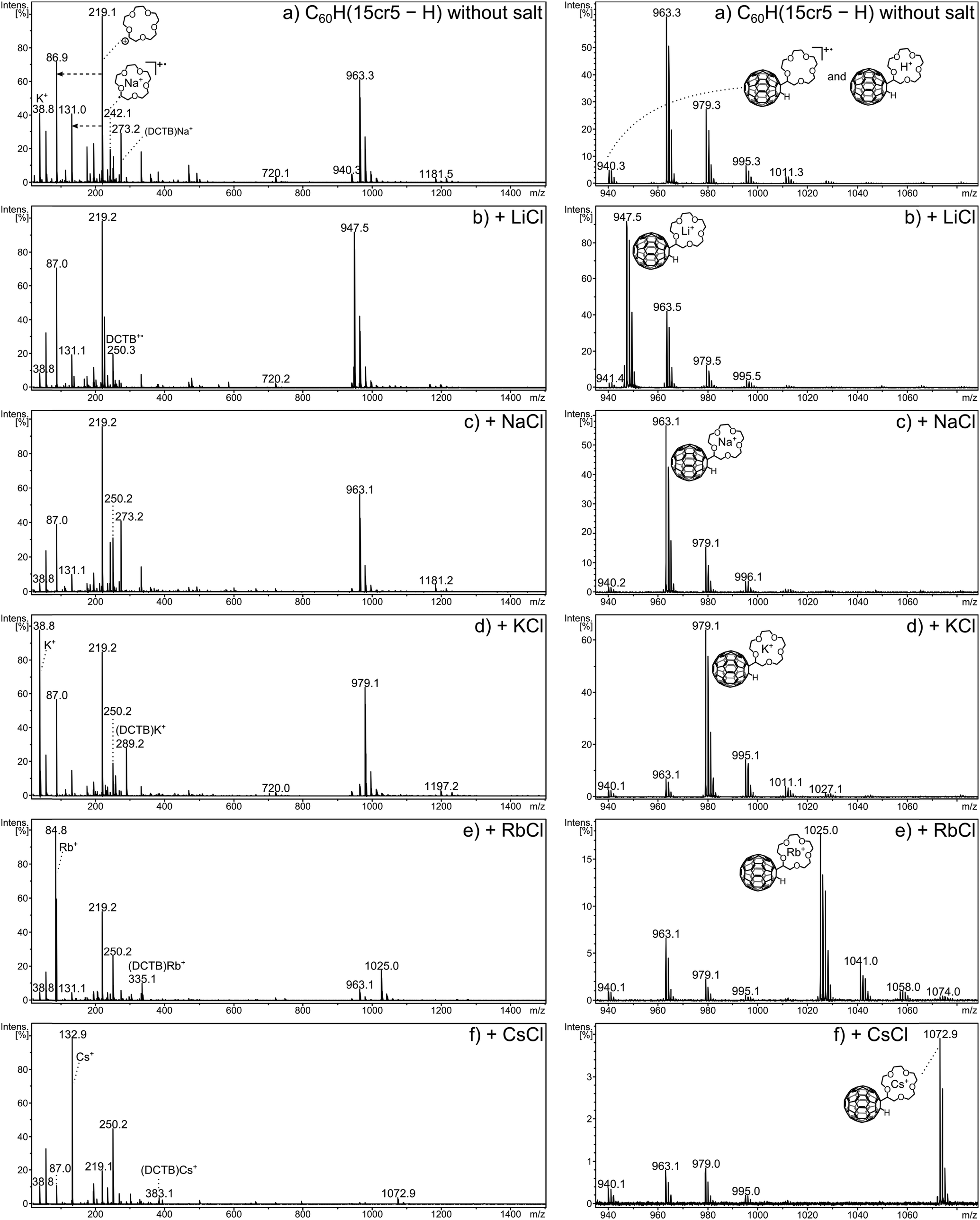 | ||
| Fig. 3 Positive-ion mode MALDI mass spectra of C60H(15cr5 − H) without added salt (a) and with each of the five alkali metal chlorides added (b–f). An enlargement of the molecular ion region is given on the right side of the respective mass spectrum on the left. | ||
A striking feature is the occurrence of a very abundant signal of the ionised crown ether ligand (15cr5 − H)+, at m/z 219 in the low mass part of the spectra. This signal is the base peak in spectra (a), (b) and (c) and remains still amongst the most intense signals in the other spectra. The signals at m/z 87 and 131 result from successive C2H4O losses from m/z 219. The enormous abundance of the m/z 219 ion is remarkable as none of the cationised crown ether adducts is likely to be the precursor of it, as a charge transfer from metal ion to crown ether is energetically impossible. Also, the cation radical of the adduct can produce this ion in only minor amounts, as the product ion analysis will show below. The neutral adduct seems to be the only feasible precursor, which upon laser activation dissociates into C60H˙ and (crown − H)˙. The (crown − H)˙ radical fragment is subsequently ionised by electron transfer reaction with the matrix cation radical, DCTB+˙. For this reaction to occur the ionisation energy of the (crown − H)˙ moiety must be not only lower than that of DCTB+ (IE = 8.5 eV),37 to allow electron transfer from (crown − H)˙ to DCTB+˙, but also lower than that of C60H˙, so that it may not be ionised in competition to (crown − H)˙.
