 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diastereoselective synthesis of novel aza-diketopiperazines via a domino cyclohydrocarbonylation/addition process†
Pierre
Regenass
,
Jean-François
Margathe
,
André
Mann
,
Jean
Suffert
,
Marcel
Hibert
,
Nicolas
Girard
* and
Dominique
Bonnet
*
Laboratoire d'Innovation Thérapeutique, UMR7200 CNRS/Université de Strasbourg, Labex Médalis, Faculté de Pharmacie, 74 route du Rhin, 67412 Illkirch, France. E-mail: nicolas.girard@unistra.fr; dominique.bonnet@unistra.fr
First published on 4th July 2014
Abstract
Herein, we report an unprecedented, short and diastereo-selective synthesis of newly reported aza-diketopiperazine (aza-DKP) scaffolds starting from amino acids. The strategy is based on a Rh(I)-catalyzed hydroformylative cyclohydrocarbonylation of allyl-substituted aza-DKP, followed by a diastereoselective functionalization of the platform. This methodology allows the synthesis of novel bicyclic and tricyclic aza-DKP scaffolds incorporating six- or seven-membered rings, with potential applications in medicinal chemistry.
The diketopiperazine (DKP) moiety found in several natural products has been extensively studied in medicinal chemistry.1 However, the corresponding aza-DKP platform remains underexplored.2 This class of heterocycles can be viewed as a constrained dipeptidomimetic DKP analogue (Fig. 1). As reported for aza-peptides,3 the replacement of one Cα-stereogenic center by a planar nitrogen atom could have a profound impact on both the chemical and biological properties of DKP and could offer new potential for drug discovery and chemical biology.
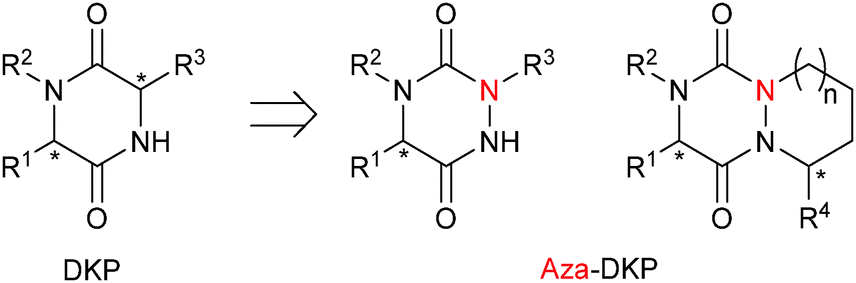 | ||
| Fig. 1 General structure of diketopiperazines (DKP) and aza-diketopiperazines (aza-DKP). | ||
Recently, we have described a convenient access to original 2,4,5-trisubstituted-1,2,4-triazine-3,6-diones, both in solution and in the solid-phase.2b In the present work, we report a diversity-oriented, efficient and stereoselective synthesis of novel bicyclic and tricyclic scaffolds 6 derived from aza-DKP. To access such structures, we have explored a strategy based on cyclohydrocarbonylation (CHC)4 of allyl aza-DKP 4, followed by an acid-catalyzed diastereoselective nucleophilic addition on the resulting enamide 5 (Scheme 1). This strategy involves for the first time the catalytic hydroformylation of a newly reported 1,2,4-triazine-3,6-dione system.2a,b The scope, limitations and diastereoselectivity of the approach have been carefully studied, resulting in the preparation of enantiomerically pure scaffolds with potential applications in medicinal chemistry.
