A prototype point-of-use assay for measuring heavy metal contamination in water using time as a quantitative readout†
Gregory G.
Lewis
,
Jessica S.
Robbins
and
Scott T.
Phillips
*
The Pennsylvania State University, 104 Chemistry Bldg., University Park, PA 16802, USA. E-mail: sphillips@psu.edu; Fax: +1-814-865-5235; Tel: +1-814-867-2502
First published on 20th November 2013
Abstract
This Communication describes a prototype quantitative paper-based assay that simultaneously measures the levels of Pb2+ and Hg2+ in water. The assay requires only measurements of time to yield a quantitative readout, and the results are independent of sample volume, humidity, and sample viscosity.
A grand challenge in the area of point-of-care and point-of-use diagnostics is the development of inexpensive quantitative assays that can be conducted by anyone, anywhere, requiring essentially no auxiliary components (e.g., pipettes, specialized readers, etc.).1 Obtaining a quantitative readout without using instruments—even battery-operated hand-held readers—is particularly challenging since typical colorimetric, fluorescent, and electrochemical assays require an electronic reader to measure the intensity of the output from the assay. We recently demonstrated a quantitative paper-based assay for active enzymes that requires measurements of time to appearance of color as the readout.2 This time-based approach circumvents the need for an external reader to obtain a quantitative result.3 Herein we substantially expand upon this time-based approach by creating an assay for inorganic ions, specifically Pb2+ and Hg2+ (Fig. 1).
 | ||
| Fig. 1 Photographs of a paper-based assay platform for simultaneous measurements of Hg2+ and Pb2+ in drinking water. The white dotted lines denote the edges of laminate that hold multiple layers of stacked, wax-patterned paper in contact with one another. The dimensions of the stacked paper are 20 mm × 30 mm × 1.8 mm (Fig. S5, ESI†). | ||
This work is important for three reasons. First, the new configuration uses aptamers and thus opens the assay strategy to new classes of analytes. The time-based approach now can be used for quantitative analysis of enzymes, inorganic ions, and small molecules (e.g., adenosine; see the relevant details in the ESI†). The assays are easily reconfigured by exchanging reagents in the device both to alter the specificity for a target analyte, and to adjust the sensitivity and dynamic range of the assay(s). Second, the strategy is expanded to multiplexed assays, which is a capability that will enable complete analysis of samples using a single aliquot of the sample and a single step by the user. Finally, the assay is simple, requiring that the user only add a sample onto the device. Pre-processing occurs within the device, and the device meters the volume of the sample to enable a quantitative readout.8 The assay platform includes reagents both for selective detection as well as signal amplification, yet no washing steps or other manipulations are required.
The assay uses a paper-based microfluidic device as a means for distributing a sample to assay regions (Fig. 1).2 The device has hydrophobic regions that define where the sample travels by capillary action through the hydrophilic regions in the paper. A few minutes after adding the sample to the center region, the “start” hydrophilic region turns green, at which point the user begins to measure the time required for the “stop” region to turn red (Fig. 1). This measurement of time—which can be accomplished in a number of ways without using an electronic timer—reflects the concentration of the analyte in the sample. The device in Fig. 1 is configured to quantify Hg2+ and Pb2+ in water simultaneously. Therefore, once the sample of water is added to the entry point, it distributes into four separate portions of the device, two of which lead to “start” regions, while the other two lead to complementary “stop” regions.
The sample distributes to the start and stop regions through five layers of wax-patterned paper that are held together via laminate (Fig. 2).2 Each layer and hydrophilic region in the device serves a unique function to facilitate the assay, with the central hydrophilic region in layer 1 operating as the collection zone where the sample is added to the device. Bead-bound glucose oxidase (GOX) and catalase in layer 2 pre-process the sample to remove up to 50 mM glucose and hydrogen peroxide that would otherwise interfere with the assay (Fig. S1–S3, ESI†).2 The beads are embedded in the fibers of the paper, therefore these enzymes remain fixed in layer 2,9 whereas the sample distributes to all hydrophilic regions of the device.
