 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The taming of oxygen: biocatalytic oxyfunctionalisations
Dirk
Holtmann
a,
Marco W.
Fraaije
b,
Isabel W. C. E.
Arends
c,
Diederik J.
Opperman
d and
Frank
Hollmann
*c
aDECHEMA Research Institute, Theodor-Heuss-Allee 25, 60486 Frankfurt am Main, Germany
bMolecular Enzymology group, Biomolecular Sciences and Biotechnology Institute, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands
cDepartment of Biotechnology, Delft University of Technology, Julianalaan 136, 2628 BL Delft, The Netherlands. E-mail: f.hollmann@tudelft.nl
dDepartment of Biotechnology, University of the Free State, Bloemfontein 9300, South Africa
First published on 23rd May 2014
Abstract
The scope and limitations of oxygenases as catalysts for preparative organic synthesis is discussed.
 Dirk Holtmann | Dirk Holtmann was born in Bremen, Germany, and has completed his diploma in chemical engineering/biotechnology in 1999. He obtained his PhD at the Otto von Guericke University of Magdeburg on the electrochemical measurement of the microbial activities in fermentations. He is group leader of the biochemical engineering group at the DECHEMA Research Institute in Frankfurt, Germany. His current research activities concentrate on biocatalysis and biotransformations, the development and evaluation of electro-enzymatic processes as well as microbial electrosynthesis. |
 Marco W. Fraaije | Marco Fraaije obtained his PhD in Biochemistry in 1998 from Wageningen University, The Netherlands. After a long-term EMBO fellowship in protein crystallography in the group of Prof. Andrea Mattevi at the University of Pavia, Italy, he moved to the University of Groningen, The Netherlands. Since 2012 he is full professor, leading a Molecular Enzymology research group. His research interests cover many areas of biocatalysis, in particular the knowledge-based discovery, engineering and redesign of oxidative enzymes. |
 Isabel W. C. E. Arends | Isabel Arends received her PhD in physical organic chemistry in 1993, from Leiden University, the Netherlands. Postdoctoral work followed with Keith Ingold at the NRC in Canada on selective liquid phase oxidations. She joined the group of Roger Sheldon at the Delft University of Technology in 1995. In 2007 she became full professor in biocatalysis at the same university. Her research focuses on redox biocatalysts, novel enzymes, and chemo/biocatalytic methods for conversion of biomass. Prof. Arends is author of over 140 publications and 17 book chapters. She is co-author of the seminal monograph "Green Chemistry and Catalysis". |
 Diederik J. Opperman | Diederik J. Opperman obtained his PhD in biochemistry from the University of the Free State (South Africa) in 2008. He then joined the research group of Manfred T. Reetz at the Max-Planck-Institut für Kohlenforschung (Germany) as a postdoctoral co-worker in directed evolution. He is currently a senior lecturer and group leader at the University of the Free State (SA) with a research focus on the structure–function relationship of biocatalysts. |
 Frank Hollmann | Frank Hollmann studied Chemistry in Bonn, Germany. He received his PhD in Bioelectrocatalysis from the ETH in Zurich, Switzerland. After a postdoctoral stay at the MPI for Coal Research in Mülheim, Germany and some years with Evonik Goldschmidt he joined the Biocatalysis and Organic Chemistry group of TU Delft in 2008 as assistant professor. His research centres on the use of enzymes for organic synthesis. Particularly, he is interested in redox biocatalysis for stereospecific reduction and oxyfunctionalization chemistry using novel biocatalysts and alternative regeneration approaches. |
Introduction
Selective oxyfunctionalisation of non-activated hydrocarbons represents a contemporary issue of organic chemistry. The inertness of most C–H bonds requires powerful oxygen transfer agents.1 High activity, however, is frequently accompanied by poor selectivity, which is in contrast to the demands of modern chemical synthesis.2Nature provides us with a class of catalysts that comprises both features: activity and selectivity: Oxygenases catalyse the introduction of oxygen atoms from molecular oxygen or hydrogen peroxide into (non-)activated C–H- and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C-bonds and to heteroatoms. Often, these oxyfunctionalisation reactions occur highly selectively. Many oxygenases are selective for only one position in structurally complex molecules and they introduce the oxygen atoms with high stereocontrol. Furthermore, overoxidation, which is a frequent problem in chemical oxyfunctionalisation, is far less observed in enzymatic oxyfunctionalisations. This unique combination of activity and selectivity stems from the embedding of reactive oxygen transfer reagents such as highly oxidised iron–oxo complexes or organic peroxides in the cavity of an enzyme's active site. The well-defined and chiral environment not only positions the starting material precisely to the ‘hot’ oxygen atom transferred (accounting for selectivity) but also stabilises transition states thereby leading to, sometimes dramatic, rate accelerations as compared to the biomimetic analogues (Scheme 1).
C-bonds and to heteroatoms. Often, these oxyfunctionalisation reactions occur highly selectively. Many oxygenases are selective for only one position in structurally complex molecules and they introduce the oxygen atoms with high stereocontrol. Furthermore, overoxidation, which is a frequent problem in chemical oxyfunctionalisation, is far less observed in enzymatic oxyfunctionalisations. This unique combination of activity and selectivity stems from the embedding of reactive oxygen transfer reagents such as highly oxidised iron–oxo complexes or organic peroxides in the cavity of an enzyme's active site. The well-defined and chiral environment not only positions the starting material precisely to the ‘hot’ oxygen atom transferred (accounting for selectivity) but also stabilises transition states thereby leading to, sometimes dramatic, rate accelerations as compared to the biomimetic analogues (Scheme 1).
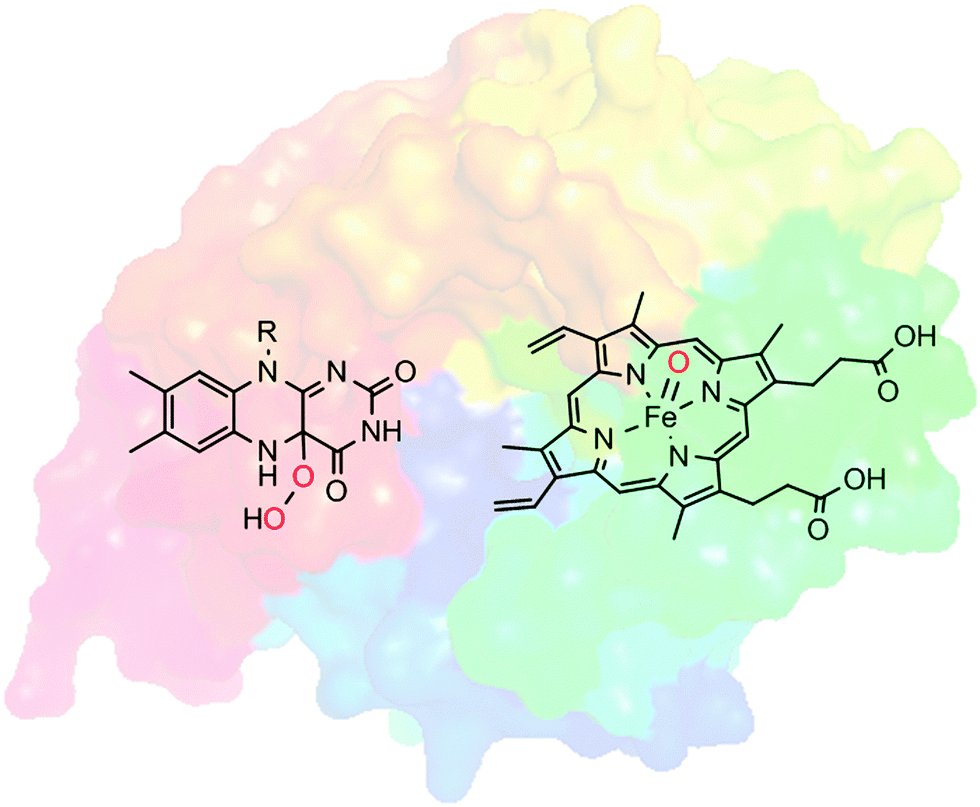 | ||
| Scheme 1 Oxygenases comprise highly reactive oxygenation agents such as (formal) FeV-oxyferryl-heme, or (hydro)peroxo-flavins embedded in the well-defined framework of an enzyme.3 | ||
Therefore, it is not very astonishing that the interest in biological oxyfunctionalisation chemistry has been steadily growing over the past decades. We are convinced that oxygenases, following the well-established hydrolases4–6 and the ever more popular dehydrogenases,5,7–10 will represent the next upcoming wave of biocatalysts used in chemical synthesis.11
The aim of this perspective is to encourage organic chemists to consider oxygenases in their synthesis planning more often. For this, we will focus on the application of oxygenases for organic synthesis, giving an overview over the rich product spectrum available already today and discussing some recent preparative examples. In particular, we will cover the most-common oxygenases, i.e. flavin-dependent monooxygenases as well as heme-dependent monooxygenases and -peroxygenases. Also some perceived and real limitations en route to becoming truly practical catalysts, together with some promising solutions will be discussed. A detailed discussion of the catalytic mechanisms would be beyond the scope of this perspective; the interested reader is referred to some excellent reviews5,6,12–14 exhaustively covering our current mechanistic understanding.
It is also worth mentioning here that continuously new oxygenases are discovered as well as existing ones are improved to match with the requirements of chemical synthesis.1,10,12,14–35 Again, an in-depth discussion of these approaches, let alone the new enzymes obtained is not possible here, but some excellent recent contributions exhaustively cover this field.
Flavin-dependent monooxygenases
Flavin-dependent monooxygenases represent an extremely diverse class of enzymes catalysing an equally diverse range of synthetically useful oxyfunctionalisation reactions (Scheme 2).28–30,33,36–40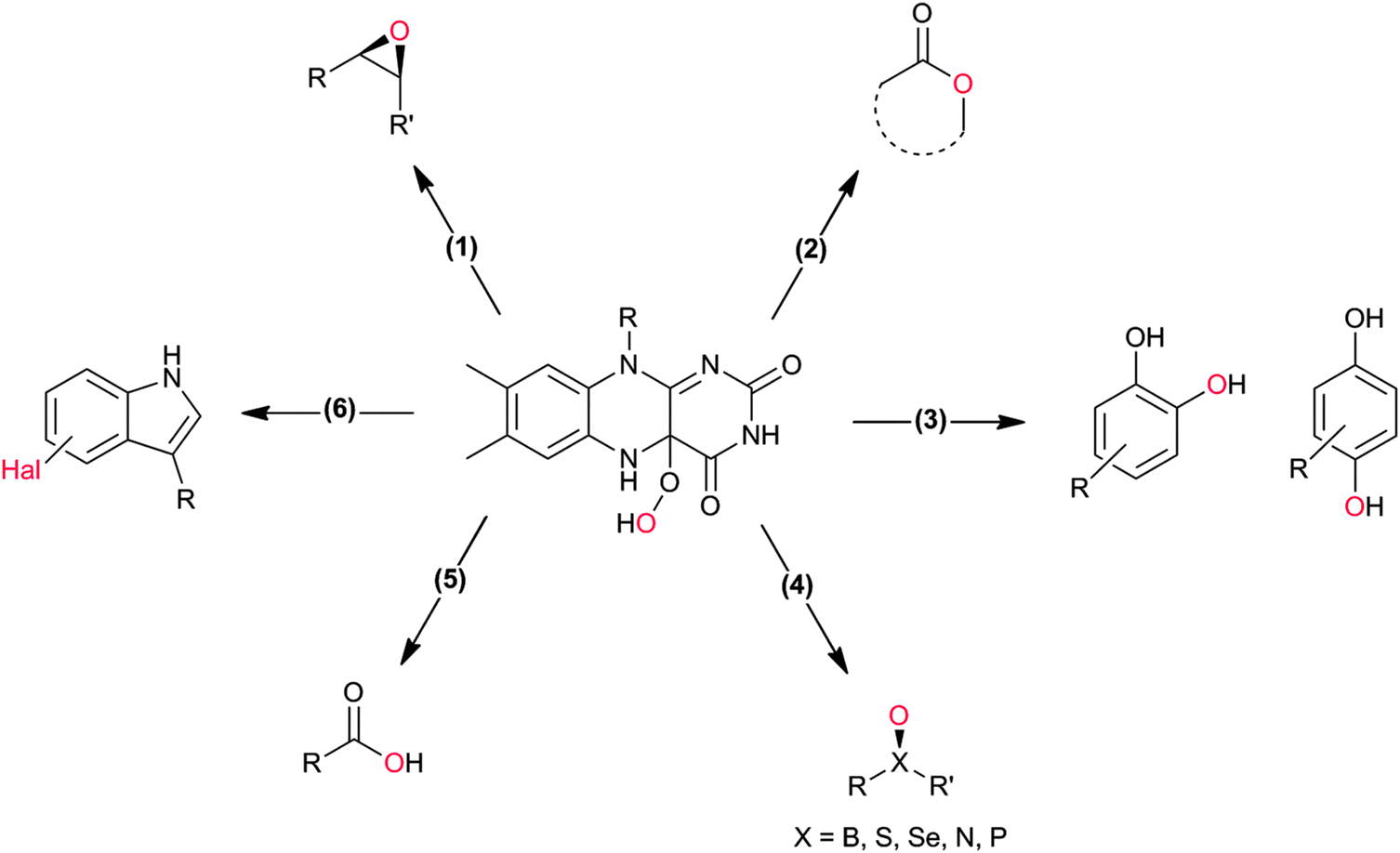 | ||
| Scheme 2 4a-(Hydro)peroxoflavin as the active species leading to the diverse reactions catalysed by flavin-dependent monooxygenases. (1) Epoxidation of CC-double bonds, (2) Baeyer–Villiger oxidation of (cyclic) ketones, (3) ortho- and para-hydroxylation of phenols, (4) heteroatom oxygenation, (5) oxidation of aldehydes, and (6) halogenation reactions. | ||
The catalytic mechanism of the flavin-dependent monooxygenases comprises an oxidative and a reductive half reaction. The catalytic cycles start with the latter by NAD(P)H-mediated reduction of the enzyme-bound flavin group. The reduced flavin interacts with molecular oxygen to form an organic peroxide (C4a-(hydro)peroxoflavin), which acts as the actual oxygen transfer agent (oxidative half reaction). The resulting C4a-hydroxyflavin eliminates water and thereby returns to the oxidized resting form (Scheme 3). Only very recently an alternative mechanism for oxygenation by a flavoprotein has been revealed.41 It was shown that the bacterial enzyme EncM, involved in the biosynthesis of the antibiotic enterocin, performs an coenzyme-independent oxygen insertion via a reactive flavin N5-oxide. In this specific example, the reducing equivalents come from the substrate itself. It remains to be seen whether this is a rare example or whether there are more flavoproteins in nature that employ such a mechanism.
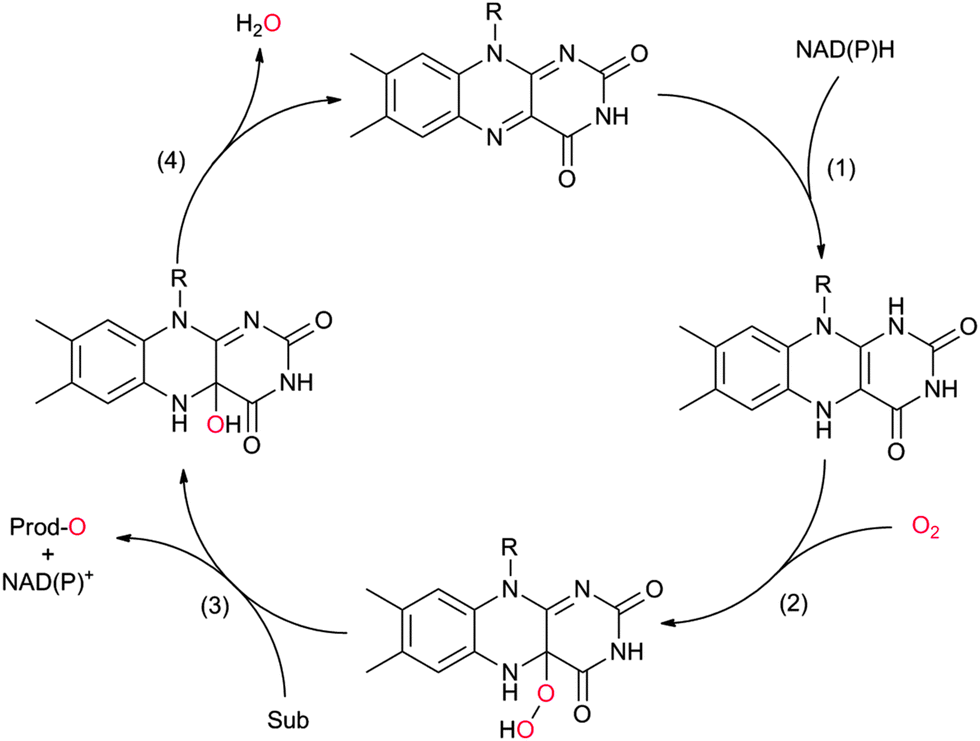 | ||
| Scheme 3 Simplified catalytic cycle of flavin-dependent monooxygenases. In the resting state of the enzyme the reduced nicotinamide cofactor (NAD(P)H) binds to the enzyme and transfers its hydride to the alloxazine moiety (1). In step (2) the reduced flavin reacts with molecular oxygen yielding the catalytically active (hydro)peroxoflavin, which, in step (3) performs the oxyfunctionalisation reaction. The resting state is regenerated after water extrusion (step (4)). Please note that for reasons of simplicity, protonation steps have been omitted from the scheme. | ||
In principle, the chemical versatility of flavins should also allow for H2O2-driven oxyfunctionalisation reactions as frequently demonstrated with N5-alkylated flavins as ‘chemical catalysts’.28 Recently, Fraaije and coworkers have demonstrated that flavoproteins can be engineered to act as peroxygenases by replacing the riboflavin cofactor in the riboflavin-binding protein with alkylated flavins. Overall the general feasibility of ‘flavoperoxygenases’ could be demonstrated (Scheme 4).42
 | ||
| Scheme 4 Simplified scheme of a ‘flavoperoxygenase’ mechanism. | ||
Organic chemists are probably most acquainted to the enzymatic Baeyer–Villiger oxidation mediated by so-called Baeyer–Villiger monooxygenases (BVMOs, Scheme 2, 2). Since some early contributions from the mid-20th century43–45 and the pioneering work by Trudgill and coworkers46,47 the scope of enzymatic Baeyer–Villiger reactions has been expanded significantly in the last two decades31,48–50 and a range of synthetically useful transformations have been reported (Table 1). Also, the first crystal structure of a BVMO51 has significantly contributed to the mechanistic understanding of BVMOs52,53 and enabled rational approaches to engineer tailored new BVMOs.54–64 Today, the enzymatic Baeyer–Villiger oxidation (in the form of whole cell biotransformation, vide infra) has reached the multi-kg scale65–68 and further scale-ups and industrial applications may be expected in the nearer future.