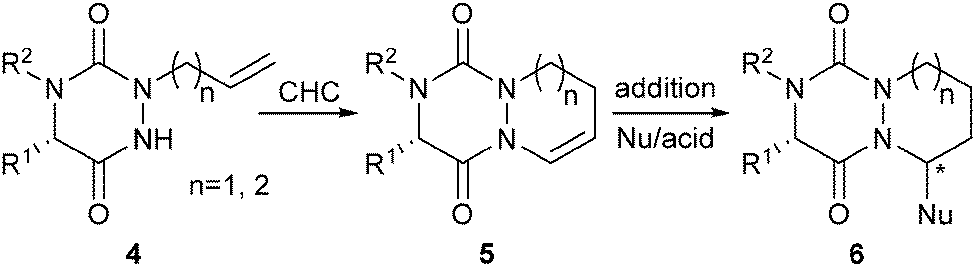 | ||
| Scheme 1 Strategy towards novel N-heterocyclic aza-DKP scaffolds 6. | ||
To investigate the applicability of the CHC reaction to aza-DKP systems, we initially prepared a set of allyl-substituted precursors 4a–g and 4k,l according to our previously described procedure.2b The amino acids were converted into amino esters which were alkylated by reductive amination. The resulting secondary amines 2a–g and 2k,l as well as the proline derivatives 2h–j and 2m were reacted with bis(trichloromethyl)carbonate (BTC) and allyl or homoallyl t-butyl carbazate 1a or 1b, obtained in one step from commercially available t-butyl carbazate (see ESI† for a detailed procedure). The crude semicarbazides 3a–m were then treated in TFA/water (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) for 1 h, resulting in the consecutive semi-carbazide deprotection and cyclization. This led to allyl derivatives 4a–m, in 27% to 77% yields from amines 2a–j, the lower yields being obtained with the most sterically hindered R1 and R2 substituents (Table 1, entries 3 and 4). Noteworthily, the preparation of aza-DKP 4i and 4m (from L-hydroxyproline) required hydroxy protection prior to semicarbazide cyclization (Table 1, entries 9 and 12). With compounds 4a–m in hand, we explored the CHC using syngas (H2/CO) in the presence of a Rh(I) catalyst.5 BiPhePhos was selected as a metal chelating agent to ensure the formation of linear rather than branched aldehydes.6 All reactions were performed under acid catalysis (pyridinium p-toluenesulfonate: PPTS or camphorsulfonic acid: CSA) to promote cyclization, if any, into enamide 5 in the same reactor.
:5) for 1 h, resulting in the consecutive semi-carbazide deprotection and cyclization. This led to allyl derivatives 4a–m, in 27% to 77% yields from amines 2a–j, the lower yields being obtained with the most sterically hindered R1 and R2 substituents (Table 1, entries 3 and 4). Noteworthily, the preparation of aza-DKP 4i and 4m (from L-hydroxyproline) required hydroxy protection prior to semicarbazide cyclization (Table 1, entries 9 and 12). With compounds 4a–m in hand, we explored the CHC using syngas (H2/CO) in the presence of a Rh(I) catalyst.5 BiPhePhos was selected as a metal chelating agent to ensure the formation of linear rather than branched aldehydes.6 All reactions were performed under acid catalysis (pyridinium p-toluenesulfonate: PPTS or camphorsulfonic acid: CSA) to promote cyclization, if any, into enamide 5 in the same reactor.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Amino acid | R1 | R2 | n | Yield 4b (%) |
Yield 5b (%) |
a iPe = isopentyl.
b Isolated yields.
c Semicarbazide 3 was obtained in THF/CH2Cl2.
d Compound 4g was obtained in TFA/water/triisopropylsilane (95/2.5/2.5, v/v/v).
e Cleavage of the benzyl protecting group was performed prior to CHC.
