 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Platinum corroles†
Abraham B.
Alemayehu
a,
Hugo
Vazquez-Lima
a,
Christine M.
Beavers
b,
Kevin J.
Gagnon
b,
Jesper
Bendix
c and
Abhik
Ghosh
*a
aDepartment of Chemistry and Center for Theoretical and Computational Chemistry, UiT – The Arctic University of Norway, 9037 Tromsø, Norway. E-mail: abhik.ghosh@uit.no
bAdvanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, CA 94720-8229, USA
cDepartment of Chemistry, University of Copenhagen, Universitetsparken 5, DK-2100 Copenhagen, Denmark
First published on 9th June 2014
Abstract
Platinum has been inserted into corroles for the first time and three oxidized PtIV(corrole˙2−)ArAr′ complexes have been structurally characterized. The Soret maxima of these complexes exhibit an unusually strong dependence on the meso-aryl substituents on the corrole, indicating aryl → corrole˙2− charge transfer character in these transitions.
As trianionic ligands with contracted N4 cores, corroles sustain a great deal of unique coordination chemistry relative to porphyrins.1 Within this area, heavy element corroles are of particular interest as optical sensors, near-IR dyes, phosphors, and organic light emitting diodes.2 The size mismatch between the corrole N4 cores and the large ionic radii of the lower oxidation states of the 5d elements, however, poses formidable challenges for metal insertion. As of today, Hf,3 W,4 Re,5 Ir,6 and Au,7 corroles have been synthesized, whereas others such as osmium and platinum corroles are still to be reported. Here we present unambiguous evidence, including multiple single-crystal X-ray analyses, for the formation of platinum corroles.
Insertion of Pt into corroles proved to be extraordinarily challenging. A large number of commercially available Pt precursors, each tested with a wide selection of solvents, failed to yield isolable Pt corrole derivatives. In the end, microwave irradiation of the commercially unavailable tetranuclear platinum acetate complex [Pt(OAc)2]4·2HOAc8 in benzonitrile at 140–150 °C for 2 hours led to low but reproducible yields (∼6%) of diamagnetic Pt(IV) corroles. Notably, aerobic conditions were critical to the success of the reaction; strict exclusion of oxygen did not result in Pt-containing products. Based on MALDI-TOF mass spectrometry and 1H NMR spectroscopy, the products could be formulated as Pt{T(p-X-Ph)C}(o/m/p-C6H4CN)(PhCN), where {T(p-X-Ph)C}3− is a meso-triarylcorrole trianion with aryl para substituents X = CF3, H, CH3, and OCH3 and the axial benzonitrile-derived aryl ligand may be bound through the o-, m-, or p-carbon, relative to the CN group (Fig. 1). Thus, C–H activation of benzonitrile resulting in a Pt(IV)-aryl center has been critical to the synthesis of stable platinum corroles. Electrospray ionization mass spectrometric studies indicated the N-bound benzonitrile ligand in these Pt(IV) complexes to be labile. Preliminary X-ray analysis of the putative Pt{T(p-CF3-Ph)C}(m-C6H4CN)(PhCN) complex indicated extensive disorder involving benzonitrile-derived ligands; despite some evidence of five-coordinate Pt{T(p-X-Ph)C}(o/m/p-C6H4CN), much of the material consisted of various regioisomers of Pt{T(p-X-Ph)C}(o/m/p-C6H4CN)2, which formally correspond to a Pt(V) oxidation state. Thus, a second C–H activation, involving the N-coordinated benzonitrile, had taken place during crystallization. Air-stable, oxidized platinum complexes Pt{T(p-X-Ph)C}(o/m/p-C6H4CN)(p-C6H4CH3) could be more reliably obtained by treating the Pt(IV) complexes with an aryl-Grignard reagent (Fig. 1). Because of the low yields, only the m-C6H4CN regioisomers were fully characterized for all the corroles, whereas the p-C6H4CN isomer could be characterized for only one of the corroles. Fortunately, three oxidized Pt corroles yielded X-ray quality crystals, affording full structural characterization.