| Substrate | Product | R | % Conv. (% yield) | Catalyst | ee (%) | Volumetric scale | Remarks | Ref. |
|---|---|---|---|---|---|---|---|---|
| SMO: styrene monooxygenase; CHMO: cyclohexanone monooxygenase; BVMO: Baeyer–Villiger monooxygenase; PAMO: phenylacetone monooxygenase; HAPMO: 4-hydroxyacetophenone monooxygenase; HbpA: 2-hydroxy biphenyl-3-monooxygenase; PheA1: phenol hydroxylase; 3HB6H: 3-hydroxy benzoate-6-hydroxylase; (D)KR: (dynamic) kinetic resolution. | ||||||||
| (1) Epoxidation reactions | ||||||||
|
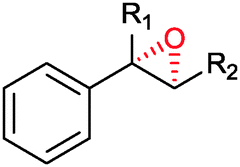
|
R1 = H, R2 = Me | (87) | SMO (rec in growing E. coli) | >99 (S) | 1 L | [S]0 = 695 mM; t = 8 h | 100 |
| R1 = H, R2 = Me | 95 (87) | SMO (crude cell extract) | 99.7 (S) | 100 mL | [S]0 = 50 mM; t = 10 h; TON(SMO) = 1844 | 114 | ||
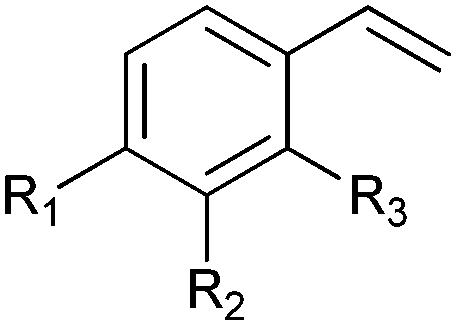
|
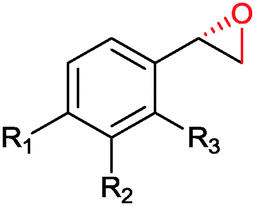
|
R1 = R2 = H, R3 = F, Cl, Br, Me | (90, 70, 31, 62) | SMO (rec in resting E. coli) | All >99.9 (S) except 62.8 (S) for Me | 1 mL | [S]0 = 200 mM; t = 24 h | 119 and 120 |
| R1 = R3 = H, R2 = Cl | 91 (73) | SMO (crude cell extract) | >99.9 (S) | 100 mL | [S]0 = 50 mM; t = 10 h; TON(SMO) = 2171 | 114 | ||
|
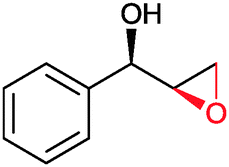
|
(50) | SMO (rec in resting E. coli) | >99 epoxide (de = 98%) | 20 mL | [S]0 = 4,5 mM; t = 2 h | 137 and 138 | |
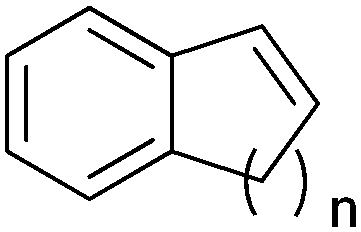
|
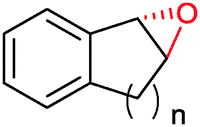
|
n = 1 | (48) | SMO (rec in growing E. coli) | 98 (S) | 1 L | [S]0 > 150 mM; t = 19 h | 112 |
| n = 2 | (53) | SMO (rec in growing E. coli) | 98.5 (S) | 1 L | [S]0 > 150 mM; t = 45 h | 112 | ||
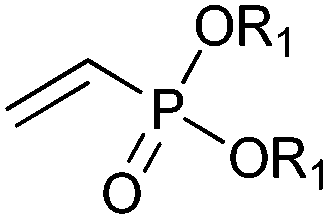
|
|
R1 = Et, Me | (40, 10) | CHMO (partially purified enzyme) | ≥98 (R) | 10 mL | [S]0 = 12 mM; t = 48 h | 139 |
| (2) Baeyer–Villiger oxidations | ||||||||
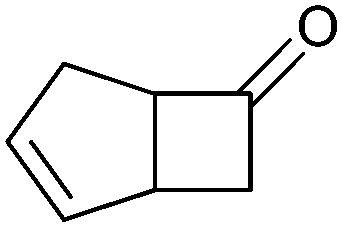
|
|
>99 (58) | CHMO (rec in growing E. coli) | 99% (1R,5S) 97 (1S,5R) | 50 L | 900 g; [S]0 > 0.1 M; t = 20 h | 65 and 66 | |
| 200 L | Fed-batch 4.5 g L−1; ([S]total = 36 mM); t = 7 h | 140 | ||||||
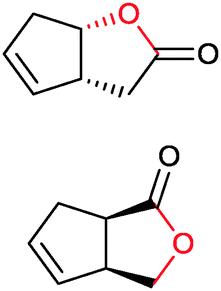
|
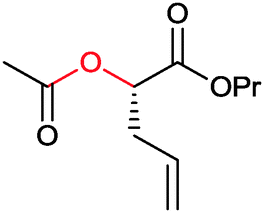
|
>99 | PAMO (partially purified enzyme) | >99 (S) | 13 mL | [S]0 > 20 mM; t = 24 h; DKR | 70 | |
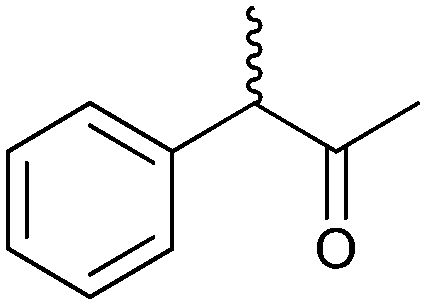
|
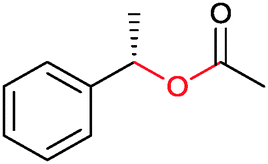
|
47 | HAPMO (partially purified enzyme) | >98% (product) E > 100 | 50 mL | KR; [S]0 = 5.4 mM | 141 | |
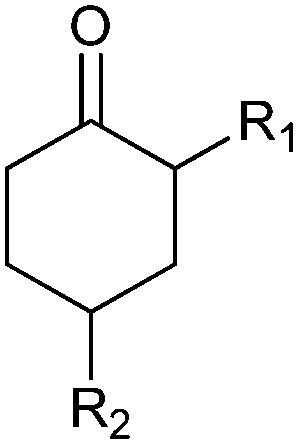
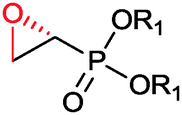
|
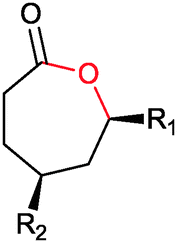
|
R1, R2 = Me, Et, nPr, iPr, nBu, allyl (independently) | 50 | CHMO (partially purified enzyme) | E > 200 | 1 mL | KR or desymmetrization; [S]0 = 2 mM; t = 2 h | 142 |
| (3) Phenol hydroxylation reactions | ||||||||
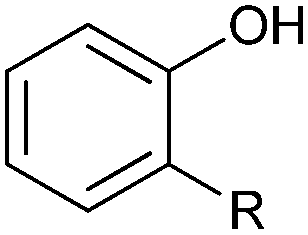
|
|
R = alkyl, aryl | >99 | HbpA (partially purified enzyme) | 10 mL | [S]0 = 160 mM; t = 600 h up to 160 mM | 85 and 86 | |
|
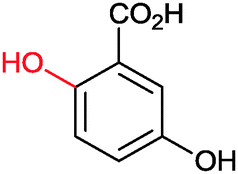
|
>99 | 3HB6H (purified enzyme) | 1 mL | [S]0 = 50 mM; t = 8 h | 92 and 143 | ||
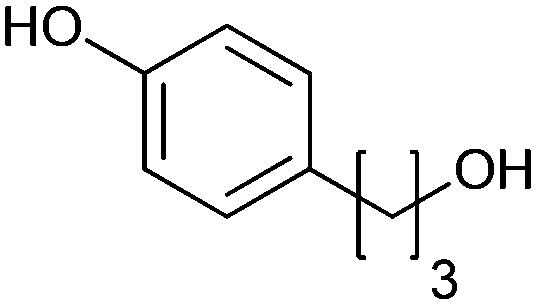
|
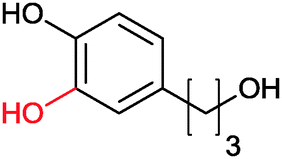
|
>99 | PheA1 (rec in growing E. coli) | 50 mL | [S]0 = 5 mM; t = 10 h | 90 | ||
| (4) Heteroatom oxygenations | ||||||||
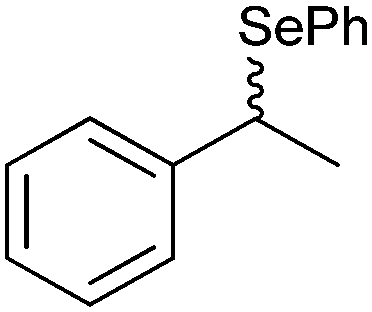
|

|
50% | PAMO (purified enzyme) | 99 (unreacted enantiomer) | 1 mL | KR; [S]0 = 10 mM; t = 24 h | 75 and 76 | |
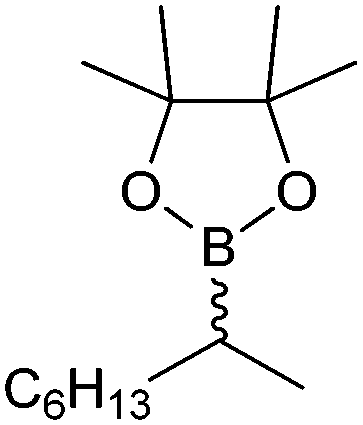
|
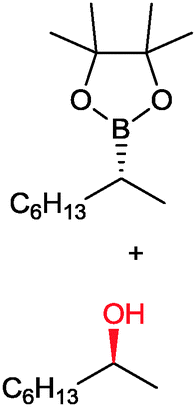
|
49 | PAMO (purified enzyme) | E = 23 | 0.5 mL | KR; [S]0 = 10 mM; t = 24 h | 79 | |
|
|
(45) | SMO (rec in resting E. coli) | 54.5 (S) | 1 mL | [S]0 = 200 mM; t = 24 h | 119 and 120 | |
A special case of BVMO-catalysed Baeyer–Villiger oxidations is the oxygenation of aldehydes (Scheme 2, 5). In most cases, BVMOs convert aldehydes into the corresponding formyl esters (following the migration rules of the chemical Baeyer–Villiger oxidation). However, recently Bisagni and coworkers reported on a novel BVMO from Dietzia sp. preferentially catalysing acid formation of e.g. profene aldehydes.69
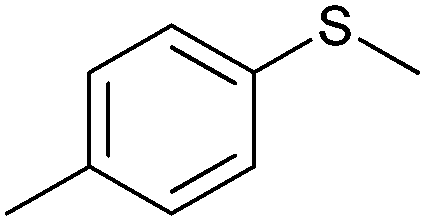
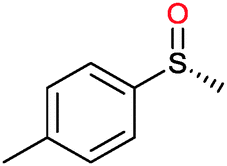
Many of the aforementioned BVMOs are also capable of heteroatom oxygenation (Scheme 2, 4). Enantiospecific sulfoxidation here clearly represents the reaction most frequently investigated.30,70–74 Next to the often high enantioselectivity of this reaction, also its high chemoselectivity is valued as the ‘overoxidation product’ i.e. the corresponding sulfone is usually not observed in enzymatic sulfoxidation reactions. Next to sulphur also other heteroatoms such as selenium,75–78 boron,77,79 or nitrogen48 can be oxygenated using flavin-dependent monooxygenases. Recently, Codexis evolved cyclohexanone monooxygenase for the selective sulfoxidation to yield enantiopure (S)-pantoprazole (Scheme 5),74,80 thereby impressively demonstrating the potential of protein engineering to tailor a monooxygenase to meet the requirements of industrial-scale synthesis.
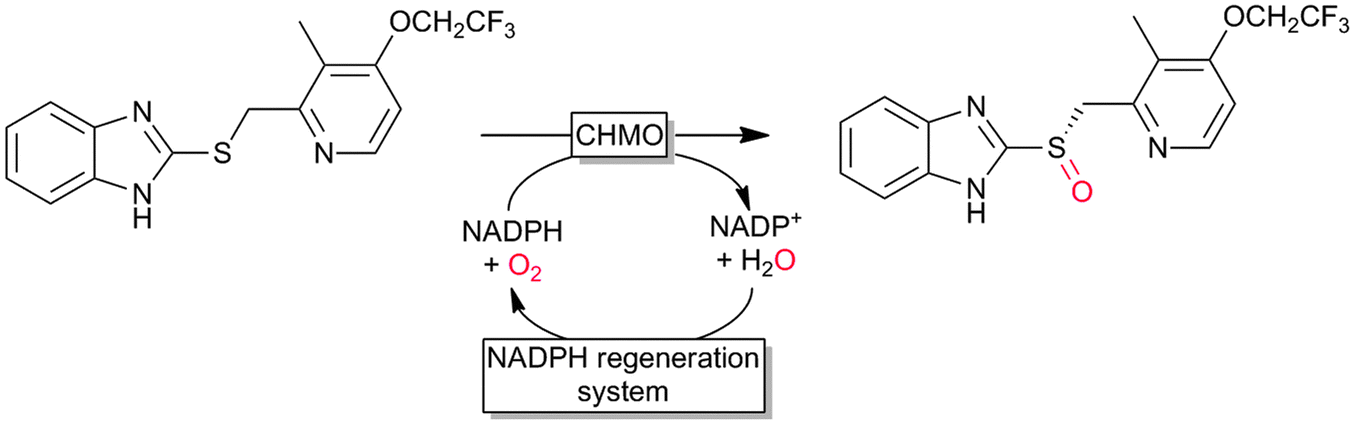 | ||
| Scheme 5 Cyclohexanone monooxygenase (CHMO)-mediated enantiospecific sulfoxidation for the synthesis of (S)-pantoprazole. | ||
Also the regioselective hydroxylation of phenols (Scheme 2, 3) either in the ortho- or para-position represents a principally highly interesting reaction for organic synthesis as the repertoire of chemical aromatic hydroxylations (apart from Dakin-, Sandmeyer- and Baeyer–Villiger-type reactions as well as boronic acid oxidations) is rather limited. However, at present also the number of phenol hydroxylases is rather restricted as well.81 Nevertheless for example, 2-hydroxy biphenyl-3-monooxygenase (HbpA) has been used at up to kg-scale synthesis of catechols.82–89 But also phenol hydroxylase (PheA1),90–93 and the benzoate hydroxylases94–96 have been reported and might exhibit some synthetic potential.
Stereoselective epoxidation of CC-double bonds (Scheme 2, 1) is an emerging field in flavoprotein-monooxygenase chemistry. Especially the so-called styrene monooxygenases are enjoying increased popularity. For example the styrene monooxygenase from Pseudomonas sp. VLB 120 has been investigated intensively by Schmid and coworkers.97–114 But also new styrene monooxygenases from other sources are constantly added to the portfolio.32,115–121
Currently, the styrene monooxygenases available appear to have a clear preference for vinyl aromatic substrates even though also aliphatic alkenes have been reported as substrates.119,120 The more severe limitation of the present portfolio is that exclusively (S)-selective styrene monooxygenases are known. Hopefully, screening of natural diversity and/or protein engineering will close this gap in the nearer future.
An interesting cascade reaction producing enantiomerically pure (S)-styrene oxide from (renewable) phenylalanine in a cascade of amino lyase, decarboxylase and styrene monooxygenase was reported recently by Nielsen and coworkers (Scheme 6).122
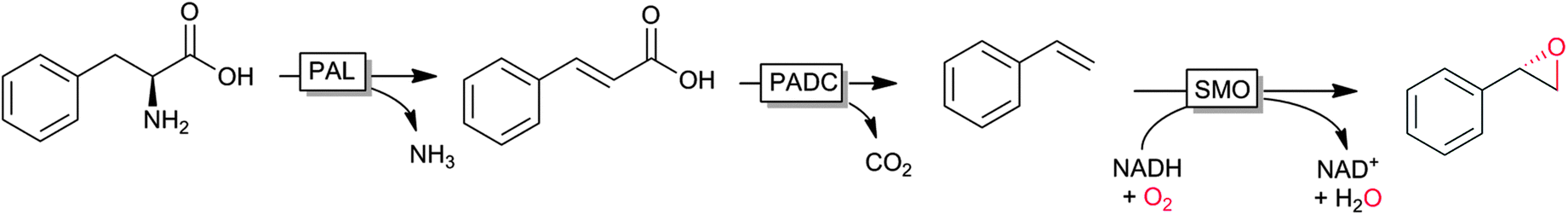 | ||
| Scheme 6 Cascade from natural phenylalanine to (S)-styrene oxide entailing phenylalanine ammonia lyase (PAL), phenoloic acid decarboxylase (PADC) and styrene monooxygenase (SMO). | ||
Also, flavoprotein monooxygenase-catalysed oxidative halogenation reactions (Scheme 2, 6) are worth mentioning here. In the stricter sense, these reactions are not oxyfunctionalisation reactions as a halogen is incorporated instead of an oxygen atom.123–133 According to our present knowledge, flavin-dependent halogenases primarily catalyse the oxidation of halogenides to hypohalous acids (HOCl, HOBr). These are guided by the protein backbone directly and exclusively to the substrate performing the electrophilic halogenation reaction. Even though, flavin-dependent halogenases seem to be limited to activated (aromatic) substrates, their exclusive regioselectivity should make them highly interesting tools not only for pharmaceutical applications. However, en route to preparative application their still comparably poor catalytic activity (kcat being in the range of <1 min−1) needs to be addressed.
Very recently, an interesting cascade of tryptophan synthase and tryptophane-7-halogenase was reported giving access to a broad range of tryptophan derivates from simple indole starting materials (Scheme 7).134
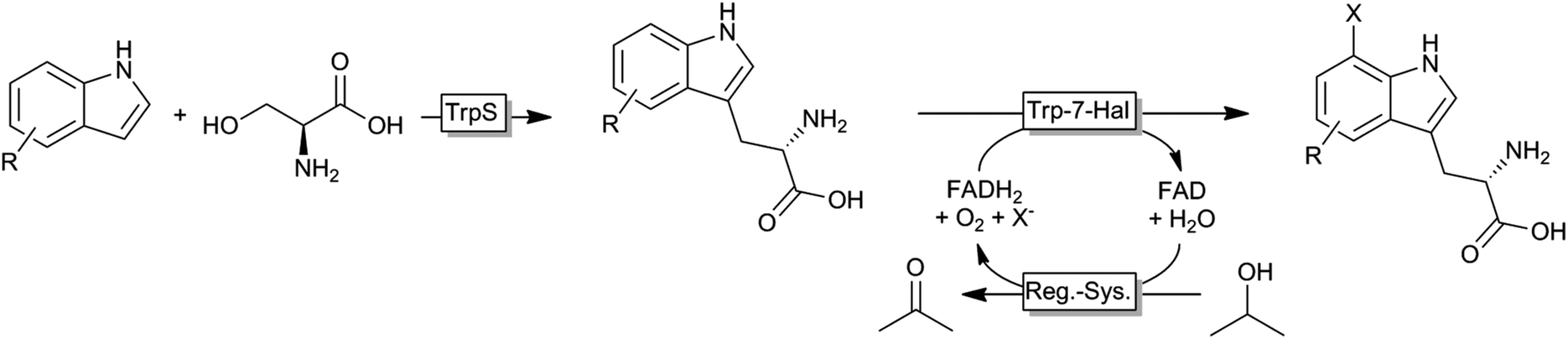 | ||
| Scheme 7 Cascade for the synthesis of unnatural halogenated tryptophanes from simple indoles. TrpS: tryptophan synthase; Trp7-Hal: tryptophan-7-halogenase; Reg.-Sys.: FADH2-regeneration system comprising a NADH:FAD oxidoreductase, NADH and an alcohol dehydrogenase. | ||
Finally, also a newly discovered long-chain alkane degrading flavoprotein monooxygenase (LadA) should be mentioned here.135,136 So far, this is the only flavin-dependent monooxygenase reported capable of hydroxylating non-activated hydrocarbons (being largely the domain of (non)-heme iron monooxygenases, vide infra). At present, only a few reports deal with this interesting enzyme and future will tell the preparative potential of LadA.