f CSA instead of PPTS.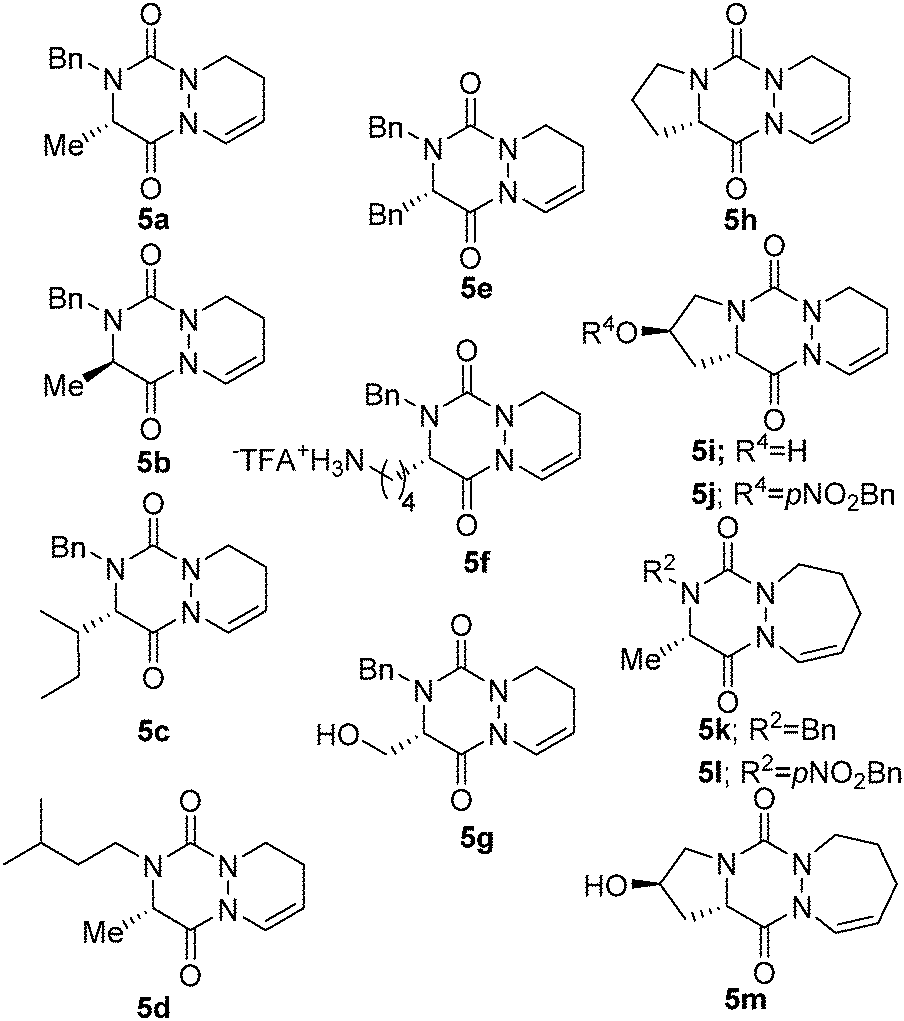
|
||||||
| 1 | L-Ala | (S)-Me | Bn | 1 | 4a (70)c | 5a (81) |
| 2 | D-Ala | (R)-Me | Bn | 1 | 4b (68)c | 5b (79) |
| 3 | L-Ile | (S)-sec-Bu | Bn | 1 | 4c (31) | 5c (81) |
| 4 | L-Ala | (S)-Me | iPea | 1 | 4d (31) | 5d (78) |
| 5 | L-Phe | (S)-Bn | Bn | 1 | 4e (49) | 5e (77) |
| 6 | L-Lys(Boc) | (S)-H2N(CH2)4 | Bn | 1 | 4f (51) | 5f (43) |
| 7 | L-Ser(tBu) | (S)-HOCH2 | Bn | 1 | 4g (45)d | 5g (57) |
| 8 | L-Pro | (S)-(CH2)3 | 1 | 4h (38)c | 5h (69) | |
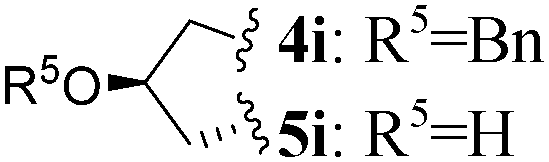
| 9 | L-Pro(OBn) |
|
1 | 4i (29)c,e | 5i (73) | |
| 10 | L-Pro(OpNO2Bn) |
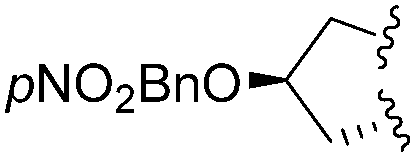
|
1 | 4j (39)c | 5j (62) | |
| 11 | L-Ala | (S)-Me | Bn | 2 | 4k (77)c | 5k (72)f |
| 12 | L-Ala | (S)-Me | pNO2-Bn | 2 | 4l (54)c | 5l (81)f |
| 13 | L-Pro(OBn) |
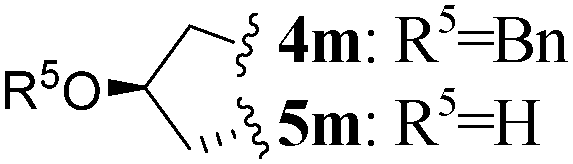
|
2 | 4m (27)c,e | 5m (61)f | |
In our first attempt, we were pleased to obtain cyclized compound 5a in an excellent 81% yield from allyl compound 4a, thus validating the CHC as a convenient and high yielding method for bicyclic aza-DKP synthesis (Table 1, entry 1).