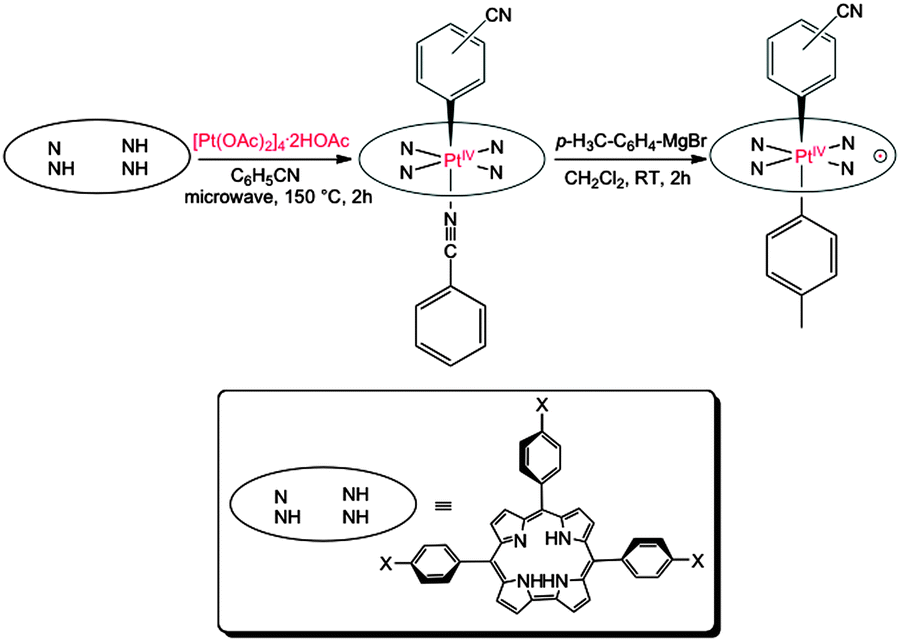 | ||
| Fig. 1 Synthesis of six-coordinate PtIV–Ar corroles and oxidized PtIV–corrole˙2−–ArAr′ derivatives. | ||
X-band EPR spectra were recorded for the oxidized Pt corroles in the solid state (Fig. 2), in solution (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH2Cl2:toluene) at RT, and in a frozen glass at 73 K obtained from the same solution. The complexes all gave strong signals with g-values ∼2.00, consistent with the ligand radical formulation Pt(corrole˙2−)ArAr′. The spectra could be simulated with fairly narrow linewidths (Lorentzian, fwhh = 3 G) and a slight g-anisotropy, but notably without any 195Pt hyperfine coupling.9 Upon dissolution in a mixture of CH2Cl2 and toluene at RT, the spectra changed profoundly (Fig. 3); the bandwidth became nearly twice as large, with concomitant appearance of fully resolved hyperfine coupling to 195Pt. Freezing of the solution at 73 K had only a slight effect on the spectrum (Fig. 3). The much narrower signals in the solid state may be interpreted as an unusually well-behaved example of “exchange narrowing”.10 This is in agreement with the crystal structures, which indicate partial corrole π-stacking with relatively short interplanar distances of about 3.4 Å. The observed g-value range of 1.997–2.011 agrees with literature values for corrole-based radical complexes.11 Although well-resolved in solution, the hyperfine coupling to Pt is too small for the radical to be Pt-centered.
:1 CH2Cl2:toluene) at RT, and in a frozen glass at 73 K obtained from the same solution. The complexes all gave strong signals with g-values ∼2.00, consistent with the ligand radical formulation Pt(corrole˙2−)ArAr′. The spectra could be simulated with fairly narrow linewidths (Lorentzian, fwhh = 3 G) and a slight g-anisotropy, but notably without any 195Pt hyperfine coupling.9 Upon dissolution in a mixture of CH2Cl2 and toluene at RT, the spectra changed profoundly (Fig. 3); the bandwidth became nearly twice as large, with concomitant appearance of fully resolved hyperfine coupling to 195Pt. Freezing of the solution at 73 K had only a slight effect on the spectrum (Fig. 3). The much narrower signals in the solid state may be interpreted as an unusually well-behaved example of “exchange narrowing”.10 This is in agreement with the crystal structures, which indicate partial corrole π-stacking with relatively short interplanar distances of about 3.4 Å. The observed g-value range of 1.997–2.011 agrees with literature values for corrole-based radical complexes.11 Although well-resolved in solution, the hyperfine coupling to Pt is too small for the radical to be Pt-centered.