A representative selection of examples ranging from analytical proof-of-concept contributions to (near) industrial scale implementations is shown in Table 1.
Overall, flavin-dependent monooxygenases have great potential for organic synthesis. They enable highly selective introduction of molecular oxygen into organic compounds. Very often, a comparable chemical route is yet unknown. Unfortunately, at present only a few of these promising catalysts are commercially available (e.g. some BVMOs).
Heme-iron-monooxygenases and -peroxygenases
Heme-iron-monooxygenases, also called P450- or CYP-monooxygenases, exhibit an even higher oxyfunctionalisation potency than most of the flavoprotein monooxygenases discussed above. P450 monooxygenases can also abstract non-activated C–H bonds such as in alkanes. Obviously, this has inspired many research groups worldwide to explore the catalytic potential of this enzyme class (Scheme 8).20,21,144–147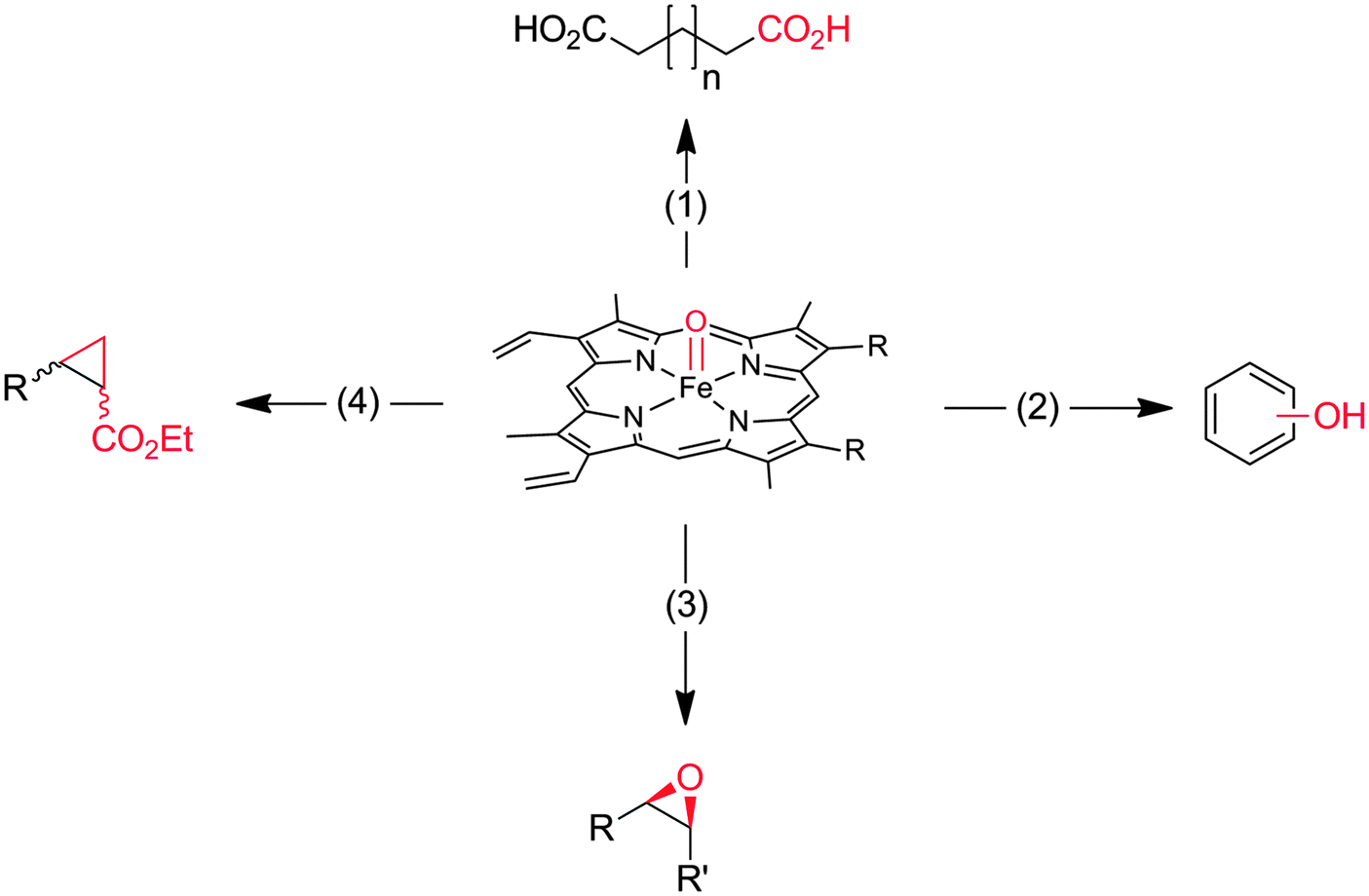 | ||
| Scheme 8 Compound I as the active species leading to the diverse reactions catalysed by P450-monooxygenases and peroxidases. (1) Hydroxylation of (non)activated C–H-bonds148–153 even of methane;154,155 (2) hydroxylation of aromatics;156 (3) epoxidation of CC-double bonds; (4) carbene transfer to CC-double bonds (actually starting from the reduced FeII stage and not being an oxyfunctionalisation reaction).157 | ||
P450 monooxygenases comprise a somewhat more complicated mechanism than flavoprotein monooxygenases. Molecular oxygen is activated by coordination to a highly oxidized (formally FeV) iron species enabling electrophilic O-transfer. To achieve this, two additional reducing equivalents are necessary, which are delivered in two individual single electron transfer steps (Scheme 9).
 | ||
| Scheme 9 Simplified catalytic cycle of P450-monooxygenases. The catalytic cycle starts with binding of the substrate to the resting (FeIII) state of the enzyme followed by single electron transfer (from NAD(P)H via the electron transport chain), O2-binding and the second electron transfer. The resulting hydroperoxo iron is dehydrated after successive protonation resulting in a (formal) FeV–oxo species (compound I) performing the O-transfer reaction. The intermediate hydroperoxo iron can also be formed directly from the resting state by addition of H2O2 (hydrogen peroxide shunt). | ||
Again, NAD(P)H serves as a source for the reducing equivalents needed. However, since NAD(P)H is an obligate hydride donor, it is not capable of interacting directly with the iron centre (being an obligate single electron acceptor). To overcome the mechanistic incompatibility of NAD(P)H and Fe3+, nature uses e.g. NAD(P)H-ferredoxin oxidoreductases as relay systems. These enzymes contain flavins, which due to their mechanistic versatility, can accept a hydride from NAD(P)H and pass on the two electrons in two subsequent steps either to the monooxygenase directly or via electron transport proteins such as Fe2S2 ferredoxin (Scheme 10).
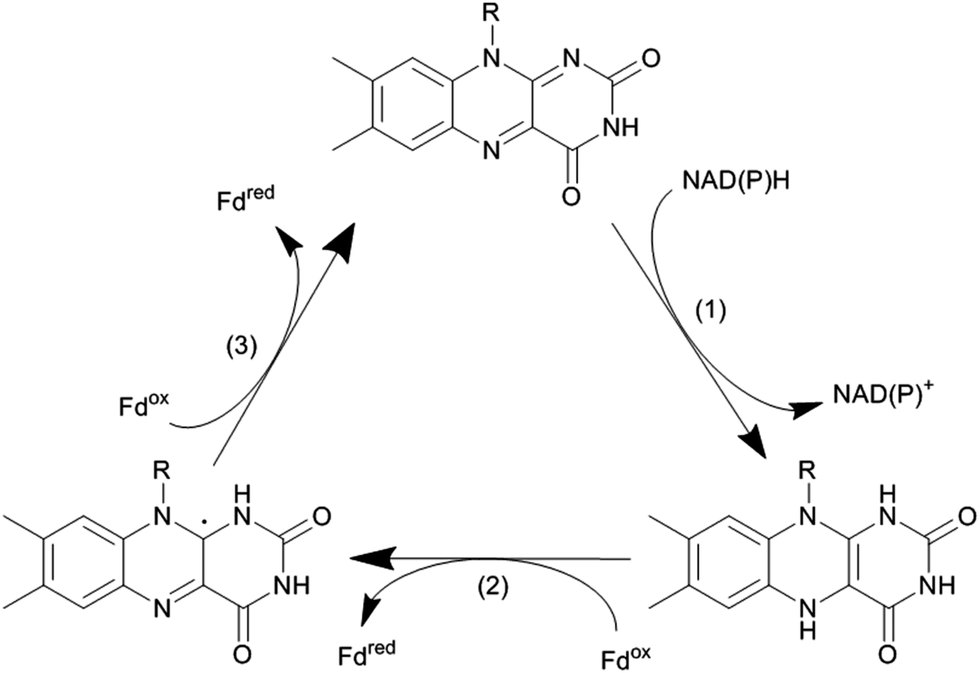 | ||
| Scheme 10 Transformation of a hydride transfer reagent (NAD(P)H) into two single electron transfer reagents (e.g. ferredoxin, Fd). NAD(P)H:ferredoxin oxidoreductase catalyse the oxidation of NAD(P)H (by hydride transfer to the enzyme-bound flavin cofactor, step (1)). The reduced flavin than successively delivers one electron each to two ferredoxins (Fd). | ||
It is worth mentioning here that some P450 monooxygenases are capable of forming the catalytically active oxyferryl species from the resting state using hydrogen peroxide (Scheme 9). However, usually, their efficiency with H2O2 is relatively poor so that oxidative inactivation of the heme-prosthetic group (and the protein backbone) dominates. In contrast, the so-called peroxygenases utilise this hydrogen peroxide shunt pathway very efficiently enabling the H2O2-driven P450-oxyfunctionalisation chemistry. In fact, it has been hypothesised that the P450 monooxygenases have evolved from H2O2-dependent ancestors.158
P450 monooxygenases have received tremendous attention due to their capability to hydroxylate non-activated C–H bonds. For example, the selective and mild hydroxylation of (cyclo)alkanes represents a focus of research. The promise here lies in the high selectivity of the enzymatic hydroxylation avoiding undesired overoxidation of the (more reactive) reaction products as frequently encountered in (industrial) oxidation methods.159 A very recent contribution by Gröger and coworkers nicely demonstrates the synthetic potential of biocatalytic oxyfunctionalisation (Scheme 11).160,161 By combining a P450-monooxygenase with an alcohol dehydrogenase, the authors achieved clean aerobic oxidation of cyclohexane to cyclohexanone with absolute selectivity. The overall low yield of the reaction (<10%) is most probably attributed to the volatility of the reagents and the non-optimised reaction conditions. Nevertheless, the high selectivity of this reaction convinces especially compared to chemical processes, where frequently by-products originating from overoxidation and ring-cleavage are observed.159
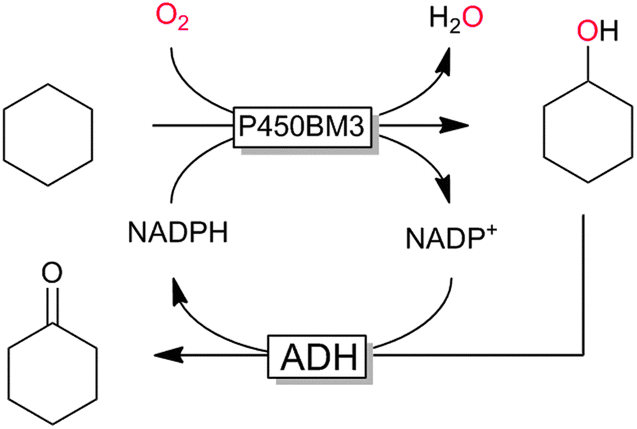 | ||
| Scheme 11 Bi-enzymatic cascade for the selective aerobic oxidation of (cyclo)alkanes to the corresponding ketones using a sequence of P450 monooxygenase-catalysed hydroxylation (here P450BM3 from Bacillus megaterium) and ADH-catalysed further oxidation to the ketone. | ||
Similar examples comprise the selective transformation of (cyclo)alkanols into the corresponding esters (e.g. cyclohexanol to ε-caprolactone).162,163
Industrial interest in P450 monooxygenases today mainly stems from the pharmaceutical industry for the generation of drug metabolites and the production of active pharmaceutical ingredients (APIs) such as steroids.26,146,147,164,165 But also selective oxyfunctionalisation of terpenes is of great interest.152,166–173
A very elegant approach was reported by Keasling and coworkers who used an engineered Saccharomyces cerevisiae overexpressing a P450 monooxygenase to produce artemisinic acid (basically from sugar) (Scheme 12).174,175
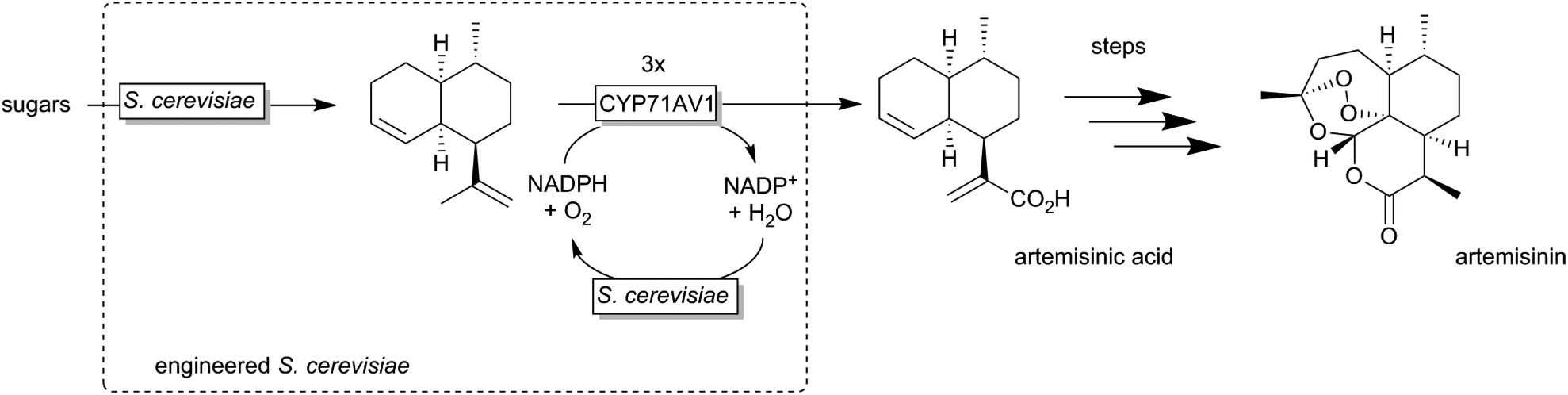 | ||
| Scheme 12 Engineered Saccharomyces cerevisiae overexpressing a P450 monooxygenase (CYP71AV1) for the selective (triple) oxygenation of amorpha-3,11-diene to artemisinic acid, a precursor for the anti-malaria drug artemisin. | ||
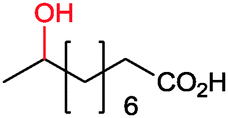
Often, P450 monooxygenase-catalysed reactions are still somewhat too slow and yield too low product titers (vide infra)26 to meet the economic requirements for bulk chemicals.176 Nevertheless, oxyfunctionalisation of fatty acids to yield α,ω-dicarboxylic acids (as polymer building blocks) has been demonstrated at appreciable product titres of more than 100 g L−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (ref. 177–180) pointing towards large-scale applicability of these biocatalysts.
(ref. 177–180) pointing towards large-scale applicability of these biocatalysts.
For a long time, methane hydroxylation was believed to be limited to non-heme iron monooxygenases and out of scope for P450-monooxygenases. Especially the higher bond-strength of the methane C–H bond as compared to higher homologues was thought to be the major reason for this.154 However, recently Reetz and coworkers have demonstrated that inert decoy molecules such as perfluorinated acids enable the P450BM3-catalysed hydroxylation of even methane.155 It is thought that the perfluorinated alkyl chain ‘fills up’ the large cavity around the active site and thereby facilitates methane binding. The same approach (decoy molecules) was also successful in enhancing P450BM3-catalysed hydroxylation of e.g. benzene.156,181 It is also interesting to note that such decoy molecules may have an influence on the stereoselectivity of P450-catalysed oxyfunctionalisation reactions.182
Next to protein engineering20,158,164,183–189 also substrate engineering may be a promising approach to improve P450-monooxygenase-catalysis as demonstrated by Griengl and coworkers.190–193 For example Beauveria bassiana cannot convert cyclopentanone whereas the corresponding N-benzoylspirooxazolidine was converted smoothly by the same organism (Scheme 13).
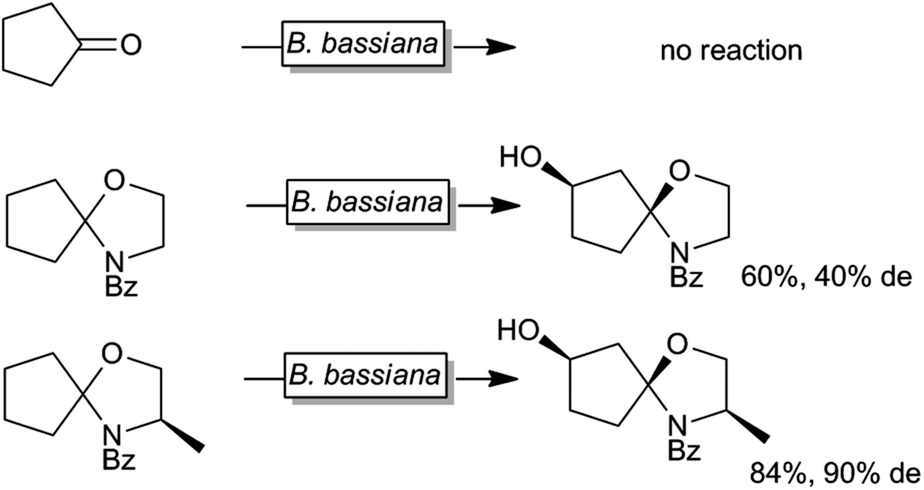 | ||
| Scheme 13 Effect of temporary attachment of an oxazolizine protection group to cyclopentanone on the biohydroxylation by B. bassiana. | ||
It was suggested that the starting material itself was not efficiently recognised by the monooxygenase. Also the nature of the temporary protecting group had an influence on the stereoselectivity of the hydroxylation reaction. So far, this approach has not found widespread and systematic investigation.
