
NZVI modified magnetic filter paper with high redox and catalytic activities for advanced water treatment technologies†
K. K. R.
Datta
a,
E.
Petala
ab,
K. J.
Datta
a,
J. A.
Perman
a,
J.
Tucek
a,
P.
Bartak
c,
M.
Otyepka
a,
G.
Zoppellaro
*a and
R.
Zboril
*a
aRegional Centre of Advanced Technologies and Materials, Department of Physical Chemistry, Faculty of Science, Palacky University in Olomouc, Slechtitelu 11, 78371 Olomouc, Czech Republic. E-mail: radek.zboril@upol.cz; giorgio.zoppellaro@upol.cz
bDepartment of Materials Science and Engineering, University of Ioannina, GR-45110 Ioannina, Greece
cRegional Centre of Advanced Technologies and Materials, Department of Analytical Chemistry, Faculty of Science, Palacky University in Olomouc, Slechtitelu 11, 78371 Olomouc, Czech Republic
First published on 25th September 2014
Abstract
The in situ synthesis of air-stable zero-valent iron nanoparticles (NZVI) embedded in cellulose fibers leads to the assembly of highly reactive magnetic filter papers. These engineered materials display a wide range of applications in the treatment of wastewater and drinking water, including chromium removal, phenol degradation, environmental bioremediation, and catalysis.
The design of nanostructured magnetic materials engineered for environmental applications represents the forefront of research in the field of applied materials sciences.1 Among others, the nanoscale zero-valent iron (NZVI) is one successful example of a highly-efficient environmentally friendly nanosystem currently used for the treatment of ground water contaminated by various inorganic/organic pollutants.2 NZVI acts through redox chemistry and/or sorption processes2a,3 and has been employed in the environmental removal of many emerging pollutants including arsenic, halogenated hydrocarbons, phosphorus, heavy metals, radio-nuclides or cyanobacteria.4 Despite being highly effective against several harmful contaminants, NZVI exhibits some physical weaknesses that still limit the spectrum of its direct application in a broader range of bioremediation scenarios mainly in the treatment of waste and drinking water. The key limitations are intimately connected to its strong tendency towards aggregation, rapid sedimentation and its oxophilicity.5 Depending on the environmental conditions (e.g., pH of soil and/or water, oxygen content), these properties may lead to a decrease in the activity of NZVI after exposure to the pollutants, a factor that can severely restrict its operational time-window.2a,6 Much research effort has been directed towards the engineering of active and long-lasting NZVI systems.2b So far, one of the most promising approaches described in the literature relies on the NZVI inclusion into host materials such as polymers,7 porous solids,8 functionalized clays,9 and graphene sheets.10 These host materials act in a way to limit the agglomeration proclivity of NZVI and, in turn, to promote their activity compared to unsupported nanoparticles. Cellulose fibers can be considered as good supports for NZVI, and may offer several advantages with respect to the currently explored host materials. For example, the common laboratory filter paper (thereafter abbreviated as FP) can deliver such an appealing alternative for the assembly of environmental friendly (nano)composites, because it combines biodegradability and flexibility into a highly cost-effective support. Cellulose polymers are known to contain oxygen rich lined microfibrils and pores, with overall structural architectures that offer anchoring sites and inner cavities for trapping metal ions as well as metal nanoparticles.1e,11 FP has been already used to host silver nanosystems, and the so-formed host–guest materials proved their efficiency as surface-enhanced Raman spectroscopy (SERS) substrates,12 sensors,13 and became key components in water treatment devices.14 To date, no reports have appeared in the literature on the use of FP as a support for the in situ synthesis of NZVI nanoparticles (NPs).
