 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Promoting charge-separation in p-type dye-sensitized solar cells using bodipy†
Jean-François
Lefebvre
,
Xue-Zhong
Sun
,
James A.
Calladine
,
Michael W.
George
and
Elizabeth A.
Gibson
*
School of Chemistry, The University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: Elizabeth.Gibson@nottingham.ac.uk; Fax: +44 115 951 3563; Tel: +44 115 951 3523
First published on 27th September 2013
Abstract
The viability of applying bodipy sensitisers to NiO-based p-type dye-sensitised solar cells (p-DSCs) has been successfully demonstrated. The triphenylamine donor–bodipy acceptor design promotes a long-lived charge-separated state which is difficult to achieve with NiO-based devices. The current was above 3 mA cm−2 and the IPCE was 28%.
Tandem dye-sensitised solar cells could provide a route to high efficiency solar energy conversion at low cost.1 Lindquist et al. proposed in 1999 that replacing the platinised counter electrode in Grätzel cells with a dye-sensitized photocathode (in series) should greatly improve the device efficiency by collecting high energy photons at the top electrode and lower energy photons at the bottom electrode.2 This method increases the light collection and reduces thermal energy losses; the theoretical efficiency may be as high as 43%. To date, however, there has not been a tandem cell reported with power conversion efficiency higher than that of a state-of-the-art TiO2-based DSC.
Since the rapid charge recombination reactions at the photocathode limit the photoelectric conversion efficiency of the p-DSCs, the main difficulty of enhancing the performance of tandem DSCs exists in raising the current generated at the photocathode to match that generated at the TiO2 anode.3 The precise reason for fast recombination is still unclear but it has been demonstrated that the dye can be engineered to promote charge-separation, thereby improving the photocurrent.1 A myriad of different dyes have been reported with TiO2, but very few have been designed for p-type materials such as NiO.4 The most efficient sensitisers for NiO have both high extinction coefficients for maximum light harvesting on thin electrodes (e.g. dyes with favourable π–π* electronic transitions5–7) and charge-transfer character to promote and maintain charge-separation (e.g. π-donor–acceptor (“push–pull”) functionality,5 or with pendant NDI or C60 electron acceptors8,9).
Boradiazaindacene (bodipy) dyes are popular in a range of applications but have been used infrequently and with moderate success in n-DSCs.10–13 However, their large extinction coefficients, electrochemical stability and tuneable absorption properties (i.e. to optically match photoanodes in tandem cells) prompted us to test them in p-DSCs. Initially, we have chosen to use a triphenylamine–thiophene motif used in one of the most successful NiO photosensitisers (“P1”) coupled to a highly substituted bodipy (Fig. 1).6 The dye was prepared by reacting 4-carboxy-4′,4′′-di(5-formyl-2-thienyl)triphenylamine with 2,4-dimethyl-3-ethylpyrrole in the presence of trifluoroacetic acid (see ESI† for details). The resulting dipyrrane was oxidised with p-chloranil before reacting with boron trifluoride etherate to give 1.
 | ||
| Fig. 1 Structure of bodipy dye 1. | ||
The optical and electrochemical properties are summarised in Table S1 (ESI†). The electronic spectrum of 1 contains absorption maxima at 365 nm (ε = 56![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 L mol−1 cm−1) and 540 nm (ε = 112000 L mol−1 cm−1, almost double that of the push–pull sensitiser P1 at λmax = 481 nm, Fig. S3 in the ESI†).14 The absorption and emission spectra are characteristic for a bodipy derivative, with narrow absorption and emission bands and a small Stokes shift when excited at 488 nm. λmax was the same in CH2Cl2 or CH3CN solution and when 1 was adsorbed onto NiO (Fig. S4 and S5, ESI†), suggesting that the electronic properties of the chromophore were unchanged on binding. However, when 1 was excited in the higher energy band (406 nm) the emission spectrum was solvent dependent: emitting from the higher energy triphenylamine (475 nm) when in CH3CN; emitting from the lower energy bodipy (560 nm) when in CH2Cl2.
