
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Vibrational dynamics and solvatochromism of the label SCN in various solvents and hemoglobin by time dependent IR and 2D-IR spectroscopy†
Luuk J. G. W.
van Wilderen‡
,
Daniela
Kern-Michler‡
,
Henrike M.
Müller-Werkmeister§
and
Jens
Bredenbeck
*
Johann Wolfgang Goethe-University, Institute of Biophysics, Max-von-Laue-Str. 1, 60438, Frankfurt am Main, Germany. E-mail: bredenbeck@biophysik.uni-frankfurt.de; Fax: +49 69 798 46423; Tel: +49 69 798 46428
First published on 1st August 2014
Abstract
We investigated the characteristics of the thiocyanate (SCN) functional group as a probe of local structural dynamics for 2D-IR spectroscopy of proteins, exploiting the dependence of vibrational frequency on the environment of the label. Steady-state and time-resolved infrared spectroscopy are performed on the model compound methylthiocyanate (MeSCN) in solvents of different polarity, and compared to data obtained on SCN as a local probe introduced as cyanylated cysteine in the protein bovine hemoglobin. The vibrational lifetime of the protein label is determined to be 37 ps, and its anharmonicity is observed to be lower than that of the model compound (which itself exhibits solvent-independent anharmonicity). The vibrational lifetime of MeSCN generally correlates with the solvent polarity, i.e. longer lifetimes in less polar solvents, with the longest lifetime being 158 ps. However, the capacity of the solvent to form hydrogen bonds complicates this simplified picture. The long lifetime of the SCN vibration is in contrast to commonly used azide labels or isotopically-labeled amide I and better suited to monitor structural rearrangements by 2D-IR spectroscopy. We present time-dependent 2D-IR data on the labeled protein which reveal an initially inhomogeneous structure around the CN oscillator. The distribution becomes homogeneous after 5 picoseconds so that spectral diffusion has effectively erased the ‘memory’ of the CN stretching frequency. Therefore, the 2D-IR data of the label incorporated in hemoglobin demonstrate how SCN can be utilized to sense rearrangements in the local structure on a picosecond timescale.
Introduction
Currently there is large interest in unnatural functional groups as site-specific vibrational probes for infrared spectroscopy of proteins.1–12 These labels are incorporated at specific target sites and provide local information on a molecule's structure, polarity, hydrogen bonding and electric field. Ideally, these labels are small, do not perturb the protein fold and, more importantly, do not affect the protein's function. Suitable candidates should furthermore have large oscillator strengths, and exhibit isolated infrared absorption frequencies (also separated from solvent absorption), which facilitate their tracking, and sensitively respond to their surroundings. For 2D-IR spectroscopy, they should in particular have long vibrational lifetimes, as the lifetime determines the time interval during which the oscillator frequency can be tracked, and report on the dynamics of the local environment. A large anharmonicity of the label is beneficial to minimize cancellation between the 0 → 1 and 1 → 2 transitions.The label we focus on is thiocyanate (SCN) which can be incorporated into proteins by post-translational chemical modification of a cysteine residue that can be placed by site-directed mutagenesis. We insert SCN in hemoglobin by cysteine cyanylation and show that it possesses the aforementioned desired properties. We show steady-state FTIR and time-dependent 2D-IR data which indicate the presence of a non-homogenous structural distribution in the surroundings of the S13C15N label. We analyze the central wavenumber, lineshape, anharmonicity and the lifetimes of the thiocyanate vibration in different environments. The results are interpreted in terms of local electric fields and solvent properties (polar vs. apolar and protic vs. aprotic). We demonstrate that the use of the SCN label in 2D-IR spectroscopy of a protein gives rich in situ dynamic information. This makes it promising for applications in proteins and other systems which do not possess intrinsic infrared structural markers and for which local information is not accessible otherwise.
We have chosen bovine hemoglobin (Hb) to incorporate the SCN label and determine its (ultrafast) infrared properties. Hb is a transporter protein that can bind a small ligand to a covalently-bound heme group (see Fig. 1), and is found in all vertebrates. Hb consists of four subunits (two α and two β monomers) that are non-covalently associated.13 Bovine Hb contains one heme per subunit, only one cysteine in each β subunit14 and no cysteine in the α subunits. Here we substitute the cysteine's SH for SCN via cyanylation that is similar to the one performed in ref. 15 and 16. Because of significant H2O absorption in the region where the cyanide stretching frequency appears, the 2D-IR spectra are collected on the isotope S13C15N-labeled protein in deuterated aqueous buffer. The protein preparation protocol described here can be easily used to build S13C15N instead of S12C14N, highlighting the simplicity of the described cyanylation procedure.
 | ||
| Fig. 1 Structures of bovine hemoglobin and methylthiocyanate. Panel A shows the secondary structure of Hb through its transparent surface (adapted from pdb-file 2QSS,17 carbon monoxide not shown). For clarity the two α- and β-subunits are colored green and grey, respectively. Each subunit contains one heme (blue), but only the β-monomers contain one cysteine (red ribbon) each. (B) Zoom on one of the two cysteines that are to be cyanylated. (C) Structure of MeSCN.18 The images were generated using Chimera.19 | ||
Ignited by the current large interest, there are already several examples of proteins with inserted thiocyanate labels: into the active site of ketosteroid isomerase (KSI),20 in a bacterial reaction center,15 modification of a S-peptide bound to the ribonuclease S-protein (RNase S),15 human aldose reductase (hALR2),15 the Ral guanine nucleotide dissociation stimulator (RalGDS) protein,21 and the Measles Virus Nucleoprotein2 to name a few. Fig. S5 (ESI†) attempts to give an overview of all IR studies on SCN labels in proteins. Despite the increasing library of labeled proteins, to the best of our knowledge, time dependent 2D-IR spectroscopy of SCN has not yet been used to measure protein dynamics. To demonstrate the capability of their 2D-IR setup to resolve very small signals, Cheatum and coworkers reported one 2D-IR time point of SCN-labeled dihydrofolate reductase.22 2D-IR spectroscopy on proteins containing different unnatural or labeled amino acids has been shown to be a promising tool to access the local structure and dynamics.12,23–26
Materials and methods
Methyl thiocyanate samples
Methyl thiocyanate and all other solvents and chemicals were purchased from Sigma Aldrich and used without further purification unless stated otherwise. D2O was purchased from euriso-top. For the measurements in buffer, 100 mM sodium phosphate buffer with pH 7.9 was used. Other used solvents are tetrahydrofuran (THF), tetrachloroethylene (PCE) and 2,2,2-trifluoroethanol (TFE). For the measurements in dimethyl sulfoxide (DMSO) the purchased solvent was dried overnight using molecular sieves.Protein sample preparation
In order to incorporate the SCN label into a protein, bovine hemoglobin (Sigma Aldrich) was used. Its tetrameric structure was verified by ultracentrifugation. SCN was introduced via cysteine cyanylation.15,16 The protocols in the literature involve protein purification by using columns, which we replaced by dialysis. The lyophilized protein was dissolved in 100 mM sodium phosphate buffer (pH 7.9) without further purification. Then, 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) was added in two-fold excess and the mixture was filled into a membrane (cut-off at 14 kDa)-covered micro dialysis capsule QuixSep (Carl Roth), and dialyzed against 1000 ml of 100 mM sodium phosphate buffer (pH 7.9) for approximately 8 hours. The dialysis solvent exhibits a faint yellow color. In the next step, an approximately 100-fold excess of potassium cyanide was added,27 which resulted in a change in the color of the protein solution from brown to red due to complexation of the heme iron. The mixture was subsequently dialyzed (overnight) against 200 ml of 100 mM sodium phosphate buffer (pH 7.9). The dialysis solvent turns bright yellow if the protein is successfully labeled. Finally, a second dialysis step of 8 hours against 800 ml of 100 mM sodium phosphate buffer (pH 7.9) is required to obtain pure protein (approximately 8 mM of labeled protein with no significant absorption of free cyanide (2094 cm−1))28,29 and no traces of yellow, which would hint at DTNB or TNB (2-nitro-5-thiobenzoate) in the dialysis solution. Cyanylation at pH > 8 can lead to cleavage of the protein at the cysteine.30,31 The sample used for further measurements is therefore investigated by MALDI MS to rule out the presence of protein fragments.Isotope-labeled protein was prepared in the same way, the only differences being that K13C15N (Sigma Aldrich; 99% 13C and 98% 15N) was used as a label, and the final dialysis step (3–4 hours) was done in 100 ml of pD-adjusted D2O-buffer.
The above protocol resulted in SCN-labeled hemoglobin that was stable under lab conditions for more than a week (evident by very minor intensity changes in the FTIR spectrum). The stability and labeling efficiency were determined by mass-spectrometry (MS-MALDI) to be >95% after two days and ∼85% after 7 days, based on the relative intensity of the β-subunit peak with/without (i.e. native) the label (see Fig. S4, ESI†).
Steady-state measurements
The FTIR spectra were recorded using a Bruker Tensor 27 equipped with an MCT detector using a flow cell32 with a sample thickness of 100 μm. A long-pass filter was used, blocking light with wavenumbers over 2500 cm−1. For the protein measurements baseline correction was necessary since the thiocyanate signal was on top of a water absorption band. This band was fitted with a 5th order polynomial and subtracted (Fig. 2, raw data in Fig. S1, ESI†). For concentration determination, UV-Vis absorption measurements were carried out on a JASCO V-670 spectrophotometer using a 1 mm quartz cuvette. | ||
| Fig. 2 Normalized FTIR absorption spectra of bovine hemoglobin (in aqueous buffer solution) with CN bound to the heme (blue), labeled with SCN (red), and S13C15N (black, in D2O buffer). All spectra are scaled to the low frequency peak and have the water background subtracted. Note that the lower panel is shifted by 80 cm−1 due to isotope labeling. | ||
Extinction coefficient determination
For the determination of the IR extinction coefficient concentration series from 5 mM to 30 mM in 5 mM steps were done for MeSCN in buffer, THF and DMSO. In PCE the concentration was varied from 10 mM to 40 mM in 5 mM steps and in TFE from 50 mM to 200 mM in 25 mM steps. For the steady-state measurements shown in Fig. 3 the MeSCN concentrations were 25 mM in buffer and 20 mM in PCE, THF and TFE and 15 mM in DMSO and for the ultrafast measurements shown in Fig. 4 and 5, 90 mM in THF, PCE, TFE and DMSO and 450 mM in buffer. | ||
| Fig. 3 Normalized absorption spectra of methylthiocyanate in different solvents (continuous lines) as well as the normalized SCN absorption in bovine hemoglobin (in H2O; continuous line with circles). The used solvents are THF – tetrahydrofuran, PCE – tetrachloroethylene, TFE – 2,2,2-trifluoro-ethanol, and DMSO – dimethyl sulfoxide. Sample details are shown in the Materials and methods section. The arrows depict the link between solvent properties and the general direction of the observed frequency shift. | ||
 | ||
| Fig. 4 Vibrational lifetimes of MeSCN and the isotope labeled protein Hb-S13C15N. Panel A/B shows the ESA and bleach amplitudes, respectively, (normalized to the maximum absorption in the raw data at 1 ps; the bleach minimum is scaled with the same normalization factor) of the MeSCN samples in different solvents after broadband infrared excitation, and of the protein in D2O buffer after narrow-band excitation (FWHM = 11 cm−1, centered at 2075 cm−1). The continuous lines result from a global fit after applying a sequential model (its species-associated difference spectra – with error bars – and associated time constants are shown in Fig. 5). Sample details are given in the Materials and methods section. | ||
 | ||
| Fig. 5 Species-associated difference spectra of MeSCN and the isotope labeled protein Hb-S13C15N. The continuous lines result from a global fit after applying a sequential model (its species-associated difference spectra – with error bars – and associated time constants are shown). The long-lived components represent the spectra that do not decay on the timescale of the experiment. The relative contribution of each time constant corresponds to the difference between two consecutive spectra. | ||
The literature UV-Vis extinction coefficient for methemoglobin with cyanide bound to the heme at 540 nm is 11 mM−1 cm−1 according to ref. 33–36. With this we calculate an extinction coefficient of 41 ± 5 M−1 cm−1 for the SCN absorption (see ESI†).
Ultrafast measurements
For the ultrafast measurements an experimental setup was used that consisted of a Ti:Sapph regenerative amplifier (3.5 mJ, 800 nm, 90 fs, 1 kHz, Tsunami-Spitfire XP combination of Spectra Physics, Newport, USA) that pumped two home-built optical parametric amplifiers (OPAs). One OPA was used to generate IR probe pulses by difference frequency generation (DFG) of the signal and idler in AgGaS2. The other OPA generated IR pump pulses with higher intensity (>4 μJ). For the ultrafast measurements on the protein the IR pump was controlled using a Fabry–Perot filter (11 cm−1 bandwidth, 7th order). The spectra were recorded on a 2 × 32 pixel mercury cadmium telluride (MCT) detector (Infrared Associates, USA). The optical resolution was 2 cm−1. The delay of the IR pump pulse, having a 54.7° polarization with respect to the IR probe pulse, was controlled by a translation stage. A chopper was used to collect pump-on and pump-off signals. The shown spectra are difference spectra between pump-on and pump-off. The instrument response was about 150 fs for broadband and about 1 ps for narrow-band excitation measurements. For the MeSCN measurements in THF, PCE, TFE and DMSO an optical pathlength of 250 μm (at 90 mM concentration) was used. For the measurement in buffer the pathlength was reduced to 50 μm (450 mM) and for the protein a pathlength of 100 μm (5 mM, D2O) was used. A global analysis was performed on the ultrafast data by using the Globe Toolbox.37Results
In this section we show by steady-state FTIR spectroscopy that we have successfully incorporated the SCN label in Hb. Because it has previously been shown that the central frequency of the CN vibration is modulated by solvent properties and CN–solvent interaction,2,3,38,39 we have collected steady-state and time-resolved infrared spectra of the model compound MeSCN. We have investigated the effect of the solvent on thiocyanate's vibrational relaxation, anharmonicity, central frequency and lineshape. To mimic the effect of different protein environments on the above properties of the thiocyanate group, we used protic and aprotic solvents with different polarities, i.e. dielectric constants. Finally, we present time dependent 2D-IR data on S13C15N-Hb.Steady state measurements
Hemoglobin (see the ESI† for details on sample characterization) does not exhibit absorption bands in the 2100–2180 cm−1 region.40 Upon binding of cyanide (CN−) to the heme a single peak appears at 2122 cm−1 (Fig. 2; blue spectrum), consistent with results of McCoy et al.,28 who showed that the reaction of potassium cyanide with hemoglobin leads to absorption at this wavenumber. Upon cyanylation (see Materials and methods for our protocol, which is simplified with respect to previously published ones) an additional peak appears at 2156 cm−1 (Fig. 2; red spectrum, see Discussion for the used background subtraction procedure). Therefore, the second peak is assigned to the absorption of the thiocyanate label, and not to free CN− which absorbs at lower frequencies (2080 cm−1).41,42 These assignments are supported by the FTIR spectra of the doubly isotope labeled S13C15N (the black spectrum), which shows a downshift of almost 80 cm−1 for both bands: the heme-bound cyanide shifts by 77 cm−1 to 2045 cm−1 and the thiocyanate absorption by 79 cm−1 to 2077 cm−1. Previous work on S13C15N-labels in ketosteroid isomerase mutants43 reports on thiocyanate absorption frequencies in a similar spectral region, i.e. 2088–2090 cm−1. The downshift has the advantage that the isotope label now absorbs in a region with less water background absorption, and even less in D2O. This is beneficial in particular for 2D-IR spectroscopy, where the excitation that generates our signal competes with direct solvent excitation.As is known from previous work on MeSCN and ethylthiocyanate in solution the SCN label can be used as a probe of the local environment,2,3,38,39 because its central frequency is sensitive to its polarity, hydrogen bonding and temperature. Therefore, we measured the model compound MeSCN in different solvents, mimicking apolar, polar and hydrogen bonding environments in proteins (Fig. 3). For comparison the protein absorption is also depicted. Fits to the absorption bands and determined extinction coefficients (see the Materials and methods section for details) are displayed in Table 1.
| Central wavenumber/cm−1 | FWHM/cm−1 | Extinction coefficient/M−1 cm−1 | Dielectric constant ε44 | |
|---|---|---|---|---|
| a The lineshape is not a simple Lorentzian (evident by the relative large error). b Calculated by averaging 5 concentration measurements, see the ESI for more details. c This is the value for pure water. | ||||
| MeSCN, buffer | 2162.4 | 10.6 (2) | 168 (7) | 80.100c |
| MeSCN, THF | 2156.9 | 7.5 (2) | 134 (7) | 7.52 |
| MeSCN, PCE | 2161.6 | 7.3 (3) | 99.7 (4) | 2.268 |
| MeSCN, TFE | 2169.5 | 19.0 (5)a | 142 (8) | 27.68 |
| MeSCN, DMSO | 2153.1 | 9.4 (3) | 150 (5) | 47.24 |
| Protein (S12C14N) | 2156.7 | 11.7 (7)a | 41 (4)b | — |
To the best of our knowledge, the values for the apolar solvent (PCE) and for the polar but aprotic solvent (DMSO) were not reported yet. PCE's central wavenumber is similar to MeSCN in aqueous buffer, which is remarkable because the buffer is highly polar. This is in contrast to the previously reported trend that non-H-bonding solvents exhibit the lowest frequencies.38
Ultrafast measurements
In the previous section we have seen that the central wavenumber changes if the polarity and hydrogen bonding properties of the solvent are changed. This is one requirement of a useful vibrational label. Its applicability for 2D-IR spectroscopy dramatically increases if the vibrational lifetime is long (in the order of tens of picoseconds) so that time-dependent changes in the local environment (i.e. solvent or protein dynamics that alter the local electric field) can be tracked as long as possible. The lifetimes of MeSCN and the label SCN inside the protein are therefore determined using ultrafast infrared pump-infrared probe spectroscopy (see Fig. 4 and 5). We focus on the spectral regions where we observe the solvent-dependent bleach of the absorption of the thiocyanate stretching vibration and its corresponding vibrationally excited state absorption (ESA). The time traces of the ESA and the bleach are depicted at the maximum and minimum signal frequencies (Fig. 4A and B, respectively). For unscaled traces the reader is referred to Fig. S2 (ESI†). The complete measured spectral window (as shown in Fig. 5) is also subjected to global analysis which, after application of a sequential model, allows us to extract species-associated difference spectra and their associated time constants,37 which are shown in Fig. 5. Note that the protein signals are significantly smaller than those of the model compound (the traces in Fig. 4 are normalized at 1 ps, but the real amplitude is better seen in Fig. S2 (ESI†) where the protein traces have been multiplied by 40 for better visibility). We find that the vibrational lifetime of S13C15N in the protein is similar to MeSCN in buffer, but shorter than MeSCN in the other used solvents. Nevertheless, the lifetime in the protein is 37 ps, making it a relatively long-lived infrared marker band (vibrational lifetimes are typically only a few picoseconds45) that could report on changes in local environment. The anharmonicity of the thiocyanate stretching frequency is obtained by fitting Lorentzians to the spectra of Fig. 5 and shown in Table 2. The anharmonicity in the protein is a bit smaller than that of MeSCN in solution but large enough for the bleach and the ESA not to significantly overlap and cancel the signal.| Life-time/ps | Bleach (FWHM)/cm−1 | Excited state (FWHM)/cm−1 | Anharmonicity/cm−1 | |