Another exciting new development in P450-chemistry is the recent finding by Arnold and coworkers that reduced (FeII) P450 monooxygenases are efficient cyclopropanation catalysts (Scheme 14), opening up new avenues for P450-chemistry.157
 | ||
| Scheme 14 Enzymatic cyclopropanation of styrene using a serine-mutant of P450BM3 (P411BM3-CIS). | ||
Table 2 gives a representative, yet naturally incomplete, overview over the diversity of P450 monooxygenase-catalysed oxyfunctionalisation reactions.
| Substrate | Product | Yield | Catalyst | ee (%) | Volumetric scale (aq only) | Remarks | Ref. |
|---|---|---|---|---|---|---|---|
| P450BM3: CPY102A1 from Bacillus megaterium; P450cam: CPY101 from Pseudomonas putida; P450pyr: from Sphingomonas sp. HXN-200; CYP152A1: from Bacillus subtilis; CYP152A2: from Clostridium acetobutylicum. | |||||||
| (1) CH bonds | |||||||
|
|
n.d. (up to 3.3 mM product) | P450BM3 (purified enzyme) | — | 1 mL | Perfluoro carboxylic acids as ‘decoy’ molecules | 155 and 181 |
|
|
n.d. (up to 67 mM product) | CYP153A6 (recombinant in E. coli) | Selectivity for 1-octanol = 94% | 1 mL | t = 24 h | 167 |
|
|
Up to 90% | BM3 variants (purified enzymes) | — | 1 mL | [S]0 = 200 μM; t = 1 h | 194 |

|
|
86% (up to 174 g L−1) | Candida tropicalis | — | 1 L | 100–200 g L−1 | 177 |
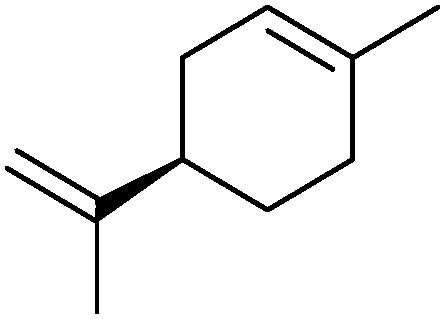
|
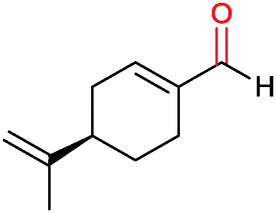
|
30% | CYP153A6 (rec. in E. coli) | — | 1 L | t = 26 h; whole-cell biocatalysts | 166 |

|
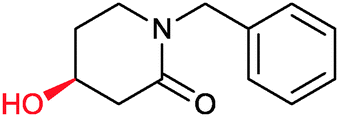
|
73% | P450 pyrvariant (rec. in E. coli) | 94% (S) exclusive regioselectivity | 0.5 mL | [S]0 = 4.5 mM; t = 9 h | 149 |
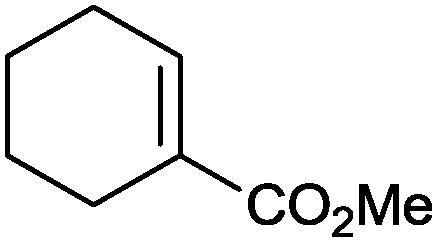
|
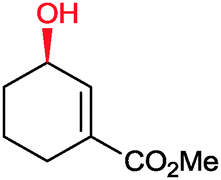
|
86% | P450BM3 variants (rec. in E. coli) | >99% (R) regioselectivity >93% | 50 mL | t = 8 h | 195 |
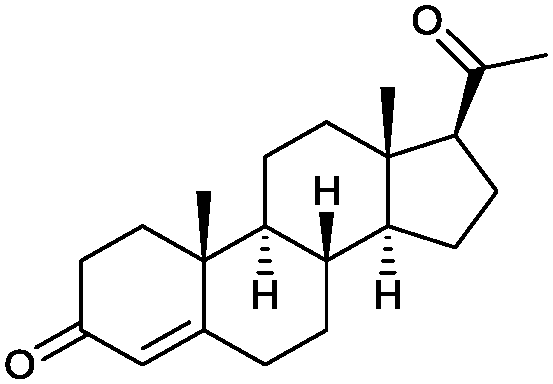
|
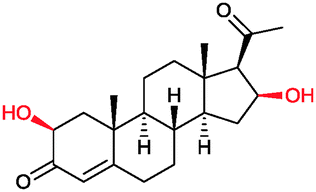
|
90% | P450BM3 variants (rec. in E. coli) | 96–97% up to exclusive regioselectivity | 100 mL | [S]0 = 1 mM; t = 24 h | 164 |
| (2) Aromatic hydroxylations | |||||||
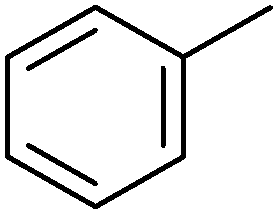
|
|
n.d. | P450BM3 variants (purified enzymes) | Up to 90% selectivity (p- and benzyl-products) | 1 mL | [S]0 = 5 mM; t = 24 h use of decoy molecules | 156 |
|
|
n.d. | Beauveria bassina | ||||
| (3) Epoxidations | |||||||
|
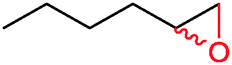
|
42% | P450BM3 variants (rec in B. subtilis 3C5N) | 27.8 (R) | 10 mL | 196 | |
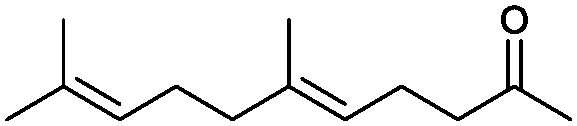
|
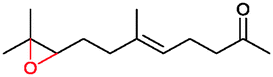
|
90% | P450BM3 variants | 97% ee (n.d.) exclusive regioselectivity | 1 mL | [S]0 = 0.2 mM | 172 |
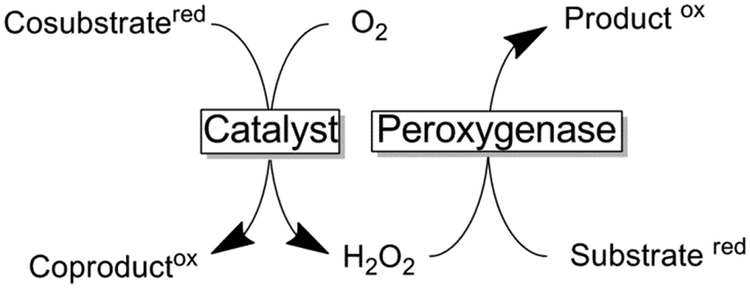
Peroxygenases entail the chemical versatility of P450 monooxygenases without being dependent on the complicated electron transport chains (vide infra). Instead of relying on reductive activation of molecular oxygen, peroxygenases utilise H2O2 to form the catalytically active oxyferryl species (hydrogen peroxide shunt in Scheme 9).197
For many decades chloroperoxidase from Caldariomyces fumago (CPO) has been the role model and only representative of this highly interesting enzyme class.198–227 However, more recently a range of new (and potentially more useful) members of this enzyme class have gained interest.228–244 For example a peroxygenase from the basidomycetous fungus Agrocybe aegerita is capable of cyclohexane hydroxylation, a reaction that has never been observed for CPO.228 If developed further, this reaction might become a viable alternative to the existing, selectivity-wise highly challenging chemical cyclohexane oxygenation reactions.159
Even though peroxygenases are most valuable for the organic chemist due to their oxygen transfer capability (yielding e.g. enantiomerically pure epoxides or sulfoxides), some other – potentially very useful – applications have been reported recently. Haloperoxidases, for example, catalyse the oxidation of halogenides and release of the corresponding hypohalogenide into the reaction medium. Recently, Le Nôtre, Scott and coworkers reported a ‘biocatalytic’ oxidative decarboxylation of natural amino acids such as phenyl alanine yielding the corresponding nitriles as the major product (Scheme 15).245 The role of the vanadium peroxidase was restricted to in situ formation of hypochloride, which mediated the oxidative decarboxylation of the α-amino acid. Compared to a stoichiometric use of bleach, this catalytic method excels by its catalytic use of chloride thereby significantly reducing salt wastes.
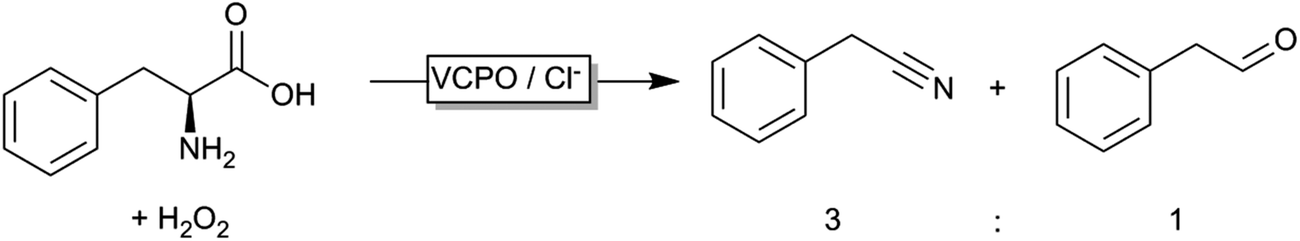 | ||
| Scheme 15 Vanadium-haloperoxidase catalysed oxidative decarboxylation of phenylalanine. | ||
Some of us have applied CPO recently for the chemoenzymatic halogenation of thymol. The hydrogen peroxide needed for the enzymatic reaction was generated by electrochemical reduction of molecular oxygen (Scheme 16) thereby providing H2O2 in sufficient amounts to sustain the catalytic cycle while minimising H2O2-related inactivation of the enzyme.
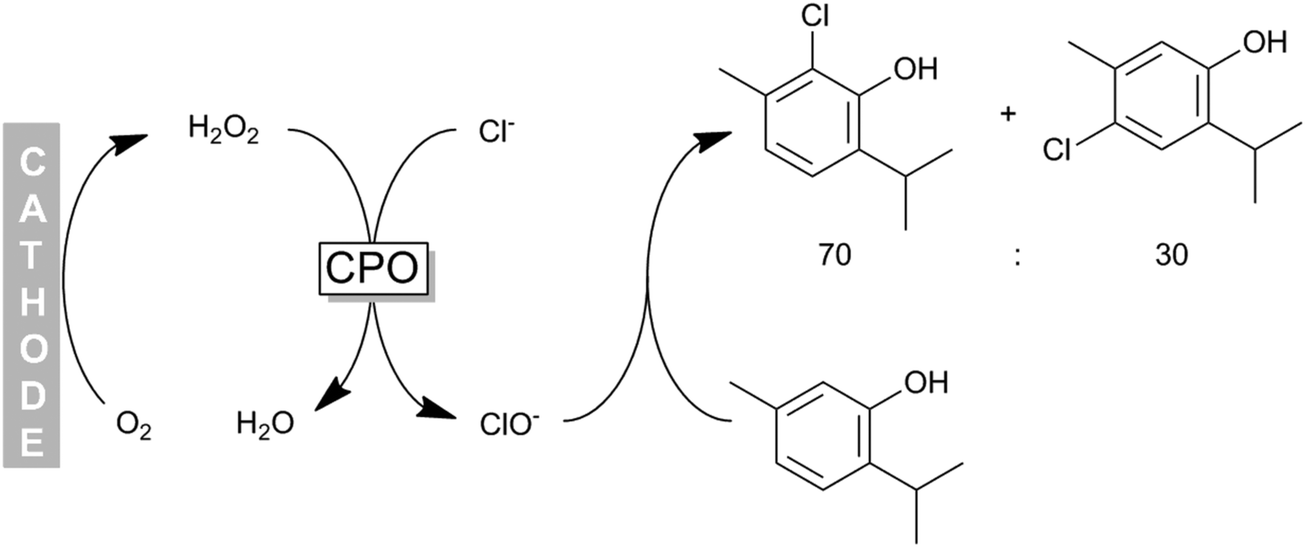 | ||
| Scheme 16 Chemoenzymatic halogenation of phenols (e.g. thymol) using CPO-generated hypochloride. H2O2 was obtained in situ from electrochemical O2 reduction. | ||
Overall, P450-enzymes (monooxygenases and peroxygenases) exhibit enormous potential for preparative organic synthesis.
Regeneration strategies
As mentioned above, reduced nicotinamides serve as universal electron donors for the reductive activation of molecular oxygen. Therefore, it is not very astonishing that the majority of published applications include NAD(P)H. Due to its fairly high costs, NAD(P)H cannot be applied in stoichiometric amounts but rather in catalytic amounts together with a suitable cofactor regeneration system (Table 3). Overall, oxidation of a simple sacrificial cosubstrate is utilised to promote monooxygenase catalysis.|
|
||||
|---|---|---|---|---|
| Regeneration enzyme | Cosubstrate | Coproduct | Cofactor | Ref. |
| FDH: formate dehydrogenase; ADH: alcohol dehydrogenase; PDH: phosphite dehydrogenase; G6PDH: glucose-6-phosphate dehydrogenase. | ||||
| FDH | Formate | CO2 | NADH | 86, 114, 173 and 251 |
| ADH | Isopropanol | Acetone | NADPH | 62 and 120 |
| PDH | Phosphite | Phosphate | NAD(P)H | 148, 252 and 253 |
| G6PDH | Glucose-6-phosphate | Glucono-6-phosphate lactone | NAD(P)H | 40 and 254–256 |

One obvious approach is to utilise the microbial metabolism to provide the reducing equivalents needed.65,97,98,100,103–105,111–113,137,138,140,246–250 The advantages of this approach are: (1) the enzyme(s) are not isolated thereby significantly reducing the catalyst preparation costs,176 the same is true for the nicotinamide cofactor, which is supplied by the microbial cell; (2) generally, enzymes within their natural environment (the microbial cell) are more stable than as isolated preparations, also the microbes can constantly replace inactivated enzymes; (3) the growth substrate (e.g. glucose) for the microorganism also serves as a sacrificial electron donor for the biotransformation reaction thereby theoretically providing the maximal amount of reducing equivalents available from complete mineralisation. These advantages explain why fermentative processes still dominate the field of biocatalytic oxyfunctionalisation chemistry. There are, however, also a range of challenges that have fostered research on the use of isolated enzymes. Amongst them are the frequently observed toxicity of the reagents to the microbial cells, transport limitations of reagents over the cell membranes, as well as issues related to undesired side reactions catalysed by the many other enzymes present in the microbial cells. Also, it is not always easy to balance the microorganism's redox needs with the demands of the desired biotransformation.
Using isolated monooxygenases circumvents some of the aforementioned challenges: by utilising isolated enzymes, the biotransformation is uncoupled from the fermentation step and therefore can be controlled more easily. Also, undesired side reactions are generally observed to a lesser extent than in whole-cell systems. The major challenge of using isolated enzymes is that the supply of the monooxygenase with reducing equivalents does not anymore come along with the microbial metabolism. Hence, the nicotinamide cofactor (in substoichiometric amounts) has to be applied together with a suitable in situ regeneration system. Table 3 gives an overview on common enzymatic NAD(P)H regeneration systems used to promote monooxygenase reactions. To estimate the efficiency of a given regeneration system the total turnover number (TTN) of the nicotinamide cofactor can be used. As a rule of thumb, TTN of greater than 1000 are generally considered to be sufficient for an economically reasonable process (even though it must be mentioned here that this very much depends on the price of the product amongst others).
Recently, so-called designer cells have gained significant attention. This approach combines the advantages of whole cell biocatalysis and the regeneration approaches outlined in Table 3 by coexpressing the production enzyme together with a suitable regeneration system and using the whole cells as catalysts.120,252 On the one hand the enzymes are protected in their natural environment and also protein-isolation and -purification is not applied. On the other hand, the cells are usually not metabolically active thereby largely eliminating toxicity issues as well as undesired side reactions. Overall, the designer-cell approach appears to be a very promising catalyst concept for future biocatalytic oxyfunctionalisation reactions.
The regeneration approaches outlined before all are based on the enzyme-coupled regeneration concept: regeneration reaction and production reaction are linked via the nicotinamide cofactor only. Hence, the cofactor regeneration consumes another substrate (cosubstrate) and produces a stoichiometric coproduct. Another interesting approach is the so-called intrasequential cascade, wherein the NAD(P)H-consuming (oxyfunctionalisation) reaction is also productively coupled to the NAD(P)H-regenerating reaction via an intermediate product (Scheme 17). Their use elegantly circumvents the need for the additional cosubstrate–coproduct couple by double transformation of only one substrate but naturally is rather limited in scope. However, the self-sufficient nature of these reactions as well as the apparent non-generation of wastes might point towards bulk-scale application such as ε-caprolactone or cyclohexanone from simple starting materials.
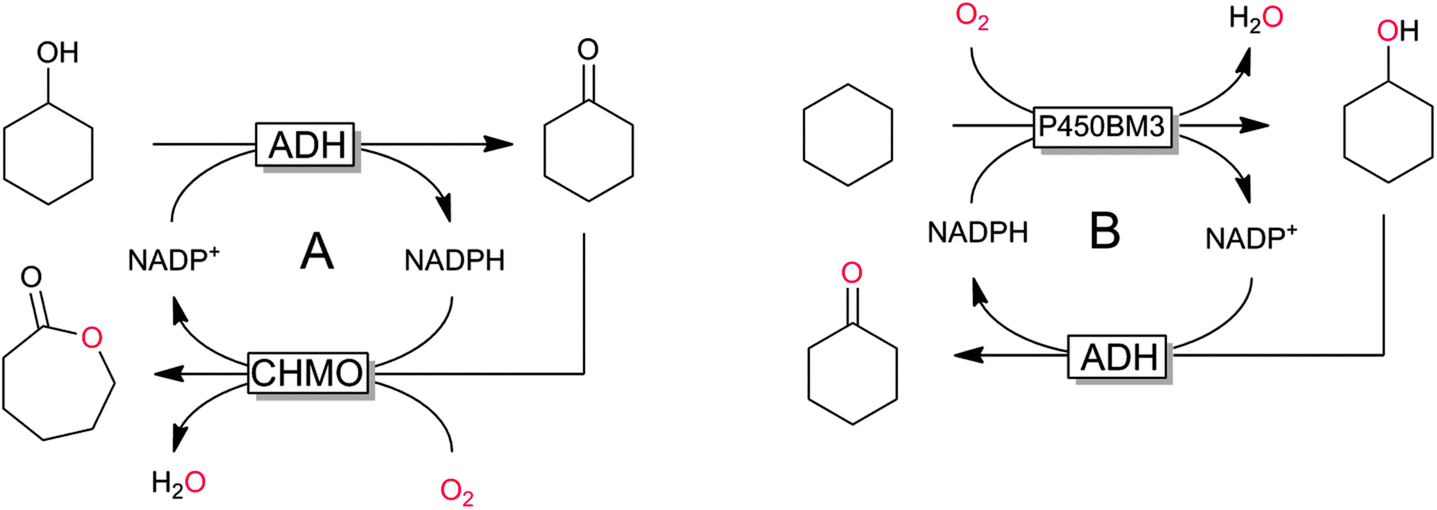 | ||
| Scheme 17 Examples of intrasequential oxyfunctionalisation sequences with the monooxygenase catalysing the second (A)162,163 or the first step (B)160,161 of the sequence. | ||
Direct regeneration approaches
Many monooxygenases rely on rather complicated, multi-enzyme electron transport chains to deliver the reducing equivalents from NAD(P)H to the flavin- or heme-prosthetic group. While this architecture makes sense for the (microbial) cell as it adds further levels of control over the cellular redox metabolism, it adds further complexity to the application of monooxygenases for chemical synthesis. Often, only the reducing equivalents are needed to sustain the monooxygenases' catalytic cycles; therefore in recent years a growing number of research efforts have been dedicated to delivering the reducing equivalents directly to the monooxygenases and circumventing the electron transport chains (Scheme 18).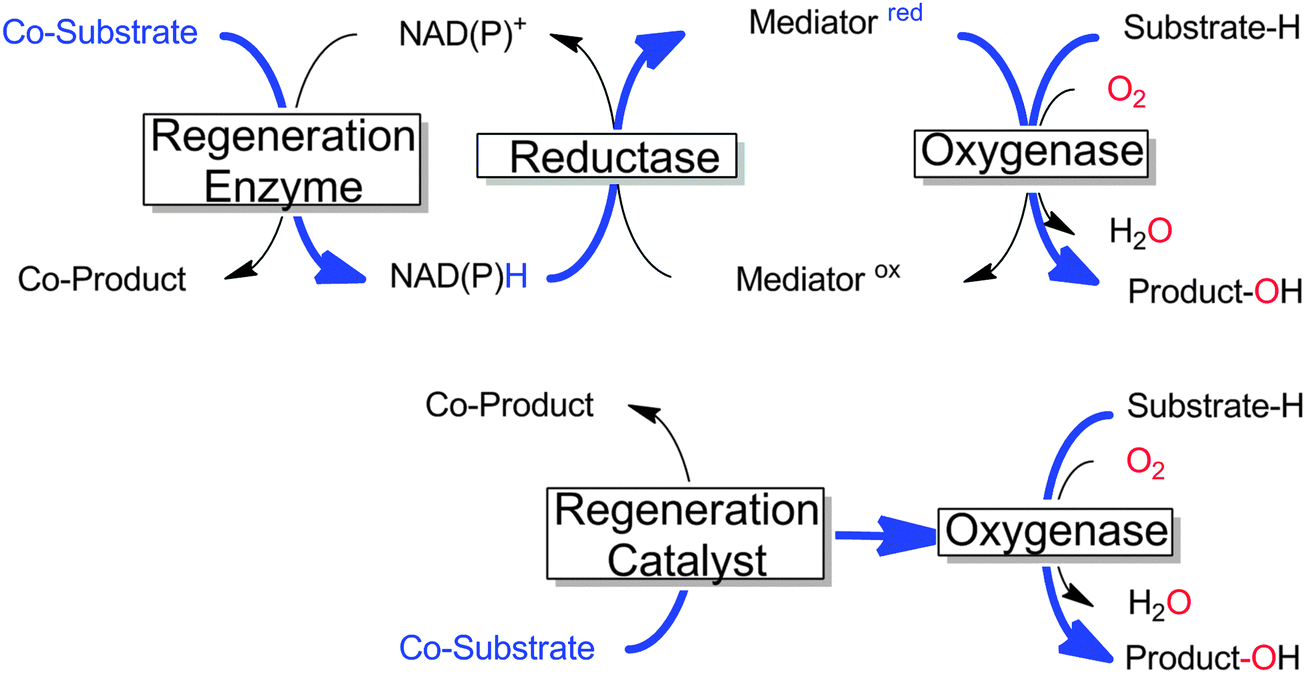 | ||
| Scheme 18 Comparison of the ‘traditional’ regeneration of multi-component monooxygenases (upper) with the direct regeneration approach (lower). The flow of reducing equivalents is shown in bold blue.11,257,258 | ||
The potential benefit of such direct regeneration approaches is that complicated electron transport chains comprising up to 2 additional enzymes and 2 additional cofactors can be substituted by only one regeneration catalyst, resulting in highly simplified (and more easily controllable) reaction schemes.