Furthermore, no nanocomposite materials merging FP and NZVI NPs have been so far tested for environmental applications. In this report, we describe the assembly of such flexible, magnetically controllable, air-stable and redox active nanocomposites, NZVI@FP, and validate their effectiveness for the fast remediation of the Cr(VI) pollutant as well as organic contaminants (e.g., phenols) from water solutions through consecutive and simple filtration processes. Our preparative methodology generates not only magnetic papers that can be manipulated and/or removed from the processed solution using an external magnet but also allows the straightforward generation of nanocomposites that can be selectively patterned with NZVI over the FP support. This approach can aid in the assembly of flexible filter-paper-devices that require unusual shapes to be operational. The preparation of NZVI nanoparticles embedded in FP consisted of a simple two-step procedure (Fig. 1a). In the first step, the impregnation of FP with the ferric chloride salt was performed by using an alcoholic (EtOH) solution of the metallic precursor, followed by air-drying FP at room temperature. Then, in situ reduction of the trapped FeCl3 with NaBH4 gave the active NZVI@FP material. The successful coating of FP with iron cations and their reduction to zero-valent iron could be easily monitored by the naked eye as shown in Fig. 1a, since the impregnation of filter paper with the ferric salt led to the formation of a bright-yellow material, which turned dark-black and responsive to an external magnet upon borohydride reduction. Ash free FP (Whatmann, Grade 40, r = 55 mm) was used as a scaffold for the NZVI guest without the need for any FP pre-treatment. From the X-ray photoelectron spectroscopic (XPS) analyses, we found that in the nanocomposite material, the NZVI nanoparticles remained highly stable in air for one month (see Fig. S1 and S2 in the ESI†) and only a minor surface oxidation of NZVI was noticed (vide infra). Furthermore, the loading of NZVI into FP could be controlled by varying the initial concentration of the ferric salt in the ethanol solution used within the initial impregnation phase. The efficacy in trapping NZVI by FP can be understood by noting that cellulose fibers contain oxygen rich moieties that are prone to interact with the surface exposed iron of the NZVI particles. The surfaces of these fibers are rough and their interconnections form pores that allow boosting further the inclusion of guest molecules into their inner spaces (Fig. 1b). The nanosized nature of the trapped NZVI particles was further validated by transmission electron microscopy (TEM) and scanning electron microscopy (SEM) that revealed the entrapment of NZVI by the cellulose fibers (Fig. 1c and d, and Fig. S3 in ESI†). The so-formed NZVI exhibited, however, quite large size distribution, in the range of 15–240 nm. The statistical analysis of the particle size using a Gaussian distribution function showed a mean diameter of ∼100 nm, with a Gaussian half-width of ∼80 nm (Fig. 1c, inset, and Fig. S4 in ESI†). The chemical nature, crystallinity and magnetic characteristics of NZVI@FP were evaluated with the aid of several techniques, in particular, X-ray diffraction (XRD), Mössbauer spectroscopy and SQUID magnetization measurements (for more details, see the ESI†). The XRD pattern of NZVI@FP showed the characteristic peak at 52.5° (2θ, deg), a signature that corresponds to the (110) diffraction of the body-centered cubic (bcc) α-Fe phase (Fig. S5 in ESI†). The (110) diffraction peak increased in intensity upon increasing the amount of the iron salt used within the initial FP immersion cycle and following NaBH4 reduction. Therefore, through such a simple synthetic procedure, it was possible to derive from the bottom-up approach the effective amount of NZVI loaded into FP (from 2.5% to 5% in weight for our tested nanocomposites). Diffraction signals due to other iron oxide phases (e.g., γ-Fe2O3/Fe3O4 and α-Fe2O3) were absent in all tested preparations. These findings suggested that the NZVI nanoparticles intimately embedded into FP were less prone to undergo fast surface-passivation at ambient air, due to the emergence of some interactions between surface exposed iron atoms and cellulose fibers. In this context, low-temperature in-field 57Fe Mössbauer spectroscopy was used to unveil such interactions and to gain better knowledge of the electronic signature of NZVI sequestered into FP. The recorded Mössbauer spectrum at T = 5 K under a 5 T external field (Bext) is shown in Fig. 2a together with the fitting-deconvolution results (red line). Three components contributed to the overall envelope, two sextets and one doublet, signatures that were found to hold for all preparations, irrespective of the loaded amount of NZVI into FP (from 2.5% to 5.0% NZVI in weight). One sextet encoded a highly effective hyperfine magnetic field, Beff, of 28.5 T and featured the typical signature of zero-valent iron (Fe0) cores (Fig. 2a, blue line), with an isomer shift (δ) of 0.11 mm s−1, and a zero quadrupole splitting (ΔEQ) value. The second magnetically split component reflected a fraction of Fe0 interacting with the sugar support (Fig. 2a, magenta line) with a Beff of 19.7 T, a δ of 0.22 mm s−1, and zero ΔEQ. From these results, the NZVI@FP system can be described as composed of nanoparticles with a pseudo core–shell configuration, having the core formed by purely zero-valent iron and the shell of zero-valent iron in nature but with altered electronic features as also derived from the room-temperature Mössbauer spectrum (for more information, see the ESI,† Fig. S6 and Table S1).
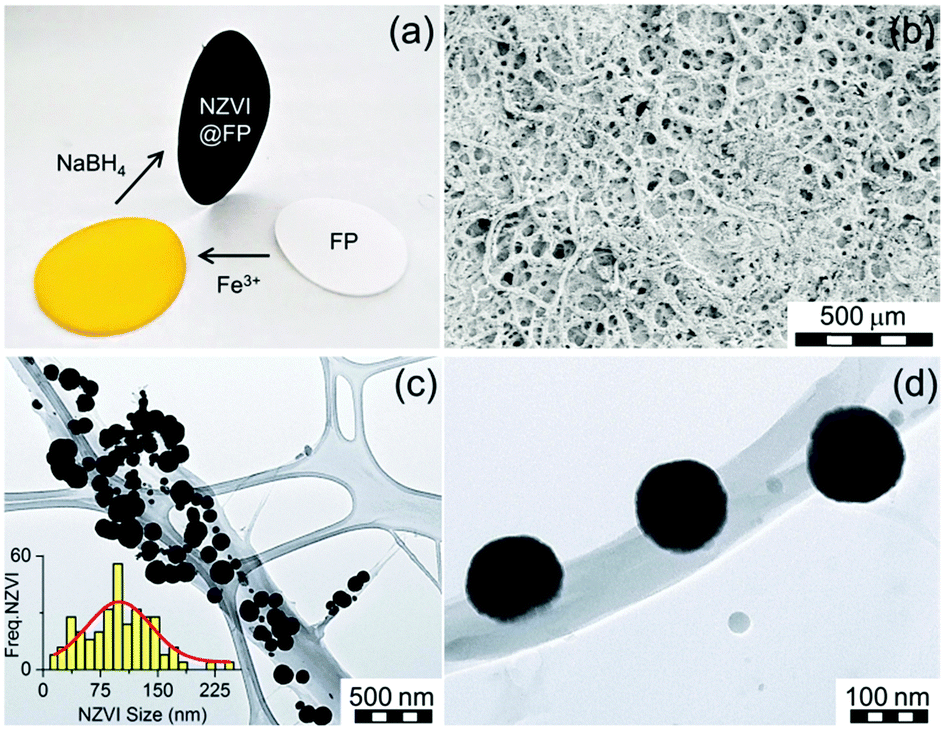 | ||
| Fig. 1 (a) Optical image shows the two step assembly of the magnetic NZVI@FP nanocomposite. (b) Scanning electron micrograph of NZVI@FP loaded with 5% in weight of NZVI. (c) TEM micrograph of NZVI entrapped over a cellulose fibre of FP. The inset shows the particle size distribution, as estimated from TEM (Freq.-NZVI vs. NZVI-size), together with the fitting analysis (red-line). (d) TEM micrograph of a magnified region of (c) revealing the intimate interaction between NZVIs and the fiber surface. | ||
 | ||
| Fig. 2 (a) In-field low-temperature (T = 5 K, Bext = 5 T) 57Fe Mössbauer spectrum of the NZVI@FP nanocomposite. The solid-lines show the three components emerging from the deconvolution analysis. (b) Comparison of the XPS spectra recorded in the Fe2p region for the NZVI@FP nanocomposite freshly prepared (red pattern) and after ageing in air for one month (black pattern). | ||
The witnessed effect is thought to originate from the intimate contacts of the surface exposed Fe0 metal with the sugar fibers, as shown in the representative TEM micrograph in Fig. 1d. The last spectral component observed, the doublet (Fig. 2a, green line), reconciled with the fraction of NZVI characterized by small sizes (∼10 nm), which produced the fast relaxation of their NZVI shells in the Mössbauer time-scale (10−8 s). The Mössbauer parameters for such component were found to fall at 0.23 mm s−1 for δ and 0.45 mm s−1 for ΔEQ. In this scenario, the external magnetic field (5 T) was still not strong enough to hinder superparamagnetic fluctuations of the shell spins. We noted that the XPS spectrum of the NZVI@FP nanocomposite freshly prepared and NZVI@FP aged in air for one month did not differ substantially from each other (Fig. 2b), but revealed in the former the presence of a low energy peak at 704.9 eV (Fig. 2b, red spectrum) that became much weaker in the latter (Fig. 2b, black spectrum). The change was accompanied by (i) the broadening of the dominant Fe2p3/2 signal (708.5 eV) with the formation of a more pronounced shoulder around 710 eV, and (ii) the broadening of the Fe2p1/2 signal at 721.8 eV. All these signatures are consistent with the mild oxidation of the zero-valent iron located on the nanoparticle surfaces (for more details, see Fig. S1 and S2 in the ESI†). In addition, we may suggest that the observed signature at 704.9 eV corresponds to the fraction of zero-valent iron in intimate contact with the sugar molecules of FP, as anticipated earlier in the Mössbauer analysis, and this is the Fe0 fraction more sensitive to undergo the oxidation process. The dual nature of the electronic behaviour associated with Fe0 in the NZVI-core and surface-exposed Fe0 located on the NZVI-shell was also confirmed by the nanocomposite's magnetic response by bulk susceptibility measurements. The magnetic behaviour of NZVI@FP with high loading (5% of NZVI loaded, % in weight) showed symmetric hysteretic behaviour and the saturation of magnetization at low field (<1 T), reaching the value of 10.7 Am2 kg−1 at room temperature, which slightly increased to 11.5 Am2 kg−1 at T = 5 K (Fig. S7a in ESI†). The observed coercivity (Bc) was found to be much higher than that reported for bulk iron (<0.1 mT), namely 42 mT and 52 mT at T = 300 K and 5 K, respectively (Table S2, ESI†), implying magnetic hardening of the nanoparticle system. In particular, as shown in Fig. S7b (ESI†), the zero-field-cooled (ZFC) magnetization curve measured upon warming under an applied field of 0.01 T showed the presence of a maximum at very low temperatures (∼30 K) corresponding to the blocking temperature of the NZVI nanoparticle shells.
The specific NZVI@FP architecture obtained from the in situ synthesis translated into the superior redox activity and proclivity for the catalysis of this material with respect to bare NZVI in the environmental remediation processes of pollutants from water solutions. Noteworthily, it also prevented the severe release into the environment of NZVI from FP, whilst maintaining effective transport properties with high flux of the fluid systems, due to the porous nature of FP. The effectiveness of NZVI@FP was tested in two scenarios: (i) reduction of the toxic hexavalent chromium into the environmentally friendlier Cr(III) and (ii) the proclivity of NZVI@FP to activate hydroxylation reactions, in particular, the in situ conversion of phenols to catechols. The results obtained for the Cr(VI) removal by simple filtration of gravity-feed solutions over the NZVI@FP nanocomposite are shown in Fig. 3a. A freshly prepared stock solution of Cr(VI) (6.5 mg L−1, 30 mL) was slowly poured through NZVI@FP (∼5 min) and the filtrate was collected and characterized by UV-Vis absorption spectroscopy to determine the residual amount of Cr(VI) (details are given in the ESI†). The observed efficiency in the Cr(VI) reduction by NZVI@FP (expressed in %) was compared with the performance obtained for the nanocomposites assembled through an ex situ approach (NZVI + FP) as well as bare FP. The NZVI + FP material was generated by simple impregnation of FP with separately and freshly synthesized NZVI nanoparticles (for details, see the Experimental section in the ESI†). It is important to underline that the final amount of loaded NZVI was found consistently lower in the case of the ex situ synthesis (NZVI + FP), about 3 wt% on average, in comparison to the highest NZVI entrapment on FP obtained from the in situ assembly (5 wt%). Looking at the process of Cr(VI) removal, the use of the NZVI@FP nanocomposite resulted in a remarkable value of 64% after just three filtration cycles (Fig. 3a). In contrast, the ex situ material (FP + NZVI) showed, in such a set-up, a decreased ability to reduce Cr(VI), and only 7.5% of hexavalent chromium could be successfully removed. Finally, the use of bare FP had, as expected, no visible effect on the reduction process of Cr(VI). While the different loading of NZVI into FP, from the in situ to the ex situ materials, may justify slight variations in the total amount of Cr(VI) removal in favour of NZVI@FP, the witnessed differences are striking and suggest that the in situ assembly process clearly generated a much better performing nanocomposite. The ability of NZVI@FP to act as a catalyst in hydroxylation reactions was tested using the simple phenol as a benchmark, which is a highly toxic organic pollutant with suspected mutagenic and carcinogenic properties,15 and by employing hydrogen peroxide as a green oxidant.
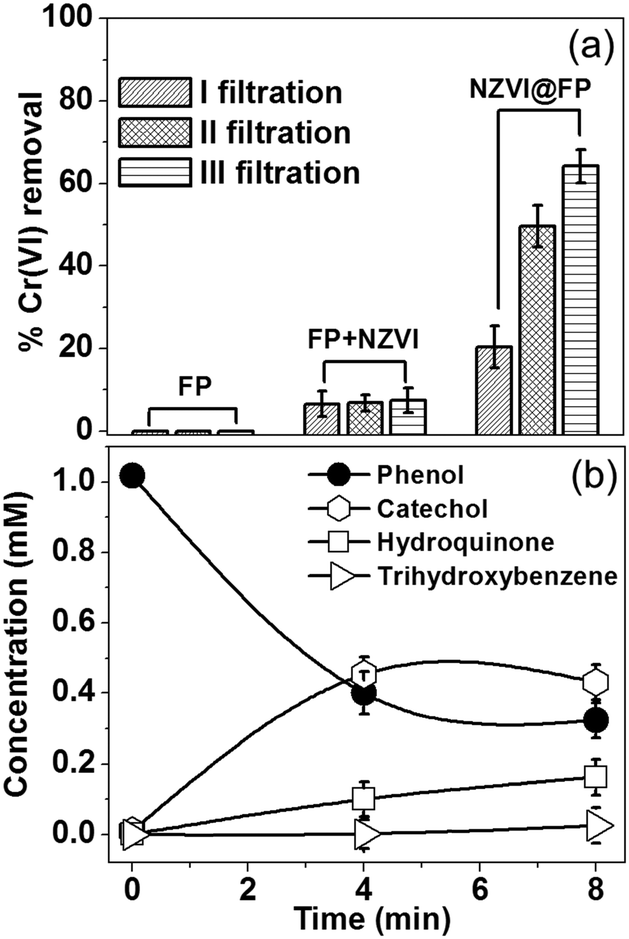 | ||
| Fig. 3 (a) The Cr(VI) removal performance of bare FP, ex situ synthesized NZVI deposited over FP (3 wt%) and the in situ synthesis of NZVI@FP (5 wt%) as evaluated from 3 filtration cycles. (b) Formation of aromatic compounds as obtained by GC-MS analysis in the phenol hydroxylation process using NZVI@FP (5 wt%) as a catalytic membrane. | ||
The possible mechanism of peroxide activation by NZVI and the subsequent hydroxylation of phenol are shown in Scheme S1 in the ESI.† There are very few reports in the literature where NZVI (supported) has shown the catalytic activity towards the hydroxylation of phenols. The reaction conditions for those materials required the use of high temperatures, low pH and prolonged times.16 We found that NZVI@FP was capable of promoting the conversion of phenols into less toxic catechols even at low temperature (40 °C), at neutral pH and after short time via simple filtration steps. Fig. 3b summarizes the experimental findings with NZVI@FP (5% wt of NZVI) used as a membrane catalyst. Nearly 70% of the initial amount of phenol was converted in 8 min, giving rise to catechol (47%), hydroquinone (18%) and trihydroxybenzene (3%) after analyses by GC-MS (Fig. S8 in ESI†). Catechol was the predominant product, thus the hydroxylation occurred preferentially in the ortho-position. When the same amount of unsupported NZVI powder was used as a catalyst, 48% of phenol conversion was achieved only after 12 h (Fig. S9 in ESI†) with indeed a more even distribution in catechol (32%) and hydroquinone (21%) formation, as found from GC-MS analyses (Fig. S10 in ESI†). Control experiments using bare FP (and hydrogen peroxide) showed, as expected, no ability to perform the hydroxylation of phenol.
In conclusion, we demonstrated that the in situ synthesis of NZVI on cellulose filter paper allowed the assembly of novel magnetically active membrane nanocomposites. The NZVI@FP nanocomposite showed high activity towards the removal of hexavalent chromium as well as an excellent catalytic ability to convert phenols into catechols, by simple filtration processes of the contaminated water solutions. We believe that this work opens the doors for the application of NZVI not only in groundwater remediation but also in the treatment of wastewater and drinking water with several superior aspects including easy handling, green synthesis, air stability, magnetic separation and high redox and catalytic efficiencies.
This work was supported by the Operational Program Research and Development for Innovations – European Regional Development Fund (CZ.1.05/2.1.00/03.0058) and by the Operational Program Education for Competitiveness – European Social Fund (CZ.1.07/2.3.00/20.0155, CZ.1.07/2.3.00/30.0004) of the Ministry of Education, Youth and Sports of the Czech Republic and by the Fund of Ministry of Health IGA MZCR NT 14060-3/2013 and by the project of the Technology Agency of the Czech Republic “Competence Centres” (TE01020218).
Notes and references
- (a) C. T. Yavuz, J. T. Mayo, W. W. Yu, A. Prakash, J. C. Falkner, S. Yean, L. L. Cong, H. J. Shipley, A. Kan, M. Tomson, D. Natelson and V. L. Colvin, Science, 2006, 314, 964 CrossRef PubMed; (b) I. Ali, Chem. Rev., 2012, 112, 5073 CrossRef CAS PubMed; (c) M. A. Shannon, P. W. Bohn, M. Elimelech, J. G. Georgiadis, B. J. Marinas and A. M. Mayes, Nature, 2008, 452, 301 CrossRef CAS PubMed; (d) C. L. Warner, R. S. Addleman, A. D. Cinson, T. C. Droubay, M. H. Engelhard, M. A. Nash, W. Yantasee and M. G. Warner, ChemSusChem, 2010, 3, 749 CrossRef CAS PubMed; (e) R. T. Olsson, M. A. S. A. Samir, G. Salazar-Alvarez, L. Belova, V. Strom, L. A. Berglund, O. Ikkala, J. Nogues and U. W. Gedde, Nat. Nanotechnol., 2010, 5, 584 CrossRef CAS PubMed.
- (a) P. G. Tratnyek and R. L. Johnson, Nano Today, 2006, 1, 44 CrossRef; (b) F. L. Fu, D. D. Dionysiou and H. Liu, J. Hazard. Mater., 2014, 267, 194 CrossRef CAS PubMed; (c) C. Noubactep, Clean: Soil, Air, Water, 2013, 41, 702 CrossRef CAS.
- (a) B. I. Kharisov, H. V. R. Dias, O. V. Kharissova, V. M. Jimenez-Perez, B. O. Perez and B. M. Flores, RSC Adv., 2012, 2, 9325 RSC; (b) C. Noubactep, S. Care and R. Crane, Water, Air, Soil Pollut., 2012, 223, 1363 CrossRef CAS PubMed.
- (a) Z. Markova, K. M. Siskova, J. Filip, J. Cuda, M. Kolar, K. Safarova, I. Medrik and R. Zboril, Environ. Sci. Technol., 2013, 47, 5285 CrossRef CAS PubMed; (b) B. Marsalek, D. Jancula, E. Marsalkova, M. Mashlan, K. Safarova, J. Tucek and R. Zboril, Environ. Sci. Technol., 2012, 46, 2316 CrossRef CAS PubMed; (c) S. Klimkova, M. Cernik, L. Lacinova, J. Filip, D. Jancik and R. Zboril, Chemosphere, 2011, 82, 1178 CrossRef CAS PubMed; (d) I. Dror, O. M. Jacov, A. Cortis and B. Berkowitz, ACS Appl. Mater. Interfaces, 2012, 4, 3416 CrossRef CAS PubMed; (e) W. L. Yan, M. A. V. Ramos, B. E. Koel and W. X. Zhang, J. Phys. Chem. C, 2012, 116, 5303 CrossRef CAS; (f) L. Sun, L. D. Zhang, C. H. Liang, Z. G. Yuan, Y. Zhang, W. Xu, J. X. Zhang and Y. Z. Chen, J. Mater. Chem., 2011, 21, 5877 RSC; (g) J. T. Nurmi, P. G. Tratnyek, V. Sarathy, D. R. Baer, J. E. Amonette, K. Pecher, C. M. Wang, J. C. Linehan, D. W. Matson, R. L. Penn and M. D. Driessen, Environ. Sci. Technol., 2005, 39, 1221 CrossRef CAS.
- D. L. Huber, Small, 2005, 1, 482 CrossRef CAS PubMed.
- X. Q. Li, D. W. Elliott and W. X. Zhang, Crit. Rev. Solid State Mater. Sci., 2006, 31, 111 CrossRef CAS.
- (a) S. M. Ponder, J. G. Darab and T. E. Mallouk, Environ. Sci. Technol., 2000, 34, 2564 CrossRef CAS; (b) Q. Huang, X. Shi, R. A. Pinto, E. J. Petersen and W. J. Weber, Environ. Sci. Technol., 2008, 42, 8884 CrossRef CAS.
- (a) L. B. Hoch, E. J. Mack, B. W. Hydutsky, J. M. Hershman, J. M. Skluzacek and T. E. Mallouk, Environ. Sci. Technol., 2008, 42, 2600 CrossRef CAS; (b) E. Petala, K. Dimos, A. Douvalis, T. Bakas, J. Tucek, R. Zbořil and M. A. Karakassides, J. Hazard. Mater., 2013, 261, 295 CrossRef CAS PubMed.
- (a) Y. Hwang, Y. C. Lee, P. D. Mines, Y. S. Huh and H. R. Andersen, Appl. Catal., B, 2014, 147, 748 CrossRef CAS PubMed; (b) P. X. Wu, S. Z. Li, L. T. Ju, N. W. Zhu, J. H. Wu, P. Li and Z. Dang, J. Hazard. Mater., 2012, 219, 283 CrossRef PubMed.
- H. Jabeen, V. Chandra, S. Jung, J. W. Lee, K. S. Kim and S. Bin Kim, Nanoscale, 2011, 3, 3583 RSC.
- (a) J. H. He, T. Kunitake and A. Nakao, Chem. Mater., 2003, 15, 4401 CrossRef CAS; (b) M. A. S. A. Samir, F. Alloin and A. Dufresne, Biomacromolecules, 2005, 6, 612 CrossRef CAS PubMed.
- (a) Y. J. Meng, Y. C. Lai, X. H. Jiang, Q. Q. Zhao and J. H. Zhan, Analyst, 2013, 138, 2090 RSC; (b) C. H. Lee, M. E. Hankus, L. Tian, P. M. Pellegrino and S. Singamaneni, Anal. Chem., 2011, 83, 8953 CrossRef CAS PubMed.
- A. Abbas, A. Brimer, J. M. Slocik, L. M. Tian, R. R. Naik and S. Singamaneni, Anal. Chem., 2013, 85, 3977 CrossRef CAS PubMed.
- (a) T. A. Dankovich and D. G. Gray, Environ. Sci. Technol., 2011, 45, 1992 CrossRef CAS PubMed; (b) R. Vyhnalkova, N. Mansur-Azzam, A. Eisenberg and T. G. M. van de Ven, Adv. Funct. Mater., 2012, 22, 4096 CrossRef CAS.
- (a) G. Busca, S. Berardinelli, C. Resini and L. Arrighi, J. Hazard. Mater., 2008, 160, 265 CrossRef CAS PubMed; (b) B. Meunier, Science, 2002, 296, 270 CrossRef CAS PubMed; (c) S. Sen Gupta, M. Stadler, C. A. Noser, A. Ghosh, B. Steinhoff, D. Lenoir, C. P. Horwitz, K. W. Schramm and T. J. Collins, Science, 2002, 296, 326 CrossRef CAS PubMed; (d) R. Prucek, M. Hermanek and R. Zboril, Appl. Catal., A, 2009, 366, 325 CrossRef CAS PubMed.
- (a) D. H. Bremner, A. E. Burgess, D. Houllemare and K. C. Namkung, Appl. Catal., B, 2006, 63, 15 CrossRef CAS PubMed; (b) H. Q. Sun, G. L. Zhou, S. Z. Liu, H. M. Ang, M. O. Tade and S. B. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 6235 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization techniques, scanning electron microscopy images, X-ray powder diffraction patterns, Mössbauer spectroscopy data and further spectral analysis, hysteresis loops, XPS data, and GC-MS data. See DOI: 10.1039/c4cc06241h |
| This journal is © The Royal Society of Chemistry 2014 |