Next, the scope and limitations of the reaction were evaluated on allylic aza-DKP 4b–m. In all cases, the expected cyclized compounds 5 were isolated in yields ranging from 43 to 81% (Table 1), thus demonstrating the efficiency of the method, regardless of the nature of R1 and R2 (Table 1, entries 3–5) or of the configuration of the starting amino acid (Table 1, entry 2). Noteworthily, CHC still occurred in reasonable yields with compounds 4f and 4g encompassing nucleophile groups at R3, which could possibly compete as a ligand for the metal (Table 1, entries 6 and 7).7
Interestingly, CHC also gave access to tricyclic L-proline-based aza-DKP 5h, 5i and 5j in good 69%, 73% and 62% yields, respectively (Table 1, entries 8–10). This scaffold is particularly appealing for medicinal chemistry as the corresponding DKP is embedded in the core of several natural product classes used in targeted cancer therapy.8
These promising results for the synthesis of six-membered rings prompted us to evaluate CHC as an entry to aza-DKP fused to a seven-membered ring. Thus, with homoallylic derivative 4k, the CHC reaction proceeded smoothly and 5k was obtained in moderate yield (34%). Then, we switched from PPTS to the more acidic CSA, which drives the reaction to completion and dramatically improves the yield (72%). This optimized procedure was also applied to the synthesis of tricyclic L-hydroxyproline-based aza-DPK 5m obtained in 61% yield.
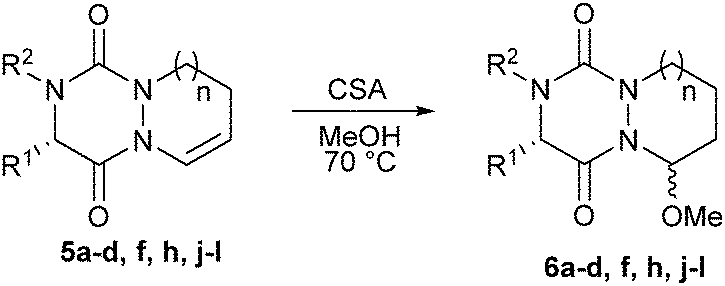
With all these novel structures in hand, we decided to investigate the functionalization of the diaza-cyclohexene and diaza-cycloheptene rings in order to extend the molecular diversity of these novel scaffolds. A first experiment was carried out by subjecting compound 5a to a CSA acid-catalyzed addition of MeOH which led to hemiaminal 6a with a high 86% yield and a good diastereoisomeric ratio (dr) of 93:7 (Table 2, entry 1). The major isomer was readily isolated by preparative HPLC and was shown to be the C9–C2 trans-isomer by X-ray diffraction analysis (Fig. 2). This result combined with the axial position of the methoxy group indicate that the nucleophilic attack of the acyl iminium intermediate is likely under stereoelectronic control.9 The out-of-plane substituents associated with the presence of stereocenters make the aza-DKP scaffold a promising platform to increase receptor–ligand interactions and to develop potentially active and selective compounds.10