Table 4 summarises some representative examples of oxyfunctionalisation reactions performed by direct regeneration.
| Product | Monooxygenase | Regeneration catalyst | Source of reducing equivalents | TTN (regeneration catalyst) | Ref. |
|---|---|---|---|---|---|
|
P450 cam | Pdx | Cathode | 0.06 | 259 |
| StyA | FAD | Cathode | Up to 700 | 101, 108 and 260 | |
| StyA | [Cp*Rh(bpy)(H2O)]2+ | NaHCO2 | 10 | 110 | |
| Myoglobin | None | Cathode | — | 261 and 262 | |

|
P450 cam | Pdx | Cathode | 0.51 | 263 |
|
P450rFP45004A1 | Co(sep) | Cathode | 0.016 | 264 |
| P450BM3 | Co(sep) | Zn | <1 | 265 and 266 | |
| P450BM3 | [Cp2Co]+ | Cathode | 0.05 | 267 | |
| P450BM3 | Co(sep) | Zn | 0.15 | 266 | |
| P450BM3 – Ru-hybrid catalyst | Sodium diethyldithiocarbamate | 140 | 153 and 268 | ||
| P450BM3 | Deazaflavin | EDTA | 7 | 269 | |
| P450BM3 | Co(sep) | n.d. | Cathode | 168 and 270–272 | |
|
P450 cin | Co(sep), phenosafranine T, neutral red, flavins | n.d. | Cathode | 273 |
It becomes clear from Table 4 that the immense potential of direct regeneration of monooxygenases still remains to be exploited. The major limitation is the regeneration catalysts, which in most cases do not appear to act as true catalysts. We believe that the reason lies in the high reactivity of most mediators with molecular oxygen. Hence, futile reoxidation of the reduced regeneration catalysts by molecular oxygen competes with the desired monooxygenase regeneration thereby lowering the productive catalytic turnover of the regeneration catalysts. Furthermore, the futile cycle leads to the generation of reactive (partially reduced) oxygen species (ROS), which impair enzyme stability and lead to undesired side reactions.
The molecular reason for this undesired side reaction probably lies in the redox properties of the regeneration catalysts used. With the exception of [Cp*Rh(bpy)(H2O)]2+ and deaza-flavin, all regeneration catalysts are obligate one electron donors; hence their reaction with the predominant triplet oxygen (3O2) is spin-allowed (and fast) whereas the reactions of the two-electron donors ([Cp*Rh(bpy)H]+ and deaza-flavinred) with 3O2 are spin-forbidden (and therefore slow). Also the natural redox mediators such as putida redoxin (Pdx) are single electron mediators and therefore prone to fast aerobic reoxidation. The same is true to some extent for reduced flavins. Even though they are traditionally denoted as two-electron donors (in the classical mechanisms) they are also able to form semiquinones e.g. during sequential reduction of FeS-redoxproteins or during fast synproportionation with oxidised flavins.274 This also explains the exceptionally high turnover numbers observed by Schmid and coworkers (Table 4).101 By using very low flavin concentrations they could efficiently shift the flavin synproportionation equilibrium away from the reactive flavin semiquinones. Furthermore the advanced electrochemical reactor setup enabled minimising the contact time of reduced flavins and molecular oxygen.
Unfortunately, so far this undesired uncoupling reaction (also observed quite frequently with the ‘traditional’ regeneration approaches) has not been addressed very much and a general awareness of this oxygen dilemma is missing.
Overall, it can be asserted that on the one hand undesired, spontaneous reduction of molecular oxygen leads to a waste of reducing equivalents and concomitant of toxic ROS. On the other hand, molecular oxygen, as an integral part of the catalytic cycle of monooxygenases, cannot be omitted form the reaction schemes, resulting in the ‘oxygen dilemma’. Practical solutions to the oxygen dilemma are urgently needed in order to make monooxygenase-catalysis truly practical for organic synthesis.
Supply with H2O2 to promote peroxygenase catalysis
The dependence of peroxygenases on simple peroxides only at first sight offers a trouble-free catalyst system to be used in organic synthesis. The major challenge of using peroxides as a source of oxygen lies in their reactivity, especially with the prosthetic heme group itself. The exact inactivation mechanism is still under debate but it is clear by now that already small concentrations of hydrogen peroxide irreversibly inactivate the heme group (most probably oxidatively).275The simplest approach to circumvent the undesired oxidative inactivation of peroxygenases is to supply H2O2 in various, small portions. This approach is, however, work-intensive and also leads to significant volume increases which will complicate downstream processing. Alternatively, organic hydroperoxides have received some attention as milder alternatives to H2O2.197 More recently, various in situ O2 reduction methods have been evaluated to generate H2O2. The principle behind is that these methods enable to generate H2O2in situ in just the right amounts to sustain peroxygenase catalysis while minimising the H2O2 related inactivation. Table 5 gives an overview of the different sources of reducing equivalents needed for O2 reduction and catalysts used.
|
|
|||
|---|---|---|---|
| Catalyst | Cosubstrate | Coproduct | Ref. |
| GOx: Glucose oxidase. | |||
| GOx | Glucose | Gluconolactone | 197, 211 and 276–279 |
| Cathode | — | — | 201 and 280–284 |
| Pd | H2 | — | 285 |
| Flavin/hν | EDTA | Ethylene diamine, CO2, formaldehyde | 228, 286 and 287 |
Each H2O2 generation method has specific advantages and disadvantages. For example, cathodic reduction of molecular oxygen represents a potentially very simple and environmentally benign approach, but also requires specialised equipment. Glucose oxidase mediated reduction of O2 is simple and easy to apply, however gluconic acid accumulates in stoichiometric amounts leading to challenges in pH control. Overall, ‘the ideal’ in situ H2O2 generation does not exist and a suitable method has to be established for every peroxygenase reaction.
How green/environmentally friendly are biocatalytic oxyfunctionalisation reactions?
Today, biocatalysis is generally accepted as a ‘green technology’. Often, one or a few of the famous Twelve Principles of Green Chemistry288 are used to substantiate the green touch of a given biocatalytic reaction. However, we are convinced that authors should be more careful with green claims as a critical evaluation of the environmental impact might actually result in nasty surprises. The Twelve Principles of Green Chemistry are a wonderful framework for the design of environmentally more friendly processes but simple adherence to some of these principles does not make a given reaction/process ‘green’. Therefore, a more quantitative (and critical) evaluation is a prerequisite en route to truly Green Chemistry. Today, full life cycle assessment, taking into account as many parameters as possible, represents the ‘golden standard’ of environmental evaluation. However, the data basis required for LCAs is enormous, making LCAs time-consuming and too expensive for academics to perform.We believe that simple, mass-based metrics such as the E-factor (E being the amount of waste generated per kg of product)289 may be a valuable tool especially for academic researchers to estimate the ‘greenness’ of a given reaction.290–293 The E-factor concept is easily understood and the calculations easily and quickly done. Of course, such a simple mass-based tool neglects important contributors to the environmental impact such as energy consumption and does not weigh the quality of the different mass flows (scarce starting materials, hazardous wastes).294,295 Nevertheless, the E-factor frequently qualitatively points into the same direction as more advanced analyses and thereby can give valuable hints to optimise a reaction.100,290,296 Therefore, we would like to encourage especially academic researchers to use this simple tool more often to critically assess the environmental impact of their reactions.
In the following, we highlight and discuss two Green Chemistry issues that – to our mind – are notoriously underestimated in the scientific literature dealing with (bio)catalytic oxyfunctionalisation chemistry: substrate loading and downstream processing.
Substrate loading has a very significant impact on the economic feasibility and environmental impact of a reaction. However, the majority of reactants of interest is poorly water soluble. Unfortunately, the general solution to this challenge is to apply the starting material in low concentrations (often in the lower millimolar range). A simple E-factor estimation however demonstrates the dimensions of the water ballast resulting from this (Fig. 1). Producing tons of contaminated waste water per kg of product cannot be the ultimate green (not to mention economical) solution.
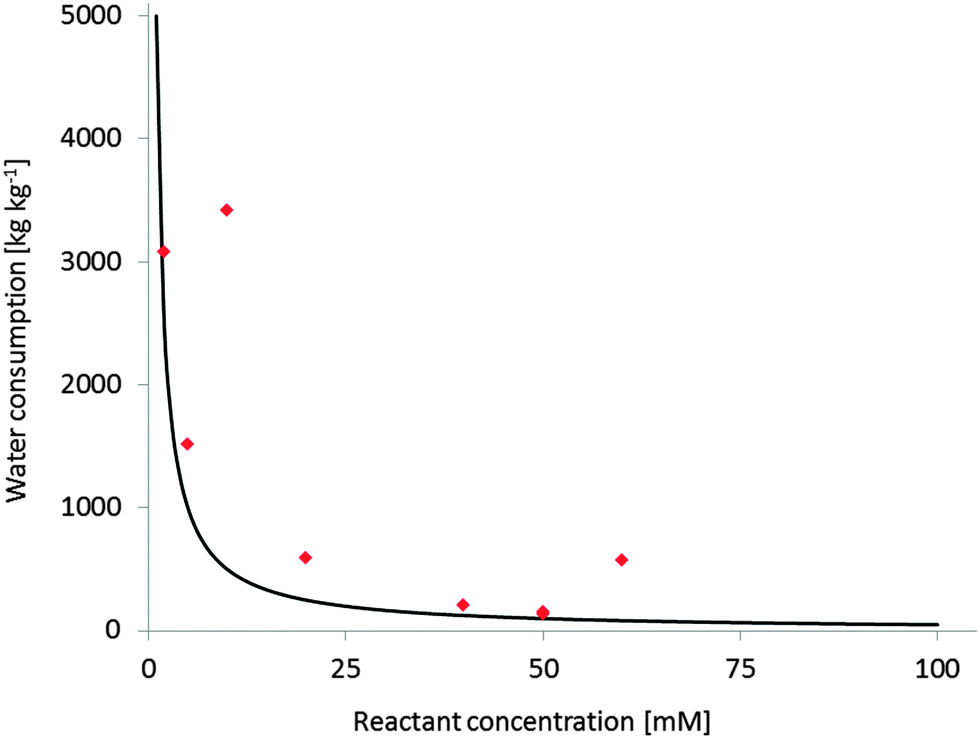 | ||
| Fig. 1 Water consumption at different reactant concentrations (expressed as kg(wastewater) per kg(product)). Black line: theoretical Water-E-factor (assumption M(product) = 200 g mol−1, full conversion); red diamonds: experimental Water-E-factors from published monooxygenase reactions,290 the examples with higher than expected E-factors are those with incomplete conversion. | ||
Increasing the overall substrate concentration can significantly reduce the water ballast of any given reaction and therefore should be more often be strived for. To cope with the poor substrate solubility, the two-liquid-phase-system (2LPS) approach seems to be a viable solution. Here, a water-immiscible apolar organic phase serves as the substrate reservoir and product sink enabling overall high payloads (Scheme 19).
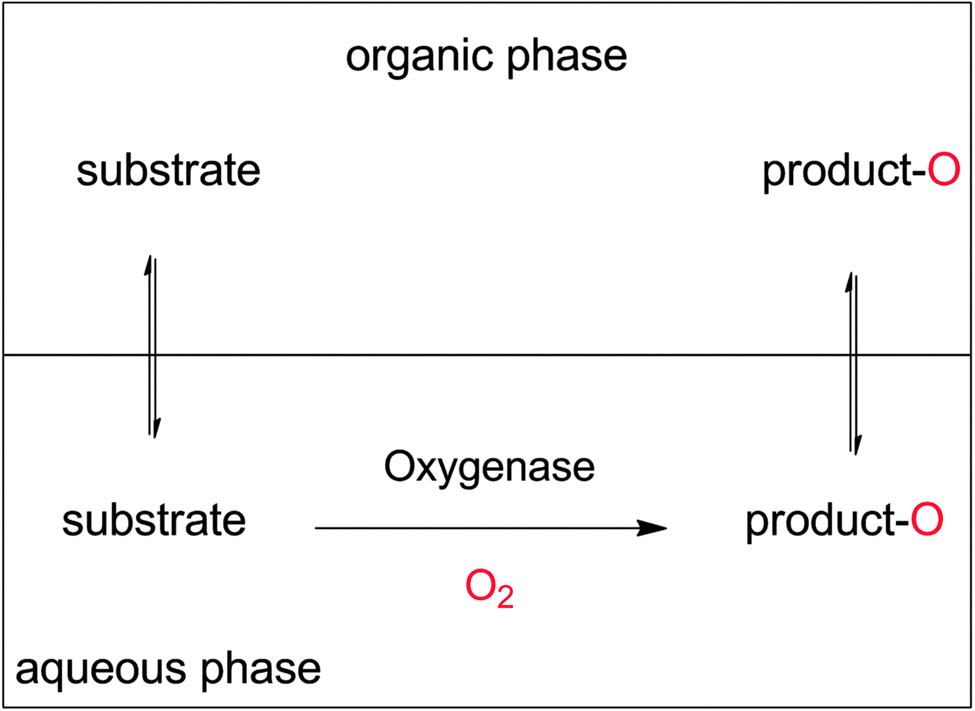 | ||
| Scheme 19 The two-liquid phase system (2LPS) approach. A hydrophobic organic phase solubilises overall high amounts of the hydrophobic reactants. From there, the substrate partitions into the aqueous, oxygenase-containing aqueous phase to be converted. The product, again, partitions between both phases and thereby is largely extracted into the organic layer. | ||
Additional benefits from the 2LPS concept may be alleviation of reactant toxicities and inhibitory effects (especially for whole cell biocatalysts),100,297 prevention of over-oxidation (e.g. of aldehydes into carboxylic acids),11,100,246,297–303 and prevention of hydrolytic degradation of e.g. epoxide or lactone products.14,111,112,114,304–306 Furthermore, high product concentrations in easily water separable organic phases will also facilitate DSP (vide infra).
Of course, the selection of the organic solvent should –next to practical issues such as partitioning coefficients and biocatalyst stability – also take its potential environmental impact into account.307–309
One issue of 2LPS arises from the rather hydrophilic character of many sacrificial electron donors used such as formates, phosphites or glucose making them incompatible with 2LPSs exhibiting high volumetric ratios of organic to aqueous phase. To solve this challenge, some of us have recently proposed using hydrophobised formates as the organic phase for biocatalytic oxyfunctionalisation reactions (Scheme 20).251
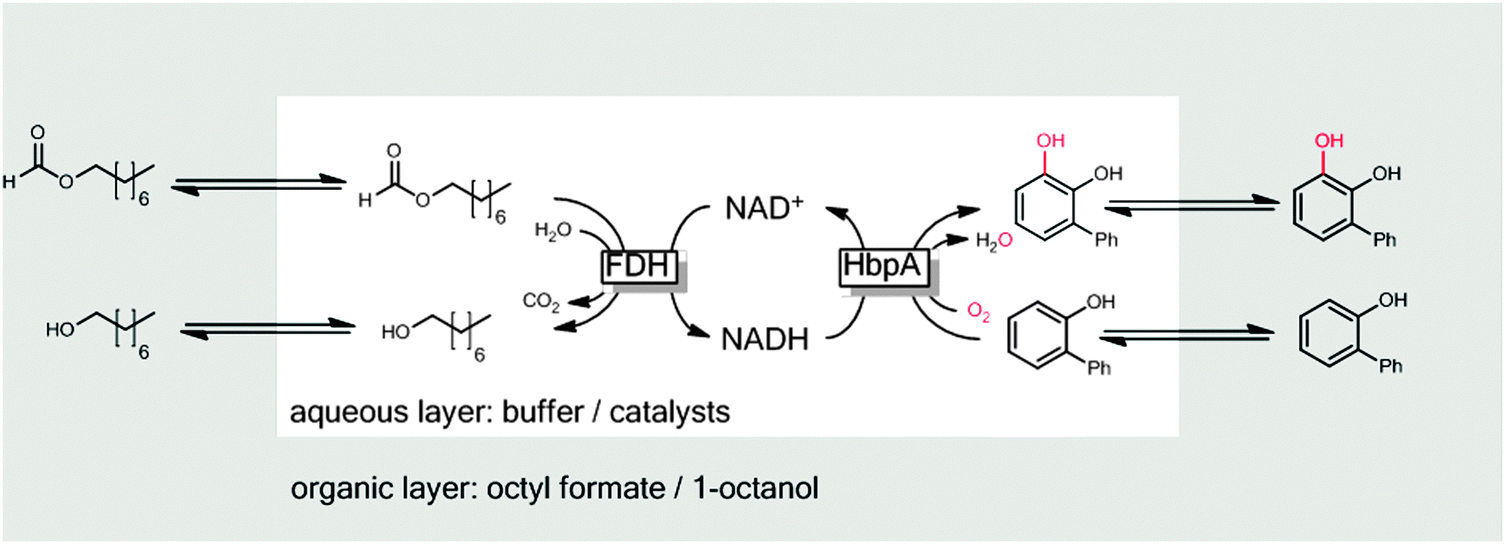 | ||
| Scheme 20 Hydrophobic formic acid esters as the reactive organic phase serving as the substrate reservoir/product sink and source of reducing equivalents for FDH-catalysed NADH regeneration. | ||
Downstream processing (DSP) is an issue that is not very frequently addressed in typical publications dealing with (bio)catalysis. However, DSP is an integral part of the production system as ‘it is no product unless it is in a bottle’ (C. Wandrey). From an environmental point-of-view, DSP can contribute very significantly to the overall environmental impact of a production system.290,310 Therefore, an increased focus on DSP issues also at an early stage of development is highly desirable. This should include ‘smart by design’ reaction systems enabling facile product isolation e.g. by filtration or 2LPSs enabling low-energy distillation DSP.