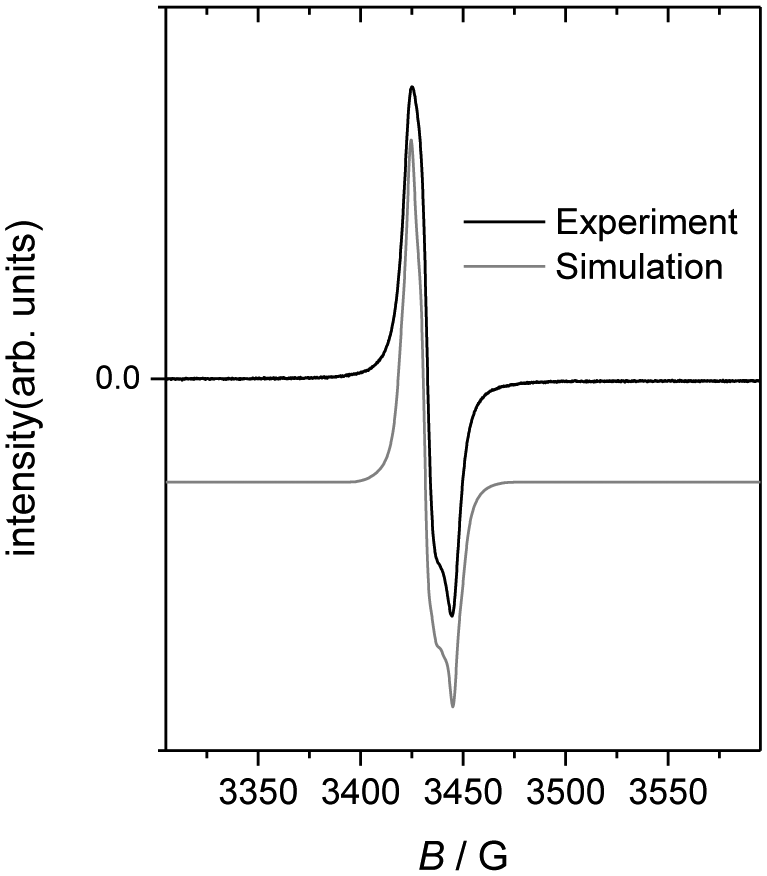 | ||
| Fig. 2 Solid state, RT X-band EPR spectrum of Pt(TPC)(m-C6H4CN)(p-C6H4CH3). Simulation offset for clarity. Simulation parameters: g = 1.999, 2.007, 2.011, value of A(195Pt) used = 0.8 × 10−3 cm−1, Lorentzian bandshape, fwhh = 3.0 G. | ||
 | ||
| Fig. 3 Left: RT, solution, X-band EPR spectrum of Pt(TPC)(m-C6H4CN)(p-C6H4CH3). Simulation offset for clarity. Simulation parameters: g = 2.005, A(195Pt) = 5.9 × 10−3 cm−1, Lorentzian bandshape, fwhh = 5.6 G. Right: frozen glass, 73 K spectrum of same solution. Simulation parameters: g = 2.005, A(195Pt) = 5.9 × 10−3 cm−1, Lorentzian bandshape, fwhh = 5.7 G. | ||
Three of the oxidized Pt complexes Pt(TPC)(m-C6H4CN)(p-C6H4CH3), Pt{T(p-CH3-Ph)C}(p-C6H4CN)(p-C6H4CH3), and Pt{T(p-CH3-Ph)C}(m-C6H4CN)(p-C6H4CH3) proved amenable to single-crystal X-ray analysis. All the structures refined well, giving R1 [I > 2σ(I)] values ∼3%. No evidence was found for counterions, positive or negative, consistent with the PtIV–corrole˙2− formulation mentioned above. Selected bond distances are listed in Table 1 and a representative thermal ellipsoid plot is shown in Fig. 4. As for many metallocorroles,12,13 the corrole macrocycles in all three complexes are almost perfectly planar. The short Pt–N distances of approximately 1.95 Å are consistent with a Pt(IV) oxidation state and the sterically constrained nature of the corrole N4 core, while the axial Pt–C distances of about 2.1 Å are typical for unconstrained PtIV–C bond distances.