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | n | Yieldb (%) | dr (trans/cis)c |
| a iPe = isopentyl. b Isolated yields. c Diastereomeric ratio was determined by 1H NMR or HPLC analysis of the crude reaction mixtures. | |||||
| 1 | (S)-Me | Bn | 1 | 6a (86) | 93:7 |
| 2 | (R)-Me | Bn | 1 | 6b (83) | 92:8 |
| 3 | (S)-sec-Bu | Bn | 1 | 6c (65) | >99:1 |
| 4 | (S)-Me | iPea | 1 | 6d (74) | 97:3 |
| 5 | (S)-H2N(CH2)4 | Bn | 1 | 6f (61) | 96:4 |
| 6 | (S)-(CH2)3 | 1 | 6h (65) | 4:96 |
|
| 7 |
|
1 | 6j (59) | 4:96 |
|
| 8 | (S)-Me | Bn | 2 | 6k (62) | 37:63 |
| 9 | (S)-Me | pNO2Bn | 2 | 6l (55) | 37:63 |
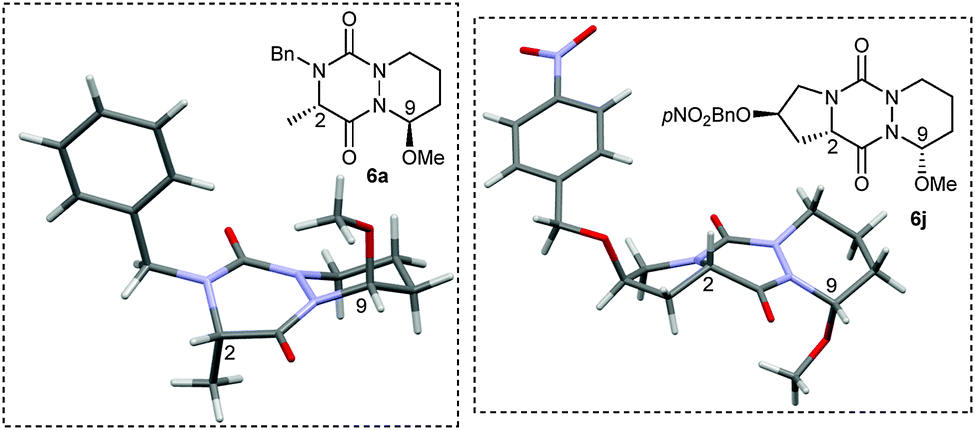 | ||
| Fig. 2 X-ray structures of compounds 6a and 6j. | ||
The diastereoselective addition reaction was then extended to various enamides. As shown in Table 2, the expected compound was obtained whatever the absolute configuration at Cα (Table 2, entry 2). The steric hindrance at R2 was found to impact the selectivity (Table 2, entry 4). In contrast, when a hindered group was introduced at R1, only one diastereomer was detected by 1H NMR and HPLC analyses of the crude material (Table 2, entry 3). The diastereoselective addition was also found compatible with the presence of a nucleophilic primary amine at R1 (Table 2, entry 5). Interestingly, when the addition was performed on tricyclic proline derivatives 5h and 5j (Table 2, entries 6 and 7), desired hemiaminals 6h and 6j were also obtained in good yields (65% and 59%, respectively) but with an inverted dr in favor of the cis-isomer (4:96), as demonstrated by X-ray diffraction analysis of 6j (Fig. 2). The inversion of dr for proline-based substrates compared to other amino acids was previously reported for the 2,5-diketopiperazine system.11,12 Finally, the addition performed on seven-membered rings 5k and 5l led to the corresponding hemiaminals 6k and 6l in still good yields (62 and 55%, respectively) but with a lower dr (37:63), likely due to greater flexibility of the seven-membered ring.13 As aforementioned for 6h and 6j, the X-ray diffraction analysis of 6l revealed that the major isomer was the C9–C2 cis-isomer.
Looking for a further improvement in the access to novel aza-DKP platforms, a domino CHC/acid-catalyzed MeOH addition sequence was envisaged (Scheme 2).14 To this end, N-allyl substituted triazinedione 4a was submitted to a CHC reaction in the presence of PPTS in MeOH/THF (10:1) and led to compound 6a in 74% yield and a good stereoselectivity (93:7). Thus, compound 6a is readily attainable in a three-step process only from simple N-benzyl amino ester 2a in a 52% overall yield. This result highlights the efficiency of our strategy to provide a rapid access to novel N-heterocyclic scaffolds.
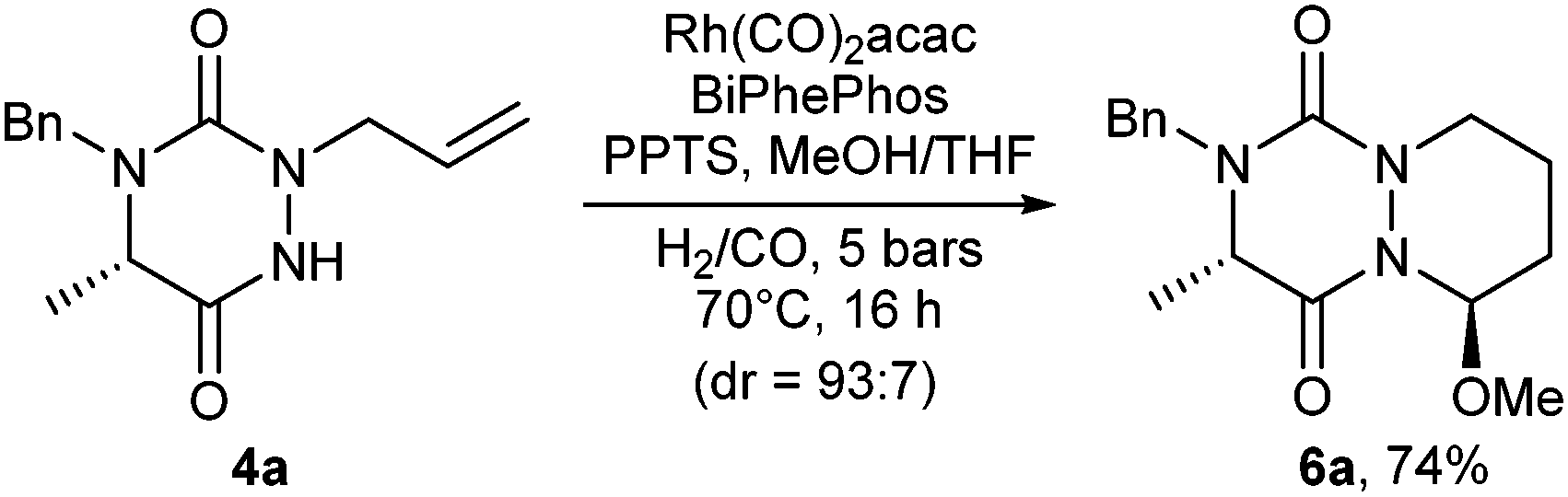 | ||
| Scheme 2 Domino cyclohydrocarbonylation/addition reaction. | ||
Finally, to further enlarge the molecular diversity of novel aza-DKP platforms and access to diversity-oriented chemical libraries, we envisaged the incorporation at C9 of functional groups able to react with commercially available building blocks. Hence, trans-isomer 6a was reacted either with TMSN3 or with TMSCN, both in the presence of BF3·OEt2 (Scheme 3).15 Thus, azide 7 was obtained in good yield (88%) and dr (>95:5). Nitrile 9 was also isolated in excellent yield (92%) but with a lower dr (68:32). Again, for both compounds, the major isomer was shown to be the C9–C2 trans-isomer (X-ray structure analysis, ESI†).
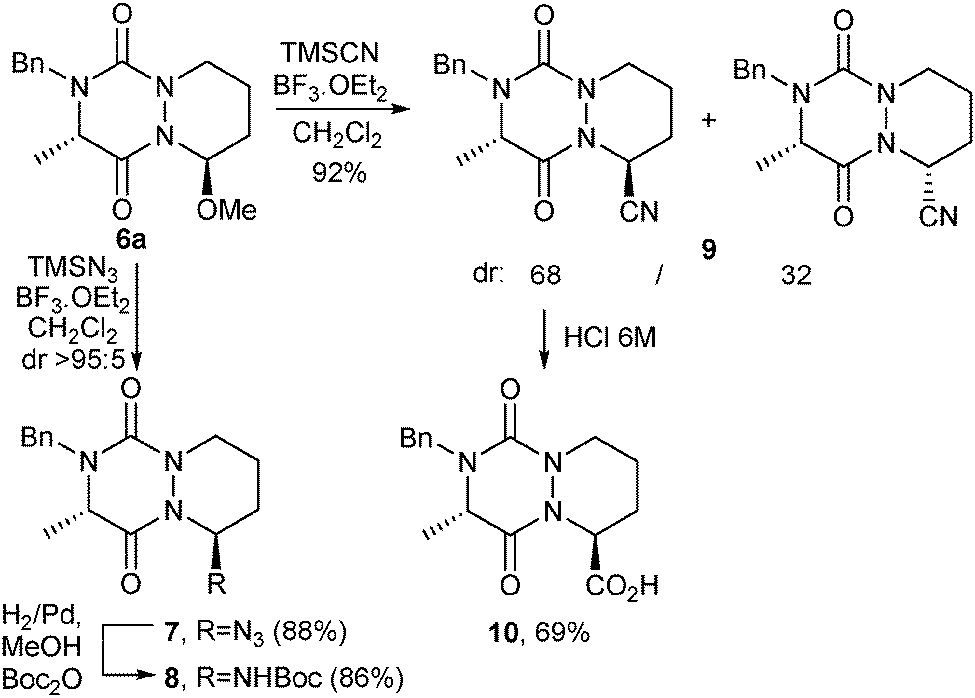 | ||
| Scheme 3 Diastereoselective functionalization of aza-DKP 6a. | ||
Besides, hydrolysis of the major isomer under acidic conditions led to carboxylic acid 10, able to react with amino building-blocks. Azide 7 was reduced with H2/Pd in the presence of di-tert-butyl dicarbonate to provide tBoc-protected compound 8 (86%). To further extend the chemical diversity of aza-diketopiperazines, compound 7 could also be engaged in Cu(I)-catalyzed azide–alkyne cycloaddition reactions.16
Starting from the amino acid pool, we have developed a diastereoselective approach for the preparation of a diverse range of N-heterocyclic scaffolds derived from aza-DKP. Indeed, this rapid and flexible method enables the efficient conversion of N-allyl substituted aza-DKP into newly reported bicyclic or tricyclic scaffolds containing six- or seven-membered rings by a domino CHC/addition sequence. A subsequent substitution at C-9 of the aza-DKP allows the diastereoselective incorporation of cyano and azido groups readily amenable, respectively, to amino or carboxylic functions which paves the way to the preparation of diversity-oriented libraries.
This work was supported by the Centre National de la Recherche Scientifique, the Université de Strasbourg (UDS) and the LABEX Medalis (ANR-10-LABX-0034). Dr Denis Heissler is kindly acknowledged for helpful discussions and for his comments on the manuscript. We are grateful to Cyril Antheaume and Barbara Schaeffer for NMR experiments (Service Commun d'Analyse, UDS).