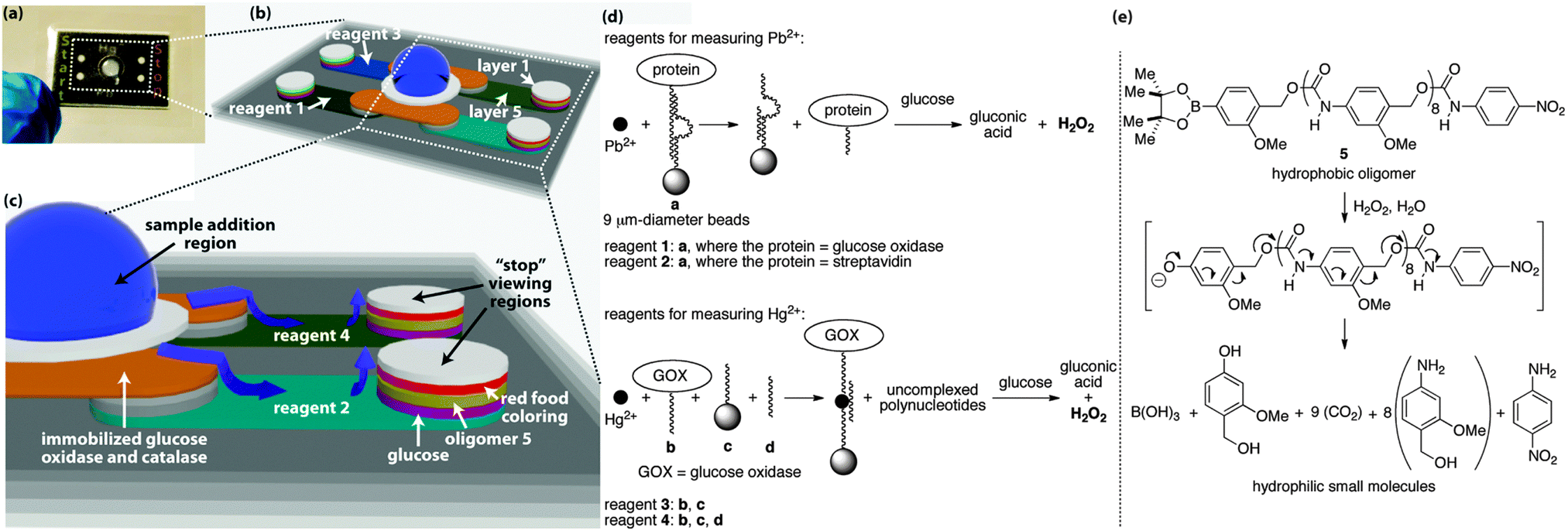 | ||
| Fig. 2 Detailed view of the assay platform (a), including the hydrophilic channels in which the sample distributes by capillary action ((b) and (c)). The grey regions in (b) and (c) correspond to paper that contains hydrophobic wax. The white and colored regions are hydrophilic paper in which the sample travels, and the blue arrows in (c) denote the direction of travel. The aptamers used in the assays are shown in (d), as are the reactions that generate hydrogen peroxide during the course of the assay. A signal amplification reaction occurs when the oligomer in (e) depolymerizes in response to the hydrogen peroxide that is generated during the detection event. The sequences of DNA in a, b, c, and d are provided in the ESI.† | ||
The 5th layer is a lateral-flow region where the sample moves away from the central sample addition region to the outer columns of hydrophilic paper. In the four lateral-flow channels, the sample re-dissolves reagents for conducting the assays, where each of the four channels contains a different set of reagents (Fig. 2d). Reagent 1 is used in the assay for Pb2+ and is a bead-bound aptamer complex consisting of two polynucleotides, one of which is a DNAzyme10 bound to a 9 μm-diameter bead that causes the complex to remain embedded in the paper.9 The second polynucleotide is covalently bound to GOX and non-covalently associated with the DNAzyme. When Pb2+ is present in the sample, it activates the DNAzyme, which cleaves the polynucleotide that is bound to GOX, thus releasing GOX. The released GOX is free to travel with the sample to the outer hydrophilic regions, while the remainder of reagent 1 stays in layer 5.