Another very promising line of research comprises so-called cascade reactions,311,312 wherein complex reaction sequences are performed without isolation of the intermediate reaction products. A very elegant cascade was reported recently by Bühler and coworkers reporting a formal terminal amination of fatty acids by combining a sequence of two hydroxylase- and one transaminase-reaction (together with the corresponding cofactor regeneration reactions) in one engineered E. coli production strain (Scheme 21).313,314
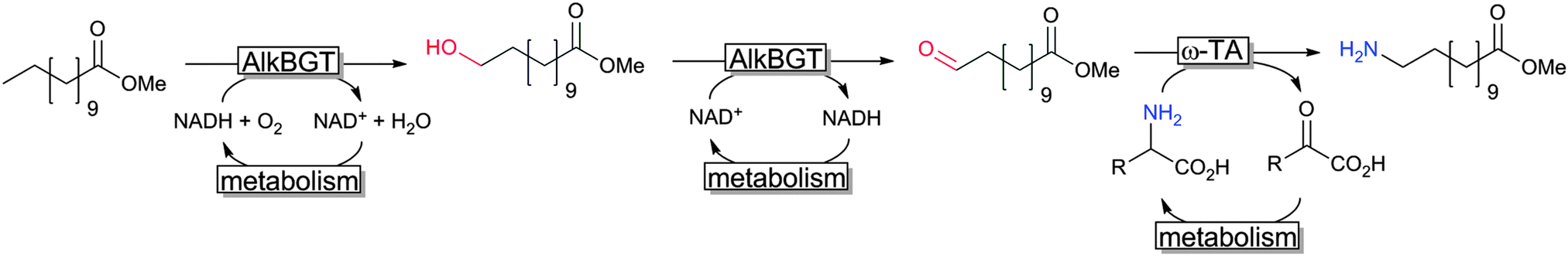 | ||
| Scheme 21 Cascade combining AlkB and ω-TA in recombinant E. coli.313,314 | ||
Today, still too many research projects on biocatalytic oxyfunctionalisation focus on the biocatalyst only while accepting low substrate loadings and completely neglecting the DSP part.
Conclusions
Biocatalysis has a lot to offer for the organic chemist. Simple and robust catalysts such as lipases for chirotechnology applications are well-established already. Oxygenases are on the go to follow them. This will significantly broaden the chemist's toolbox for selective oxidation/oxyfunctionalisation reactions. Especially the high selectivity of enzymes will enable shorter synthesis routes and yield higher quality products. Another interesting feature of biocatalysis (in general) is its potential for environmentally more benign syntheses. However, being biobased alone does not make an enzymatic route green, a more self-critical evaluation of contributors is highly desirable. In this respect, already simple metrics such as the E-factor may be valuable and simple tools.Acknowledgements
This work was supported by the European Union through CMST COST Action CM1003 (Biological oxidation reactions – mechanisms and design of new catalysts).References
- E. Roduner, W. Kaim, B. Sarkar, V. B. Urlacher, J. Pleiss, R. Gläser, W.-D. Einicke, G. A. Sprenger, U. Beifuß, E. Klemm, C. Liebner, H. Hieronymus, S.-F. Hsu, B. Plietker and S. Laschat, ChemCatChem, 2013, 5, 82–112 CrossRef CAS.
- H. Mayr and A. R. Ofial, Angew. Chem., Int. Ed., 2006, 45, 1844–1854 CrossRef CAS PubMed.
- R. B. Silverman, The Organic Chemistry of Enzyme-Catalyzed Reactions, Academic Press, San Diego, 2000 Search PubMed.
- U. Bornscheuer and R. Kazlauskas, Hydrolases in Organic Synthesis, Wiley-VCH, Weinheim, 2006 Search PubMed.
- Enzyme Catalysis in Organic Synthesis, ed. K. Drauz, H. Groeger and O. May, Wiley-VCH, Weinheim, 2012 Search PubMed.
- K. Faber, Biotransformations in Organic Chemistry, Springer, Berlin, 2011 Search PubMed.
- W. Kroutil, H. Mang, K. Edegger and K. Faber, Curr. Opin. Chem. Biol., 2004, 8, 120–126 CrossRef CAS PubMed.
- W. Kroutil, H. Mang, K. Edegger and K. Faber, Adv. Synth. Catal., 2004, 346, 125–142 CrossRef CAS.
- B. M. Nestl, S. C. Hammer, B. A. Nebel and B. Hauer, Angew. Chem., Int. Ed., 2014, 53, 3070–3095 CrossRef CAS PubMed.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed.
- F. Hollmann, I. W. C. E. Arends, K. Buehler, A. Schallmey and B. Bühler, Green Chem., 2011, 13, 226–265 RSC.
- L. M. Blank, B. E. Ebert, K. Buehler and B. Bühler, Antioxid. Redox Signaling, 2010, 13, 349–394 CrossRef CAS PubMed.
- E. G. Funhoff and J. B. Van Beilen, Biocatal. Biotransform., 2007, 25, 186–193 CrossRef CAS.
- J. B. van Beilen, W. A. Duetz, A. Schmid and B. Witholt, Trends Biotechnol., 2003, 21, 170–177 CrossRef CAS.
- R. J. Kazlauskas and U. T. Bornscheuer, Nat. Chem. Biol., 2009, 5, 526–529 CrossRef CAS PubMed.
- M. T. Reetz, J. Am. Chem. Soc., 2013, 135, 12480–12496 CrossRef CAS PubMed.
- M. T. Reetz, Tetrahedron, 2012, 68, 7530–7548 CrossRef CAS PubMed.
- M. T. Reetz, Angew. Chem., Int. Ed., 2011, 50, 138–174 CrossRef CAS PubMed.
- J. C. Lewis, S. M. Mantovani, Y. Fu, C. D. Snow, R. S. Komor, C.-H. Wong and F. H. Arnold, ChemBioChem, 2011, 11, 2502–2505 CrossRef PubMed.
- S. T. Jung, R. Lauchli and F. H. Arnold, Curr. Opin. Biotechnol., 2011, 22, 809–817 CrossRef CAS PubMed.
- V. B. Urlacher and M. Girhard, Trends Biotechnol., 2012, 30, 26–36 CrossRef CAS PubMed.
- V. B. Urlacher and S. Eiben, Trends Biotechnol., 2006, 24, 324–330 CrossRef CAS PubMed.
- V. Urlacher and R. D. Schmid, Curr. Opin. Biotechnol., 2002, 13, 557–564 CrossRef CAS.
- L. G. Otten, F. Hollmann and I. W. C. E. Arends, Trends Biotechnol., 2010, 28, 46–54 CrossRef CAS PubMed.
- D. J. Leak, R. A. Sheldon, J. M. Woodley and P. Adlercreutz, Biocatal. Biotransform., 2009, 27, 1–26 CrossRef CAS.
- M. K. Julsing, S. Cornelissen, B. Bühler and A. Schmid, Curr. Opin. Chem. Biol., 2008, 12, 177–186 CrossRef CAS PubMed.
- W. Dijkman, G. Gonzalo, A. Mattevi and M. Fraaije, Appl. Microbiol. Biotechnol., 2013, 97, 5177–5188 CrossRef CAS PubMed.
- G. de Gonzalo and M. W. Fraaije, ChemCatChem, 2012, 5, 403–415 CrossRef.
- D. E. Torres Pazmiño, M. Winkler, A. Glieder and M. W. Fraaije, J. Biotechnol., 2010, 146, 9–24 CrossRef PubMed.
- G. de Gonzalo, M. D. Mihovilovic and M. W. Fraaije, ChemBioChem, 2010, 11, 2208–2231 CrossRef CAS PubMed.
- W. J. H. van Berkel, N. M. Kamerbeek and M. W. Fraaije, J. Biotechnol., 2006, 124, 670–689 CrossRef CAS PubMed.
- S. Montersino, D. Tischler, G. T. Gassner and W. J. H. v. Berkel, Adv. Synth. Catal., 2011, 353, 2301–2319 CrossRef CAS.
- N. M. Kamerbeek, D. B. Janssen, W. J. H. van Berkel and M. W. Fraaije, Adv. Synth. Catal., 2003, 345, 667–678 CrossRef CAS.
- M. Hofrichter, R. Ullrich, M. J. Pecyna, C. Liers and T. Lundell, Appl. Microbiol. Biotechnol., 2010, 87, 871–897 CrossRef CAS PubMed.
- M. Hofrichter and R. Ullrich, Appl. Microbiol. Biotechnol., 2006, 71, 276–288 CrossRef CAS PubMed.
- K. Balke, M. Kadow, H. Mallin, S. Sass and U. T. Bornscheuer, Org. Biomol. Chem., 2012, 10, 6249–6265 CAS.
- A. Riebel, G. de Gonzalo and M. W. Fraaije, J. Mol. Catal. B: Enzym., 2013, 88, 20–25 CrossRef CAS PubMed.
- H. L. van Beek, G. d. Gonzalo and M. W. Fraaije, Chem. Commun., 2012, 48, 3288–3290 RSC.
- D. E. Torres Pazmiño, H. M. Dudek and M. W. Fraaije, Curr. Opin. Chem. Biol., 2010, 14, 138–144 CrossRef PubMed.
- G. de Gonzalo, G. Ottolina, F. Zambianchi, M. W. Fraaije and G. Carrea, J. Mol. Catal. B: Enzym., 2006, 39, 91–97 CrossRef CAS PubMed.
- R. Teufel, A. Miyanaga, Q. Michaudel, F. Stull, G. Louie, J. P. Noel, P. S. Baran, B. Palfey and B. S. Moore, Nature, 2013, 503, 552–556 CrossRef CAS PubMed.
- G. de Gonzalo, C. Smit, J. F. Jin, A. J. Minnaard and M. W. Fraaije, Chem. Commun., 2011, 47, 11050–11052 RSC.
- G. E. Turfitt, Biochem. J., 1948, 42, 376–383 CAS.
- J. Fried, R. W. Thoma and A. Klingsberg, J. Am. Chem. Soc., 1953, 75, 5764–5765 CrossRef CAS.
- D. H. Peterson, S. H. Eppstein, P. D. Meister, H. C. Murray, H. M. Leigh, A. Weintraub and L. M. Reineke, J. Am. Chem. Soc., 1953, 75, 5768–5769 CrossRef CAS.
- M. Griffin and P. W. Trudgill, Eur. J. Biochem., 1976, 63, 199–209 CrossRef CAS PubMed.
- N. A. Donoghue, D. B. Norris and P. W. Trudgill, Eur. J. Biochem., 1976, 63, 175–192 CrossRef CAS PubMed.
- C. T. Walsh and Y. C. J. Chen, Angew. Chem., Int. Ed., 1988, 27, 333–343 CrossRef.
- M. D. Mihovilovic, B. Muller and P. Stanetty, Eur. J. Org. Chem., 2002, 3711–3730 CrossRef CAS.
- J. D. Stewart, Curr. Org. Chem., 1998, 2, 195–216 CAS.
- E. Malito, A. Alfieri, M. W. Fraaije and A. Mattevi, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13157–13162 CrossRef CAS PubMed.
- A. Alfieri, E. Malito, R. Orru, M. W. Fraaije and A. Mattevi, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6572–6577 CrossRef CAS PubMed.
- R. Baron, C. Riley, P. Chenprakhon, K. Thotsaporn, R. T. Winter, A. Alfieri, F. Forneris, W. J. H. van Berkel, P. Chaiyen, M. W. Fraaije, A. Mattevi and J. A. McCammon, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10603–10608 CrossRef CAS PubMed.
- A. Kirschner and U. T. Bornscheuer, Appl. Microbiol. Biotechnol., 2008, 81, 465–472 CrossRef CAS PubMed.
- I. Polyak, M. T. Reetz and W. Thiel, J. Phys. Chem. B, 2013, 117, 4993–5001 CrossRef CAS PubMed.
- L. P. Parra, R. Agudo and M. T. Reetz, ChemBioChem, 2013, 14, 2301–2309 CrossRef CAS PubMed.
- Z.-G. Zhang, G.-D. Roiban, J. P. Acevedo, I. Polyak and M. T. Reetz, Adv. Synth. Catal., 2013, 355, 99–106 CrossRef CAS.
- Z.-G. Zhang, L. P. Parra and M. T. Reetz, Chem. – Eur. J., 2012, 18, 10160–10172 CrossRef CAS PubMed.
- S. Wu, J. P. Acevedo and M. T. Reetz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2775–2780 CrossRef CAS PubMed.
- D. J. Opperman and M. T. Reetz, ChemBioChem, 2010, 11, 2589–2596 CrossRef CAS PubMed.
- M. T. Reetz and S. Wu, J. Am. Chem. Soc., 2009, 131, 15424–15432 CrossRef CAS PubMed.
- F. Schulz, F. Leca, F. Hollmann and M. T. Reetz, Beilstein J. Org. Chem., 2005, 1, 10, DOI:10.1186/1860-5397-1-10.
- M. Bocola, F. Schulz, F. Leca, A. Vogel, M. W. Fraaije and M. T. Reetz, Adv. Synth. Catal., 2005, 347, 979–986 CrossRef CAS.
- M. T. Reetz, B. Brunner, T. Schneider, F. Schulz, C. M. Clouthier and M. M. Kayser, Angew. Chem., Int. Ed., 2004, 43, 4075–4078 CrossRef CAS PubMed.
- I. Hilker, M. C. Gutierrez, R. Furstoss, J. Ward, R. Wohlgemuth and V. Alphand, Nat. Protoc., 2008, 3, 546–554 CrossRef CAS PubMed.
- I. Hilker, R. Wohlgemuth, V. Alphand and R. Furstoss, Biotechnol. Bioeng., 2005, 92, 702–710 CrossRef CAS PubMed.
- W. H. Lee, J. B. Park, K. Park, M. D. Kim and J. H. Seo, Appl. Microbiol. Biotechnol., 2007, 76, 329–338 CrossRef CAS PubMed.
- E. H. Doo, W. H. Lee, H. S. Seo, J. H. Seo and J. B. Park, J. Biotechnol., 2009, 142, 164–169 CrossRef CAS PubMed.
- S. Bisagni, B. Summers, S. Kara, R. Hatti-Kaul, G. Grogan, G. Mamo and F. Hollmann, Top. Catal., 2014, 57, 366–375 CrossRef CAS PubMed.
- C. N. Jensen, J. Cartwright, J. Ward, S. Hart, J. P. Turkenburg, S. T. Ali, M. J. Allen and G. Grogan, ChemBioChem, 2012, 13, 872–878 CrossRef CAS PubMed.
- C. Rodriguez, G. de Gonzalo and V. Gotor, J. Mol. Catal. B: Enzym., 2011, 74, 138–143 CrossRef PubMed.
- A. Rioz-Martinez, M. Kopacz, G. de Gonzalo, D. E. Torres Pazmino, V. Gotor and M. W. Fraaije, Org. Biomol. Chem., 2011, 9, 1337–1341 CAS.
- V. Alphand and R. Wohlgemuth, Curr. Org. Chem., 2011, 14, 1928–1965 CrossRef.
- J. Zhu, Y. K. Bong, S. J. Collier, M. Vogel, M. J. Nazor, D. Smith, S. Song, M. D. Clay, B. Mijts and X. Zhang, Int. Patent Application to Codexis Inc., 2011 Search PubMed.
- P. B. Brondani, N. M. A. F. Guilmoto, H. M. Dudek, M. W. Fraaije and L. H. Andrade, Tetrahedron, 2012, 68, 10431–10436 CrossRef CAS PubMed.
- L. H. Andrade, E. C. Pedrozo, H. G. Leite and P. B. Brondani, J. Mol. Catal. B: Enzym., 2011, 73, 63–66 CrossRef CAS PubMed.
- J. A. Latham, B. P. Branchaud, Y. C. J. Chen and C. Walsh, J. Chem. Soc., Chem. Commun., 1986, 528–530 RSC.
- B. P. Branchaud and C. T. Walsh, J. Am. Chem. Soc., 1985, 107, 2153–2161 CrossRef CAS.
- P. B. Brondani, G. de Gonzalo, M. W. Fraaije and L. H. Andrade, Adv. Synth. Catal., 2011, 353, 2169–2173 CrossRef CAS.
- Y. K. Bong, M. D. Clay, S. J. Collier, B. Mijts, M. Vogel, X. Zhang, J. Zhu, J. Nazor, D. Smith and S. Song, Codexis, 2011.
- M. J. H. Moonen, M. W. Fraaije, I. M. C. M. Rietjens, C. Laane and W. J. H. van Berkel, Adv. Synth. Catal., 2002, 344, 1023–1035 CrossRef CAS.
- A. Meyer, M. Held, A. Schmid, H. P. E. Kohler and B. Witholt, Biotechnol. Bioeng., 2003, 81, 518–524 CrossRef CAS PubMed.
- A. Meyer, M. Wursten, A. Schmid, H. P. E. Kohler and B. Witholt, J. Biol. Chem., 2002, 277, 34161–34167 CrossRef CAS PubMed.
- A. Meyer, A. Schmid, M. Held, A. H. Westphal, M. Rothlisberger, H. P. E. Kohler, W. J. H. van Berkel and B. Witholt, J. Biol. Chem., 2002, 277, 5575–5582 CrossRef CAS PubMed.
- J. Lutz, V. V. Mozhaev, Y. L. Khmelnitsky, B. Witholt and A. Schmid, J. Mol. Catal. B: Enzym., 2002, 19–20, 177–187 CrossRef CAS.
- A. Schmid, I. Vereyken, M. Held and B. Witholt, J. Mol. Catal. B: Enzym., 2001, 11, 455–462 CrossRef CAS.
- A. Schmid, H. P. E. Kohler and K. H. Engesser, J. Mol. Catal. B: Enzym., 1998, 5, 311–316 CrossRef CAS.
- M. Held, W. Suske, A. Schmid, K. H. Engesser, H. P. E. Kohler, B. Witholt and M. G. Wubbolts, J. Mol. Catal. B: Enzym., 1998, 5, 87–93 CrossRef CAS.
- W. A. Suske, M. Held, A. Schmid, T. Fleischmann, M. G. Wubbolts and H.-P. E. Kohler, J. Biol. Chem., 1997, 272, 24257–24265 CrossRef CAS PubMed.
- E. Orenes-Pinero, F. Garcia-Carmona and A. Sanchez-Ferrer, Food Chem., 2013, 139, 377–383 CrossRef CAS PubMed.
- L. Saa, A. Jaureguibeitia, E. Largo, M. J. Llama and J. L. Serra, Appl. Microbiol. Biotechnol., 2010, 86, 201–211 CrossRef CAS PubMed.
- U. Kirchner, A. H. Westphal, R. Muller and W. J. H. van Berkel, J. Biol. Chem., 2003, 278, 47545–47553 CrossRef CAS PubMed.
- F. M. Duffner, U. Kirchner, M. P. Bauer and R. Müller, Gene, 2000, 256, 215–221 CrossRef CAS.
- J. Sucharitakul, C. Tongsook, D. Pakotiprapha, W. J. H. van Berkel and P. Chaiyen, J. Biol. Chem., 2013, 288, 35210–35221 CrossRef CAS PubMed.