Notes and references
- For recent reviews on DKP, see: (a) C. Prasad, Peptides, 1995, 16, 151–164 CrossRef CAS; (b) M. B. Martins and I. Carvalho, Tetrahedron, 2007, 63, 9923–9932 CrossRef CAS PubMed; (c) J. F. Gonzalez, I. Ortin, E. de la Cuesta and J. C. Menendez, Chem. Soc. Rev., 2012, 41, 6902–6915 RSC; (d) A. D. Borthwick, Chem. Rev., 2012, 112, 3641–3716 CrossRef CAS PubMed.
- (a) C. B. Bourguet, C. Proulx, S. Klocek, D. Sabatino and W. D. Lubell, J. Pept. Sci., 2010, 16, 284–296 CrossRef CAS PubMed; (b) D. Bonnet, J. F. Margathe, S. Radford, E. Pflimlin, S. Riche, P. Doman, M. Hibert and A. Ganesan, ACS Comb. Sci., 2012, 14, 323–334 CrossRef CAS PubMed.
- (a) A. Zega, Curr. Med. Chem., 2005, 12, 589–597 CAS; (b) H. J. Lee, I. A. Ahn, S. Ro, K. H. Choi, Y. S. Choi and K. B. Lee, J. Pept. Res., 2000, 56, 35–46 CrossRef CAS.
- (a) I. Ojima, M. Tzamarioudaki and M. Eguchi, J. Org. Chem., 1995, 60, 7078–7079 CrossRef CAS . For reviews on CHC, see: ; (b) B. Breit and W. Seiche, Synthesis, 2001, 1–36 CrossRef CAS PubMed; (c) W.-H. Chiou, S.-Y. Lee and I. Ojima, Can. J. Chem., 2005, 83, 681–692 CrossRef CAS; (d) G. Varchi and I. Ojima, Curr. Org. Chem., 2006, 10, 1341–1362 CrossRef CAS.
- E. Airiau, T. Spangenberg, N. Girard, A. Schoenfelder, J. Salvadori, M. Taddei and A. Mann, Chem. – Eur. J., 2008, 14, 10938–10948 CrossRef CAS PubMed.
- G. D. Cuny and S. L. Buchwald, J. Am. Chem. Soc., 1993, 115, 2066–2068 CrossRef CAS.
- L. L. W. Cheung, G. Vasapollo and H. Alper, Adv. Synth. Catal., 2012, 354, 2019–2022 CrossRef CAS.
- S. K. Rabindran, D. D. Ross, L. A. Doyle, W. Yang and L. M. Greenberger, Cancer Res., 2000, 60, 47–50 CAS.
- P. Deslongchamps, Stereoelectronic Effects in Organic Chemistry, Pergamon, New York, 1983, ch. 6 Search PubMed.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- A. Siwicka, K. Wojtasiewicz, B. Rosiek, A. Leniewski, J. K. Maurin and Z. Czarnocki, Tetrahedron: Asymmetry, 2005, 16, 975–993 CrossRef CAS PubMed.
- J. Baek, S. Y. Kang, C. Im and Y. S. Park, Eur. J. Org. Chem., 2014, 2780–2789 CrossRef CAS.
- (a) N. Zill, A. Schoenfelder, N. Girard, M. Taddei and A. Mann, J. Org. Chem., 2012, 77, 2246–2253 CrossRef CAS PubMed; (b) A. J. Pearson and Y. Kwak, Tetrahedron Lett., 2005, 46, 3407–3410 CrossRef CAS PubMed.
- E. Airiau, N. Girard, M. Pizzeti, J. Salvadori, M. Taddei and A. Mann, J. Org. Chem., 2010, 75, 8670–8673 CrossRef CAS PubMed.
- S. Röper, R. Wartchow and H. M. R. Hoffmann, Org. Lett., 2002, 4, 3179–3182 CrossRef PubMed.
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS; (b) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed; (c) P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and analytical data for all the compounds. Crystal structures of 5a, 5i, 5k, 6a, 7, trans-isomer of 9 and cis-isomers of 6l and 6j. CCDC 1001769–1001776. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc03660c |
| This journal is © The Royal Society of Chemistry 2014 |