At the outer columns, the sample travels vertically towards the top of the paper-based device. As it passes through layers of paper, the sample encounters additional reagents that were pre-deposited and dried into the layers before the device was assembled. In layer 4, the sample re-dissolves glucose, which is processed by the liberated GOX to generate hydrogen peroxide. Layer 3 contains a hydrophobic poly(benzyl carbamate) oligomer (5) that reacts with hydrogen peroxide via the aryl boronate on one end of the oligomer to initiate a continuous head-to-tail depolymerization reaction (Fig. 2e).7 This depolymerization process changes the wetting properties of layer 3 from hydrophobic back to hydrophilic and allows the sample to travel through this layer with a rate that depends on the concentration of hydrogen peroxide that is generated from the aptamer detection event. The 2nd layer contains dried food coloring, which, once re-dissolved, is carried to the top layer where it becomes visible in the appropriate “start” or “stop” region.
The Pb2+ assay also contains reagent 2 in the lateral-flow channel opposite to reagent 1. Reagent 2 is exactly the same as reagent 1, with the exception that GOX is replaced with streptavidin. Streptavidin will not generate hydrogen peroxide when it encounters glucose, therefore a sample containing Pb2+ will take longer to travel to the viewing region when it encounters reagent 2 than when it encounters reagent 1. This difference in sample transport time is dependent on the concentration of Pb2+ in the sample, thus providing the basis for the time-based measurement and quantitative assay. Moreover, this type of measurement, based on relative sample transport time, normalizes the assays for effects of humidity and sample viscosity that would normally complicate a quantitative measurement.2
The Hg2+ assay occurs on the same device simultaneously with the Pb2+ assay, but requires a different set of reagents in layer 5 than the Pb2+ assay (i.e., reagents 3 and 4, Fig. 2d). Reagent 4 consists of three components: polynucleotide b bound to GOX, polynucleotide c bound to a 9 μm-diameter bead to affix c to the paper, and polynucleotide d, which brings b, c, d, and Hg2+ together into a four-component non-covalent complex when Hg2+ is present in the sample.11 This complex prevents the GOX from traveling with the sample through the remainder of the device, and thus minimizes the quantity of hydrogen peroxide that is generated in layer 4 to cause depolymerization in layer 3.
Reagent 3 in the opposite lateral-flow region to reagent 4 contains only polynucleotides b and c, and thus is incapable of forming the four-component complex in the presence of Hg2+.11 In this channel, all of the GOX is capable of generating hydrogen peroxide in layer 4. Hence, the sample travels faster through the region containing reagent 3 than reagent 4, thus reagent 3 leads to the “start” region and reagent 4 to the “stop” region.
Using a device that contains only two channels rather than four (i.e., one channel leading to a “start” region and the other to a “stop” region; Fig. S1, ESI†), we established dose-dependent responses for Pb2+ and Hg2+ (Fig. S4, ESI†).12 The limit-of-detection13 for the Hg2+ assay is 4 nM (1 ppb) and the dynamic range extends up to 25 nM (7 ppb), while the complimentary numbers for the Pb2+ assay are 4 nM (1 ppb) up to 50 nM (17 ppb). Both of these assays are sufficiently sensitive to measure the World Health Organization's recommended upper limits for these ions in drinking water (i.e., 22 nM (6 ppb) for Hg2+ and 30 nM (10 ppb) for Pb2+).14
Based on these results in two-channel devices, we then implemented the four-channel design depicted in Fig. 1 with the goal of simultaneously quantifying Pb2+ and Hg2+ using one sample of drinking water. The assay times obtained in the four-channel devices are identical to those obtained in the two-channel versions, indicating that the calibration curves in Fig. S4 (ESI†) can be applied to the four-channel device in Fig. 1. Moreover, Fig. S6 (ESI†) reveals that the multiplexed assays in the four-channel devices are selective for the desired analytes. For example, assay times for samples containing either 100 nM Pb2+; 100 nM of Pb2+ and Hg2+; or 100 nM of Pb2+, Hg2+, Cd2+, and Zn2+, all gave identical measurement times for Pb2+, as expected based on the known selectivity of the aptamer for Pb2+. Likewise, the measurement time for a sample containing 100 nM Hg2+ was identical to times obtained using samples containing Pb2+ or a combination of Pb2+, Hg2+, Cd2+, and Zn2+ (Fig. S6, ESI†).
Finally, the assay platform provides consistent quantitative measurements even when lake water is used rather than de-ionized (DI) water. A case in point is shown in Fig. S6 (ESI†) for lake water that was spiked with three different concentrations of Pb2+. Comparison of measurement times between DI water and lake water for 100 nM Pb2+, in particular, reveals that the assay is unaffected by contaminants in lake water.
The assay strategy described in this Communication provides the basis for quantitative time-based assays using aptamers as recognition moieties. This strategy is effective under laboratory conditions when using a watch as a timing mechanism. Under these conditions, the assays require measurement times that range from seconds to ∼10 min (depending on the concentration of the analyte), with overall assay times (from addition of sample to the paper device to when the last “stop” region turns color) requiring only ∼30 min. Further tests are needed, however, to determine whether this early stage strategy provides quantitative or semi-quantitative results for a range of users, under a variety of operating conditions, and when using samples of varying complexity. Future studies extending beyond the current prototype also will (i) explore new configurations of the assay platform to simplify its assembly, (ii) determine which additives best stabilize DNAzymes and other oligonucleotides on paper,15 and (iii) optimize the quantity of oligonucleotides needed to balance cost16 with the desired sensitivity and dynamic range for an assay.
This work was supported by NSF (CHE-1150969) and Louis Martarano. S.T.P acknowledges support from the Alfred P. Sloan Research Fellows program.
Notes and references
- M. Urdea, L. A. Penny, S. S. Olmsted, M. Y. Giovanni, P. Kaspar, A. Sheperd, P. Wilson, C. A. Dahl, S. Buchsbaum, G. Moeller and D. C. Hay Burgess, Nature, 2006, 444, 73 Search PubMed.
- G. G. Lewis, J. S. Robbins and S. T. Phillips, Anal. Chem., 2013, 85, 10432 CAS.
- A handful of unconventional outputs have been suggested recently including measurements based on (i) the distance that a fluid, color, or object travels during an assay;4 (ii) the number of regions that turn color on a device;5 or (iii) the time required for a signal to appear during an assay6,7.
- M. Zhong, C. Y. Lee, C. A. Croushore and J. V. Sweedler, Lab Chip, 2012, 12, 2037 CAS; J. Wu, S. Balasubramanian, D. Kagan, K. M. Manesh, S. Campuzano and J. Wang, Nat. Commun., 2010, 1, 36 Search PubMed; D. Chatterjee, D. S. Mansfield, N. G. Anderson, S. Subedi and A. T. Woolley, Anal. Chem., 2012, 84, 7057 Search PubMed; Y. Song, Y. Zhang, P. E. Bernard, J. M. Reuben, N. T. Ueno, R. B. Arlinghaus, Y. Zu and L. Qin, Nat. Commun., 2012, 3, 1283 Search PubMed; X. Yang, J. Kanter, N. Z. Piety, M. S. Benton, S. M. Vignes and S. S. Shevkoplyas, Lab Chip, 2013, 13, 1464 Search PubMed; M. P. Allen, A. DeLizza, U. Ramel, H. Jeong and P. Singh, Clin. Chem., 1990, 36, 1591 Search PubMed; D. M. Cate, W. Dungchai, J. C. Cunningham, J. Volckens and C. S. Henry, Lab Chip, 2013, 13, 2397 Search PubMed; W. Dungchai, Y. Sameenoi, O. Chailapakul, J. Volckens and C. S. Henry, Analyst, 2013, 138, 6766 Search PubMed.