| Pt{T(p-CH3-Ph)C}(p-C6H4CN)(p-C6H4CH3) | Pt(TPC)(m-C6H4CN)(p-C6H4CH3) | Pt{T(p-CH3-Ph)C}(m-C6H4CN)(p-C6H4CH3) | |||
|---|---|---|---|---|---|
| Pt1–N1 | 1.948(2) | Pt1–N1 | 1.942(3) | Pt1–N1 | 1.951(2) |
| Pt1–N2 | 1.9698(19) | Pt1–N2 | 1.965(3) | Pt1–N2 | 1.973(2) |
| Pt1–N3 | 1.9693(19) | Pt1–N3 | 1.970(3) | Pt1–N3 | 1.976(2) |
| Pt1–N4 | 1.9470(19) | Pt1–N4 | 1.945(3) | Pt1–N4 | 1.944(2) |
| Pt1–Cax(41) | 2.119(2) | Pt1–Cax(38) | 2.119(3) | Pt1–Cax(41) | 2.128(2) |
| Pt1–Cax(48) | 2.129(2) | Pt1–Cax(45) | 2.114(3) | Pt1–Cax(48) | 2.123(3) |
 | ||
| Fig. 4 Thermal ellipsoid plot for Pt(TPC)(m-C6H4CN)(p-C6H4CH3) with ellipsoid probabilities at 40%. H-atoms and solvent molecules have been omitted for clarity. | ||
Fig. 5 depicts key results from DFT calculations on the model complexes Pt(corrole)(Ph)(PhCN) (Cs) and Pt(corrole)Ph2 (C2v). The optimized structural parameters are in excellent agreement with those observed experimentally (Table 1). To a first approximation, the spin density of Pt(corrole)Ph2 corresponds to a corrole b1 radical (in terms of C2v irreps), which resembles a porphyrin a2u-type radical,14i.e., the spin density is largely localized on the three meso carbons and the four nitrogens and to a lesser extent on the direct Cβ–Cβ linkage. The Pt does not carry a significant amount of spin density, thereby ruling out any degree of Pt(V) character. Closer examination of Fig. 5 indicates that the corrole carries only about two-thirds of the total molecular spin density; the remaining one-third of the spin density is evenly divided between the two formally anionic phenyl ipso carbons. Thus, the HOMO is not a pure corrole b1 HOMO, but has a certain amount of aryl character as well.
 | ||
| Fig. 5 Selected BP86-D3/STO-TZ2P results on the model complexes Pt(corrole)(Ph)(PhCN) (Cs) and Pt(corrole)Ph2 (C2v): (a) distances (black, Å) and Mulliken spin populations (blue); (b) spin density plots for Pt(corrole)Ph2: top and side views; (c) the HOMO of Pt(corrole)Ph2: top and side views. | ||
Table 2 presents redox potentials for Pt{T(p-X-Ph)C}(m-C6H4CN)(PhCN), the oxidized Pt{T(p-X-Ph)C}(m-C6H4CN)(p-P)C} complexes in CH2Cl2. Note that, whereas the first oxidation potentials vary little among the different classes of metallocorroles, the reduction potentials vary considerably. Compared with Cu corroles, the Pt(IV) and Au(III) corroles undergo reduction at considerably substantially more negative potentials.7c Thus, Pt(IV) and Au(III) corroles exhibit relatively large electrochemical “HOMO–LUMO gaps” (i.e., the algebraic difference between the first oxidation and reduction potentials) – ∼1.4 eV for Pt(IV) and ∼2.2 eV for Au(III). These two metal ions appear to be strongly stabilized by the trianionic corrole ligands, which would explain the resistance to reduction. In contrast, the oxidized Pt{T(p-X-Ph)C}(m-C6H4CN)(p-C6H4CH3) complexes, like Cu triarylcorroles, are readily reduced at approximately 0.1 ± 0.1 V (vs. SCE), as expected on the basis of their corrole˙2− radical character.17
| Complex | X | E 1/2ox1 | E 1/2ox2 | E 1/2red1 | E 1/2red2 | E 1/2ox1 − E1/2red1 |
|---|---|---|---|---|---|---|
| a Irreversible feature is noted at −1.43 eV. | ||||||
| Cu{T(p-X-P)C}16 | CF3 | 0.89 | — | −0.08 | — | 0.97 |
| H | 0.76 | — | −0.20 | — | 0.96 | |
| CH3 | 0.70 | — | −0.23 | — | 0.93 | |
| OCH3 | 0.65 | — | −0.24 | — | 0.89 | |
| Au{T(p-X-P)C}7c | CF3 | 0.94 | — | −1.29 | — | 2.23 |
| H | 0.80 | 1.35 | −1.38 | — | 2.18 | |
| CH3 | 0.78 | 1.35 | −1.42 | — | 2.20 | |
| OCH3 | 0.76 | 1.32 | −1.57 | — | 2.33 | |