- B. Entsch and W. J. H. Vanberkel, FASEB J., 1995, 9, 476–483 CAS.
- S. Montersino and W. J. H. van Berkel, Biochim. Biophys. Acta, Proteins Proteomics, 2012, 1824, 433–442 CrossRef CAS PubMed.
- R. Gross, K. Buehler and A. Schmid, Biotechnol. Bioeng., 2013, 110, 424–436 CrossRef CAS PubMed.
- M. K. Julsing, D. Kuhn, A. Schmid and B. Bühler, Biotechnol. Bioeng., 2012, 109, 1109–1119 CrossRef CAS PubMed.
- R. Ruinatscha, R. Karande, K. Buehler and A. Schmid, Molecules, 2011, 16, 5975–5988 CrossRef CAS PubMed.
- D. Kuhn, M. A. Kholiq, E. Heinzle, B. Bühler and A. Schmid, Green Chem., 2010, 12, 815–827 RSC.
- R. Ruinatscha, C. Dusny, K. Buehler and A. Schmid, Adv. Synth. Catal., 2009, 351, 2505–2515 CrossRef CAS.
- R. Gross, K. Lang, K. Bühler and A. Schmid, Biotechnol. Bioeng., 2009, 105, 705–717 Search PubMed.
- B. Buehler, J. B. Park, L. M. Blank and A. Schmid, Appl. Environ. Microbiol., 2008, 74, 1436–1446 CrossRef CAS PubMed.
- L. M. Blank, B. E. Ebert, B. Bühler and A. Schmid, Biotechnol. Bioeng., 2008, 100, 1050–1065 CrossRef CAS PubMed.
- J. B. Park, B. Bühler, S. Panke, B. Witholt and A. Schmid, Biotechnol. Bioeng., 2007, 98, 1219–1229 CrossRef CAS PubMed.
- J. B. Park, B. Bühler, T. Habicher, B. Hauer, S. Panke, B. Witholt and A. Schmid, Biotechnol. Bioeng., 2006, 95, 501–512 CrossRef CAS PubMed.
- K. A. Feenstra, K. Hofstetter, R. Bosch, A. Schmid, J. N. M. Commandeur and N. P. E. Vermeulen, Biophys. J., 2006, 91, 3206–3216 CrossRef CAS PubMed.
- F. Hollmann, K. Hofstetter, T. Habicher, B. Hauer and A. Schmid, J. Am. Chem. Soc., 2005, 127, 6540–6541 CrossRef CAS PubMed.
- K. Otto, K. Hofstetter, M. Rothlisberger, B. Witholt and A. Schmid, J. Bacteriol., 2004, 186, 5292–5302 CrossRef CAS PubMed.
- F. Hollmann, P. C. Lin, B. Witholt and A. Schmid, J. Am. Chem. Soc., 2003, 125, 8209–8217 CrossRef CAS PubMed.
- S. Panke, M. Held, M. G. Wubbolts, B. Witholt and A. Schmid, Biotechnol. Bioeng., 2002, 80, 33–41 CrossRef CAS PubMed.
- A. Schmid, K. Hofstetter, H.-J. Feiten, F. Hollmann and B. Witholt, Adv. Synth. Catal., 2001, 343, 732–737 CrossRef CAS.
- S. Panke, M. G. Wubbolts, A. Schmid and B. Witholt, Biotechnol. Bioeng., 2000, 69, 91–100 CrossRef CAS.
- K. Hofstetter, J. Lutz, I. Lang, B. Witholt and A. Schmid, Angew. Chem., Int. Ed., 2004, 43, 2163–2166 CrossRef CAS PubMed.
- D. Tischler, M. Schlömann, W. J. H. van Berkel and G. T. Gassner, FEBS Lett., 2013, 587, 3848–3852 CrossRef CAS PubMed.
- D. Tischler, J. A. D. Groning, S. R. Kaschabek and M. Schlömann, Appl. Biochem. Biotechnol., 2012, 167, 931–944 CrossRef CAS PubMed.
- D. Tischler, R. Kermer, J. A. D. Groning, S. R. Kaschabek, W. J. H. van Berkel and M. Schlömann, J. Bacteriol., 2010, 192, 5220–5227 CrossRef CAS PubMed.
- D. Tischler, D. Eulberg, S. Lakner, S. R. Kaschabek, W. J. H. van Berkel and M. Schlömann, J. Bacteriol., 2009, 191, 4996–5009 CrossRef CAS PubMed.
- H. Toda, R. Imae, T. Komio and N. Itoh, Appl. Microbiol. Biotechnol., 2012, 96, 407–418 CrossRef CAS PubMed.
- H. Toda, R. Imae and N. Itoh, Tetrahedron: Asymmetry, 2012, 23, 1542–1549 CrossRef CAS PubMed.
- E. W. van Hellemond, D. B. Janssen and M. W. Fraaije, Appl. Environ. Microbiol., 2007, 73, 5832–5839 CrossRef CAS PubMed.
- R. McKenna, S. Pugh, B. Thompson and D. R. Nielsen, Biotechnol. J., 2013, 8, 1465–1475 CAS.
- K.-H. van Pee, Curr. Org. Chem., 2012, 16, 2583–2597 CrossRef CAS.
- K. H. van Pee, ChemBioChem, 2011, 12, 681–683 CrossRef CAS PubMed.
- A. Lang, S. Polnick, T. Nicke, P. William, E. P. Patallo, J. H. Naismith and K. H. van Pee, Angew. Chem., Int. Ed., 2011, 50, 2951–2953 CrossRef CAS PubMed.
- X. F. Zhu, W. De Laurentis, K. Leang, J. Herrmann, K. Lhiefeld, K. H. van Pee and J. H. Naismith, J. Mol. Biol., 2009, 391, 74–85 CrossRef CAS PubMed.
- S. Flecks, E. R. Patallo, X. F. Zhu, A. J. Ernyei, G. Seifert, A. Schneider, C. J. Dong, J. H. Naismith and K. H. van Pee, Angew. Chem., Int. Ed., 2008, 47, 9533–9536 CrossRef CAS PubMed.
- X. P. Chen and K. H. van Pee, Acta Biochim. Biophys. Sin., 2008, 40, 183–193 CrossRef CAS PubMed.
- K. H. van Pee and E. P. Patallo, Appl. Microbiol. Biotechnol., 2006, 70, 631–641 CrossRef PubMed.
- C. Seibold, H. Schnerr, J. Rumpf, A. Kunzendorf, C. Hatscher, T. Wage, A. J. Ernyei, C. J. Dong, J. H. Naismith and K. H. van Pee, Biocatal. Biotransform., 2006, 24, 401–408 CrossRef CAS.
- S. Unversucht, F. Hollmann, A. Schmid and K.-H. van Pée, Adv. Synth. Catal., 2005, 347, 1163–1167 CrossRef CAS.
- C. J. Dong, S. Flecks, S. Unversucht, C. Haupt, K. H. van Pee and J. H. Naismith, Science, 2005, 309, 2216–2219 CrossRef CAS PubMed.
- S. Keller, T. Wage, K. Hohaus, M. Hölzer, E. Eichhorn and K.-H. van Pée, Angew. Chem., Int. Ed., 2000, 39, 2300–2302 CrossRef CAS.
- M. Frese, P. H. Guzowska, H. Voß and N. Sewald, ChemCatChem, 2014, 6, 1270–1276 CAS.
- Y. Dong, J. Yan, H. Du, M. Chen, T. Ma and L. Feng, Appl. Microbiol. Biotechnol., 2012, 94, 1019–1029 CrossRef CAS PubMed.
- L. Li, X. Liu, W. Yang, F. Xu, W. Wang, L. Feng, M. Bartlam, L. Wang and Z. Rao, J. Mol. Biol., 2008, 376, 453–465 CrossRef CAS PubMed.
- H. Lin, J. Qiao, Y. Liu and Z. L. Wu, J. Mol. Catal. B: Enzym., 2011, 67, 236–241 CrossRef PubMed.
- H. Lin, Y. Liu and Z.-L. Wu, Tetrahedron: Asymmetry, 2011, 22, 134–137 CrossRef CAS PubMed.
- S. Colonna, N. Gaggero, G. Carrea, G. Ottolina, P. Pasta and F. Zambianchi, Tetrahedron Lett., 2002, 43, 1797–1799 CrossRef CAS.
- C. V. F. Baldwin, R. Wohlgemuth and J. M. Woodley, Org. Process Res. Dev., 2008, 12, 660–665 CrossRef CAS.
- K. Geitner, A. Kirschner, J. Rehdorf, M. Schmidt, M. D. Mihovilovic and U. T. Bornscheuer, Tetrahedron: Asymmetry, 2007, 18, 892–895 CrossRef CAS PubMed.
- J. D. Stewart, K. W. Reed, C. A. Martinez, J. Zhu, G. Chen and M. M. Kayser, J. Am. Chem. Soc., 1998, 120, 3541–3548 CrossRef CAS.
- S. Kara, D. Spickermann, J. H. Schrittwieser, C. Leggewie, W. J. H. Van Berkel, I. W. C. E. Arends and F. Hollmann, Green Chem., 2013, 15, 330–335 RSC.
- C. J. C. Whitehouse, S. G. Bell and L.-L. Wong, Chem. Soc. Rev., 2012, 41, 1218–1260 RSC.
- R. Fasan, ACS Catal., 2012, 2, 647–666 CrossRef CAS.
- E. O'Reilly, V. Kohler, S. L. Flitsch and N. J. Turner, Chem. Commun., 2011, 47, 2490–2501 RSC.
- G. Grogan, Curr. Opin. Chem. Biol., 2011, 15, 241–248 CrossRef CAS PubMed.
- H. Watanabe, H. Hirakawa and T. Nagamune, ChemCatChem, 2013, 5, 3835–3840 CrossRef CAS.
- S. Q. Pham, P. Gao and Z. Li, Biotechnol. Bioeng., 2013, 110, 363–373 CrossRef CAS PubMed.
- S. H. Lee, Y.-C. Kwon, D.-M. Kim and C. B. Park, Biotechnol. Bioeng., 2013, 110, 383–390 CrossRef CAS PubMed.
- M. Girhard, E. Kunigk, S. Tihovsky, V. V. Shumyantseva and V. B. Urlacher, Biotechnol. Appl. Biochem., 2013, 60, 111–118 CrossRef CAS PubMed.
- D. Zehentgruber, V. B. Urlacher and S. Lütz, J. Mol. Catal. B: Enzym., 2012, 84, 62–64 CrossRef CAS PubMed.
- N.-H. Tran, N. Huynh, G. Chavez, A. Nguyen, S. Dwaraknath, T.-A. Nguyen, M. Nguyen and L. Cheruzel, J. Inorg. Biochem., 2012, 115, 50–56 CrossRef CAS PubMed.
- M. M. Chen, P. S. Coelho and F. H. Arnold, Adv. Synth. Catal., 2012, 354, 964–968 CrossRef CAS.
- F. E. Zilly, J. P. Acevedo, W. Augustyniak, A. Deege, U. W. Häusig and M. T. Reetz, Angew. Chem., Int. Ed., 2011, 50, 2720–2724 CrossRef CAS PubMed.
- O. Shoji, T. Kunimatsu, N. Kawakami and Y. Watanabe, Angew. Chem., Int. Ed., 2013, 52, 6606–6610 CrossRef CAS PubMed.
- P. S. Coelho, Z. J. Wang, M. E. Ener, S. A. Baril, A. Kannan, F. H. Arnold and E. M. Brustad, Nat. Chem. Biol., 2013, 9, 485–487 CrossRef CAS PubMed.
- H. Joo, Z. L. Lin and F. H. Arnold, Nature, 1999, 399, 670–673 CrossRef CAS PubMed.
- M. T. Musser, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- S. Staudt, E. Burda, C. Giese, C. A. Müller, J. Marienhagen, U. Schwaneberg, W. Hummel, K. Drauz and H. Gröger, Angew. Chem., Int. Ed., 2013, 52, 2359–2363 CrossRef CAS PubMed.
- C. A. Müller, B. Akkapurathu, T. Winkler, S. Staudt, W. Hummel, H. Gröger and U. Schwaneberg, Adv. Synth. Catal., 2013, 355, 1787–1798 CrossRef.
- A. J. Willetts, C. J. Knowles, M. S. Levitt, S. M. Roberts, H. Sandey and N. F. Shipston, J. Chem. Soc., Perkin Trans. 1, 1991, 1608–1610 RSC.
- H. Mallin, H. Wulf and U. T. Bornscheuer, Enzyme Microb. Technol., 2013, 53, 283–287 CrossRef CAS PubMed.
- S. Kille, F. E. Zilly, J. P. Acevedo and M. T. Reetz, Nat. Chem., 2011, 3, 738–743 CrossRef CAS PubMed.
- D. Zehentgruber, F. Hannemann, S. Bleif, R. Bernhardt and S. Lütz, ChemBioChem, 2010, 11, 713–721 CrossRef CAS PubMed.
- S. Cornelissen, M. K. Julsing, J. Volmer, O. Riechert, A. Schmid and B. Buhler, Biotechnol. Bioeng., 2013, 110, 1282–1292 CrossRef CAS PubMed.
- R. Gudiminchi, C. Randall, D. Opperman, O. Olaofe, S. L. Harrison, J. Albertyn and M. Smit, Appl. Microbiol. Biotechnol., 2012, 96, 1507–1516 CrossRef CAS PubMed.
- H. Schewe, D. Holtmann and J. Schrader, Appl. Microbiol. Biotechnol., 2009, 83, 849–857 CrossRef CAS PubMed.
- H. Schewe, B.-A. Kaup and J. Schrader, Appl. Microbiol. Biotechnol., 2008, 78, 55–65 CrossRef CAS PubMed.
- M.-A. Mirata, M. Wüst, A. Mosandl and J. Schrader, J. Agric. Food Chem., 2008, 56, 3287–3296 CrossRef CAS PubMed.
- M. Girhard, K. Machida, M. Itoh, R. D. Schmid, A. Arisawa and V. B. Urlacher, Microb. Cell Fact., 2009, 8, 12 CrossRef PubMed.
- Y. Watanabe, S. Laschat, M. Budde, O. Affolter, Y. Shimada and V. B. Urlacher, Tetrahedron, 2007, 63, 9413–9422 CrossRef CAS PubMed.
- S. C. Maurer, H. Schulze, R. D. Schmid and V. B. Urlacher, Adv. Synth. Catal., 2003, 345, 802–810 CrossRef CAS.
- J. A. Dietrich, Y. Yoshikuni, K. J. Fisher, F. X. Woolard, D. Ockey, D. J. McPhee, N. S. Renninger, M. C. Y. Chang, D. Baker and J. D. Keasling, ACS Chem. Biol., 2009, 4, 261–267 CrossRef CAS PubMed.
- D. K. Ro, E. M. Paradise, M. Ouellet, K. J. Fisher, K. L. Newman, J. M. Ndungu, K. A. Ho, R. A. Eachus, T. S. Ham, J. Kirby, M. C. Y. Chang, S. T. Withers, Y. Shiba, R. Sarpong and J. D. Keasling, Nature, 2006, 440, 940–943 CrossRef CAS PubMed.
- P. r. Tufvesson, J. Lima-Ramos, M. Nordblad and J. M. Woodley, Org. Process Res. Dev., 2010, 15, 266–274 CrossRef.
- W. H. Lu, J. E. Ness, W. C. Xie, X. Y. Zhang, J. Minshull and R. A. Gross, J. Am. Chem. Soc., 2010, 132, 15451–15455 CrossRef CAS PubMed.
- S. Zibek, S. Huf, W. Wagner, T. Hirth and S. Rupp, Chem. Ing. Tech., 2009, 81, 1797–1808 CrossRef CAS.
- W. H. Eschenfeldt, Y. Y. Zhang, H. Samaha, L. Stols, L. D. Eirich, C. R. Wilson and M. I. Donnelly, Appl. Environ. Microbiol., 2003, 69, 5992–5999 CrossRef CAS.
- S. C. Liu, C. Li, X. C. Fang and Z. A. Cao, Enzyme Microb. Technol., 2004, 34, 73–77 CrossRef CAS PubMed.
- N. Kawakami, O. Shoji and Y. Watanabe, Angew. Chem., Int. Ed., 2011, 50, 5315–5318 CrossRef CAS PubMed.
- T. Fujishiro, O. Shoji, N. Kawakami, T. Watanabe, H. Sugimoto, Y. Shiro and Y. Watanabe, Chem. – Asian J., 2012, 7, 2286–2293 CrossRef CAS PubMed.
- A. Rentmeister, T. R. Brown, C. D. Snow, M. N. Carbone and F. H. Arnold, ChemCatChem, 2011, 3, 1065–1071 CrossRef CAS.
- C. A. Tracewell and F. H. Arnold, Curr. Opin. Chem. Biol., 2009, 13, 3–9 CrossRef CAS PubMed.
- J. C. Lewis and F. H. Arnold, Chimia, 2009, 63, 309–312 CrossRef CAS.
- P. Meinhold, M. W. Peters, A. Hartwick, A. R. Hernandez and F. H. Arnold, Adv. Synth. Catal., 2006, 348, 763–772 CrossRef CAS.
- M. W. Peters, P. Meinhold, A. Glieder and F. H. Arnold, J. Am. Chem. Soc., 2003, 125, 13442–13450 CrossRef CAS PubMed.
- P. C. Cirino and F. H. Arnold, Angew. Chem., Int. Ed., 2003, 42, 3299–3301 CrossRef CAS PubMed.
- A. Glieder, E. T. Farinas and F. H. Arnold, Nat. Biotechnol., 2002, 20, 1135–1139 CrossRef CAS PubMed.
- D. F. Munzer, H. Griengl, A. Moumtzi, R. Saf, T. Terzani and A. de Raadt, Eur. J. Org. Chem., 2005, 793–796 CrossRef.
- A. de Raadt and H. Griengl, Curr. Opin. Biotechnol., 2002, 13, 537–542 CrossRef CAS.
- A. de Raadt, B. Fetz, H. Griengl, M. F. Klingler, B. Krenn, K. Mereiter, D. F. Munzer, P. Plachota, H. Weber and R. Saf, Tetrahedron, 2001, 57, 8151–8157 CrossRef CAS.
- G. Braunegg, A. de Raadt, S. Feichtenhofer, H. Griengl, I. Kopper, A. Lehmann and H. J. Weber, Angew. Chem., Int. Ed., 1999, 38, 2763–2766 CrossRef CAS.
- E. Weber, A. Seifert, M. Antonovici, C. Geinitz, J. Pleiss and V. B. Urlacher, Chem. Commun., 2011, 47, 944–946 RSC.
- R. Agudo, G.-D. Roiban and M. T. Reetz, ChemBioChem, 2012, 13, 1465–1473 CrossRef CAS PubMed.
- A. Siriphongphaew, P. Pisnupong, J. Wongkongkatep, P. Inprakhon, A. Vangnai, K. Honda, H. Ohtake, J. Kato, J. Ogawa, S. Shimizu, V. Urlacher, R. Schmid and T. Pongtharangkul, Appl. Microbiol. Biotechnol., 2012, 95, 357–367 CrossRef CAS PubMed.
- F. van Rantwijk and R. A. Sheldon, Curr. Opin. Biotechnol., 2000, 11, 554–564 CrossRef CAS.
- M. Hofrichter and R. Ullrich, Curr. Opin. Chem. Biol., 2014, 19, 116–125 CrossRef CAS PubMed.
- M. Pešić, C. López, G. Álvaro and J. López-Santín, J. Mol. Catal. B: Enzym., 2012, 84, 144–151 CrossRef PubMed.