- G. G. Lewis, M. J. DiTucci and S. T. Phillips, Angew. Chem., Int. Ed., 2012, 51, 12707 CAS; W. Leung, C. P. Chan, T. H. Rainer, M. Ip, G. W. H. Cautherley and R. Renneberg, J. Immunol. Methods, 2008, 336, 30 Search PubMed; S. C. Lou, C. Patel, S. Ching and J. Gordon, Clin. Chem., 1993, 39, 619 Search PubMed; J.-H. Cho and S.-H. Paek, Biotechnol. Bioeng., 2001, 75, 725 Search PubMed; T. Huynh, B. Sun, L. Li, K. P. Nichols, J. L. Koyner and R. F. Ismagilov, J. Am. Chem. Soc., 2013, 135, 14775 Search PubMed.
- M. S. Baker and S. T. Phillips, J. Am. Chem. Soc., 2011, 133, 5170 CAS; D. Aili, M. Mager, D. Roche and M. M. Stevens, Nano Lett., 2011, 11, 1401 Search PubMed.
- G. G. Lewis, J. S. Robbins and S. T. Phillips, Macromolecules, 2013, 46, 5177 CAS.
- So long as the minimum volume of sample is added to wet the device completely.2 This volume is ∼40 μL for the device in Fig. 1.
- H. Liu, Y. Xiang, Y. Lu and R. M. Crooks, Angew. Chem., Int. Ed., 2012, 51, 6925 CAS.
- Z. Wang, J. H. Lee and Y. Lu, Adv. Mater., 2008, 20, 3263 CAS; Y. Xiang and Y. Lu, Chem. Commun., 2013, 49, 585 Search PubMed.
- X. Xue, F. Wang and X. Liu, J. Am. Chem. Soc., 2008, 130, 3244 CAS.
- Examples of other paper-based assays for detecting heavy metals include: M. M. Mentele, J. Cunnignham, K. Koehler, J. Volckens and C. S. Henry, Anal. Chem., 2012, 84, 4474 CAS; M. Zhang, L. Ge, S. Ge, M. Yan, J. Yu, J. Huang and S. Liu, Biosens. Bioelectron., 2013, 41, 544 Search PubMed; C. Yuan, K. Zhang, Z. Zhang and S. Wang, Anal. Chem., 2012, 84, 9792 Search PubMed; Z. Nie, C. A. Nijhuis, J. Gong, X. Chen, A. Kumachev, A. W. Martinez, M. Narovlyansky and G. M. Whitesides, Lab Chip, 2010, 10, 477 Search PubMed.
- The limit of detection was defined as 3× the standard deviation of the blank divided by the slope of the linear dynamic region.
- Guidelines for Drinking-water Quality, World Health Organization, Geneva, Switzerland, 4th edn, 2011 Search PubMed.
- A number of strategies are available for stabilizing oligonucleotides, including (i) immobilizing the oligonucleotide onto a solid support ( D. P. Wernette, C. B. Swearingen, D. M. Cropek, Y. Lu, J. V. Sweedler and P. W. Bohn, Analyst, 2006, 131, 41 CAS ), (ii) drying the oligonucleotide in the presence of trehalose ( B. Shirkey, N. J. McMaster, S. C. Smith, D. J. Wright, H. Rodriguez, P. Jaruga, M. Birincioglu, R. F. Helm and M. Potts, Nucleic Acids Res., 2003, 31, 2995 Search PubMed ), (iii) storing the oligonucleotides as dry reagents ( J. Bonnet, M. Colotte, D. Coudy, V. Couallier, J. Portier, B. Morin and S. Tuffet, Nucleic Acids Res., 2010, 38, 1531 Search PubMed ), and (iv) freeze drying the oligonucleotide ( T. K. Armstrong and T. J. Anchordoquy, J. Pharm. Sci., 2004, 93, 2698 Search PubMed ).
- A breakdown of materials costs for the current prototype assays are provided in Table S16, ESI†.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures, experimental procedures, and tables of data. See DOI: 10.1039/c3cc47698g |
| This journal is © The Royal Society of Chemistry 2014 |