|---|---|---|---|---|
| MeSCN, buffer | 0.7 | 2163 (11) | 2138 (10) | 25 |
| 32 | 2164 (9) | 2138 (10) | 26 | |
| MeSCN, THF | 0.6 | 2158 (5) | 2133 (7) | 25 |
| 106 | 2158 (6) | 2133 (7) | 25 | |
| MeSCN, PCE | 4.8 | 2162 (5) | 2137 (10) | 25 |
| 158 | 2162 (6) | 2138 (8) | 24 | |
| MeSCN, TFE | 56 | 2170 (15) | 2146 (17) | 24 |
| MeSCN, DMSO | 1.2 | 2152 (9) | 2128 (7) | 24 |
| 78 | 2153 (8) | 2128 (8) | 25 | |
| Protein (S13C15N) | 37 | 2077 (10) | 2056 (13) | 21 |
Spectral diffusion in hemoglobin
The protein label is followed by 2D-IR spectroscopy as a function of delay time (Fig. 6). The wavenumber of the narrow-band infrared pump pulse is scanned and the resulting probe spectrum is collected for each time delay. The 2D-IR spectra reveal more details than can be observed in time-dependent IR spectra with only the band maximum excited (Fig. 5F). To better visualize the differences at different pump frequencies, cuts through the 2D-IR spectra at different times are shown (Fig. 7). The cuts clearly show spectral diffusion: with increasing waiting time the central bleach wavenumber in each cut converges to the band maximum at 2077 cm−1. These results are discussed in terms of the local field that the CN vibration senses, both in the model compound and the protein. | ||
| Fig. 6 2D-IR spectra of Hb-S13C15N at different delays between the narrow-band IR pump (FWHM = 10.3 cm−1; 1 ps duration) and probe pulses. All panels share the same color scale, and the contours are equidistant (2.9 μOD steps). The measurement at −20 ps is subtracted as the background, and in addition the average overall pixels in each 2D-IR-spectrum is subtracted as well to correct for a transient heating signal (evident in the red spectrum of Fig. 5F). The black line represents the diagonal, and the colored arrows at 2 ps correspond to the pump wavenumbers 2070 cm−1, 2074 cm−1 and 2078 cm−1 of the spectral cuts through the data depicted in Fig. 7. | ||
 | ||
| Fig. 7 Spectral diffusion of the S13C15N label in Hb. Spectral slices at selected time points of the 2D-IR spectra that are shown in Fig. 6. All panels share the same legend, and both the data (circles) and their corresponding fit (continuous line; resulting from a simultaneous fit with 2 Lorentzians) are shown at different pump wavelengths and delay times. The average of each spectrum is set as the baseline. The vertical dashed colored lines are guide to the eye. | ||
Discussion
Steady state spectroscopic properties of MeSCN: influences on the extinction coefficient and central frequency of SCN
In this section we discuss the steady-state spectroscopic properties of MeSCN in different solvents and examine the influence of the solvent's polarity and hydrogen-bonding properties (i.e. solvatochromism). First we focus on the extinction coefficient, then on the linewidth and the central frequencies.A quick look at Table 1 that summarizes the extinction coefficients leads to the conclusion that they are not or only mildly dependent on the solvent's polarity and hydrogen-bonding capacity. A similar observation has been made by Maienschein-Cline et al. for a different set of solvents.38 The extinction coefficient of MeSCN in aqueous buffer is consistent with the value reported previously,38 but our value for TFE is slightly different (142 M−1 cm−1 instead of 163.6 M−1 cm−1). The results for the absorption in THF, often used as a model system for the protein interior because of its similar dielectric constant, agree with those obtained by McMahon et al.39 To further check if there is a strong relationship between the extinction coefficient of MeSCN and the solvent polarity and hydrogen-bonding capabilities we also measured in DMSO, which is a polar solvent that cannot form hydrogen bonds. The extinction coefficient obtained for DMSO is similar as for the apolar, aprotic THF and the only slightly less polar, protic TFE. It is lower than for highly polar and protic aqueous buffer. The extinction coefficient for apolar PCE is remarkably low. An even lower value has been reported for CCl4 (79 M−1 cm−1).38
In order to examine the spectra measured in buffer a baseline correction needs to be applied, because the spectra of the SCN absorption band are superimposed on a water band which is fitted by a 5th order polynomial function and subtracted as baseline. Although the central frequency is hardly affected by this correction, this procedure may affect the observed lineshape, which is the reason why we refrain from an extensive lineshape analysis. Referring to the ultrafast data, which do not require a potentially distorting background correction, we can however conclude that the linewidth is broader in H-bonding solvents than in aprotic solvents (see the values for FWHM in Table 2). Both steady-state and ultrafast data (see Tables 1 and 2) show TFE to have the broadest linewidth. The comparison between aqueous buffer and PCE, both having almost the same central frequency, shows the absorption in the aprotic PCE to be significantly narrower.
The shifts in central frequency reported in Table 1 cannot be explained by solvent polarity alone, for example, MeSCN in aqueous buffer (ε = 80.1; all ε values come from ref. 44) and PCE (ε = 2.268) absorb almost at the same wavenumber. Hydrogen bonding has to be taken into account as an additional factor. This can be achieved using empirical solvatochromic scales such as the Kamlet–Taft solvatochromic relationship (depending on three parameters α, β, and π* characterizing the solvent's hydrogen bond donor acidities, hydrogen bond acceptor basicities and solvent dipolarity/polarizability46,47) which we successfully used to fit the central wavenumber of MeSCN in both protic and aprotic solvents (see Fig. S6, ESI†). However, the empirical Kamlet–Taft relationship which has been derived for bulk solvents cannot directly be transferred to a protein environment and a strict molecular interpretation is not possible. We therefore discuss the solvatochromic shift in terms of the vibrational Stark effect,3,50 keeping in mind that hydrogen bonding will cause additional effects. The shift in the central wavenumber of a solute with the solvent polarity can be described using the Onsager reaction field theory.51,52 This approach models the electric field of the solute's dipole and its interaction with the solvent. The solute-induced polarization of the solvent generates an electric field that is sensed by the solute and causes a solvatochromic wavenumber shift 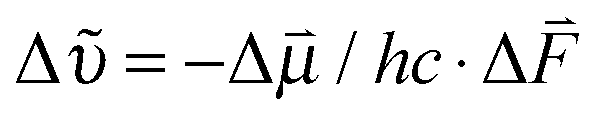 through the vibrational Stark effect.
through the vibrational Stark effect.  is the change in the dipole moment of the oscillator caused by vibrational excitation (often expressed as the Stark tuning rate
is the change in the dipole moment of the oscillator caused by vibrational excitation (often expressed as the Stark tuning rate  in cm−1 (V m−1)−1) and
in cm−1 (V m−1)−1) and  is the change in the field acting on the oscillator. In the Onsager theory this field (in V m−1) is given by:
is the change in the field acting on the oscillator. In the Onsager theory this field (in V m−1) is given by:

 is the permanent dipole moment of the solute in vacuo
is the permanent dipole moment of the solute in vacuo 18 and a is the Onsager spherical cavity radius (aMeSCN = NA·MW/ρ = 1.19 × 10−28 m3, NA is Avogadro's constant, MW the molecular weight and ρ the density in g m−3) in which the solute resides.51 Because the change in the dipole moment of the solute caused by vibrational excitation is typically small (at least two orders of magnitude smaller than in the case of electronic transitions, where changes of several Debye are typical),52 the treatment of the solvatochromic shift of vibrational transitions used here assumes that the reaction field is not changed by vibrational excitation.53 We apply a linear fit (see Fig. 8) of the central wavenumber as a function of the solvent field
18 and a is the Onsager spherical cavity radius (aMeSCN = NA·MW/ρ = 1.19 × 10−28 m3, NA is Avogadro's constant, MW the molecular weight and ρ the density in g m−3) in which the solute resides.51 Because the change in the dipole moment of the solute caused by vibrational excitation is typically small (at least two orders of magnitude smaller than in the case of electronic transitions, where changes of several Debye are typical),52 the treatment of the solvatochromic shift of vibrational transitions used here assumes that the reaction field is not changed by vibrational excitation.53 We apply a linear fit (see Fig. 8) of the central wavenumber as a function of the solvent field  to obtain
to obtain ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) 0 = 2165(2) cm−1 and a Stark tuning rate of
0 = 2165(2) cm−1 and a Stark tuning rate of  for the aprotic solvents reported here together with those reported in previous publications. A comparison of the ratio
for the aprotic solvents reported here together with those reported in previous publications. A comparison of the ratio  gives 5.88 cm−1 (MV cm−1 nm3)−1 for MeSCN vs. 5.14 cm−1 (MV cm−1 nm3)−1 for EtSCN,3 respectively, which corresponds reasonably. These ratios are in the same range as those for several (amino-)benzonitriles: 4.12–7.65 cm−1 (MV cm−1 nm3)−1.52
gives 5.88 cm−1 (MV cm−1 nm3)−1 for MeSCN vs. 5.14 cm−1 (MV cm−1 nm3)−1 for EtSCN,3 respectively, which corresponds reasonably. These ratios are in the same range as those for several (amino-)benzonitriles: 4.12–7.65 cm−1 (MV cm−1 nm3)−1.52
 | ||
| Fig. 8 Central wavenumber of the nitrile group in MeSCN in different solvents (left) and selected proteins (right). Left: central wavenumber of MeSCN's nitrile group in protic (open symbols) and aprotic (solid symbols) solvents as a function of the calculated solvent field FOnsager. For aprotic solvents a linear fit (black line, R2 = 0.76) is shown. The measured central wavenumber of SCN in Hb is depicted as a green line. The data points symbolized by circles are measured in this work, the data in squares are taken from ref. 38 with the exception of DMF (dimethylformamide), which has been taken from ref. 22. Right: central wavenumber of thiocyanate incorporated into proteins for which the orientation is known from X-ray data (data points 1, 2, 5, 11 and 12 are taken from ref. 48; 3, 4, 6, 7 and 8 from ref. 21; and 9 and 10 from ref. 49). An additional figure with SCN central frequencies in proteins summarized from the literature is shown in Fig. S5 (ESI†). | ||
The central wavenumber in DMSO is to our knowledge the lowest reported for MeSCN yet. For all protic, hydrogen bonding solvents we observe a blue shift with respect to the value predicted by Onsager theory. Recent DFT calculations by Cho and coworkers for CN in a hydrogen bonding environment have shown that if only the field component along the cyanide bond is taken into account, the observed solvatochromic shifts (for MeSCN with H2O clusters) cannot be explained.54 However, this approach has previously given good results for the modeling of HOD in D2O.55 For CN it is proposed by Cho and coworkers that additional electric field components (caused by additional water molecules placed in the vicinity of the CN bond) need to be included which could cause a blue-shift of the CN stretching frequency (which we also observe experimentally in the polar buffer and TFE solvents, see Fig. 3). They show that the angle of the formed hydrogen bond determines the direction of the frequency shift (i.e. a linear geometry leads to a blue shift, a distorted one to a red shift) with respect to the non-hydrogen bonded case. In our analysis (see Fig. 8) we always observe a blue shift for hydrogen bonding solvents with respect to the value predicted by Onsager theory. A linear geometry (the C and N atoms of MeSCN and the H atom of the solvent lie on a line) seems therefore likely. A red shift with respect to the Onsager value in H-bonding solvents has not been reported so far. Such a red shift with respect to the non-H-bonding situation might occur in a protein environment, where the geometry of the protein enforces the formation of a distorted H-bond. However, in the solvents analyzed here we generally observe two opposing trends – higher polarity as reflected by higher ε causes a solvatochromic shift to lower wavenumbers and the presence of H-bonds, which is usually correlated with higher ε causes a shift to higher wavenumbers. In protic solvents, we therefore expect partial cancellation of the two influencing factors, as reflected e.g. by the approximately equal wavenumber of MeSCN in aqueous buffer and PCE.
Protein label: steady state spectroscopic properties and local environment
The absorption spectra of Hb show that the SCN label appears about 35 cm−1 upshifted with respect to the heme-bound cyanide (Fig. 2). Upon cyanylation, the absorption band of the heme-bound cyanide does not change at all, indicating that the label does not cause structural changes at the binding site. The intensity of the two bands is similar in the 13C15N sample, but a bit lower for the thiocyanate in the 12C14N sample, which we attribute to lower labeling yield. The aforementioned baseline subtraction may also induce artificial peak intensity differences, in particular, as the isotopologues lie on different parts of the water background. Because the observed heme-bound peak does not increase with higher KCN concentration, the heme groups are taken to be saturated with CN− (see ESI†). The SCN peak does not alter its relative intensity with higher KCN concentration during the labeling reaction either, which could be explained by the maximum labeling efficiency reached, or by a fully labeled protein. In order to determine the labeling efficiency mass spectra are collected on the native and labeled protein (see Fig. S4, ESI†), which show a negligible amount of unlabeled protein in the labeled samples, and we are therefore confident that we have obtained a completely labeled protein. In our best samples the bands of heme-bound CN and thiocyanate are approximately equally intense, implying that the oscillator strength of the label is about twice that of the heme bound cyanide (there are four heme groups and two cyanylated cysteines).The absorption of SCN in the protein is significantly broadened (compared to MeSCN in THF), but because the protein absorption is superimposed on a water background it is difficult to reliably determine the linewidth from FTIR spectroscopy. The broader lineshape of SCN in the protein is caused by inhomogeneous broadening, i.e. SCN interacting with the local protein environment, as can be seen in the 2D-IR spectra (Fig. 6, see Discussion on spectral diffusion below). Interestingly, the estimated extinction coefficient at the band maximum of SCN in the protein is about two times smaller than that of MeSCN in CCl4. The extinction coefficient of a thiocyanate group in its present protein environment seems therefore to be significantly smaller than that of MeSCN in the investigated solvents and the one reported by Fafarman et al. (120 M−1 cm−1) for the S-Peptide bound to RNase S at 80 K.3
A closer look at the left part of Fig. 8 shows that the frequency of the SCN label in Hb (i.e. the nitrile vibration) is similar to that of MeSCN in a solvent that has a high solvent field (i.e. where the green line intersects the black line) and therefore a high dielectric constant. In addition, because the recorded central wavenumber lies below or on the Stark-tuning rate line of the apolar solvents (the black line), it is likely to assume that the label is not hydrogen bonded in the protein (all hydrogen-bonding solvents lie above the black line), although the calculations by Cho et al. suggest that this could also be caused by a distorted H-bond geometry leading to a red shift.54 However, the native crystal structure does not indicate any likely hydrogen bond donors. A discussion of the location of the cysteine S–H in native bovine Hb is given in a previous report,56 which explains that the measured high S–H stretching frequency is caused by S–H residing in a non-polar pocket resulting in the absence of a hydrogen bond to other residues or the solvent. Nevertheless, the S–H group is still accessible to reactants such as DTNB, which we used for cysteine cyanylation or PMB which has been used to test the reactivity of the cysteines.56 This corresponds to the crystal structure showing cysteine as part of the surface with the side chain pointing inwards in the proximity of charged Arg and Asp residues. The SCN probe investigated here is likely situated at the same location, which is in line with the low wavenumber we observe.
Looking at the right part of Fig. 8, where the central frequencies of SCN in different protein(-mutant)s are shown, three regions are evident by the blue, green and red colors, each corresponding to a number of hydrogen-bond partners as determined by crystallography. It is striking to see that our central frequency for SCN in Hb lies in the same region as the thiocyanate vibrations for which the absence of a hydrogen bond is confirmed by X-ray crystallography. The low frequency indicates a high field at the thiocyanate, similar to the field for MeSCN in highly polar solvents. As shown in Fig. S5 (ESI†), reviewing a large number of examples,2,15,21,22,39,48,49,57 the wavenumber of the current label is among the lowest reported so far. The fact that the central frequency of the label in the protein is rather low due to the possible absence of a hydrogen bond has previously been observed as well.48,49,57 The central frequency in the RNase S complex (2156.2 cm−1) (Numbers 9 and 10 in Fig. 8) is explained by prevention of a thiocyanate–water H-bond upon complex formation.49 The low wavenumber in the M150C-CN mutant of ketosteroid isomerase (2153.2–2154.0 cm−1, depending on the mutant and the ligand) (Numbers 11 and 12 in Fig. 8) is according to the authors also caused by a non-hydrogen-bonded thiocyanate group.48
Polarity of the surroundings and hydrogen bonding determine the lifetime of the CN vibration
We determined the vibrational lifetime of the CN vibration in different solvents as well as in hemoglobin by global analysis (Fig. 5) to range from 32 ps to 158 ps. These long lifetimes are comparable to different proline-SCN derivatives that have been reported to have lifetimes of 45–75 ps in D2O and CHCl3, with the slower lifetimes in the latter solvent.58 The spectral features show large similarities for all solvents, although differences in baseline dynamics are seen, which cause fast components in THF and buffer solvents (a consequence of minor direct solvent excitation). Similarly, the 4.8 ps component in PCE and the 1.2 ps component in DMSO are attributed to solvent dynamics and are therefore not discussed further (the spectral changes are very minor with respect to the slower components). As a trend observed in the data less polar solvents lead to longer vibrational lifetimes (τDMSO < τTHF < τPCE, Table 2). The latter correlation is consistent with previous reports59,60 that vibrational relaxation is faster in more polar solvents, which has been attributed to stronger modulation of the vibrational energy levels.59,61,62 Furthermore, vibrational relaxation in protic TFE is faster than in aprotic DMSO, although the latter has a higher dielectric constant. This suggests an additional speed-up of vibrational relaxation by hydrogen bonding. Accordingly, we observe the fastest vibrational relaxation in aqueous buffer. Our observations are consistent with previous studies on smaller solutes (SCN−, N3− and [N(CN)2]−), where a speed-up of a factor 2–3 has been observed when the solvent is changed from polar, non-hydrogen bonding to polar, hydrogen bonding.63,64 Broader linewidths are observed in hydrogen-bonding solvents, which in addition cause shorter lifetimes.64 Such a correlation between lifetimes and linewidths has been reported by Ohta and Tominaga, who attributed it to the fact that enhanced modulation of the vibrational energy levels caused by interactions between the oscillator and its surroundings lead to both a broader linewidth and a shorter lifetime. The lifetime and linewidth of our protein label (Fig. 5F) is very similar to that in buffer. A sub-picosecond signal as seen in buffer in Fig. 5A (due to direct solvent excitation) is not found in the protein measurement because of the much lower excitation level of the solvent when using narrow-band excitation (excitation at the main bleach with FWHM = 11 cm−1, corresponding to a pulse duration of 1 ps). As discussed in the context of the low central wavenumber, we exclude the existence of a hydrogen bond. However, the label seems to experience a high field whose fluctuations could also cause modulation of the vibrational energy levels and thus rapid vibrational relaxation as observed for MeSCN in buffer. Attachment of the label to the protein backbone and van der Waals contacts to the protein surroundings are likely to open additional relaxation channels. The observed anharmonicities (the wavenumber difference of about 25 cm−1 between the excited state and the bleach) are identical within the experimental error for all solvents. Only the protein's anharmonicity is slightly lower (21 cm−1 or 22 cm−1 when corrected for isotopic shift), which might be caused by the local environment of the label inside the protein. We can however not exclude the difference caused by the protein backbone to which the label is connected to as opposed to a single methyl group for MeSCN.Spectral diffusion of SCN in hemoglobin
The collected time dependent 2D-IR spectra in Fig. 6 show that the shape of the bleach signal changes as a function of time. To highlight the changes spectral cuts are made at the colored positions (Fig. 7). In the case of a homogenously broadened band the signal shape does not change as a function of time. Regardless of the pump wavenumber, the signal would look the same in all cuts and only decrease in size with time.65 In contrast, inhomogeneously broadened spectra lead to varying spectral shapes as a function of delay time and pump wavenumber (i.e. spectral diffusion). The 2D-IR signal would change from a lineshape that extends along the diagonal to a signal where only the amplitude but not the lineshape varies with the pump wavenumber if spectral diffusion is completed. The selected time slices for different pump wavenumbers show that initially different bleach positions are observed (at 2 ps). However, a single bleach position remains at 5 ps, irrespective of the used pump wavenumber (i.e. spectral diffusion has occurred within 5 ps). To show that spectral diffusion occurs at all pump frequencies, more pump frequencies are plotted in Fig. S3 (ESI†). There is no clear evidence of spectral diffusion in the excited state (a similar effect is also observed for 13C15N− in D2O66). The available S/N ratio does not allow a center line slope analysis (a measure of the frequency fluctuation correlation function45). A more powerful pump pulse67 than our current few 100 nJ will certainly lead to higher S/N ratios and enable a more detailed analysis of the frequency fluctuations. Note that the observed dynamics is an intrinsic protein property (which may depend on temperature and solvent properties). Therefore, our measurements show that hemoglobin in buffer exhibits ultrafast rearrangements at room temperature that are reflected by the observed spectral diffusion. The protein obviously is not a static molecular framework, but it dynamically interacts with its surroundings and continuously reorganizes its structure, which can be sensed by the here employed CN vibration. In some special cases it may even be possible to probe the protein dynamics without the need of cyanylation, it has been demonstrated by Hamm and coworkers that the (very polarizable) S–H stretching vibration of a cysteine significantly gains in oscillator strength if it is involved in a strong (protected) hydrogen bond.68 Without this interaction, however, the oscillator strength is too weak to be observed. In contrast to our results they observe very slow spectral diffusion (diffusion time of 12 ps in porcine hemoglobin) because of the hydrogen-bond induced structural rigidity within the helix in which the cysteine is located, ultimately providing a different type of information that is dominated by the fluctuations of the hydrogen bond.Conclusions
We have shown that the incorporation of a SCN label inside a protein can give rich dynamic information about the direct surroundings of the unnatural amino acid. Our results on hemoglobin, which contains a cyanylated cysteine with S13C15N in each of its two β-subunits, show that the label experiences a strong field in the proximity of the charged, solvent exposed surface and does not possess a hydrogen bond. Evidence for this comes from the fact that the central frequency of the thiocyanate stretching vibration is rather low. In addition, the observed linewidth is relatively broad and the lifetime rather short (both compared to apolar solvents) albeit still long for a vibrational label (37 ps). Depending on the positioning of the SCN label, even longer lifetimes might occur and allow extended observation of protein dynamics, as we observed a great variability of MeSCN vibrational lifetimes ranging from 32 ps in aqueous buffer to 158 ps in PCE. We have demonstrated the use of the isotope label S13C15N in the protein that functions as a local probe inside a protein to report on local structural dynamics. The use of the isotope facilitates 2D-IR spectroscopy of the CN stretching vibration in D2O because of the higher solvent transmission at the wavenumber of the isotopologue. Time resolved 2D-IR spectra show spectral diffusion on a few picosecond timescale. The initial inhomogeneity (at delays <5 ps) reflects the dynamic heterogeneous local environment of the label inside the protein. To investigate the cause of spectral diffusion of a label more closely, further studies would be required. In particular, it has been shown that comparing experimental observation to molecular dynamics simulations can assist in identifying the types of motion that are probed by spectral diffusion.68–71 Future experiments with higher pump power and photon echo implementation of 2D-IR will allow for a higher signal to noise ratio. At longer delays, the low extinction coefficient of SCN is overcompensated by the long vibrational lifetime. The combination of a long vibrational lifetime and its high sensitivity for local electric fields make the SCN label therefore a useful and versatile probe for 2D-IR spectroscopy of protein dynamics.Acknowledgements
The authors thank Ute Bahr and Marion Bäumlisberger of the Institute of Pharmaceutical Chemistry, Goethe University Frankfurt for performing the MALDI-mass spectrometry measurements, Vitali Vogel of the Institute of Biophysics (Goethe University Frankfurt) for the analytical ultracentrifuge measurements, Sabrina Oesteritz and Georg Wille for helping with the development of the protocol for protein preparation, Stephen D. Fried and Steven G. Boxer for helpful discussion, and the referees for the constructive dialogue. J. B. thanks the Alexander von Humboldt Foundation for a Sofja Kovalevskaja award.Notes and references
- H. Kim and M. Cho, Chem. Rev., 2013, 113, 5817–5847 CrossRef CAS PubMed.
- C. G. Bischak, S. Longhi, D. M. Snead, S. Costanzo, E. Terrer and C. H. Londergan, Biophys. J., 2010, 99, 1676–1683 CrossRef CAS PubMed.
- A. T. Fafarman, P. A. Sigala, D. Herschlag and S. G. Boxer, J. Am. Chem. Soc., 2010, 132, 12811–12813 CrossRef CAS PubMed.
- S. D. Fried, S. Bagchi and S. G. Boxer, J. Am. Chem. Soc., 2013, 135, 11181–11192 CrossRef CAS PubMed.
- H. M. Müller-Werkmeister, Y.-L. Li, E.-B. W. Lerch, D. Bigourd and J. Bredenbeck, Angew. Chem., Int. Ed., 2013, 52, 6214–6217 CrossRef PubMed.
- C. G. Bazewicz, M. T. Liskov, K. J. Hines and S. H. Brewer, J. Phys. Chem. B, 2013, 117, 8987–8993 CrossRef CAS PubMed.
- J. T. King, E. J. Arthur, C. L. Brooks and K. J. Kubarych, J. Am. Chem. Soc., 2014, 136, 188–194 CrossRef CAS PubMed.
- H. M. Müller-Werkmeister and J. Bredenbeck, Phys. Chem. Chem. Phys., 2014, 16, 3261–3266 RSC.
- H. Taskent-Sezgin, J. Chung, P. S. Banerjee, S. Nagarajan, R. B. Dyer, I. Carrico and D. P. Raleigh, Angew. Chem., Int. Ed., 2010, 49, 7473–7475 CrossRef CAS PubMed.
- S. Ye, E. Zaitseva, G. Caltabiano, G. F. X. Schertler, T. P. Sakmar, X. Deupi and R. Vogel, Nature, 2010, 464, 1386–1389 CrossRef CAS PubMed.
- J. Zimmermann, M. C. Thielges, Y. J. Seo, P. E. Dawson and F. E. Romesberg, Angew. Chem., Int. Ed., 2011, 50, 8333–8337 CrossRef CAS PubMed.
- R. Bloem, K. Koziol, S. A. Waldauer, B. Buchli, R. Walser, B. Samatanga, I. Jelesarov and P. Hamm, J. Phys. Chem. B, 2012, 116, 13705–13712 CrossRef CAS PubMed.
- E. Antonini and M. Brunori, Annu. Rev. Biochem., 1970, 39, 977–1042 CrossRef CAS PubMed.
- M. F. Perutz, G. Fermi, C. Poyart, J. Pagnier and J. Kister, J. Mol. Biol., 1993, 233, 536–545 CrossRef CAS PubMed.
- A. T. Fafarman, L. J. Webb, J. I. Chuang and S. G. Boxer, J. Am. Chem. Soc., 2006, 128, 13356–13357 CrossRef CAS PubMed.
- J. L. Wood and N. Catsimpoolas, J. Biol. Chem., 1963, 238, 2887–2888 CAS.
- R. Aranda, H. Cai, C. E. Worley, E. J. Levin, R. Li, J. S. Olson, G. N. Phillips and M. P. Richards, Proteins, 2009, 75, 217–230 CrossRef CAS PubMed.
- S. Nakagawa, S. Takahashi, T. Kojima and C. C. Lin, J. Chem. Phys., 1965, 43, 3583 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- P. A. Sigala, A. T. Fafarman, P. E. Bogard, S. G. Boxer and D. Herschlag, J. Am. Chem. Soc., 2007, 129, 12104–12105 CrossRef CAS PubMed.
- A. J. Stafford, D. L. Ensign and L. J. Webb, J. Phys. Chem. B, 2010, 114, 15331–15344 CrossRef CAS PubMed.
- W. Rock, Y.-L. Li, P. Pagano and C. M. Cheatum, J. Phys. Chem. A, 2013, 117, 6073–6083 CrossRef CAS PubMed.
- J. T. King and K. J. Kubarych, J. Am. Chem. Soc., 2012, 134, 18705–18712 CrossRef CAS PubMed.
- A. Remorino, I. V. Korendovych, Y. Wu, W. F. DeGrado and R. M. Hochstrasser, Science, 2011, 332, 1206–1209 CrossRef CAS PubMed.
- A. M. Woys, S. S. Mukherjee, D. R. Skoff, S. D. Moran and M. T. Zanni, J. Phys. Chem. B, 2013, 117, 5009–5018 CrossRef CAS PubMed.
- S. Bagchi, S. G. Boxer and M. D. Fayer, J. Phys. Chem. B, 2012, 116, 4034–4042 CrossRef CAS PubMed.
- C. Franzini, G. Cattozzo and M. Besozzi, J. Clin. Chem. Clin. Biochem., 1987, 25, 183–184 CAS.
- S. McCoy and W. Caughey, Biochemistry, 1970, 9, 2387–2393 CrossRef CAS.
- K. S. Reddy, T. Yonetani, A. Tsuneshige, B. Chance, B. Kushkuley, S. S. Stavrov and J. M. Vanderkooi, Biochemistry, 1996, 35, 5562–5570 CrossRef CAS PubMed.
- N. Catsimpoolas and J. L. Wood, J. Biol. Chem., 1966, 241, 1790–1796 CAS.
- Y. Degani and A. Patchornik, Biochemistry, 1974, 13, 1–11 CrossRef CAS.
- J. Bredenbeck and P. Hamm, Rev. Sci. Instrum., 2003, 74, 3188 CrossRef CAS PubMed.
- A. Zwart, A. Buursma, E. J. van Kampen and W. G. Zijlstra, Clin. Chem., 1984, 30, 373–379 CAS.
- A. Zwart, Clin. Chem., 1993, 39, 1570–1572 CAS.
- T. Stigbrand, Scand. J. Clin. Lab. Invest., 1967, 20, 252–254 CrossRef.
- I. Wootton and W. R. Blevin, Lancet, 1964, 284, 434–436 CrossRef.
- L. J. G. W. van Wilderen, C. N. Lincoln and J. J. van Thor, PLoS One, 2011, 6, e17373 CAS.
- M. G. Maienschein-Cline and C. H. Londergan, J. Phys. Chem. A, 2007, 111, 10020–10025 CrossRef CAS PubMed.
- H. A. McMahon, K. N. Alfieri, K. A. A. Clark and C. H. Londergan, J. Phys. Chem. Lett., 2010, 1, 850–855 CAS.
- J. O. Alben and W. S. Caughey, Biochemistry, 1968, 7, 175–183 CrossRef CAS.
- K.-K. Lee, K.-H. Park, D. Kwon, J.-H. Choi, H. Son, S. Park and M. Cho, J. Chem. Phys., 2011, 134, 64506 CrossRef PubMed.
- F. A. Miller and C. H. Wilkins, Anal. Chem., 1952, 24, 1253–1294 CrossRef CAS.
- S. K. Jha, M. Ji, K. J. Gaffney and S. G. Boxer, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16612–16617 CrossRef CAS PubMed.
- CRC Handbook of chemistry and physics, A ready-reference book of chemical and physical data, ed. D. R. Lide, CRC Press, Boca Raton Fla. [u.a.], 77th edn, 1996 Search PubMed.
- P. Hamm, Concepts and methods of 2D infrared spectroscopy, Cambridge University Press, Cambridge, 2011 Search PubMed.
- M. J. Kamlet, J. L. M. Abboud, M. H. Abraham and R. W. Taft, J. Org. Chem., 1983, 48, 2877–2887 CrossRef CAS.
- M. J. Kamlet, J. L. M. Abboud and R. W. Taft, in Progress in Physical Organic Chemistry, ed. R. W. Taft, John Wiley & Sons, Inc, Hoboken, NJ, USA, 1981, vol. 13, pp. 485–630 Search PubMed.
- A. T. Fafarman, P. A. Sigala, J. P. Schwans, T. D. Fenn, D. Herschlag and S. G. Boxer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E299 CrossRef CAS PubMed.
- A. T. Fafarman and S. G. Boxer, J. Phys. Chem. B, 2010, 114, 13536–13544 CrossRef CAS PubMed.
- A. Chattopadhyay and S. G. Boxer, J. Am. Chem. Soc., 1995, 117, 1449–1450 CrossRef CAS.
- L. Onsager, J. Am. Chem. Soc., 1936, 58, 1486–1493 CrossRef CAS.
- N. M. Levinson, S. D. Fried and S. G. Boxer, J. Phys. Chem. B, 2012, 116, 10470–10476 CrossRef CAS PubMed.
- The treatment frequently used for electronic transitions (J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, Boston, US, 3rd edn, 2006 Search PubMed and see equation 6.11) takes the change in electronic solvent polarization into account which is caused by the change of the solute's dipole moment due to excitation. Therefore, the index of refraction of the solvent enters the respective equation. In the current study on vibrational excitations this is not the case, because the vibrational excitation's influence on the solvent is considered negligible. Our equation incorporates the index of refraction of the solute to obtain the actual dipole moment of the solute in the solvent from the dipole moment's gas phase value (see ref. 51).
- J.-H. Choi, K.-I. Oh, H. Lee, C. Lee and M. Cho, J. Chem. Phys., 2008, 128, 134506 CrossRef PubMed.
- S. A. Corcelli, C. P. Lawrence and J. L. Skinner, J. Chem. Phys., 2004, 120, 8107–8117 CrossRef CAS PubMed.
- P. P. Moh, F. G. Fiamingo and J. O. Alben, Biochemistry, 1987, 26, 6243–6249 CrossRef CAS.
- K. N. Alfieri, A. R. Vienneau and C. H. Londergan, Biochemistry, 2011, 50, 11097–11108 CrossRef CAS PubMed.
- K.-H. Park, J. Jeon, Y. Park, S. Lee, H.-J. Kwon, C. Joo, S. Park, H. Han and M. Cho, J. Phys. Chem. Lett., 2013, 4, 2105–2110 CAS.
- H. J. Bakker, P. C. M. Planken and A. Lagendijk, Nature, 1990, 347, 745–747 CrossRef CAS.
- H. J. Bakker, J. Chem. Phys., 1993, 98, 8496 CrossRef CAS PubMed.
- H. J. Bakker, J. Chem. Phys., 2004, 121, 10088 CrossRef CAS PubMed.
- E. L. Sibert, S. G. Ramesh and T. S. Gulmen, J. Phys. Chem. A, 2008, 112, 11291–11305 CrossRef CAS PubMed.
- K. Dahl, G. M. Sando, D. M. Fox, T. E. Sutto and J. C. Owrutsky, J. Chem. Phys., 2005, 123, 84504 CrossRef PubMed.
- K. Ohta and K. Tominaga, Chem. Phys. Lett., 2006, 429, 136–140 CrossRef CAS PubMed.
- S. Woutersen and P. Hamm, J. Phys.: Condens. Matter, 2002, 14, R1035 CrossRef CAS.
- M. Koziński, S. Garrett-Roe and P. Hamm, Chem. Phys., 2007, 341, 5–10 CrossRef PubMed.
- H. Bian, J. Li, X. Wen, Z. Sun, J. Song, W. Zhuang and J. Zheng, J. Phys. Chem. A, 2011, 115, 3357–3365 CrossRef CAS PubMed.
- M. Koziński, S. Garrett-Roe and P. Hamm, J. Phys. Chem. B, 2008, 112, 7645–7650 CrossRef PubMed.
- J. D. Eaves, J. J. Loparo, C. J. Fecko, S. T. Roberts, A. Tokmakoff and P. L. Geissler, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13019–13022 CrossRef CAS PubMed.
- J.-H. Choi, K. Kwak and M. Cho, J. Phys. Chem. B, 2013, 117, 15462–15478 CrossRef CAS PubMed.
- M. W. Lee, J. K. Carr, M. Göllner, P. Hamm and M. Meuwly, J. Chem. Phys., 2013, 139, 54506 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp01498g |
| ‡ These authors contributed equally. |
| § Current address: University of Toronto, Chemistry Department, 80 St. George Street, M5S 3H4, Toronto, Canada. |
| This journal is © the Owner Societies 2014 |