- C. Li, L. Wang, Y. Jiang, M. Hu, S. Li and Q. Zhai, Appl. Biochem. Biotechnol., 2011, 165, 1691–1707 CrossRef CAS PubMed.
- T. Krieg, S. Huttmann, K.-M. Mangold, J. Schrader and D. Holtmann, Green Chem., 2011, 13, 2686–2689 RSC.
- S. Águila, R. Vazquez-Duhalt, C. Covarrubias, G. Pecchi and J. B. Alderete, J. Mol. Catal. B: Enzym., 2011, 70, 81–87 CrossRef PubMed.
- V. Yazbik and M. Ansorge-Schumacher, Process Biochem., 2010, 45, 279–283 CrossRef CAS PubMed.
- G. Díaz-Díaz, M. C. Blanco-López, M. J. Lobo-Castañón, A. J. Miranda-Ordieres and P. Tuñón-Blanco, J. Mol. Catal. B: Enzym., 2010, 66, 332–336 CrossRef PubMed.
- W. Wang, Y. Xu, D. I. C. Wang and Z. Li, J. Am. Chem. Soc., 2009, 131, 12892–12893 CrossRef CAS PubMed.
- C. Roberge, D. Amos, D. Pollard and P. Devine, J. Mol. Catal. B: Enzym., 2009, 56, 41–45 CrossRef CAS PubMed.
- R. Renirie, C. Pierlot, R. Wever and J. M. Aubry, J. Mol. Catal. B: Enzym., 2009, 56, 259–264 CrossRef CAS PubMed.
- D. I. Perez, F. van Rantwijk and R. A. Sheldon, Adv. Synth. Catal., 2009, 351, 2133–2139 CrossRef CAS.
- H. M. de Hoog, M. Nallani, J. Cornelissen, A. E. Rowan, R. J. M. Nolte and I. Arends, Org. Biomol. Chem., 2009, 7, 4604–4610 CAS.
- B.-A. Kaup, K. Ehrich, M. Pescheck and J. Schrader, Biotechnol. Bioeng., 2008, 99, 491–498 CrossRef CAS PubMed.
- D. Jung, C. Streb and M. Hartmann, Microporous Mesoporous Mater., 2008, 113, 523–529 CrossRef CAS PubMed.
- C. E. Grey, F. Rundbäck and P. Adlercreutz, J. Biotechnol., 2008, 135, 196–201 CrossRef CAS PubMed.
- S. Aguila, R. Vazquez-Duhalt, R. Tinoco, M. Rivera, G. Pecchi and J. B. Alderete, Green Chem., 2008, 10, 647–653 RSC.
- B. A. Kaup, U. Piantini, M. Wust and J. Schrader, Appl. Microbiol. Biotechnol., 2007, 73, 1087–1096 CrossRef CAS PubMed.
- C. E. Grey, M. Hedström and P. Adlercreutz, ChemBioChem, 2007, 8, 1055–1062 CrossRef CAS PubMed.
- J.-B. Park and D. S. Clark, Biotechnol. Bioeng., 2006, 94, 189–192 CrossRef CAS PubMed.
- X. Yi, M. Mroczko, K. M. Manoj, X. Wang and L. P. Hager, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12412–12417 CrossRef CAS.
- E. Kiljunen and L. T. Kanerva, Tetrahedron: Asymmetry, 1999, 10, 3529–3535 CrossRef CAS.
- S. Hu and L. P. Hager, J. Am. Chem. Soc., 1999, 121, 872–873 CrossRef CAS.
- L. P. Hager, F. J. Lakner and A. Basavapathruni, J. Mol. Catal. B: Enzym., 1998, 5, 95–101 CrossRef CAS.
- S. Aoun and M. Baboulene, J. Mol. Catal. B: Enzym., 1998, 4, 101–109 CrossRef CAS.
- F. J. Lakner, K. P. Cain and L. P. Hager, J. Am. Chem. Soc., 1997, 119, 443–444 CrossRef CAS.
- F. J. Lakner and L. P. Hager, J. Org. Chem., 1996, 61, 3923–3925 CrossRef CAS.
- A. Zaks and D. R. Dodds, J. Am. Chem. Soc., 1995, 117, 10419–10424 CrossRef CAS.
- E. J. Allain, L. P. Hager, L. Deng and E. N. Jacobsen, J. Am. Chem. Soc., 1993, 115, 4415–4416 CrossRef CAS.
- J. H. Dawson and M. Sono, J. Am. Chem. Soc., 1987, 87, 1255–1276 CAS.
- J. Geigert, D. J. Dalietos, S. L. Neidleman, T. D. Lee and J. Wadsworth, Biochem. Biophys. Res. Commun., 1983, 114, 1104–1108 CrossRef CAS.
- E. Churakova, M. Kluge, R. Ullrich, I. Arends, M. Hofrichter and F. Hollmann, Angew. Chem., Int. Ed., 2011, 50, 10716–10719 CrossRef CAS PubMed.
- X. Wang, S. Peter, R. Ullrich, M. Hofrichter and J. T. Groves, Angew. Chem., Int. Ed., 2013, 52, 9238–9241 CrossRef CAS PubMed.
- K. Piontek, E. Strittmatter, R. Ullrich, G. Gröbe, M. J. Pecyna, M. Kluge, K. Scheibner, M. Hofrichter and D. A. Plattner, J. Biol. Chem., 2013 DOI:10.1074/jbc.M113.514521.
- M. Kluge, R. Ullrich, K. Scheibner and M. Hofrichter, Green Chem., 2012, 14, 440–446 RSC.
- S. Peter, M. Kinne, X. S. Wang, R. Ullrich, G. Kayser, J. T. Groves and M. Hofrichter, FEBS J., 2011, 278, 3667–3675 CrossRef CAS PubMed.
- A. Gutierrez, E. D. Babot, R. Ullrich, M. Hofrichter, A. T. Martinez and J. C. del Rio, Arch. Biochem. Biophys., 2011, 514, 33–43 CrossRef CAS PubMed.
- K. Barková, M. Kinne, R. Ullrich, L. Hennig, A. Fuchs and M. Hofrichter, Tetrahedron, 2011, 67, 4874–4878 CrossRef PubMed.
- M. M. Vdovenko, R. Ullrich, M. Hofrichter and I. Y. Sakharov, Appl. Biochem. Microbiol., 2010, 46, 65–68 CrossRef CAS.
- K. Piontek, R. Ullrich, C. Liers, K. Diederichs, D. A. Plattner and M. Hofrichter, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2010, 66, 693–698 CrossRef CAS PubMed.
- M. Kinne, C. Zeisig, R. Ullrich, G. Kayser, K. E. Hammel and M. Hofrichter, Biochem. Biophys. Res. Commun., 2010, 397, 18–21 CrossRef CAS PubMed.
- E. Aranda, R. Ullrich and M. Hofrichter, Biodegradation, 2010, 21, 267–281 CrossRef CAS PubMed.
- R. Ullrich, C. Liers, S. Schimpke and M. Hofrichter, Biotechnol. J., 2009, 4, 1619–1626 CAS.
- M. J. Pecyna, R. Ullrich, B. Bittner, A. Clemens, K. Scheibner, R. Schubert and M. Hofrichter, Appl. Microbiol. Biotechnol., 2009, 84, 885–897 CrossRef CAS PubMed.
- M. Kinne, M. Poraj-Kobielska, S. A. Ralph, R. Ullrich, M. Hofrichter and K. E. Hammel, J. Biol. Chem., 2009, 284, 29343–29349 CrossRef CAS PubMed.
- M. Kinne, M. Poraj-Kobielska, E. Aranda, R. Ullrich, K. E. Hammel, K. Scheibner and M. Hofrichter, Bioorg. Med. Chem. Lett., 2009, 19, 3085–3087 CrossRef CAS PubMed.
- M. Kinne, R. Ullrich, K. E. Hammel, K. Scheibner and M. Hofrichter, Tetrahedron Lett., 2008, 49, 5950–5953 CrossRef CAS PubMed.
- R. Ullrich and M. Hofrichter, Cell. Mol. Life Sci., 2007, 64, 271–293 CrossRef CAS PubMed.
- A. But, J. Le Nôtre, E. L. Scott, R. Wever and J. P. M. Sanders, ChemSusChem, 2012, 5, 1199–1202 CrossRef CAS PubMed.
- M. Schrewe, M. K. Julsing, B. Bühler and A. Schmid, Chem. Soc. Rev., 2013, 42, 6346–6377 RSC.
- L. M. Blank, G. Ionidis, B. E. Ebert, B. Bühler and A. Schmid, FEBS J., 2008, 275, 5173–5190 CrossRef CAS PubMed.
- D. Meyer, B. Witholt and A. Schmid, Appl. Environ. Microbiol., 2005, 71, 6624–6632 CrossRef CAS PubMed.
- M. J. Fink, F. Rudroff and M. D. Mihovilovic, Bioorg. Med. Chem. Lett., 2011, 21, 6135–6138 CrossRef CAS PubMed.
- M. D. Mihovilovic, P. Kapitan and P. Kapitanova, ChemSusChem, 2008, 1, 143–148 CrossRef CAS PubMed.
- E. Churakova, B. Tomaszewski, K. Buehler, A. Schmid, I. W. C. E. Arends and F. Hollmann, Top. Catal., 2014, 57, 385–391 CrossRef CAS.
- D. E. Torres Pazmiño, A. Riebel, J. d. Lange, F. Rudroff, M. D. Mihovilovic and M. W. Fraaije, ChemBioChem, 2009, 10, 2595–2598 CrossRef PubMed.
- D. E. Torres Pazmiño, R. Snajdrova, B.-J. Baas, M. Ghobrial, M. D. Mihovilovic and M. W. Fraaije, Angew. Chem., Int. Ed., 2008, 47, 2307–2310 CrossRef.
- A. Rioz-Martínez, G. de Gonzalo, T. P. D. E. M. W. Fraaije and V. Gotor, Eur. J. Org. Chem., 2009, 2526–2532 CrossRef.
- E. Beneventi, G. Ottolina, G. Carrea, W. Panzeri, G. Fronza and P. C. K. Lau, J. Mol. Catal. B: Enzym., 2009, 58, 164–168 CrossRef CAS PubMed.
- C. Rodriguez, G. de Gonzalo, M. W. Fraaije and V. Gotor, Tetrahedron: Asymmetry, 2007, 18, 1338–1344 CrossRef CAS PubMed.
- M. Mifsud, S. Gargiulo, S. Iborra, I. W. C. E. Arends, F. Hollmann and A. Corma, Nat. Commun., 2014, 5 DOI:10.1038/ncomms4145.
- F. Hollmann, I. W. C. E. Arends and K. Buehler, ChemCatChem, 2010, 2, 762–782 CrossRef CAS.
- M. P. Mayhew, V. Reipa, M. J. Holden and V. L. Vilker, Biotechnol. Prog., 2000, 16, 610–616 CrossRef CAS PubMed.
- D. Lim, Y. H. Kim, J. C. Joo and Y. J. Yoo, Enzyme Microb. Technol., 2010, 47, 313–321 CrossRef CAS PubMed.
- A. Vaze, M. Parizo and J. F. Rusling, Langmuir, 2004, 20, 10943–10948 CrossRef CAS PubMed.
- B. Munge, C. Estavillo, J. B. Schenkman and J. F. Rusling, ChemBioChem, 2003, 4, 82–89 CrossRef CAS PubMed.
- V. Reipa, M. P. Mayhew and V. L. Vilker, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 13554–13558 CrossRef CAS.
- K. M. Faulkner, M. S. Shet, C. W. Fisher and R. W. Estabrook, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7705–7709 CrossRef CAS.
- L. Zhao, G. Güven, Y. Li and U. Schwaneberg, Appl. Microbiol. Biotechnol., 2011, 91, 989–999 CrossRef CAS PubMed.
- U. Schwaneberg, D. Appel, J. Schmitt and R. D. Schmid, J. Biotechnol., 2000, 84, 249–257 CrossRef CAS.
- A. K. Udit, F. H. Arnold and H. B. Gray, J. Inorg. Biochem., 2004, 98, 1547–1550 CrossRef CAS PubMed.
- N.-H. Tran, N. Huynh, T. Bui, Y. Nguyen, P. Huynh, M. E. Cooper and L. E. Cheruzel, Chem. Commun., 2011, 47, 11936–11938 RSC.
- F. E. Zilly, A. Taglieber, F. Schulz, F. Hollmann and M. T. Reetz, Chem. Commun., 2009, 7152–7154 RSC.
- F. W. Ströhle, S. Z. Cekic, A. O. Magnusson, U. Schwaneberg, D. Roccatano, J. Schrader and D. Holtmann, J. Mol. Catal. B: Enzym., 2013, 88, 47–51 CrossRef PubMed.
- C. Ley, H. Schewe, F. W. Ströhle, A. J. Ruff, U. Schwaneberg, J. Schrader and D. Holtmann, J. Mol. Catal. B: Enzym., 2013, 92, 71–78 CrossRef CAS PubMed.
- D. Holtmann, K.-M. Mangold and J. Schrader, Biotechnol. Lett., 2009, 31, 765–770 CrossRef CAS PubMed.
- S. Z. Çekiç, D. Holtmann, G. Güven, K.-M. Mangold, U. Schwaneberg and J. Schrader, Electrochem. Commun., 2010, 12, 1547–1550 CrossRef PubMed.
- V. Massey, J. Biol. Chem., 1994, 269, 22459–22462 CAS.
- B. Valderrama, M. Ayala and R. Vazquez-Duhalt, Chem. Biol., 2002, 9, 555–565 CrossRef CAS.
- R. Narayanan, G. Y. Zhu and P. Wang, J. Biotechnol., 2007, 128, 86–92 CrossRef CAS PubMed.
- A. Borole, S. Dai, C. L. Cheng, M. Rodriguez and B. H. Davison, Appl. Biochem. Biotechnol., 2004, 113, 273–285 CrossRef.
- F. van de Velde, N. D. Lourenço, M. Bakker, F. van Rantwijk and R. A. Sheldon, Biotechnol. Bioeng., 2000, 69, 286–291 CrossRef CAS.
- A. D. Ryabov, Y. N. Firsova, V. N. Goral, E. S. Ryabova, A. N. Shevelkova, L. L. Troitskaya, T. V. Demeschik and V. I. Sokolov, Chem. – Eur. J., 1998, 4, 806–813 CrossRef CAS.
- K. Lee and S.-H. Moon, J. Biotechnol., 2003, 102, 261–268 CrossRef CAS.
- C. E. La Rotta, E. D'Elia and E. P. S. Bon, Electron. J. Biotechnol., 2007, 10, 24–37 Search PubMed.
- C. Kohlmann, L. Greiner, W. Leitner, C. Wandrey and S. Lütz, Chem. – Eur. J., 2009, 15, 11692–11700 CrossRef CAS PubMed.
- C. Kohlmann and S. Lütz, Eng. Life Sci., 2006, 6, 170–174 CrossRef CAS.
- L. Getrey, T. Krieg, F. Hollmann, J. Schrader and D. Holtmann, Green Chem., 2014, 16, 1104–1108 RSC.
- S. K. Karmee, C. Roosen, C. Kohlmann, S. Lütz, L. Greiner and W. Leitner, Green Chem., 2009, 11, 1052–1055 RSC.
- E. Churakova, I. W. C. E. Arends and F. Hollmann, ChemCatChem, 2013, 5, 565–568 CrossRef CAS.
- D. I. Perez, M. Mifsud Grau, I. W. C. E. Arends and F. Hollmann, Chem. Commun., 2009, 6848–6850 RSC.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- R. A. Sheldon, Chem. Commun., 2008, 3352–3365 RSC.
- Y. Ni, D. Holtmann and F. Hollmann, ChemCatChem, 2014, 6, 930–943 CrossRef CAS.
- M. Eissen, Chem. Educ. Res. Pract., 2012, 13, 103–111 RSC.
- E. Heinzle, A. Biwer, M. Eissen and M. A. Kholiq, Chem. Ing. Tech., 2006, 78, 301–305 CrossRef CAS.
- M. Eissen and J. O. Metzger, Chem. – Eur. J., 2002, 8, 3580–3585 CrossRef CAS.
- F. G. Calvo-Flores, ChemSusChem, 2009, 2, 905–919 CrossRef CAS PubMed.
- M. Ribeiro and A. Machado, Green Chem. Lett. Rev., 2013, 6, 1–18 CrossRef CAS.
- D. Ravelli, S. Protti, P. Neri, M. Fagnoni and A. Albini, Green Chem., 2011, 13, 1876–1884 RSC.
- B. Buhler and A. Schmid, J. Biotechnol., 2004, 113, 183–210 CrossRef PubMed.
- B. Buhler, I. Bollhalder, B. Hauer, B. Witholt and A. Schmid, Biotechnol. Bioeng., 2003, 82, 833–842 CrossRef CAS PubMed.
- B. Buhler, I. Bollhalder, B. Hauer, B. Witholt and A. Schmid, Biotechnol. Bioeng., 2003, 81, 683–694 CrossRef CAS PubMed.
- B. Buhler, B. Witholt, B. Hauer and A. Schmid, Appl. Environ. Microbiol., 2002, 68, 560–568 CrossRef CAS.
- R. Gandolfi, K. Cavenago, R. Gualandris, J. V. Sinisterra Gago and F. Molinari, Process Biochem., 2004, 39, 749–753 CrossRef.
- R. Villa, A. Romano, R. Gandolfi, J. V. Sinisterra Gago and F. Molinari, Tetrahedron Lett., 2002, 43, 6059–6061 CrossRef CAS.
- Y. Ni, P.-L. Hagedoorn, J.-H. Xu, I. W. C. E. Arends and F. Hollmann, Chem. Commun., 2012, 48, 12056–12058 RSC.
- P. Könst, S. Kara, S. Kochius, D. Holtmann, I. W. C. E. Arends, R. Ludwig and F. Hollmann, ChemCatChem, 2013, 5, 3027–3032 CrossRef.
- S. Kara, D. Spickermann, J. H. Schrittwieser, A. Weckbecker, C. Leggewie, I. W. C. E. Arends and F. Hollmann, ACS Catal., 2013, 3, 2436–2439 CrossRef CAS.
- A. Díaz-Rodríguez, J. Iglesias-Fernández, C. Rovira and V. Gotor-Fernández, ChemCatChem, 2013, 6, 977–980 CrossRef.
- P. G. Jessop, Green Chem., 2011, 13, 1391–1398 RSC.
- C. Capello, U. Fischer and K. Hungerbuhler, Green Chem., 2007, 9, 927–934 RSC.
- R. A. Sheldon, Green Chem., 2005, 7, 267–278 RSC.
- J. Schrittwieser, F. Coccia, S. Kara, B. Grischek, W. Kroutil, N. d'Alessandro and F. Hollmann, Green Chem., 2013, 15, 3318–3331 RSC.
- J. H. Schrittwieser, J. Sattler, V. Resch, F. G. Mutti and W. Kroutil, Curr. Opin. Chem. Biol., 2011, 15, 249–256 CrossRef CAS PubMed.
- E. Ricca, B. Brucher and J. H. Schrittwieser, Adv. Synth. Catal., 2011, 353, 2239–2262 CrossRef CAS.
- M. Schrewe, N. Ladkau, B. Bühler and A. Schmid, Adv. Synth. Catal., 2013, 355, 1693–1697 CrossRef CAS.
- M. Schrewe, A. O. Magnusson, C. Willrodt, B. Bühler and A. Schmid, Adv. Synth. Catal., 2011, 353, 3485–3495 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |