 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-pressure crystallographic and spectroscopic studies on two molecular dithienylethene switches†
Christopher H.
Woodall
a,
Simon K.
Brayshaw
a,
Stefanie
Schiffers
a,
David R.
Allan
b,
Simon
Parsons
c,
Rafael
Valiente
d and
Paul R.
Raithby
*a
aDepartment of Chemistry, Bath University, BA2 7AY, UK. E-mail: p.r.raithby@bath.ac.uk
bStation I19, Diamond Light Source, Didcot, Oxfordshire, OX11 0QX, UK
cSchool of Chemistry, Joseph Black Building, West Mains Road, Edinburgh, Scotland EH9 3JJ, UK
dApplied Physics Department, Faculty of Science, University of Cantabria, 39005 Santander, Spain
First published on 15th November 2013
Abstract
Single crystals of the dithienylethene compounds, 1,2-bis(2-methylbenzothiophen-3-yl)perfluorocyclopentene 1 and 1,2-bis(2,5-dimethylthiophen-3-yl)perfluorocyclopentene 2 undergo pressure-induced single-crystal to single-crystal phase transitions between 4.45–5.35 GPa and 4.15–5.70 GPa, respectively. For 1, there is a smooth reduction in unit-cell volume of ~20% from ambient pressure to 4.45 GPa, followed by a dramatic reduction in volume that coincides with a 7.7% increase in the b axis length. Above the pressure of 5.38 GPa a smooth volume reduction continues. In contrast, for 2, there is a continuous change in unit-cell volume with an observed space group change from C2/c to P21/c, between the pressures of 4.15 and 5.70 GPa. In the crystals of 1 between 4.45 and 5.38 GPa adjacent molecules slide over each other and the dominant stacking interaction changes from a thiophene⋯thiophene interaction at 4.45 GPa to a benzothiophene⋯benzothiophene interaction at 5.38 GPa and, within each molecule, the benzothiophene groups show a significant reorientation at the phase transition. In 2 there is a loss of molecular symmetry, concomitant with the change in space group, at the phase transition with the asymmetric unit changing from containing half a unique molecule to two independent molecules. The molecules show significant reorientations of their ring systems. The nature of the observed transition in 1 was investigated using solid-state computational methods to prove the superior thermodynamic stability of the high-pressure phase to the lower pressure phase at pressures above 5.38 GPa. Solid state UV-Vis spectroscopy of 1, over the pressure range from ambient to 15.4 GPa showed that the compound displayed piezochromism with a significant red shift in the π–π* absorption band and a colour change in the crystal from colourless to red with increasing pressure.
Introduction
Over the last two decades research into diarylethenes (DAEs) has shown that these molecules have enormous promise in the field of molecular electronics with potential applications in the areas of optical data storage, optical switching in electronic devices and more recently, as light driven actuators.1–10 Such interest stems from the ability of DAEs to readily undergo a reversible photochromic electrocyclisation reaction between an open and closed form, upon irradiation with different wavelengths of light (Scheme 1).11,12 | ||
| Scheme 1 Generic ring closure reaction upon irradiation with UV or visible light of a dithienylethene. | ||
Because of the unique solid-state properties of DAEs, a better understanding of their behaviour under a variety of non-ambient conditions is of particular importance. Previous studies have suggested that the solid-state photochromism of DAEs is dependent on the distances between the C12–C32 reactive carbon atoms in the open form (Scheme 1). It has been suggested that compounds with a distance greater than 4 Å between these two atoms display no or limited solid-state photochromism.13
Hydrostatic high-pressure studies on a range of crystalline materials have shown that pressure is an effective tool for altering conformational geometries of compounds.14 These studies have been primarily focused on small organics such as amino-acids15–21 or compounds of pharmaceutical,22–27 military28,29 or geophysical interest. A range of unprecedented and fascinating behaviour such as phase transitions, piezochromism,30 changes in magnetic behaviour31–33 and complex spin crossover transitions34,35 have been discovered as a result. More recently, high-pressure has also been demonstrated to affect the solid state reactivity of anthracene-based compounds resulting in the inhibition of a previously reactive complex.36 The interesting results obtained from these high-pressure studies prompted us to investigate the conformational behaviour and solid-state reactivity of two dithienylethenes (DTEs), using high-pressure single crystal crystallography together with high-pressure UV-Vis spectroscopy and computational techniques in order to understand the nature of the transformations that take place.
Two DTE molecules were selected for study at high-pressure from the extensive collection of DTE compounds in the literature (Fig. 1). The complexes were chosen primarily for their suitability for high-pressure single-crystal diffraction experiments; i.e. the compounds afford high quality, well diffracting crystals and crystallise in crystal systems with higher symmetry than triclinic. Variation in molecular structure and crystal packing environment between the two compounds was also deemed to be of interest to facilitate a comparison of how the two crystal structures behaved under increasing hydrostatic pressure.
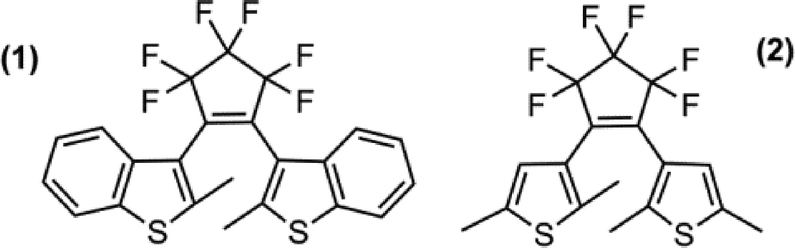 | ||
| Fig. 1 Chemical structure diagrams of 1,2-bis(2-methylbenzothiophen-3-yl)perfluorocyclopentene 1 and 1,2-bis(2,5-dimethylthiophen-3-yl)perfluorocyclopentene 2. | ||
The compounds are based on the dithienylethene molecular switching unit, varying only in the functionality of the thiophene group. In 1 the 4- and 5-positions of the thiophene ring form the linking carbon atoms in a benzothiophene moiety while the 5-position in 2 is occupied by a methyl group (Fig. 1). The structure of 1 had not been reported previously although its photochromic properties had been investigated in solution.37 Compound 1 was known to be photochemically inactive in the crystalline phase, a property associated with a C12–C32 distance greater than 4.0 Å. However, the closely related 6-nitro derivative had been studied in the crystalline state and found to exhibit dichroism.37 The structure of 2 has been reported previously. At 150 K, it crystallising in space group P21/c,10 and has been shown to be photochemically active in the solid-state.
Results and discussion
The structures of 1 and 2 were initially determined at room-temperature under ambient pressure. 1 was found to crystallise in the monoclinic space group P21/n, while 2 crystallised in the monoclinic space group C2/c. This latter result contrasts to the previously reported space group of P21/c for 2,10 and it was found that the compound undergoes a phase transition between 150 and 293 K. Thus, the structure of 2 reported here represents a new room-temperature phase for 2.An analysis of the crystal packing in the two structures at ambient pressure and temperature shows that both structures are dominated by graphitic packing though C–H⋯π and π⋯ π interactions while C–H⋯F interactions are also present. 2 packs in a herringbone fashion whilst 1 forms anti-parallel columns of molecules that extend through the crystal structure. Packing efficiency calculations show that the two compounds have packing coefficients (1 = 72%, 2 = 68%) at opposite ends of the range for organic materials, under ambient conditions, reported by Kitaigorodskii (65–77%).38
Compound 1 was studied over the range of 0.00–8.90 GPa (Table 1) and 2 from 0.00–9.80 GPa (Table 2). As would be expected, both compounds display a reduction in unit-cell volume over the pressure ranges studied. However, 1 displays a marked reduction in volume between the pressures of 4.45 GPa and 5.38 GPa (Fig. 2). The region is also associated with a sudden expansion of the b axis by 7.72% and contraction of the a and c axes by 7.27 and 8.57%, respectively. Such behaviour is suggestive of a iso-symmetric structural phase transition or a transition between phases which have the same space group.39 This phenomenon remains rare in molecular solids although it has been observed in high-pressure studies of a number of systems.30,40–42 It is also notable that the crystals become progressively redder in colour as pressure is increased.
| Pressure (GPa) | Ambient | 0.86 | 2.09 | 2.55 | 3.38 | 4.08 | 4.45 |
|---|---|---|---|---|---|---|---|
| Space group | P21/n | P21/n | P21/n | P21/n | P21/n | P21/n | P21/n |
| a, b, c (Å) | 11.4369(6) | 11.262(6) | 11.096(2) | 11.053(2) | 10.991(6) | 10.954(2) | 10.930(2) |
| 15.5450(6) | 14.9356(12) | 14.494(3) | 14.338(3) | 14.1391(8) | 14.032(3) | 13.961(3) | |
| 12.8179(6) | 12.497(4) | 12.262(3) | 12.136(2) | 12.033(4) | 11.962(2) | 11.922(2) | |
| β (°) | 113.755(6) | 113.73(6) | 113.85(3) | 113.76(3) | 113.83(5) | 113.77(3) | 113.76(3) |
| V (Å3) | 2085.41(17) | 1924.3(13) | 1803.7(6) | 1760.4(6) | 1710.6(10) | 1682.7(6) | 1665.0(6) |
| Z/Z′ | 4/1 | 4/1 | 4/1 | 4/1 | 4/1 | 4/1 | 4/1 |
| Pressure (GPa) | 5.38 | 5.50 | 5.60 | 6.56 | 7.40 | 8.90 |
|---|---|---|---|---|---|---|
| Space group | P21/n | P21/n | P21/n | P21/n | P21/n | P21/n |
| a, b, c (Å) | 10.135(9) | 10.118(18) | 10.117(18) | 10.052(2) | 9.985(2) | 9.894(2) |
| 15.0384(16) | 15.012(3) | 15.004(3) | 14.932(3) | 14.855(3) | 14.736(3) | |
| 10.900(7) | 10.877(11) | 10.869(11) | 10.789(2) | 10.722(2) | 10.588(2) | |
| β (°) | 107.52(9) | 107.53(16) | 107.52(16) | 107.39(3) | 107.35(3) | 107.21(3) |
| V (Å3) | 1584.3(17) | 1575(3) | 1573(3) | 1545.4(5) | 1517.9(5) | 1474.6(5) |
| Z/Z′ | 4/1 | 4/1 | 4/1 | 4/1 | 4/1 | 4/1 |
| Pressure (GPa) | Ambient | 0.71 | 1.02 | 2.52 | 3.64 | 4.15 |
|---|---|---|---|---|---|---|
| Space group | C2/c | C2/c | C2/c | C2/c | C2/c | C2/c |
| a, b, c (Å) | 20.5687(10) | 19.673(3) | 19.325(6) | 18.427(5) | 18.0508(11) | 17.874(8) |
| 8.8331(2) | 8.6717(8) | 8.6243(7) | 8.4721(6) | 8.3848(8) | 8.3360(7) | |
| 11.4381(5) | 11.0787(9) | 10.9544(7) | 10.6330(7) | 10.551(23) | 10.524(13) | |
| β (°) | 122.177(6) | 120.594(13) | 120.113(14) | 118.512(15) | 118.132(23) | 117.9(2) |
| V (Å3) | 1758.95(12) | 1626.8(4) | 1579.3(6) | 1458.6(5) | 1408.2(1) | 1386.9(8) |
| Z/Z′ | 4/1/2 | 4/1/2 | 4/1/2 | 4/1/2 | 4/1/2 | 4/1/2 |
| Pressure (GPa) | 5.70 | 6.70 | 7.40 | 8.55 | 9.50 | 9.80 |
|---|---|---|---|---|---|---|
| Space group | P21/c | P21/c | P21/c | P21/c | P21/c | P21/c |
| a, b, c (Å) | 10.506(2) | 10.4886(10) | 10.4663(8) | 10.4211(7) | 10.377(2) | 10.3834(5) |
| 16.364(3) | 16.3135(16) | 16.2590(12) | 16.1206(12) | 16.060(3) | 16.0279(8) | |
| 15.514(3) | 15.5602(96) | 15.4270(72) | 15.1590(68) | 15.031(3) | 15.0219(45) | |
| β (°) | 98.95(3) | 99.081(16) | 99.409(12) | 99.830(11) | 100.44(3) | 100.443(7) |
| V (Å3) | 2634.7(9) | 2629.1(15) | 2589.9(11) | 2509.3(11) | 2463.5(9) | 2458.6(7) |
| Z/Z′ | 8/2 | 8/2 | 8/2 | 8/2 | 8/2 | 8/2 |
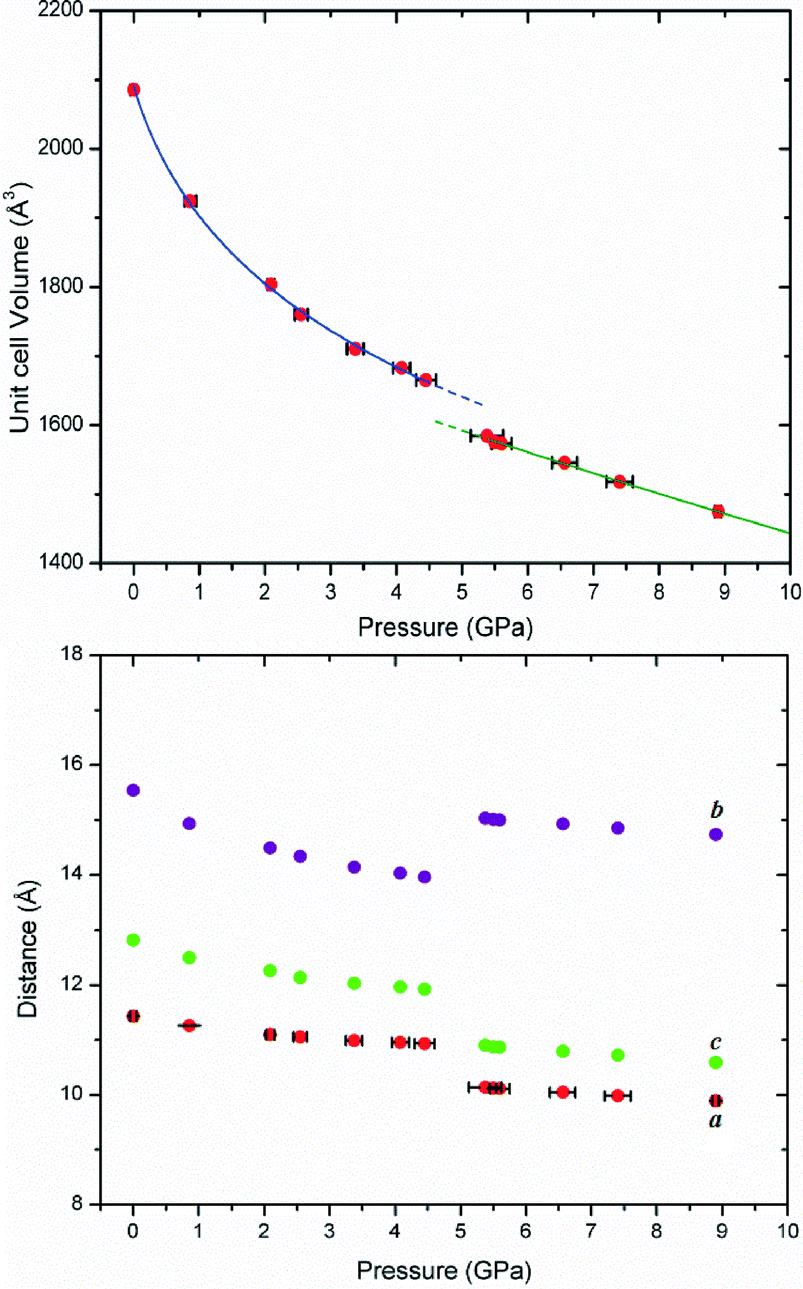 | ||
| Fig. 2 Compound 1. Top) Unit cell volume under compression. A 3rd order BMEOS has been fitted before (blue) and after (green) the phase transition. Bottom) Unit cell parameters of 1. | ||
Compound 2 does not display any dramatic changes in lattice parameters or unit-cell volume, displaying a steady reduction in volume, compressing by 30% at 9.80 GPa (Table 2). It does undergo a space group transformation, from the ambient C2/c cell to a primitive cell of P21/c between 4.15 and 5.70 GPa. The fitting of an equation of state (3rd order Birch–Murnaghan for 1; Murnaghan for 2)43 to the unit cell parameters of both compounds demonstrates that the materials have bulk moduli softer than observed for hydrogen bonded organic molecular materials.44,45 This is perhaps not surprising since there is no significant intermolecular hydrogen bonding in either compound because of the absence of hydrogen bond acceptors. Compound 1 has a B0 of 7.42 GPa, while 2 has a smaller value of 6.15 GPa consistent with the lower packing coefficient. Both values were measured specifically for the low pressure phases below 5.38 GPa for 1 and below 5.70 GPa for 2, respectively.
Unit cell content analysis of 1
At the transition between 4.45 and 5.38 GPa, 1 displays significant structural rearrangement both in terms of molecular conformation and intermolecular interactions. Fig. 3 illustrates how two molecules of 1 slide over one another between 4.45 and 5.38 GPa. The easiest way to quantify the transition is through examination of the thiophene⋯thiophene interaction that occurs between molecules related by the 1 − x, −y, 1 − z symmetry operation. It is clear that the interplanar interaction changes from a thiophene⋯thiophene interaction at 4.45 GPa to a benzothiophene⋯benzothiophene interaction above the pressure of 5.38 GPa. Measurement of the distance between the rings before and after the transition indicates a significant reduction in interplanar distance of 0.3 Å that continues to reduce as pressure is increased. Visual inspection of Hirshfeld surfaces46–50 and fingerprint plots indicates significant changes in the unit cell with an alteration upon transition of the C–H⋯π interactions to C–H⋯π and π⋯π interactions (Fig. 4).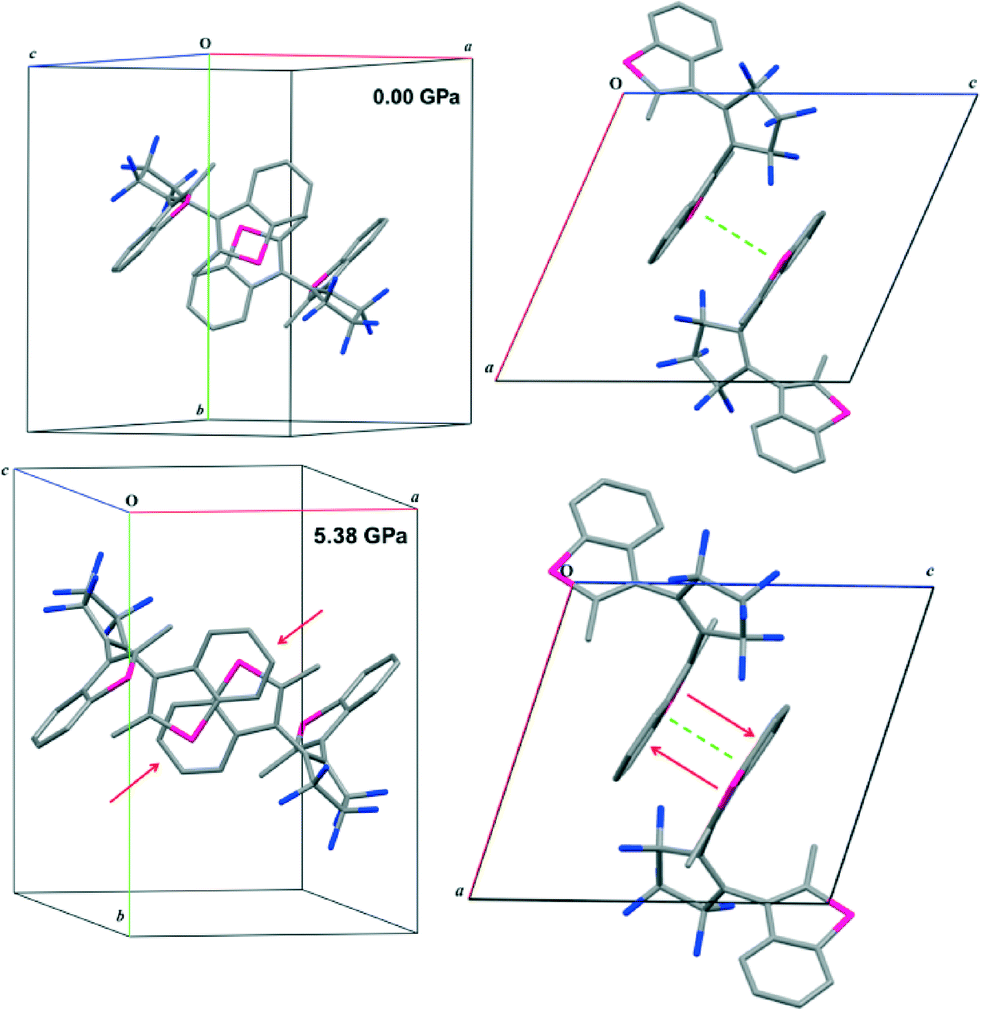 | ||
| Fig. 3 Top) Molecules of 1 at 0.00 GPa demonstrating the packing interaction between them, the dashed green line indicates the measured distance between the planes of the benzothiophene rings. Bottom) 2 molecules of 1 at 5.38 GPa. Red arrows indicate the movement within the molecules upon compression. | ||
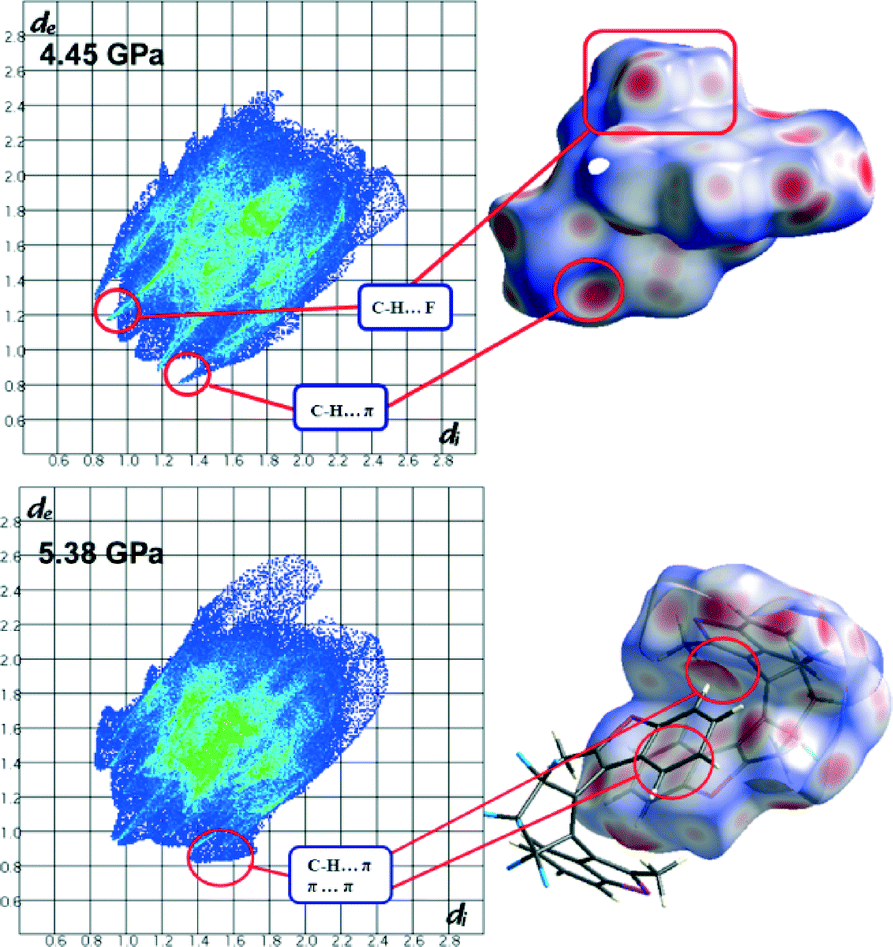 | ||
| Fig. 4 Left) Fingerprint plots of the de and di properties of 1 at 4.45 GPa and 5.38 GPa. Right) Hirshfeld surfaces of 1 with the dnorm property mapped with significant interactions and their changes with pressure highlighted. | ||
In addition to changes in the intermolecular interactions, significant changes in the molecular geometry also occur. The easiest way to quantify these geometric changes is to place a plane through the backbone of the molecule (Fig. 5) and measure the changes in torsion angles and distances relative to this plane. Measurement of the torsion angle between the benzothiophene rings and the plane demonstrate a large deviation in angle upon the phase transition, changing from 128.1(10)° and 114.8(10)° to 96.5(10)° and 90.2(10)° indicating the movement of the benzothiophene rings to a more perpendicular orientation relative to the plane. Interestingly, the carbon atoms associated with the photo-induced ring closure reaction, the C12 and C32 atoms, show movement relative to one another. As discussed earlier the distance between these atoms have been linked to solid state reactivity, the distance between the atoms between ambient and 4.45 GPa reduces consistently from 4.346(3) Å to 4.242(10) Å, however, upon the transition the distance dramatically increases to 4.383(10) Å before continuing to contract as pressure is increased above 5.38 GPa.
