
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Long-lived charge separation in a rigid pentiptycene bis(crown ether)–Li+@C60 host–guest complex†
Mustafa
Supur
a,
Yuki
Kawashima
a,
Ying-Xian
Ma
b,
Kei
Ohkubo
a,
Chuan-Feng
Chen
*b and
Shunichi
Fukuzumi
*a
aDepartment of Material and Life Science, Graduate School of Engineering, Osaka University, ALCA, Japan Science and Technology Agency (JST), Suita, Osaka 565-0871, Japan. E-mail: fukuzumi@chem.eng.osaka-u.ac.jp; Fax: +81-6-6879-7370; Tel: +81-6-6879-7368
bBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Molecular Recognition and Function, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: cchen@iccas.ac.cn
First published on 31st October 2014
Abstract
We report long-lived charge separation in a highly rigid host–guest complex of pentiptycene bis(crown ether) and Li+@C60, in which the pentiptycene framework is actively involved as an electron donor in a photoinduced electron-transfer process to the excited states of Li+@C60 through a rigid distance in the complex.
As the essential process of solar energy conversion in photosynthesis, charge separation with a long lifetime in the photosynthetic reaction centre (PRC) of photosystem II is realized by the arrangement of the electron donor and acceptor units through a rigid distance established by the weak interactions within the protein environment.1 By the spatial alignment of redox units, fast charge recombination is prevented. Constructing a rigid distance between the electron donor and acceptor moieties, assembled via weak interactions in the supramolecular model systems, is still a challenge because in most of these systems, donor and acceptor units interact directly through weak interactions, e.g. π–π interactions, in which no practical electron-transfer distance exists.2 In the case of indirect combination of donor and acceptor through a spacer group, e.g. metal–ligand axial coordination, the lack of rigidity is an obstacle for establishing a solid electron-transfer distance,3 providing a slow charge recombination. There is still a demand for rigid supramolecular systems in which the electron donor and acceptor are set in fixed positions as in the natural PRC.
In this study, we proposed a new host–guest complex consisting of a rigid pentipycene-based bis(crown ether) host (PBCE) and Li+@C60 guest. PBCE hosts the fullerene cage via the cavity between two benzo-crown ether moieties (hosting site) while an electron-transfer process from the pentiptycene unit (electron-donating site), which is spatially positioned to the Li+@C60, occurs after the photoexcitation (Fig. 1).
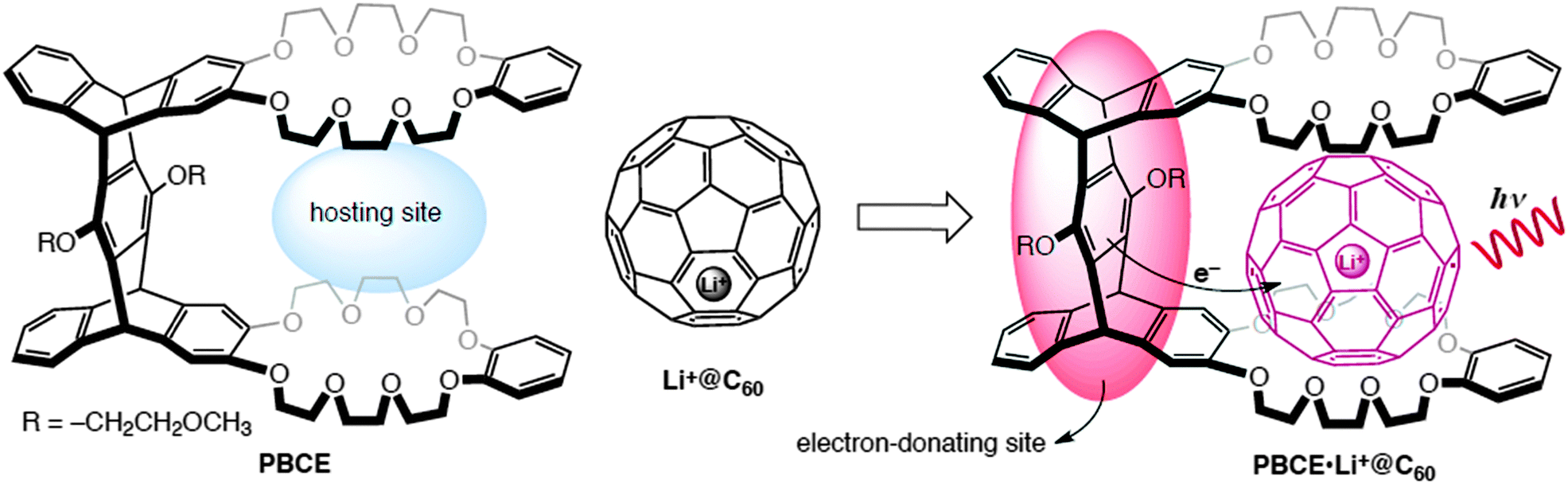 | ||
| Fig. 1 Molecular structures of the corresponding compounds and proposed host–guest complex with the description of the complexation and the electron-transfer mechanism upon the excitation of Li+@C60. | ||
Pentiptycene is a good choice due to its three-dimensional rigid structure formed by a unique combination of five planar arenes with trigonal bicyclo[2.2.2]octane bridges, and it has found numerous applications in polymer and supramolecular chemistry.4 The most notable use of pentiptycene for the former was its integration in to the highly conjugated backbones of emissive polymers or oligomers to minimize the interchain π-stacking, thereby enhancing the fluorescence.5 Supramolecular complexes involving pentiptycene frameworks have been utilized in molecular devices, such as rotors, bevel gears and so on.6
Host–guest complexes with pentiptycene scaffolds have been mostly developed after their functionalisation with crown ether moieties.7 The first reported example of PBCE is where it is effectively hosting the cyclobis(paraquat-p-phenylene) guest through benzo-crown ethers.8 After that, the trans arrangement of PBCE and pentiptycene mono(crown ether) (PMCE) were reported to form stable 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:1 complexes with paraquat derivatives, respectively.9 Recently, benzo-crown ethers, acting as macrocycles in a PBCE-based rotaxane, were shown to oscillate simultaneously between ammonium and triazolium stations on a dumbbell molecule by acid–base stimulus.10 As a result, while the pentiptycene frame conserves the rigidity of PBCE the tensile crown ethers can host molecular guests with large sizes. The electron-donating feature of pentiptycene has yet to be utilized in a photoinduced electron-transfer process in host–guest complexes of pentiptycenes.
:1 and 1:1 complexes with paraquat derivatives, respectively.9 Recently, benzo-crown ethers, acting as macrocycles in a PBCE-based rotaxane, were shown to oscillate simultaneously between ammonium and triazolium stations on a dumbbell molecule by acid–base stimulus.10 As a result, while the pentiptycene frame conserves the rigidity of PBCE the tensile crown ethers can host molecular guests with large sizes. The electron-donating feature of pentiptycene has yet to be utilized in a photoinduced electron-transfer process in host–guest complexes of pentiptycenes.
Crown ethers with appropriate sizes have been demonstrated to associate with pristine11 and metal-containing fullerenes12 through n–π interactions. In a previous study, Li+@C60 was reported to associate with π-electron donors bearing crown-ether moieties.13 Endohedral metal ions of fullerenes also assist binding with the electron-rich crown ethers.12,13 While preserving the photophysical features of pristine C60, Li+@C60 has a remarkably higher one-electron reduction potential.14 This enables efficient photoinduced electron transfer from the pentiptycene scaffold of PBCE to the excited states of Li+@C60 (Fig. 1).
The complex formation was examined by UV-vis spectroscopy. Upon the addition of PBCE to a benzonitrile (PhCN) solution of Li+@C60, a steady decrease at 334 nm was observed while the absorption at around 300 and 450 nm increased (Fig. 2). Finite decreases and slight blue shifts of the absorption bands of Li+@C60 were also noted at 539 and 596 nm during the titration with PBCE (Fig. S1, ESI†). A plot of absorbance at 334 nm versus the molar ratio of added PBCE to the total concentration gives the stoichiometry of the complexation. The plot gives an intersection at 0.5, indicating the formation of a 1:1 complex (PBCE˙Li+@C60) as illustrated in Fig. 1. The association constant (K) for PBCE˙Li+@C60 was determined using eqn (1),13
| (α−1 − 1)−1 = K([PBCE] − α[Li+@C60]0) | (1) |
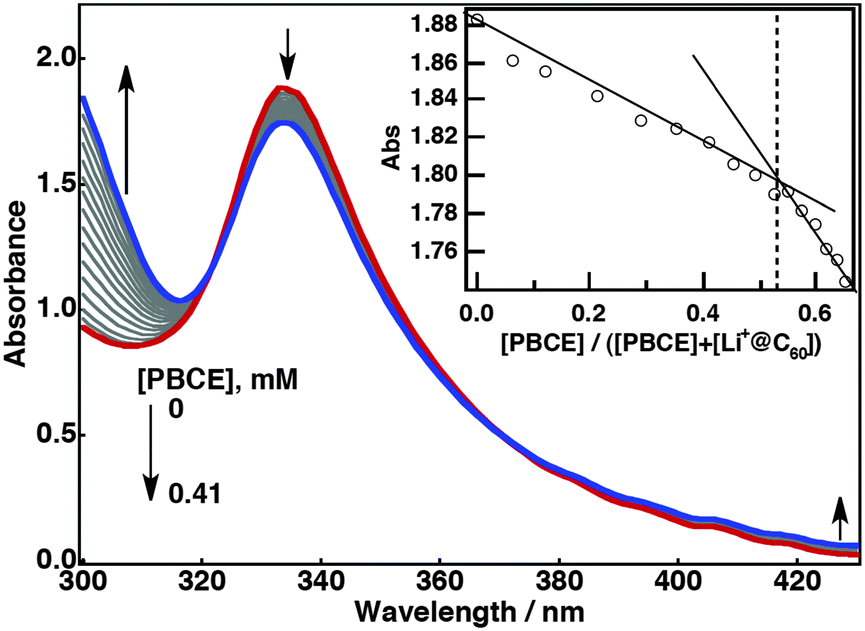 | ||
| Fig. 2 Absorption spectral changes during the titration of 0.23 mM Li+@C60 with 6.3 mM PBCE in PhCN at room temperature. Inset: Plot of absorbance at 334 nm versus [PBCE]/([PBCE] + [Li+@C60]) indicating the stoichiometry of the complex. | ||
The titration of pristine C60 with PBCE did not give any significant absorption change, exhibiting the indispensable effect of the Li ion inside the fullerene cage on the binding of Li+@C60 to the electron-rich crown ethers (Fig. S3, ESI†).
The complexation between PBCE and Li+@C60 was scrutinized in light of computational studies based on density functional (DFT) methods at the B3LYP/6-311G(d,p) level of theory. In the DFT-optimized structure, Li+@C60 is located close to the benzo-crown ethers, by which the complexation takes place in the cavity of PBCE whereas its overall distance with the pentiptycene frame is conserved (Fig. 3).15,16
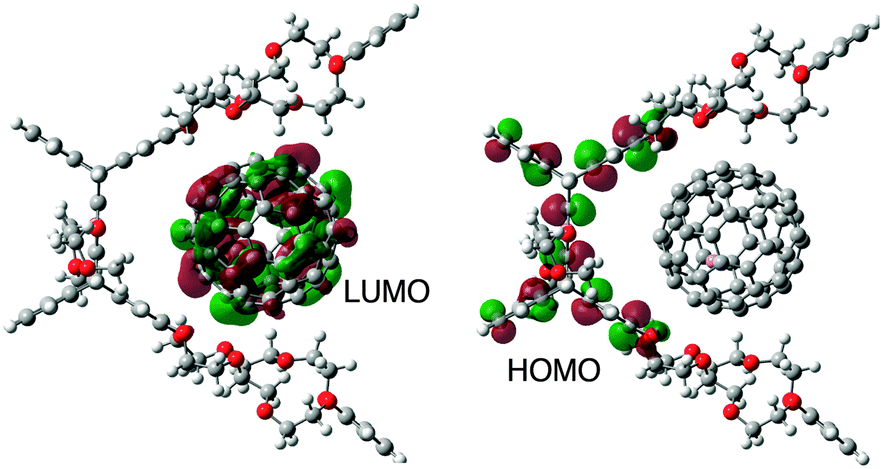 | ||
| Fig. 3 Optimized structure of the PBCE˙Li+@C60 complex showing its frontier HOMO and LUMO calculated by DFT at the B3LYP/6-311G(d,p) level of theory. | ||
While the LUMO of the complex is located on the fullerene cage, the HOMO of the complex is on the pentiptycene unit, indicating that the charge separation between pentiptycene body and Li+@C60 is feasible (Fig. 3). The distribution of the frontier orbitals of the complex reveals the dual character of PBCE, in which the complexation and the electron-transfer process with the guest molecule take place at different sites (i.e. crown ethers and pentiptycene frame).
The driving forces for the photoinduced electron transfer in PhCN via the singlet (1Li+@C60*, 1.94 eV)14a and the triplet excited states (3Li+@C60*, 1.53 eV)14a of Li+@C60 were calculated as 0.91 and 0.50 eV, respectively (see the ESI† for details).
The photoinduced electron-transfer process in the PBCE˙Li+@C60 complex was monitored by the appearance of a transient absorption at 1035 nm due the radical anion of Li+@C60 (Li+@C60˙−) upon photoexcitation of Li+@C60 at 355 nm (Fig. 4). The PBCE radical cation (λmax(PBCE˙+) = 346 nm) was only partially observed because of the overlap with the ground state absorption of Li+@C60 at around 340 nm (Fig. S6 and S7, ESI†). The charge-separated state has a triplet spin character because electron transfer occurs via3Li+@C60* as observed in the nanosecond transient absorption spectra (Fig. S8, ESI†).17 Supramolecular charge separation takes place with a rate constant (kCS) of 2.3 × 106 s−1 (0.43 μs) and the lifetime of the charge-separated state (τCS) was estimated to be 167 μs from the first-order decay of the transient absorbance at 1035 nm (insets, Fig. 4).18 In addition, photoinduced charge separation was confirmed by the electron paramagnetic resonance (EPR) measurements, in which the EPR spectrum of the radical ion was observed (g = 2.0007) in PhCN glass at 77 K (Fig. S10, ESI†). The quantum yield of charge separation (ΦCS) was evaluated to be 0.96 for PBCE˙Li+@C60 in PhCN by using the comparative method (see the ESI† for details).
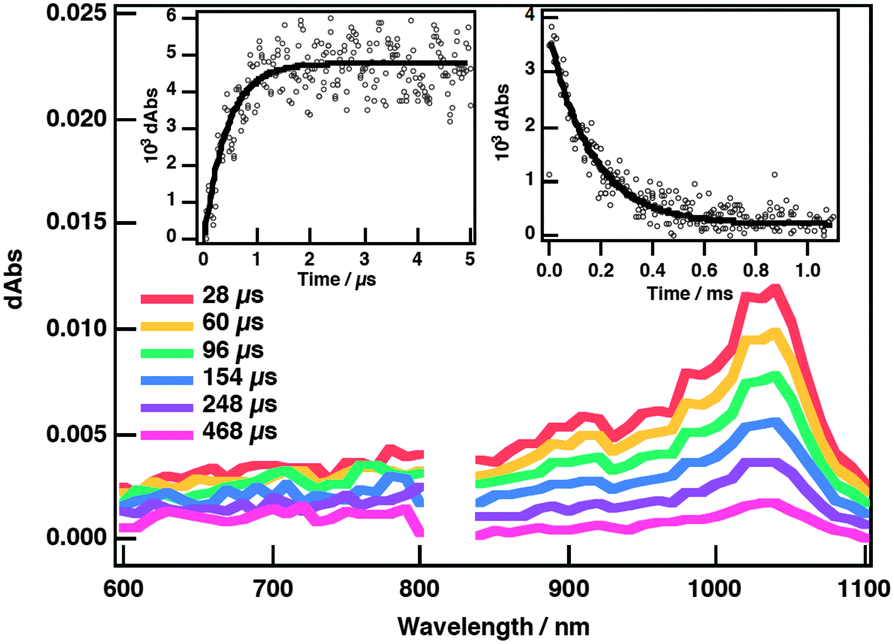 | ||
| Fig. 4 Nanosecond transient absorption spectra of PBCE and Li+@C60 in deaerated PhCN at selected time delays. Inset: Time profile of the nanosecond transient spectra of 0.660 mM PBCE with 0.020 mM Li+@C60 at 1035 nm (λexc = 355 nm). | ||
In summary, a supramolecular host–guest complex consisting of a rigid host establishing a fixed electron-transfer distance with the electron-accepting guest through a separate hosting site was formed. Efficient photoinduced electron transfer from the electron-donating pentiptycene moiety to the Li+@C60 moiety was achieved upon photoexcitation of Li+@C60 in this complex to afford the long-lived charge-separated state.
This work was supported by an Advanced Low Carbon Technology Research and Development (ALCA) program from the Japan Science Technology Agency (JST) to S.F.; a Grant-in-Aid (No. 26620154 and 26288037 to K.O.) and JSPS fellowships to M.S. (PD) and to Y.K. (25·727, DC1) from MEXT, Japan; the National Natural Science Foundation of China (21332008, 91127009), and the National Basic Research Program (2011CB932501) to C.-F.C.
Notes and references
- (a) B. Loll, J. Kern, W. Saenger, A. Zouni and L. Biesiadka, Nature, 2005, 438, 1040 CrossRef CAS PubMed; (b) N. Nelson and C. F. Yocum, Annu. Rev. Plant Biol., 2006, 57, 521 CrossRef CAS PubMed.
- (a) B. Grimm, J. Santos, B. M. Illescas, A. Muñoz, D. M. Guldi and N. Martín, J. Am. Chem. Soc., 2010, 132, 17387 CrossRef CAS PubMed; (b) M. Supur and S. Fukuzumi, J. Phys. Chem. C, 2012, 116, 23274 CrossRef CAS.
- (a) S. Fukuzumi, T. Honda, K. Ohkubo and T. Kojima, Dalton Trans., 2009, 3880 RSC; (b) S. K. Das, B. Song, A. Mahler, V. N. Nesterov, A. K. Wilson, O. Ito and F. D'souza, J. Phys. Chem. C, 2014, 118, 3994 CrossRef CAS.
- (a) J.-S. Yang and J.-L. Yan, Chem. Commun., 2008, 1501 RSC; (b) J. H. Chong and M. J. MacLachlan, Chem. Soc. Rev., 2009, 38, 3301 RSC; (c) Y. Jiang and C.-F. Chen, Eur. J. Org. Chem., 2011, 6377 CrossRef CAS.
- (a) J.-S. Yang and T. M. Swager, J. Am. Chem. Soc., 1998, 120, 5321 CrossRef CAS; (b) J.-S. Yang, J.-L. Yan, C.-K. Lin, C.-Y. Chen, Z.-Y. Xie and C.-H. Chen, Angew. Chem., Int. Ed., 2009, 48, 9936 CrossRef CAS PubMed; (c) C.-J. Lin, S. K. Kundu, C.-K. Lin and J.-S. Yang, Chem. – Eur. J., 2014, 20, 14826 CrossRef CAS PubMed.
- (a) C.-H. Yang, Ch. Prabhakar, S.-L. Huang, Y.-C. Lin, W. S. Tan, N. C. Misra, W.-T. Sun and J.-S. Yang, Org. Lett., 2011, 13, 5632 CrossRef CAS PubMed; (b) W.-T. Sun, S.-L. Huang, H.-H. Yao, I.-C. Chen, Y.-C. Lin and J.-S. Yang, Org. Lett., 2012, 14, 4154 CrossRef CAS PubMed.
- Y. Han, Z. Meng, Y.-X. Ma and C.-F. Chen, Acc. Chem. Res., 2014, 47, 2026 CrossRef CAS PubMed.
- J. Cao, Y. Jiang, J.-M. Zhao and C.-F. Chen, Chem. Commun., 2009, 1987 RSC.
- (a) Y.-X. Ma, Y. Han, J. Cao and C.-F. Chen, Org. Biomol. Chem., 2013, 11, 8183 RSC; (b) J. Cao, H.-Y. Lu, J.-F. Xiang and C.-F. Chen, Chem. Commun., 2010, 46, 3586 RSC; (c) J. Cao, H.-Y. Lu, X.-J. You, Q.-Y. Zheng and C.-F. Chen, Org. Lett., 2009, 11, 4446 CrossRef CAS PubMed.
- Y.-X. Ma, Z. Meng and C.-F. Chen, Org. Lett., 2014, 16, 1860 CrossRef CAS PubMed.
- S. Bhattacharya, A. Sharma, S. K. Nayak, S. Chottopadhyay and A. K. Mukherjee, J. Phys. Chem. B, 2003, 107, 4213 CrossRef CAS.
- T. Tsuchiya, H. Kurihara, K. Sato, T. Wakahara, T. Akasaka, T. Shimizu, N. Kamigata, N. Mizorogi and S. Nagase, Chem. Commun., 2006, 3585 RSC.
- M. Supur, Y. Kawashima, K. R. Larsen, K. Ohkubo, J. O. Jeppesen and S. Fukuzumi, Chem. – Eur. J., 2014, 20, 13976 CrossRef CAS PubMed.
- (a) Y. Kawashima, K. Ohkubo and S. Fukuzumi, J. Phys. Chem. A, 2012, 116, 8942 CrossRef CAS PubMed; (b) S. Fukuzumi, K. Ohkubo, F. D'Souza and J. L. Sessler, Chem. Commun., 2012, 48, 9801 RSC.
- In the absence of a fullerene guest, the crown ethers fold inward showing their flexibility (Fig. S4, ESI†).10.
- Complexation of Li+@C60 with PBCE through one of the benzo-crown ethers outside the host cavity was also proposed. The heat of formation value for this association was calculated by DFT to be −54 kJ mol−1, whereas that of complexation inside the cavity through both benzo-crown ethers was found to be −58 kJ mol−1, indicating that complexation with Li+@C60 inside the cavity is more favourable (Fig. S5, ESI†).
- Complexation of Li+@C60 with crown ethers promotes the intersystem crossing, which restrains the charge separation via1Li+@C60* (Fig. S9, ESI†) as also observed in a previous report.13.
- Considering the concentrations of PBCE (0.66 mM) and Li+@C60 (0.02 mM) in Fig. 4 and the K value for the association, 90% of the Li+@C60 undergoes complexation with PBCE in the hosting cavity, which shields the excited state of the Li+@C60 guest from the intermolecular interactions with the other complex or the free host molecules. Therefore, we estimate that the charge separation may involve only 10% of intermolecular electron-transfer process in this case.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cc07795d |
| This journal is © The Royal Society of Chemistry 2014 |