| Pt{T(p-X-Ph)C}(m-C6H4CN)(PhCN) | CF3 | 0.72 | 1.40 | — | ||
| H | 0.63 | 1.29 | −0.83 | — | 1.46 | |
| CH3 | 0.57 | 1.25 | −0.84 | — | 1.41 | |
| OCH3 | 0.53 | 1.14 | −0.85 | — | 1.38 | |
| Pt{T(p-X-Ph)C}(m-C6H4CN)(p-C6H4CH3) | CF3 | 0.97 | — | 0.21 | −0.77 | 0.76 |
| H | 0.88 | — | 0.09 | −0.82 | 0.79 | |
| CH3 | 0.79 | — | 0.03 | −0.86 | 0.76 | |
| OCH3 | 0.74 | — | 0.01 | −0.87 | 0.73 | |
A number of metallocorrole families such as Cu, MnCl and FeCl15meso-triarylcorroles exhibit strongly substituent-sensitive electronic absorption spectra, with the Soret maximum shifting sensitively as a function of substituents on the meso-aryl groups. These have been analyzed for copper triarylcorroles with TDDFT calculations and ascribed to so-called hyper character, i.e., phenyl-to-metal charge transfer (CT) character mixing into the Soret transitions.17 For CrO, MoO, Ag and Au triarylcorroles on the other hand the Soret maxima are relatively independent of substituents on the meso-aryl groups.7c,18 Against this backdrop, the Soret maxima of Pt corroles were found to be substituent-sensitive or -insensitive, depending on the overall oxidation level of the complexes. As shown in Fig. 6 and Table 3, the Pt(IV) complexes Pt{T(p-X-Ph)C}(m-C6H4CN)(PhCN) exhibit substituent-insensitive Soret maxima, whereas the oxidized Pt{T(p-X-Ph)C}(m-C6H4CN)(p-C6H4CH3) series exhibits some of the strongest meso substituent effects observed for metallocorroles. These observations suggest that meso-aryl substituent sensitivity occurs precisely in those cases where the corrole has substantial corrole˙2− character; this is the case for Cu, FeCl, MnCl and the oxidized Pt corroles. For the other metallocorroles, where the corrole is relatively innocent, the Soret maxima are substituent-insensitive.
 | ||
| Fig. 6 Electronic absorption spectra of the Pt(IV) (top) and oxidized Pt(IV) (bottom) series. | ||
| Complex | p-Substituent | |||
|---|---|---|---|---|
| CF3 | H | CH3 | OCH3 | |
| Cu[T(p-X-Ph)] | 407 | 410 | 418 | 434 |
| Au[T(p-X-Ph)] | 419 | 418 | 420 | 420 |
| Pt{T(p-X-Ph)C}(m-C6H4CN)(PhCN) | 430 | 426 | 427 | 427 |
| Pt{T(p-X-Ph)C}(m-C6H4CN)(p-C6H4CH3) | 443 | 453 | 460 | 475 |
Compared with other 5d metallocorroles such as Au and Ir corroles, which are comparatively unreactive, platinum corroles have long been of interest on account of their potential for significant axial reactivity vis-à-vis small-molecule activation. Here we have presented the first unambiguous proof of platinum insertion into the corrole macrocycle, including three single-crystal X-ray structures. Two series of complexes have been prepared in low yields: the six-coordinate Pt(IV) series Pt{T(p-X-Ph)C}(m-C6H4CN)(PhCN) and the oxidized series Pt{T(p-X-Ph)C}(m-C6H4CN)(p-C6H4CH3). Ongoing research on Pt corroles focuses on developing higher-yielding syntheses and on detailed studies of C–H activation and other reactivity. Whether the compounds exhibit significant biological activity, particularly anticancer activity, remains an exciting question for the future.
This work was supported by the Research Council of Norway (AG), the Danish Research Council (JB) and the Advanced Light Source, Berkeley. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Notes and references
- J. H. Palmer, Struct. Bonding, 2012, 142, 49–89 CrossRef CAS.
- I. Aviv-Harel and Z. Gross, Chem. – Eur. J., 2009, 15, 8382–8394 CrossRef CAS PubMed.
- R. Padilla, H. L. Buckley, A. L. Ward and J. Arnold, Chem. Commun., 2014, 50, 2922–2924 RSC.
- I. Nigel-Etinger, I. Goldberg and Z. Gross, Inorg. Chem., 2012, 51, 1983–1985 CrossRef CAS PubMed.