The ionisation energies (IEs) of the free crown ether molecules have been established by photoelectron spectroscopy and amount to 9.3 eV for 12cr4, 9.6 eV for 15cr5 and 9.7 eV for 18cr6.46,47 These values are considerably higher than the IE of DCTB and what can be assumed for the IE of C60H˙. The latter is not known, but can be expected to be lower than the IE of C60 (IE = 7.6 eV). Unfortunately, the ionisation energies of the crown ether radicals are unknown and the proposed mechanism has to remain speculation. However, considering the C–H bond, the IE of the [M − H]˙ radical is often considerably lower than the IE of the corresponding organic molecule M.48 Therefore, it can be safely assumed that the IE of the crown ether radical is clearly lower than the IE of DCTB, so that DCTB+˙ ions are in fact capable of ionising the crown ether radical.
As a result of aging, the crown ether adducts show a certain degree of oxygen addition to C60. The ease by which fullerene derivatives can oxidise at ambient conditions has been studied recently using C60H2 as example.32 Following the signal of MNa+ at m/z 963 in Fig. 3a, the signals at m/z 979, m/z 995 and m/z 1011 are most likely caused by this oxidation. Therefore, the signal at m/z 979 represents predominantly (if not exclusively) the sodium adduct of M with oxygen addition (MONa+) rather than K+ addition to the crown ether adduct (MK+). The mass difference between sodium (MNa+) and a potassium (MK+) adduct amounts to 16 mass units, which also accounts for the addition of an oxygen to the sodium adduct (MONa+). The same situation exists for the mass difference of a lithium (MLi+) to a sodium (MNa+) adduct. Unfortunately the instrumental resolution is not sufficiently high to distinguish these isobaric ions. As a consequence the oxidation prevents the accurate quantitation of Na+ adducts following Li+ adducts and of K+ adducts following Na+ adducts. However, if the addition of the heavier alkali cation is of sizable proportion, it will be noticed.
The dominating ion formation process (Fig. 3a) is, however, the Na+ addition to the neutral molecule, even though no sodium was added. If the alkali metal is deliberately added, the attachment of the respective metal cation is the most prominent process in each case with C60H(15cr5 − H). In the low mass region the bare metal cation signal gains more importance with increasing size and overtakes the ionised radical of the crown ether as the most intense signal (in that region) from K+ onwards.
In the present experiments, metalated ions can in principle be formed via two major pathways, including gas phase metal ion attachment following and/or accompanying the desorption process or these ions may be pre-formed and deposited on the target from solution. Both pathways cannot be clearly distinguished in the present experiments and may also change in their relative importance depending on the crown ether adduct and the metal ion. The ion distribution of the MS1 spectrum is thus influenced by a multitude of competing processes and, therefore, not suited to evaluate the bond strength between metal cation and crown ether C60 adduct. However, the careful comparison of the ion distributions obtained for the three crown ether adducts without and with addition of the five metal cations allows several observations to be made that relate to the interplay of metal cation and the crown ether adduct. Of the three crown-adducts C60H(12cr4 − H) shows the highest abundance of the real molecular ion, the radical cation. Electron transfer of the analyte with matrix ions effectively competes with metal cation attachment as ion formation process. This is particularly evident when no salt is added, here the radical cation exceeds the abundance of the sodiated ions (Fig. 4a). For the adducts with the 15cr5 and 18cr6 ligands, the sodiated adduct exceeds the radical cation (Fig. 4b and c, respectively). Evidently for the bigger-sized crowns, sodiation even with only minute amount of the omnipresent Na+ is much more attractive than the electron transfer reaction. The addition of Li+ and Na+ to C60H(12cr4 − H) reduces the radical cation. The addition of K+ and larger cations results in sizable amounts of the radical cation, indicating that electron transfer ionisation efficiently competes with cation attachment even when the cation (K+, Rb+ and Cs+) is added in excess. Throughout the experiments with added salt, the sodium ion attachment is very efficient with C60H(12cr4 − H). The metal ion adduct is always formed with the added metal. But only in the case of Li+ is it the most dominant ion and even then it is accompanied by a sizeable Na+ attachment. For the other alkali salts (KCl, RbCl and CsCl), the Na+ addition prevails as the most prominent process, which is remarkable indeed taking into account the low abundance of the Na+ impurity. Considering that Li+ possesses a higher bond strength to 12cr4,21 the enormous abundance of the Na+ adduct is even more remarkable. Evidently, bond strength is not the essential factor determining the observed distribution. A partial pre-formation of the Na+ adduct may have already occurred during the sample preparation. In solution, the selectivity of the crown ethers is controlled by a competition between solvation and complexation of the metal ion.17–19,21
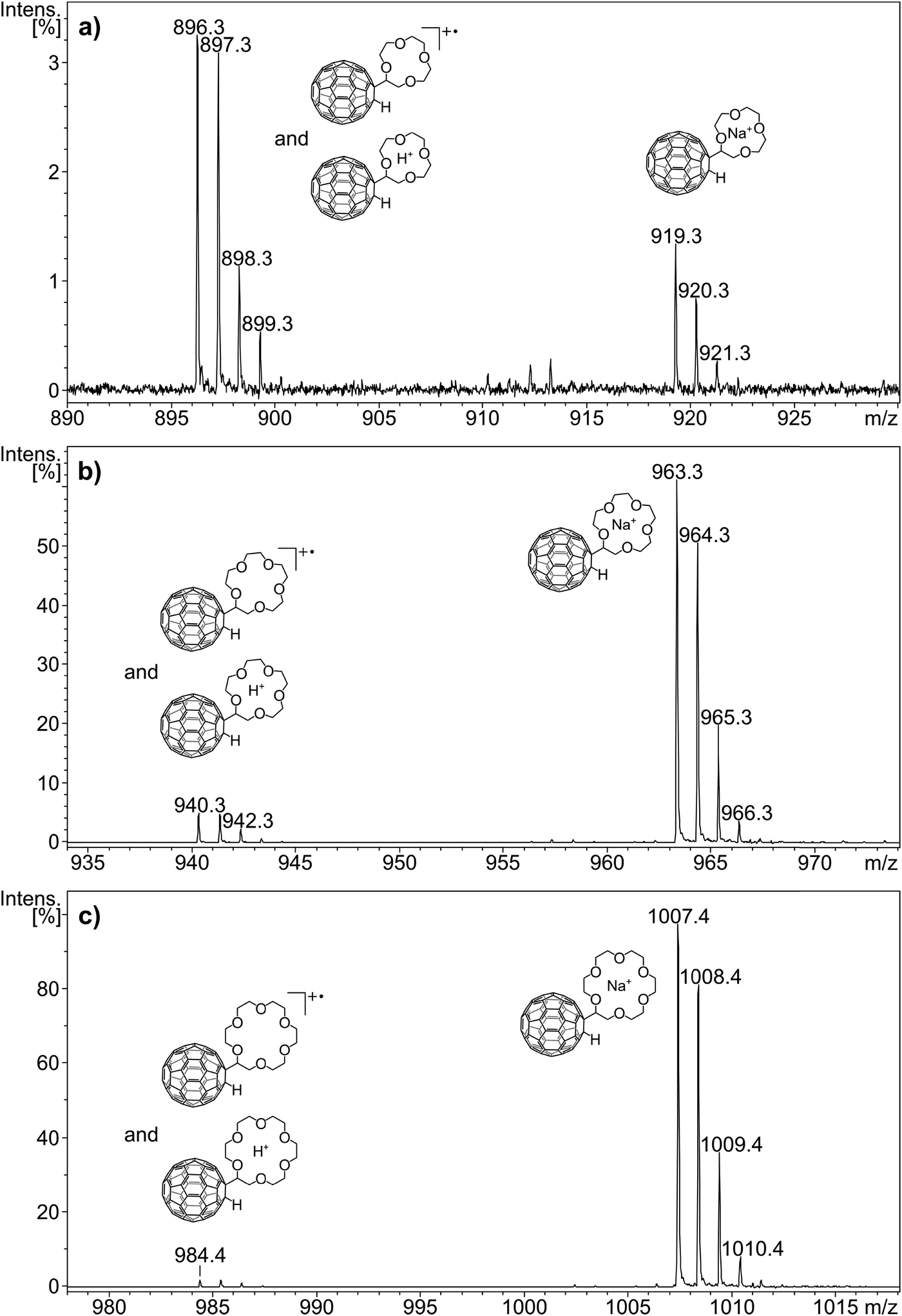 | ||
| Fig. 4 Positive-ion mode MALDI mass spectra of C60H(12cr4 − H), C60H(15cr5 − H) and C60H(18cr6 − H) without alkali metal chloride addition. The isotope patterns of the respective radical cations (m/z 896, 940 and 984) reveal the presence of minor amounts of protonated ions. The peaks at m/z 919, 963 and 1007 belong to the sodium adduct of the respective crown ether–C60 adduct. | ||
The adducts with the larger crowns, C60H(15cr5 − H) and C60H(18cr6 − H), show the attachment of the added salt metal cation as the most prominent process, with the following exceptions. Firstly, in the LiCl doping experiment, the Na+ addition with C60H(18cr6 − H) is even more abundantly occurring than with C60H(12cr4 − H). Secondly, for C60H(15cr5 − H) the Na+ addition competes to some extent with the added K+, Rb+ and Cs+. Thirdly, the observation that the Rb+ and Cs+ addition to C60H(18cr6 − H) is accompanied by K+ addition is remarkable. This may even be the case for Cs+ addition to C60H(15cr5 − H). For C60H(18cr6 − H) the K+ addition is small but clearly detectable, amounting to several per cent of the Rb+/Cs+ adduct. Since the abundance of a K+ impurity is expected to be even lower than for Na+, the origin of the K+ adduct may not be a competing gas phase reaction, but may be produced during sample preparation. In solution, 18cr6 has a pronounced selectivity for the K+ cation,21 which may have led to the formation of the K+ adduct on the target. Similarly, the Na+ addition mentioned above to C60H(15cr5 − H) and C60H(18cr6 − H) may have occurred in solution.
The low mass region shows for all crown ether adducts particularly prominent signals of the free metal cation for K+, Rb+ and Cs+. The ionised former crown ether radical, i.e. the cation (crown − H)+ (m/z 175 and m/z 219, respectively), dominates in all spectra of C60H(12cr4 − H) and C60H(15cr5 − H), indicating that electron transfer is more efficient than metal cation uptake. For C60H(18cr6 − H), however, the metalated crown ether radical, starting with Na+ addition, is more prominently produced while the crown cation becomes less important. It seems more likely to assume that the crown ether radical experiences metalation after the dissociation of the neutral adduct rather than representing the dissociation of the metalated analyte (loss of ˙C60H). The dramatic increase of the cationised crown fragment occurs only for C60H(18cr6 − H) which would mean that the fullerene–crown bond becomes considerably weaker than in the case of C60H(12cr4 − H) and C60H(15cr5 − H). There is, however, no structural reason that would lead to a considerable weakening of that bond. It seems reasonable to assume that the rate for cation addition exceeds the rate of electron transfer for 18cr6, which is reversed for the other crown ethers.
Post-source decay
The post-source decay (PSD) experiment, details of which are given in the ESI† (Fig. S3) and in the Experimental section, can be referred to as MS2 experiment or as tandem mass spectrometry, as it represents the recording of the product ions of a chosen precursor following its dissociations. Examples are shown in Fig. 5. Three examples were chosen as they represent a particular fragmentation behaviour which is typical for the ions under investigation. Fig. 5a depicts the dissociation of the unmetalated radical cation, C60H(15cr5 − H)+˙. The dissociation pattern shows with m/z 748 (C60HC2H3+˙) and m/z 720 (C60+˙) a partial and complete loss of the crown ether unit. This clearly reveals, as mentioned earlier, that the charge is located on the C60 unit. Only a small signal for the charged crown ether fragment is observed. This charge distribution is essentially in accordance with the ionisation energies of C60 (7.6 eV)44,45vs. crown ether (9.3–9.7 eV).46,47 In striking contrast is the decay behaviour of the sodiated crown ether adduct C60H(15cr5 − H)Na+, displayed in Fig. 5b. In line with the sodiation of the crown, the C60 remains neutral and is released upon dissociation, thus not producing any fullerene containing product ions. The sodiated crown − H radical ion, produced by C60H˙ loss, is accompanied by several losses of CH2O and C2H2. This dissociation pattern is similar to what has been established for the decay of metalated crown ethers.49 The differences are probably due to the fact that the observed fragments result from the metalated radical (crown − H)M+˙ rather than the intact crown ether. We note that any of these additional fragment ions is still retaining the Na+ within the decaying crown. Finally, the bare Na+ ion is detected as a product ion that may result from the sodiated precursor ion and any of the other daughter ions. As a final type of dissociation the Cs+ cationised crown ether adduct is displayed (Fig. 5c). Here, the same general decay as observed for the Na+ adduct is taking place, however, there is only one successive decay product after the fullerene loss and the bare metal ion is clearly more pronounced. Both these latter features indicate that the Cs+ ion is much more weakly chelated by the crown in comparison with Na+.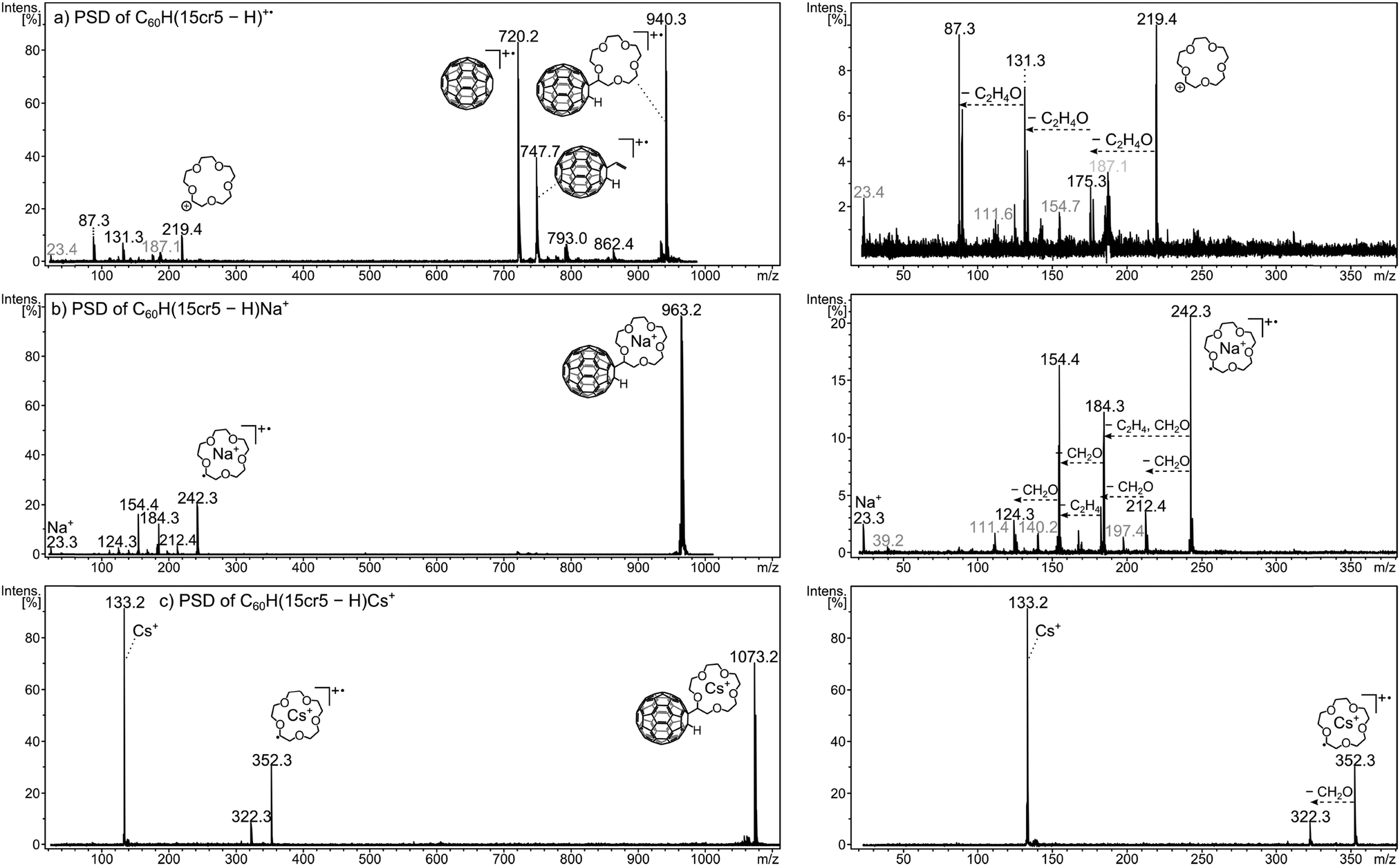 | ||
| Fig. 5 PSD mass spectra of (a) C60H(15cr5 − H)+˙ (m/z 940), (b) C60H(15cr5 − H)Na+ (m/z 963) and (c) C60H(15cr5 − H)Cs+ (m/z 1073). An enlargement of the low mass region of each spectrum is shown on the right side, containing the crown ether ion, its fragments and the bare alkali metal ion. The peak annotations shown in grey correspond to product ions generated by fragmentation of C60H(15cr5 − H)Na+ in (a) and C60H(15cr5 − H)K+ in (b) which were inadvertently included in the isolation due to the ineffectiveness of the ion gate. | ||
The dissociation of the precursor ion is caused by the internal energy content of the ion and by collisional activation that may occur during the flight.50 The dissociation pattern shows a profound dependency on the laser power variation. That is the laser activation influences the internal energy content of the ion50 and thus influences the decay behaviour of the ion. PSD spectra were recorded at varying laser power settings to account for the deviation in product ion abundances. The resulting fragmentation dynamics (Fig. 6) is used to evaluate the relative affinity of the metal ion to the crown ether adduct. The abundance of the precursor ion could not be taken into account, as its isotope pattern was distorted at higher laser power and for some measurements the laser power in the first segment, i.e. the high mass end, had to be lowered in order to gain a sufficient signal for calibration. Therefore, to evaluate the cation–crown ether bond strength the variation of the ratio of three different types of ions relative to all PSD fragment ions were considered. Firstly, the abundance ratio of the intact, metalated crown ether fragment, (crown − H)M+˙ is examined. A high ratio up to relatively high laser power settings may indicate a strong interaction between the metal cation and the crown. Secondly, the abundance of all the ions generated by CH2O and C2H2 losses from the cationised crown ether radical is considered. As all these still contain the metal, they indicate stability of the metal-crown interaction. Therefore, an increasing ratio of these ions also indicates pronounced stability of the metal-crown bond strength. And thirdly, the bare metal ion relative to all fragment ions is representative of the dissociated bond between metal and crown or crown fragments, indicating loss of the metal from its organic host. Therefore, an increase of this ratio is indicative of the more efficiently occurring separation of the cation from the crown. The ion intensity ratios are plotted against the laser power given in arbitrary units. It is evident that the variation of the laser power causes remarkable variations of the ion abundances. Clearly the laser power can be taken as a reflection of the internal energy content. The dissociation behaviour of lithiated species could not be considered, as the Li+ ion lies below the detection mass range of the product ion analysis.
 | ||
| Fig. 6 Results of the PSD experiments for the three crown ether–C60 adducts with Na+, K+, Rb+ and Cs+. The intensity of each fragment ion species, namely (cr − H)M+˙, its fragments and the bare alkali metal ion M+, divided by the sum of intensities of all fragment ions is plotted against the applied laser power. As each experiment was repeated at least three times, the arithmetic average is used with the error bars representing the standard deviation. The polynomial fit is of no mathematical relevance and is only used to guide the eye. | ||
The energy-dependent dissociations show for C60H(12cr4 − H)Na+ an almost constant ratio of (12cr4 − H)Na+˙-fragments while the sodiated, intact crown ether radical (12cr4 − H)Na+˙ decreases and the free metal ion increases as the laser power rises. The ratio of (12cr4 − H)Na+˙-fragments is, compared to the adducts of the larger metals, the highest for this crown ether. Descent of the intact crown ether and rise of the free M+ metal cation cross each other at relatively increased laser power. These features indicate a comparatively strong interaction of Na+ and C60H(12cr4 − H). Potassium binds somewhat less efficiently, while Rb+ and Cs+ show the weakest bonding of all systems, with only the free M+ dominating and with the metalated species being of only minute abundance. Going to the larger crown ether adducts the same order is retained with stronger bonding of smaller ions.
Going from C60H(12cr4 − H) to C60H(15cr5 − H), an increase in bond strength is clearly evident for all metals. For C60H(15cr5 − H)Na+ the free metal ion remains minute while the fragments of (15cr5 − H)Na+˙ increase with the laser power. The overall appearance is a representation of stability of the bonding between Na+ and the crown. For K+ with C60H(15cr5 − H) less K+ release is evident than with C60H(12cr4 − H). Drastic improvement of the bonding of Rb and Cs to C60H(15cr5 − H) is observed. However, the metal ion release seems still the major process, while fragments of the crown ether still containing the metal remain minute. Going on further to C60H(18cr6 − H), only very marginal changes are observed. While K+ and Rb+ appear to be slightly stronger attached to C60H(18cr6 − H), the bond to Cs+ seems to break at lower laser power than for C60H(15cr5 − H). The PSD behaviour of C60H(15cr5 − H)M+ and C60H(18cr6 − H)M+ is very similar, despite the fact that the cation–crown bond in C60H(18cr6 − H)M+ is expected to be stronger.21,25 Clearly, the PSD approach is not sensitive enough to distinguish between the larger adducts, while the distinct behaviour of C60H(12cr4 − H)M+ is evident.
Summary
The ion formation of directly linked crown ether–fullerene conjugates has been studied by MALDI and ESI mass spectrometry. Metalation is efficiently achieved by both ionisation methods. MALDI, with the electron transfer matrix DCTB, produces also the radical anion and cation which are not observed by ESI. ESI allows the pronounced formation of anions by deprotonation of the acidic derivatives, which is only a minor process in MALDI. Protonation has been of minor importance with only minute occurrence in MALDI.The MALDI mass spectra with added alkali metals indicate the importance of interactions that are not dominated by the bond strength between metal cation and crown. This is exemplified by the efficient inclusion of metals that are only present in very low amounts and with lower bond strength.
In contrast, the fragmentation dynamics of the selected metalated adducts are clearly dominated by the order of bond strengths established for the various metal cation–crown ether combinations.21,24,25 The trend of increasing bond strength with decreasing cation and increasing crown ether size is evident with C60H(12cr4 − H)M+ and C60H(15cr5 − H)M+. The fact that no trend was observed when going from the C60H(15cr5 − H)M+ to C60H(18cr6 − H)M+ is attributed to insufficient sensitivity of the PSD method.
Acknowledgements
The authors thank the Deutsche Forschungsgemeinschaft (DFG) – SFB 953 “Synthetic Carbon Allotropes” for financial support.References
- S. R. Wilson and Y. Wu, J. Chem. Soc., Chem. Commun., 1993, 9, 784–786 RSC.
- T. Drewello, H. Frauendorf, R. Herzschuh, A. A. Goryunkov, S. H. Strauss and O. V. Boltalina, Chem. Phys. Lett., 2005, 405, 93–96 CrossRef CAS PubMed.
- M. P. Barrow, X. Feng, J. I. Wallace, O. V. Boltalina, R. Taylor, P. J. Derrick and T. Drewello, Chem. Phys. Lett., 2000, 330, 267–274 CrossRef CAS.
- M. D. Tzirakis and M. Orfanopoulos, Angew. Chem., Int. Ed., 2010, 49, 5891–5893 CrossRef CAS PubMed.
- M. D. Tzirakis and M. Orfanopoulos, Encyclopedia of Radicals in Chemistry, Biology and Materials, Polymers and Materials, John Wiley & Sons Ltd, 2012, vol. 4, pp. 2171–2195 Search PubMed.
- M. D. Tzirakis and M. Orfanopoulos, Chem. Rev., 2013, 113, 5262–5321 CrossRef CAS PubMed.
- J. B. Fenn, Angew. Chem., Int. Ed., 2003, 42, 3871–3894 CrossRef CAS PubMed.
- F. Hillenkamp and M. Karas, Int. J. Mass Spectrom., 2000, 200, 71–77 CrossRef CAS.
- P. J. Fagan, P. J. Krusic, D. H. Evans, S. A. Lerke and E. Johnston, J. Am. Chem. Soc., 1992, 114, 9697–9699 CrossRef CAS.
- M. E. Niyazymbetov, D. H. Evans, S. A. Lerke, P. A. Cahill and C. C. Henderson, J. Phys. Chem., 1994, 98, 13093–13098 CrossRef CAS.
- G. Van Lier, B. Safi and P. Geerlings, J. Chem. Soc., Perkin Trans. 2, 1998, 349–354 RSC.
- B. Jousselme, G. Sonmez and F. Wudl, J. Mater. Chem., 2006, 16, 3478–3482 RSC.
- I. V. Kuvychko, N. B. Shustova, S. M. Avdoshenko, A. A. Popov, S. H. Strauss and O. V. Boltalina, Chem. – Eur. J., 2011, 17, 8799–8802 CrossRef CAS PubMed.
- D. Fati, Y. V. Vasil'ev, N. K. Wachter, R. Taylor and T. Drewello, Int. J. Mass Spectrom., 2003, 229, 3–10 CrossRef CAS.
- V. E. Frankevich, M. Dashtiev, R. Zenobi, T. Kitagawa, Y. Lee, Y. Murata, T. Yamazaki, Y. Gao, K. Komatsu and J. M. Oliva, Phys. Chem. Chem. Phys., 2005, 7, 1036–1042 RSC.
- J.-F. Greisch and E. De Pauw, J. Mass Spectrom., 2007, 42, 304–311 CrossRef CAS PubMed.
- J. S. Brodbelt, Int. J. Mass Spectrom., 2000, 200, 57–69 CrossRef CAS.
- C.-C. Liou and J. S. Brodbelt, J. Am. Soc. Mass Spectrom., 1992, 3, 543–548 CrossRef CAS.
- S. Maleknia and J. Brodbelt, J. Am. Chem. Soc., 1992, 114, 4295–4298 CrossRef CAS.
- M. B. More, D. Ray and P. B. Armentrout, J. Am. Chem. Soc., 1999, 121, 417–423 CrossRef CAS.
- P. B. Armentrout, Int. J. Mass Spectrom., 1999, 193, 227–240 CrossRef CAS.
- M. T. Rodgers and P. B. Armentrout, Mass Spectrom. Rev., 2000, 19, 215–247 CrossRef CAS.
- I.-H. Chu, H. Zhang and D. V. Dearden, J. Am. Chem. Soc., 1993, 115, 5736–5744 CrossRef CAS.
- P. B. Armentrout, C. A. Austin and M. T. Rodgers, Int. J. Mass Spectrom., 2012, 330–332, 16–26 CrossRef CAS PubMed.
- P. B. Armentrout, C. A. Austin and M. T. Rodgers, J. Phys. Chem. A, 2014 DOI:10.1021/jp4116172.
- S. Lee, T. Wyttenbach, G. von Helden and M. T. Bowers, J. Am. Chem. Soc., 1995, 117, 10159–10160 CrossRef CAS.
- B. Martínez-Haya, P. Hurtado, A. R. Hortal, J. D. Steill, J. Oomens and P. J. Merkling, J. Phys. Chem. A, 2009, 113, 7748–7752 CrossRef PubMed.
- B. Martínez-Haya, P. Hurtado, A. R. Hortal, S. Hamad, J. D. Steill and J. Oomens, J. Phys. Chem. A, 2010, 114, 7048–7054 CrossRef PubMed.
- C. M. Choi, H. J. Kim, J. H. Lee, W. J. Shin, T. O. Yoon, N. J. Kim and J. Heo, J. Phys. Chem. A, 2009, 113, 8343–8350 CrossRef CAS PubMed.
- C. M. Choi, D. H. Choi, J. Heo, N. J. Kim and S. K. Kim, Angew. Chem., Int. Ed., 2012, 51, 7297–7300 CrossRef CAS PubMed.
- C. A. Schalley, Int. J. Mass Spectrom., 2000, 194, 11–39 CrossRef CAS.
- L. C. Nye, M. D. Tzirakis, M. Orfanopoulos and T. Drewello, Chem. Phys. Lett., 2010, 499, 209–212 CrossRef CAS PubMed.
- B. Spengler, J. Mass Spectrom., 1997, 32, 1019–1036 CrossRef CAS.
- D. Suckau, A. Resemann, M. Schuerenberg, P. Hufnagel, J. Franzen and A. Holle, Anal. Bioanal. Chem., 2003, 376, 952–965 CrossRef CAS PubMed.
- L. Ulmer, J. Mattay, H. G. Torres-Garcia and H. Luftmann, Eur. J. Mass Spectrom., 2000, 6, 49–52 CrossRef CAS.
- A. V. Streletskii, I. N. Ioffe, S. G. Kotsiris, M. P. Barrow, T. Drewello, S. H. Strauss and O. V. Boltalina, J. Phys. Chem. A, 2005, 109, 714–719 CrossRef CAS PubMed.
- Y. V. Vasil'ev, O. G. Khvostenko, A. V. Streletskii, O. V. Boltalina, S. G. Kotsiris and T. Drewello, J. Phys. Chem. A, 2006, 110, 5967–5972 CrossRef PubMed.
- G. Bottari, C. Dammann, T. Torres and T. Drewello, J. Am. Soc. Mass Spectrom., 2013, 24, 1413–1419 CrossRef CAS PubMed.
- I. V. Kuvychko, A. V. Streletskii, N. B. Shustova, K. Seppelt, T. Drewello, A. A. Popov, S. H. Strauss and O. V. Boltalina, J. Am. Chem. Soc., 2010, 132, 6443–6462 CrossRef CAS PubMed.
- I. V. Kuvychko, A. V. Streletskii, A. A. Popov, S. G. Kotsiris, T. Drewello, S. H. Strauss and O. V. Boltalina, Chem. – Eur. J., 2005, 11, 5426–5436 CrossRef CAS PubMed.
- D. Fati, V. Leeman, Y. V. Vasil'ev, T. Drewello, B. Leyh and H. Hungerbühler, J. Am. Soc. Mass Spectrom., 2002, 13, 1448–1458 CrossRef CAS.
- T. Brown, N. L. Clipston, N. Simjee, H. Luftmann, H. Hungerbühler and T. Drewello, Int. J. Mass Spectrom., 2001, 210/211, 249–263 CrossRef CAS.
- J. Li, K. Duerr, M. S. von Gernler, N. Jux, I. Ivanović-Burmazović and T. Drewello, Int. J. Mass Spectrom., 2013, 354–355, 406–413 CrossRef CAS PubMed.
- A. V. Pogulay, R. R. Abzalimov, S. K. Nasibullaev, A. S. Lobach, T. Drewello and Y. V. Vasil'ev, Int. J. Mass Spectrom., 2004, 233, 165–172 CrossRef CAS PubMed.
- J. de Vries, H. Steger, B. Kamke, C. Menzel, B. Weisser, W. Kamke and I. V. Hertel, Chem. Phys. Lett., 1992, 188, 159–162 CrossRef CAS.
- M. Kajitani, A. Sugimori, N. Sato, K. Seki, H. Inokuchi and Y. Harada, Bull. Chem. Soc. Jpn., 1979, 52, 2199–2201 CrossRef CAS.
- A. D. Baker, G. H. Armen and S. Funaro, J. Chem. Soc., Dalton Trans., 1983, 2519–2523 RSC.
- J. M. Williams and W. H. Hamill, J. Chem. Phys., 1968, 49, 4467–4477 CrossRef CAS PubMed.
- T. L. Selby, C. Wesdemiotis and R. P. Lattimer, J. Am. Soc. Mass Spectrom., 1994, 5, 1081–1092 CrossRef CAS.
- Z. Szilágyi, J. E. Varney, P. J. Derrick and K. Vékey, Rapid Commun. Mass Spectrom., 1998, 12, 489–492 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp02374a |
| ‡ Current address: Department of Surgery and Cancer, Faculty of Medicine, Imperial College London, Sir Alexander Fleming Building, London SW7 2AZ, UK. |
| § Current address: Institute of Chemistry and Biological Chemistry, Zurich University of Applied Sciences, Einsiedlerstrasse 31, CH-8820 Wädenswil, Switzerland. |
| This journal is © the Owner Societies 2014 |