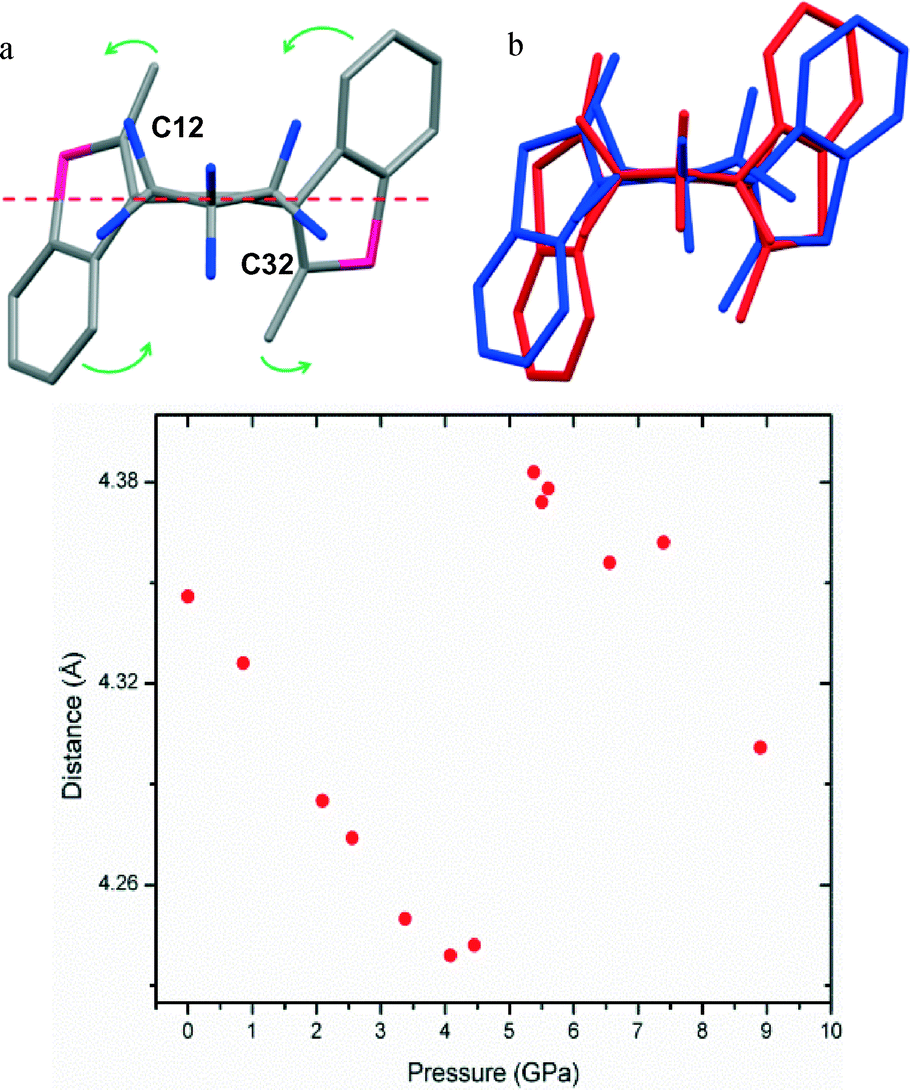 | ||
| Fig. 5 a) Molecule of 1, C13–C1–C5–C33 plane marked with red dash, green arrows indicate molecular movement of atoms relative to the plane. b) Molecule of 1 at 4.45 GPa (blue) and at 5.38 GPa (red) overlaid to demonstrate the changes in conformation in the unit cell between the two phases. c) Intramolecular distance between C12 and C32 carbon atoms. | ||
Using the “molecular overlay” facility in the Mercury software package51 it is possible to calculate the differences between the molecules at different pressures. When the ambient pressure structure is compared with that at 4.45 GPa the root-mean-square deviation (RMSD) value for the overlay of the two molecules is 0.181, while that between the ambient pressure structure with that at 5.38 GPa the RMSD value increases to 0.441. However, the biggest RMSD value is 0.5656 between the structures at 4.45 GPa and at 5.38 GPa reflecting the significant conformational changes across the phase transition.
Unit cell content analysis of 2
Compound 2 undergoes a space group transformation from C2/c (Form-I) to P21/c (Form-II) between 4.15 and 5.70 GPa. The transition results in the loss of a 2-fold rotation axis thereby affording a doubling of the b axis. The cell transforms to the new P21/c setting, quadrupling the number of molecules in the asymmetric unit from 1/2 to two complete molecules and concomitant with a doubling of unit cell volume. The new phase is a previously unreported phase of 2. It is apparent that there are several changes in molecular conformation in the new phase when compared to the ambient phase. Comparison of the two phases suggests that the loss of symmetry is due to multiple small and subtle changes, occurring in the hexafluorocyclopentene (HFCP) ring in combination with conformational distortions of the thiophene rings. The significant distortion in the HFCP ring is shown in Fig. 6. Comparison of the HFCP ring before and after the phase transition shows that there is a puckering of the ring parallel to the direction of the a axis at the higher pressure. Such behaviour can be expected to occur in situations involving lowered temperature or increased pressure, due to the reduced flexibility of the HFCP ring, in particular the C3 carbon and the fluorine atoms. The authors have found that it is common for these atoms to be disordered in structures of DTEs because of the flexibility of the HFCP ring but the disorder problem can be overcome by collecting data at lower temperature, resolving the group to a single position. The application of pressure has a similar effect, restraining the flexibility of the HFCP ring to one position and reducing thermal motion as has been observed in other systems.52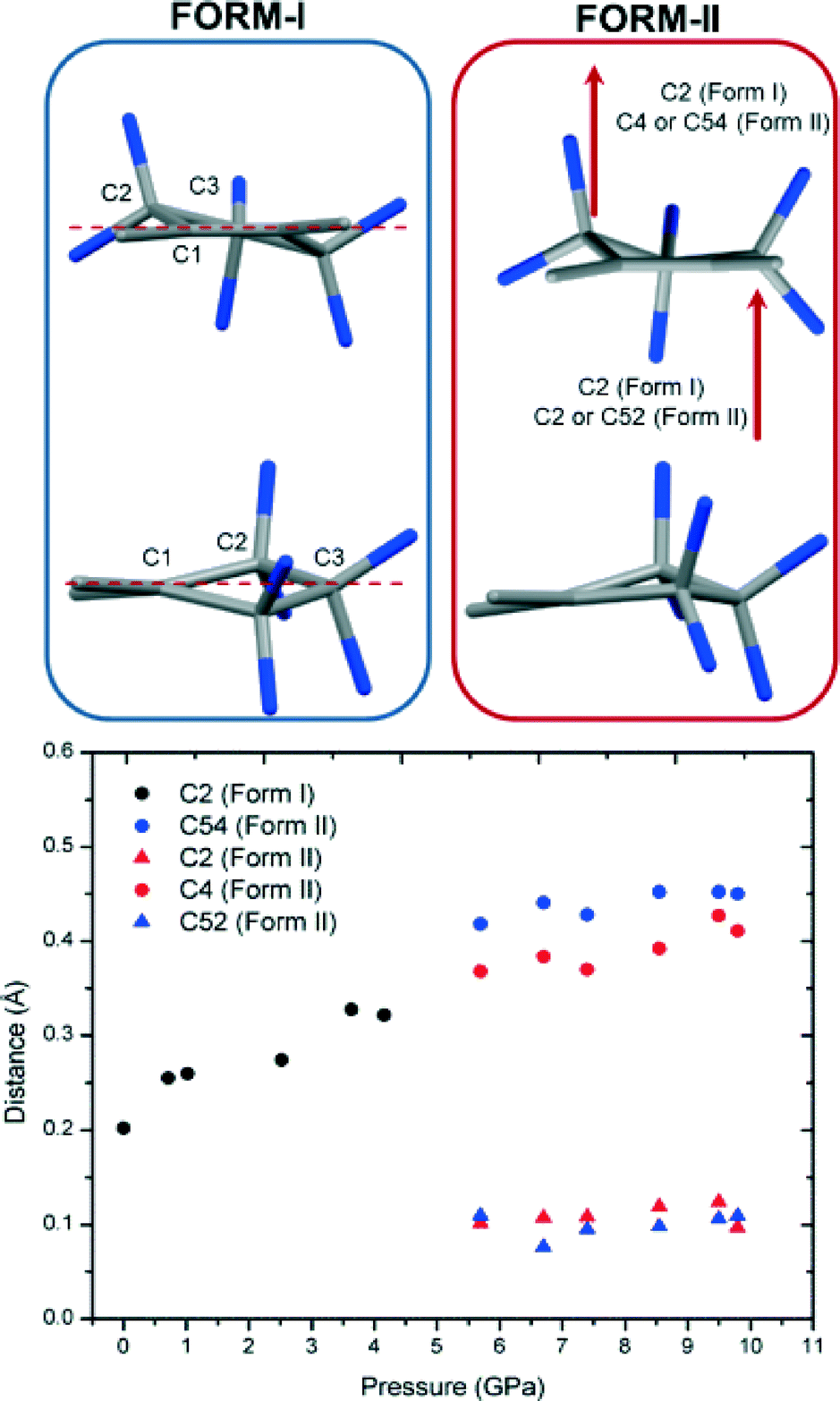 | ||
| Fig. 6 Compound 2: left) HFCP ring in Form-I. Right) HFCP ring in Form-II, with all other atoms omitted for clarity, red arrows indicate distortion. Bottom) Analysis of the displacement of noted atoms from a plane (C1–C3–C1 in Form-I; C1–C3–C4 and C51–C53–C54 of Form-II) in the HFCP ring in Form-I and Form-II. | ||
The subtle conformational difference between the structures on either side of the phase transition is illustrated in Fig. 7. The distortion of the rings is clearly apparent, indicated by the green arrows, but it is difficult to quantify the changes in terms of molecular parameters altered because of the complex geometry of the molecule. All the minor changes combine to force a lowering of the crystal symmetry. Further detailed analysis may be found in the ESI.† Changes in distances between the C12–C32, C62–C82 carbon atoms are of interest due to their relation to the photochromic behaviour of the material and it can be seen that there is a significant reduction in the distance in a similar manner to that observed in 1 from 3.581(2) Å to 3.20(2) Å between ambient and 9.80 GPa, however, it is clear that there is no relaxation of the value over the phase transition as observed in 1.
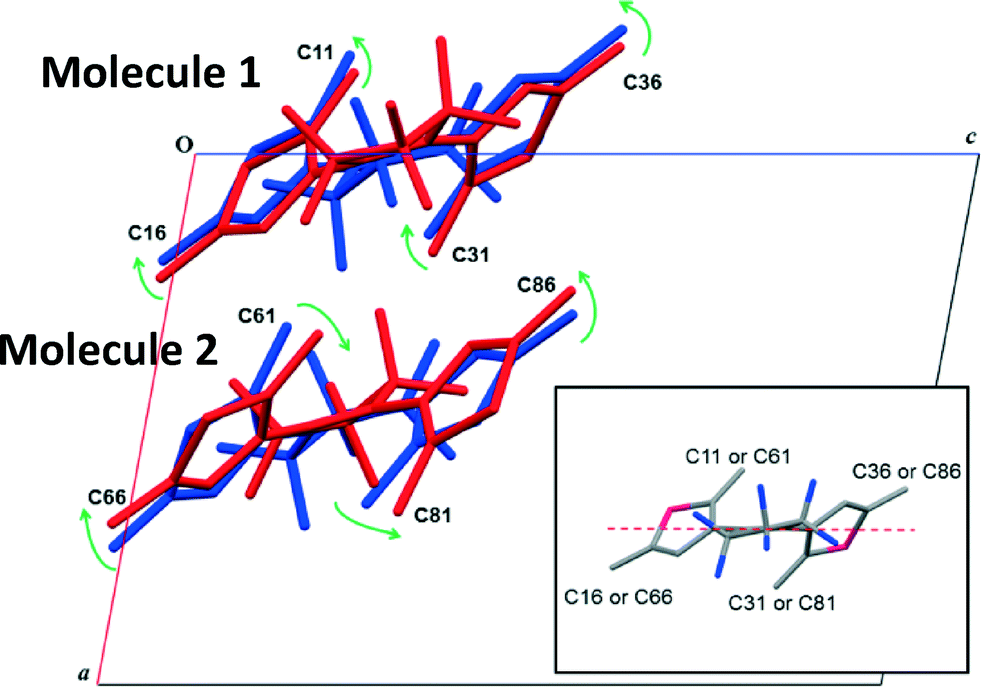 | ||
| Fig. 7 Distortion of molecules in Form-II relative to the plane displayed in the inset. Colour indicates molecules in the same asymmetric unit. Inset) Plane used to measure relative molecular changes in a molecule of 2. | ||
CLP analysis
Computational methods were employed in order to further understand the changes occurring within the crystal structure of compound 1 using the PIXEL module of the CLP package.53,54 As pressure increases, the overall lattice energy of the system increases (Fig. 8). The lattice energy can be broken down into individual components of coulombic, polarisation, dispersive and repulsive interaction energy, as is shown in Table 3 and shows that the increase in lattice energy is largely due to the increasing value of the repulsive component of total energy. There appears to be no significant reduction in the packing energy between 4.45 GPa and 5.38 GPa as one might expect if a more energy favourable packing arrangement had been achieved.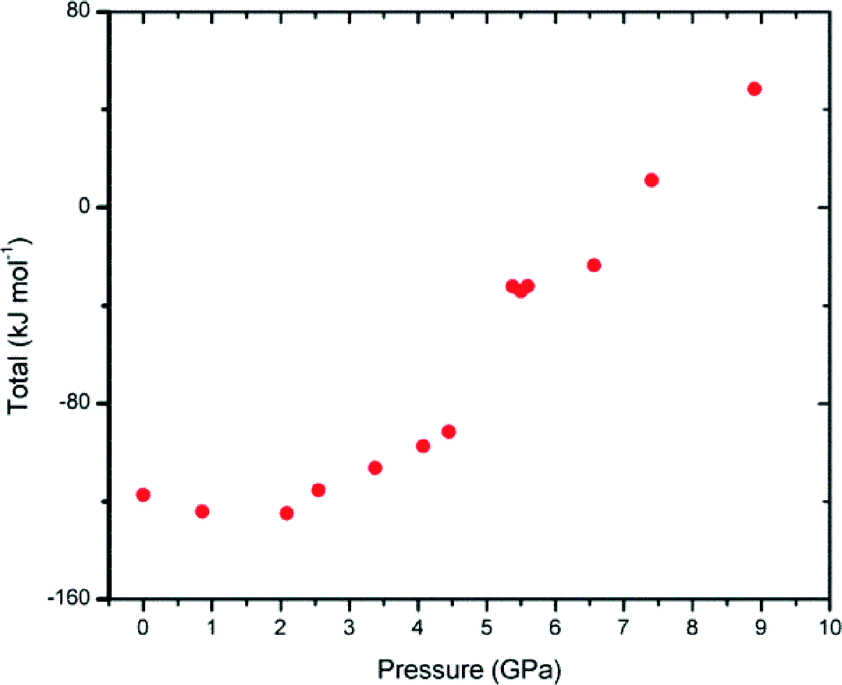 | ||
| Fig. 8 Total lattice energy as calculated by the PIXEL method of the CLP programme suite. Right) Combined coulombic (black), polarisation (red), dispersion (green) and repulsive energies generated by the PIXEL program of the CLP program suite. | ||
| Ambient | 0.86 | 2.09 | 2.55 | 3.38 | 4.08 | 4.45 | 5.38 | 5.50 | 5.60 | 6.56 | 7.40 | 8.90 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E col | −37.1 | −61.6 | −102.3 | −120.4 | −145.4 | −160.4 | −170.9 | −218.3 | −228.5 | −232.2 | −258.1 | −287.1 | −336.5 |
| E pol | −17.8 | −32.8 | −55.4 | −65.1 | −79.3 | −87.6 | −94.8 | −139.2 | −150.7 | −156.1 | −178.5 | −199.2 | −229.4 |
| E disp | −155.3 | −201.8 | −252.5 | −272.9 | −298.5 | −312.4 | −323.1 | −372.1 | −377.3 | −378.4 | −399.1 | −417.2 | −454.7 |
| E rep | 92.9 | 172.0 | 285.4 | 343.0 | 417.0 | 462.9 | 497.3 | 697.5 | 722.2 | 734.5 | 812.1 | 914.7 | 1069.1 |
| E tot | −117.3 | −124.3 | −124.9 | −115.4 | −106.3 | −97.5 | −91.5 | −32.2 | −34.2 | −32.1 | −23.6 | 11.3 | 48.5 |
| U adj | −117.3 | −110.8 | −106.1 | −94.5 | −82.6 | −71.8 | −62.5 | 24.0 | 27.9 | 33.8 | 40.2 | 84.7 | 132.1 |
| H ‡ | −117.3 | 138.3 | 461.5 | 581.3 | 786.6 | 961.9 | 1053.0 | 1306.8 | 1332.4 | 1360.2 | 1566.5 | 1775.8 | 2108.0 |
The PIXEL method only accounts for intermolecular interactions within the crystal lattice and does not take into account the large conformational changes that have occurred within the individual molecules. Such changes must be taken into account to gain a realistic estimate of the total lattice energy. Fortunately no further calculations are required as GAUSSIAN 09 calculates the total molecular energy when creating the electron density map for the PIXEL calculation. Therefore, the adjusted total lattice enthalpy (Uadj) is calculated by subtracting the energy difference due to the conformational change relative to that of the ambient structure from the PIXEL calculated total lattice energy; the results of which are given in Table 3. It is clear from the calculations that the conformational changes within the crystal structure do not have a stabilising or negative energetic effect on the total lattice energy. For 1, an increase in lattice energy between the pressure of 4.45 and 5.38 GPa is observed of approximately +86.5 kJ mol−1 (62.5 kJ mol−1 + 24.0 kJ mol−1). A contribution of 56 kJ mol−1 is due to the large conformational changes suggesting that both the changes in crystal packing and conformation result in a less energetically favourable structure in the new high-pressure phase. Previous studies have demonstrated that new conformations sometimes result to relieve strain that occurs between intermolecular interactions such as hydrogen bonding or stacking interactions at higher pressure as demonstrated by salicylaldoxime55 resulting in a dramatic decrease in lattice energy. Other transitions, such as those seen in serine, appear to be driven by the formation of more stable conformations as well as a substantial decrease in unit cell volume19 while the behaviour of salicylamide can be said to be driven by a combination of factors including a reduction in volume; strength increases in specific interactions and favourable entropy.56 It appears that it is more likely that the transition described for 1 is driven by lower molecular volume and the more efficient packing observed in the high-pressure form.18,19,45 Using the values of (Uadj) the values of enthalpy (H‡) can be calculated using the equation H = (Uadj) + Pv where P = pressure (Pa) and v = molar volume (mol m−3) the results of which are also given in Fig. 9. The lattice enthalpy becomes increasingly positive, dominated by the Pv term. The enthalpy for the two phases may be examined individually using linear lines of best fit.
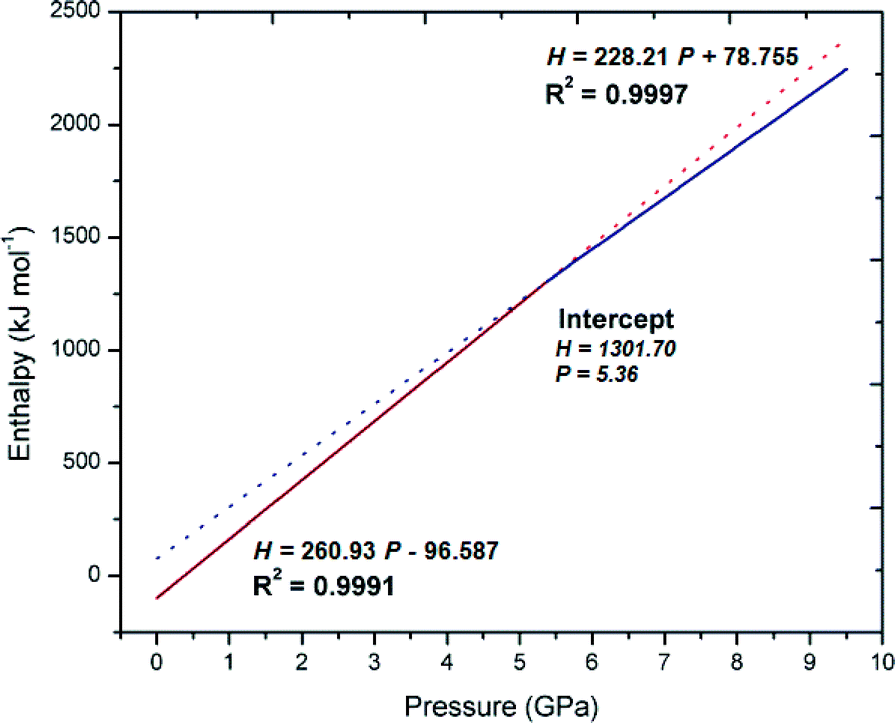 | ||
| Fig. 9 Linear lines of fit of the lattice enthalpy of 1 before (red) and after (blue) the phase transition with the point of intercept displayed. | ||
Fig. 9 displays a linear line of fit to Form-I. The line is extrapolated to higher pressures where it is clear that the data points of Form-II sit on or below the line of best fit as pressure increases above 5.38 GPa. If the experimentally obtained value of H‡ is compared to that estimated from the line of fit at 5.38 GPa, the lowest Form-II data point, a −0.4 kJ mol−1 discrepancy is observed, however, there is a definite change in gradient of the lines as shown clearly on the right of Fig. 9, something that is consistent with other high-pressure induced transitions of this sort.20 At the highest pressure observed of 8.90 GPa, the discrepancy between the two forms has reached the value of −111.69 kJ mol−1 suggesting that Form-II is significantly more stable at higher pressures than Form-I. In contrast the phase transition for 2 may be attributed to the loss of symmetry that occurs as the molecule is forced to occupy smaller volume with the previous disorder being resolved to multiple crystallographic sites.
Spectroscopy
High-pressure UV-Vis studies were hampered by a combination of factors including sample fragility and sensitivity to the light source used in the measurements rendering the study of 2 impossible. However, high-pressure spectra for 1 were measured successfully from ambient pressure through to 15.4 GPa, and produced good quality spectra presented in Fig. 10. Compound 1 was not initially chromic under ambient conditions37 and it was deemed of interest to see if piezochromism could be induced with pressure. | ||
| Fig. 10 Solid state UV-Vis spectroscopy of a single crystal of 1 with pressure. Bottom) images of 1 at varying pressures. | ||
The form of the spectra remained consistent throughout the pressure ranges measured although there is a marked change in intensity, between 3.5 and 6.3 GPa, in the region of the crystallographically identified phase transition. There was a significant colour change with the crystal changing from colourless to red with increasing pressure, and returning back to colourless upon relaxation of pressure as shown in Fig. 10. This establishes that there is a piezochromic effect with increasing pressure. Similar behaviour has been observed for a series of aromatic organic compounds known as bianthrones.21,22 In these complexes a significant red shift is observed in absorption band associated with the π–π* transition at 450 nm. It seems highly probable that a similar effect is observed here for 1 due to the aromatic nature of the compound.
Conclusions
Two crystalline DTE compounds have had their high-pressure behaviour investigated by the application of hydrostatic high-pressure single-crystal X-ray crystallographic and UV/visible spectroscopic methods. The findings have been supported by PIXEL and GAUSSIAN computational studies. The two compounds exhibit fascinating but different behaviour upon the application of high-pressures.1,2-Bis(2-methylbenzothiophen-3-yl)perfluorocyclopentene 1 exhibits a phase change over the pressure range 4.45–5.35 GPa concomitant with a dramatic reduction in the unit-cell volume of 20% and an increase in the b-axis length by 7.7%. This phase change is accompanied by a gradual change in colour of the crystals from colourless to red. At the molecular level, the phase change is coincident with a change in crystal packing, where adjacent molecules slide over each other so that the dominant stacking interaction changes from a π⋯π thiophene⋯thiophene interaction to a benzothiophene⋯benzothiophene interaction. The high-pressure UV/Visible spectroscopy shows a relatively smooth red shift associated with π⋯π* transitions across the pressure range from 0.00–15.4 GPa. The colour change is reversible upon reduction in the pressure consistent with a piezochromic effect. Computational studies show that the phase transition is not the result of a preferred realignment of the molecules with increasing pressure but is the result of a more favourable Pv dominated enthalpy term that affords a more thermodynamically stable crystal structure at higher pressures.
In contrast, 1,2-bis(2,5-dimethylthiophen-3-yl)perfluorocyclopentene 2 shows a more modest reduction in the volume of the P21/c unit-cell until 4.15 GPa, then a phase transition, with a transformation to a C2/c unit cell at 5.70 GPa. The space group change results in a loss of symmetry and the contents of the asymmetric unit changing from half a unique molecule to two independent molecules. The molecules show significant reorientation of their ring systems with increasing pressure but no major change in intermolecular interactions.
It is apparent that the high-pressure behaviour of the DTE molecules is dependent both on the molecular conformations and crystal packing, and also on the overall thermodynamic properties of the crystalline materials. The main difference in behaviour in the two molecules studied may be related to the difference in substituents on the thiophene rings in this case and their abilities to form π-stacking interactions. However, since both materials studied undergo single-crystal to single-crystal phase transitions implies that DTEs are a fascinating class of materials that are worthy of further high-pressure investigations because of their potential use as piezochromic sensors.
Experimental
General
The synthesis of 1 and 2 were based upon the methods reported by Irie et al. and Lucas et al., respectively.10,57–59 All chemicals were purchased from commercial sources and used as received. All reactions were performed under an atmosphere of dry nitrogen, however, subsequent workup was performed in air. NMR spectra were collected on a Bruker AV500 spectrometer (500.1 MHz for 1H, 470.6 MHz for 19F and 125.8 MHz for 13C) unless otherwise stated.Crystallography
High-pressure single crystal X-ray diffraction experiments were performed on a 4-circle Rigaku Saturn CCD diffractometer at station I19, Diamond Light Source, UK. A Merrill-Bassett diamond anvil cell (DAC) was used for the high-pressure measurements using Boehler-Almax diamonds with 600 μm culets. Laser cut tungsten was used as gasket material. Gasket holes were drilled using a BETSA electric discharge drill. Loading of the cell for all samples was performed using 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 methanol–ethanol mix as a hydrostatic medium, using ruby spheres as the pressure calibrant. Pressure calibration was performed via the ruby fluorescence method.60
:1 methanol–ethanol mix as a hydrostatic medium, using ruby spheres as the pressure calibrant. Pressure calibration was performed via the ruby fluorescence method.60
High-pressure data were integrated using the APEX 2 software suite. Shielding of the diffraction pattern by the DAC was dealt by the generation of dynamic masks using an external program.61 Datasets were merged using XPREP and a multi-scan absorption correction was performed using SADABS.62 No attempt at structural solution was made due to the low completeness of the high pressure datasets due to shielding of the DAC however data was refined against a previously determined room-temperature structure by full-matrix least squares on F2 using SHELXL-97 with exception of the new P21/c form where the structure was solved using SHELXS before further refinement.63 All C–F, C–C and C–S distances in the structure were restrained to the values of the room-temperature structure, on the assumption that such interactions are relatively resilient to compression. The majority of atoms were refined isotropically due to the low completeness of the data however were possible sulphur atoms were made anisotropic. The program CRYSTALS was also utilised during the process of data analysis to identify and remove anomalous reflections associated with overlap between sample reflections and those of the diamonds or gasket rings.64
Generation of Hirshfeld surfaces and fingerprint plots was performed using Crystal Explorer.65 Equations of state were fitted using EOSfit.43
PIXEL calculations
The final single-crystal structure obtained from refinement of the diffraction data were used to calculate the molecular electron densities of 2 at each pressure using the program GAUSSIAN 09 with the MP2/6-31G** basis set.66 H-atom distances were set to the standard neutron values in all calculations (C–H = 1.083 Å). The calculated electron density was used to evaluate the packing energies using the PIXEL method as provided in the CLP program suite to provide an estimate of the total packing energy as well as a breakdown of the packing energy into its coulombic, polarisation, dispersion and repulsion components.67Spectroscopy
Room-temperature optical absorption measurements at high-pressure were performed in custom-made setup that can be described as follows:The detection setup was equipped with a photomultiplier (Hamamatsu R928S) for measurements in the near UV-VIS range. The modulated monochromatic light (f = 137 Hz) from a deuterium or tungsten lamp was dispersed with an Acton Research Corporation SpectraPro-300i monochromator, and focused on the sample with a reflective microscope objective. The transmitted light was collected with another reflective microscope objective and the signal was synchronously detected with a lock-in amplifier (Stanford Research SR830). High-pressure measurements were carried out in a gasketed membrane DAC with spectroscopic paraffin oil (Merck) as transmitting media. The hydrostatic cavity had a diameter of 200 μm performed in the pre-indented Inconel gaskets with a sparkling machine (BETSA). The pressure was measured through the R-line shift of ruby chips introduced in the hydrostatic cavity. The ruby luminescence was excited with the 568 nm line of a Coherent I-302 Kr+-laser.
Acknowledgements
We are grateful to the EPSRC for financial support and for studentships to CHW and SS (EP/F021151 and EP/D072859), to the University of Bath for support for SKB, and Diamond Light Source Ltd for the award of beamtime.Notes and references
- M. Irie, T. Fukaminato, T. Sasaki, N. Tamai and T. Kawai, Nature, 2002, 420, 759–760 CrossRef CAS PubMed.
- S. Kobatake, S. Takami, H. Muto, T. Ishikawa and M. Irie, Nature, 2007, 446, 778–781 CrossRef CAS PubMed.
- F. Terao, M. Morimoto and M. Irie, Angew. Chem., Int. Ed., 2012, 51, 901–904 CrossRef CAS PubMed.
- M. Irie, S. Kobatake and M. Horichi, Science, 2001, 291, 1769–1772 CrossRef CAS PubMed.
- S. Kobatake, Y. Matsumoto and M. Irie, Angew. Chem., Int. Ed., 2005, 44, 2148–2151 CrossRef CAS PubMed.
- M. Morimoto and M. Irie, Chem. Commun., 2005, 3895–3905 RSC.
- T. Kodani, K. Matsuda, T. Yamada, S. Kobatake and M. Irie, J. Am. Chem. Soc., 2000, 122, 9631–9637 CrossRef CAS.
- M. Morimoto and M. Irie, J. Am. Chem. Soc., 2010, 132, 14172–14178 CrossRef CAS PubMed.
- S. Takami, L. Kuroki and M. Irie, J. Am. Chem. Soc., 2007, 129, 7319–7326 CrossRef CAS PubMed.
- T. Yamada, S. Kobatake, K. Muto and M. Irie, J. Am. Chem. Soc., 2000, 122, 1589–1592 CrossRef CAS.
- M. Irie, Chem. Rev., 2000, 100, 1683–1684 CrossRef CAS PubMed.
- M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed.
- S. Kobatake, K. Uchida, E. Tsuchida and M. Irie, Chem. Commun., 2002, 2804–2805 RSC.
- E. V. Boldyreva, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 218–231 CrossRef CAS PubMed.
- S. A. Moggach, W. G. Marshall and S. Parsons, Acta Crystallogr., Sect. B: Struct. Sci., 2006, 62, 815–825 Search PubMed.
- S. A. Moggach, S. Parsons and P. A. Wood, Crystallogr. Rev., 2008, 14, 143–183 CrossRef CAS.
- E. V. Boldyreva, Phase Transitions, 2009, 82, 303–321 CrossRef CAS.
- R. D. L. Johnstone, D. Francis, A. R. Lennie, W. G. Marshall, S. A. Moggach, S. Parsons, E. Pidcock and J. E. Warren, CrystEngComm, 2008, 10, 1758–1769 RSC.
- P. A. Wood, D. Francis, W. G. Marshall, S. A. Moggach, S. Parsons, E. Pidcock and A. L. Rohl, CrystEngComm, 2008, 10, 1154–1166 RSC.
- N. P. Funnell, A. Dawson, D. Francis, A. R. Lennie, W. G. Marshall, S. A. Moggach, J. E. Warren and S. Parsons, CrystEngComm, 2010, 12, 2573–2583 RSC.
- N. A. Tumanov, E. V. Boldyreva, B. A. Kolesov, A. V. Kurnosov and R. Q. Cabrera, Acta Crystallogr., Sect. B: Struct. Sci., 2010, 66, 458–471 CAS.
- E. V. Boldyreva, T. P. Shakhtschneider, M. A. Vasilchenko, H. Ahsbahs and H. Uchtmann, Acta Crystallogr., Sect. B: Struct. Sci., 2000, 56, 299–309 Search PubMed.
- H. Sowa and E. Boldyreva, J. Therm. Anal. Calorim., 2002, 68, 437–452 CrossRef.
- F. P. A. Fabbiani, D. R. Allan, W. I. F. David, S. A. Moggach, S. Parsons and C. R. Pulham, CrystEngComm, 2004, 6, 504–511 RSC.
- F. P. A. Fabbiani, D. R. Allan, S. Parsons and C. R. Pulham, CrystEngComm, 2005, 7, 179–186 RSC.
- F. P. A. Fabbiani, D. R. Allan, W. I. F. David, A. J. Davidson, A. R. Lennie, S. Parsons, C. R. Pulham and J. E. Warren, Cryst. Growth Des., 2007, 7, 1115–1124 CAS.
- F. P. A. Fabbiani, D. C. Levendis, G. Buth, W. F. Kuhs, N. Shankland and H. Sowa, CrystEngComm, 2010, 12, 2354–2360 RSC.
- F. P. A. Fabbiani and C. R. Pulham, Chem. Soc. Rev., 2006, 35, 932–942 RSC.
- D. I. A. Millar, W. G. Marshall, I. D. H. Oswald and C. R. Pulham, Crystallogr. Rev., 2010, 16, 115–132 CrossRef CAS.
- K. W. Galloway, S. A. Moggach, P. Parois, A. R. Lennie, J. E. Warren, E. K. Brechin, R. D. Peacock, R. Valiente, J. Gonzalez, F. Rodriguez, S. Parsons and M. Murrie, CrystEngComm, 2010, 12, 2516–2519 RSC.
- L. Vincent, Recent Advances in Crystallography, ed. J. B. Benedict, Croatia, 2012, ch. 2 Search PubMed.
- H. L. S. Wong, D. R. Allan, N. R. Champness, J. McMaster, M. Schroeder and A. J. Blake, Angew. Chem., Int. Ed., 2013, 52, 5093–5095 CrossRef CAS PubMed.
- A. Prescimone, C. J. Milios, S. Moggach, J. E. Warren, A. R. Lennie, J. Sanchez-Benitez, K. Kamenev, R. Bircher, M. Murrie, S. Parsons and E. K. Brechin, Angew. Chem., Int. Ed., 2008, 47, 2828–2831 CrossRef CAS PubMed.
- H. J. Shepherd, S. Bonnet, P. Guionneau, S. Bedoui, G. Garbarino, W. Nicolazzi, A. Bousseksou and G. Molnar, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84 Search PubMed.
- H. J. Shepherd, T. Palamarciuc, P. Rosa, P. Guionneau, G. Molnar, J.-F. Letard and A. Bousseksou, Angew. Chem., Int. Ed., 2012, 51, 3910–3914 CrossRef CAS PubMed.
- T. Salzillo, I. Bilotti, R. G. Della Valle, E. Venuti and A. Brillante, J. Am. Chem. Soc., 2012, 134, 17671–17679 CrossRef CAS PubMed.
- S. Kobatake, M. Yamada, T. Yamada and M. Irie, J. Am. Chem. Soc., 1999, 121, 8450–8456 CrossRef CAS.
- A. I. Kitaigorodskii, Organic Chemical Crystallography, Consultants Bureau, New York, 1961 Search PubMed.
- A. G. Christy, Acta Crystallogr., Sect. B: Struct. Sci., 1995, 51, 753–757 CrossRef.
- D. R. Allan, A. J. Blake, D. Huang, T. J. Prior and M. Schroeder, Chem. Commun., 2006, 4081–4083 RSC.
- D. R. Allan and R. J. Nelmes, J. Phys.: Condens. Matter, 1996, 8, 2337–2363 CrossRef.
- F. Datchi, S. Ninet, M. Gauthier, A. M. Saitta, B. Canny and F. Decremps, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73 Search PubMed.
- R. Angel, EOSFIT version 5.2, ( 2002) Search PubMed.
- P. A. Wood, R. S. Forgan, A. R. Lennie, S. Parsons, E. Pidcock, P. A. Tasker and J. E. Warren, CrystEngComm, 2008, 10, 239–251 RSC.
- N. P. Funnell, A. Dawson, W. G. Marshall and S. Parsons, CrystEngComm, 2013, 15, 1047–1060 RSC.
- J. J. McKinnon, A. S. Mitchell and M. A. Spackman, Chem.–Eur. J., 1998, 4, 2136–2141 CrossRef CAS.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627–668 CrossRef PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- P. A. Wood, J. J. McKinnon, S. Parsons, E. Pidcock and M. A. Spackman, CrystEngComm, 2008, 10, 368–376 CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- Y. Okano, B. Zhou, H. Tanaka, T. Adachi, Y. Ohishi, M. Takata, S. Aoyagi, E. Nishibori, M. Sakata, A. Kobayashi and H. Kobayashi, J. Am. Chem. Soc., 2009, 131, 7169–7174 CrossRef CAS PubMed.
- A. Gavezzotti, Mol. Phys., 2008, 106, 1473–1485 CrossRef CAS.
- A. Gavezzotti, J. Phys. Chem. B, 2003, 107, 2344–2353 CrossRef CAS.
- P. A. Wood, R. S. Forgan, D. Henderson, S. Parsons, E. Pidcock, P. A. Tasker and J. E. Warren, Acta Crystallogr., Sect. B: Struct. Sci., 2006, 62, 1099–1111 CAS.
- R. D. L. Johnstone, A. R. Lennie, S. F. Parker, S. Parsons, E. Pidcock, P. R. Richardson, J. E. Warren and P. A. Wood, CrystEngComm, 2010, 12, 1065–1078 RSC.
- J. Lehn, Chem.–Eur. J., 1995, 275–284 Search PubMed.
- J. Lehn and S. Kawai, Chem.–Eur. J., 1995, 1, 285 CrossRef.
- L. N. Lucas, J. J. D. de Jong, J. H. van Esch, R. M. Kellogg and B. L. Feringa, Eur. J. Org. Chem., 2003, 155–166 CrossRef CAS.
- G. J. Piermarini, S. Block, J. D. Barnett and R. A. Forman, J. Appl. Phys., 1975, 46, 2774–2780 CrossRef CAS.
- S. Parsons, D. Allan, A. Dawson and M. Ruf, J. Appl. Crystallogr., 2003, 37, 410–416 Search PubMed.
- APEX 2, Saint-Plus, XPREP and SADABS, ( 2005) Bruker AXS Inc, Madison, Wisconsin, USA Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- P. W. Betterridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, Crystal Explorer (Version 3.0), ( 2012), University of Western Australia Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03, Gaussian, Inc., Wallingford, CT, 2003 Search PubMed.
- A. Gavezzotti, New J. Chem., 2011, 35, 1360–1368 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details of all X-ray diffraction and spectroscopic experimental studies. CCDC 961251–961276. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ce41933a |
| This journal is © The Royal Society of Chemistry 2014 |