- M. K. Tse, Z. Zhang, T. C. W. Mak and K. S. Chan, J. Chem. Soc., Chem. Commun., 1998, 1199–1200 RSC.
- (a) J. H. Palmer, M. W. Day, A. D. Wilson, L. M. Henling, Z. Gross and H. B. Gray, J. Am. Chem. Soc., 2008, 130, 7786–7787 CrossRef CAS PubMed; (b) J. H. Palmer, A. C. Durrell, Z. Gross, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2010, 132, 9230–9231 CrossRef CAS PubMed.
- (a) A. Alemayehu and A. Ghosh, J. Porphyrins Phthalocyanines, 2011, 15, 106–110 CrossRef CAS; (b) E. Rabinovitch, I. Goldberg and Z. Gross, Chem. – Eur. J., 2011, 17, 12294–12301 CrossRef PubMed; (c) K. E. Thomas, A. B. Alemayehu, J. Conradie, C. Beavers and A. Ghosh, Inorg. Chem., 2011, 50, 12844–12851 CrossRef CAS PubMed; (d) K. E. Thomas, C. M. Beavers and A. Ghosh, Mol. Phys., 2012, 110, 2439–2444 CrossRef CAS.
- M. Basato, A. Biffis, G. Martinati, C. Tubaro, A. Venzo, P. Ganis and F. Benetollo, Inorg. Chim. Acta, 2003, 355, 399–403 CrossRef CAS.
- The value of A(195Pt) used in Fig. 2 (0.8 × 10−4 cm−1) is an upper limit beyond which the skewness of the large amplitude peaks becomes incorrect, resulting in shoulders on the outside of the peaks.
- (a) D. Kivelson, J. Chem. Phys., 1957, 27, 1087–1098 CrossRef CAS PubMed; (b) T. Halpern, S. M. McKoskey and J. A. McMillan, J. Chem. Phys., 1970, 52, 3526–3529 CrossRef CAS PubMed; (c) E. F. Strother, H. A. Farach and C. P. Poole, Phys. Rev. A: At., Mol., Opt. Phys., 1971, 4, 2079–2087 CrossRef.
- (a) J. H. Palmer, A. Mahammed, K. M. Lancaster, Z. Gross and H. B. Gray, Inorg. Chem., 2009, 48, 9308–9315 CrossRef CAS PubMed; (b) B. Ramdhanie, J. Telser, A. Caneschi, L. N. Zakharov, A. L. Rheingold and D. P. Goldberg, J. Am. Chem. Soc., 2004, 126, 2515–2525 CrossRef CAS PubMed.
- Review: K. E. Thomas, A. Alemayehu, J. Conradie, C. M. Beavers and A. Ghosh, Acc. Chem. Res., 2012, 45, 1203–1214 CrossRef CAS PubMed.
- Copper corroles are nonplanar and an exception to this generalization: (a) A. B. Alemayehu, E. Gonzalez, L.-K. Hansen and A. Ghosh, Inorg. Chem., 2009, 48, 7794–7799 CrossRef CAS PubMed; (b) A. B. Alemayehu, L.-K. Hansen and A. Ghosh, Inorg. Chem., 2010, 49, 7608–7610 CrossRef CAS PubMed.
- A. Ghosh, T. Wondimagegn and A. B. J. Parusel, J. Am. Chem. Soc., 2000, 122, 5100–5104 CrossRef CAS.
- A. Ghosh and E. Steene, J. Inorg. Biochem., 2002, 91, 423–436 CrossRef CAS.
- (a) I. H. Wasbotten, T. Wondimagegn and A. Ghosh, J. Am. Chem. Soc., 2002, 124, 8104–8116 CrossRef CAS PubMed; (b) Z. Ou, J. Shao, H. Zhao, K. Ohkubo, I. H. Wasbotten, S. Fukuzumi, A. Ghosh and K. M. Kadish, J. Porphyrins Phthalocyanines, 2004, 8, 1236–1247 CrossRef CAS.
- A. B. Alemayehu, J. Conradie and A. Ghosh, Eur. J. Inorg. Chem., 2011, 1857–1864 CrossRef CAS.
- I. Johansen, H.-K. Norheim, S. Larsen, A. B. Alemayehu, J. Conradie and A. Ghosh, J. Porphyrins Phthalocyanines, 2011, 15, 1335–1344 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedures, spectra and X-ray structure determinations. CCDC 995762–995764. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc02548b |
| This journal is © The Royal Society of Chemistry 2014 |