000 L mol−1 cm−1) and 540 nm (ε = 112000 L mol−1 cm−1, almost double that of the push–pull sensitiser P1 at λmax = 481 nm, Fig. S3 in the ESI†).14 The absorption and emission spectra are characteristic for a bodipy derivative, with narrow absorption and emission bands and a small Stokes shift when excited at 488 nm. λmax was the same in CH2Cl2 or CH3CN solution and when 1 was adsorbed onto NiO (Fig. S4 and S5, ESI†), suggesting that the electronic properties of the chromophore were unchanged on binding. However, when 1 was excited in the higher energy band (406 nm) the emission spectrum was solvent dependent: emitting from the higher energy triphenylamine (475 nm) when in CH3CN; emitting from the lower energy bodipy (560 nm) when in CH2Cl2.
The electrochemical properties of 1 were determined by cyclic and square wave voltammetry and calibrated vs. FeCp2+/FeCp2 (Fig. S13 and S14, ESI†). A reversible reduction process at −1.46 V and two closely spaced irreversible oxidation processes at 0.65 and 0.75 V were observed. The estimated driving forces‡ for both injection (ΔGinj) and regeneration (ΔGreg) are both negative, indicating that 1 is a suitable NiO sensitiser.15,16 DFT calculations were also performed on 1 to further explore the geometry and electronic properties. E(D*/D)− = 0.81 V, is suitably positive to allow for efficient charge-separation between geometry and electronic properties. The results supported experimental evidence that the bodipy was decoupled from the triphenylamine–thiophene donor and the calculated optimised geometry placed the two at an angle of 84 ± 0.4°. Fig. S16 (ESI†) shows the electronic distribution in the frontier orbitals: the HOMO level of 1 is located on the triphenylamine; two degenerate HOMO − 1(−2) orbitals and two degenerate LUMO(+1) levels are located on the bodipy. TD-DFT suggested that the lowest energy electronic transitions were bodipy-localised π–π* in nature, with some contribution from a charge shift from the HOMO to the LUMO.
NiO p-DSCs were assembled with 1 as the sensitiser and a I3−/I− redox shuttle (see ESI† for details). The photocurrent density–photovoltage curve and spectral response of a typical cell are given in Fig. 2. The cell parameters were JSC = 3.15 mA cm−2, VOC = 79 mV, FF = 0.31, η = 0.08%. The maximum incident photon-to-current conversion efficiency (IPCE) was 28%. These values are superior to related triphenylamine-based dyes reported previously,16,17 but fall short of those achieved with the most successful dye, P1, (JSC = 5.48 mA cm−2, IPCE = 64%).6 The photocurrents obtained with our bodipy-based dyes are extremely encouraging compared to bodipy-sensitised TiO2 devices.10,18,19 TiO2-DSCs are usually more efficient than p-DSCs, but to our knowledge the highest IPCE for a bodipy-sensitised n-DSC is less than 30%.11 This suggests that the bodipy chromophore may be better suited to the NiO system than conventional TiO2-DSCs. Further improvements in efficiency are likely to come from broadening the absorption band by increasing the electronic coupling between the π-linker and the bodipy and extending the conjugation by functionalising the dye periphery.
 | ||
| Fig. 2 J–V curve of 1-sensitised NiO solar cell. Inset: comparison of IPCE (line + symbol) and absorption spectrum (line) of 1-sensitised NiO solar cell to confirm the dye is contributing to the photocurrent. An absorbance of 2 means the light harvesting efficiency is ca. 99%. | ||
To probe the charge-transfer dynamics of this new dye–NiO system, we performed ps- and ns-transient absorption spectroscopy (TA) both with 1 in CH2Cl2 and adsorbed on NiO. Excitation of 1 in solution (Fig. 3 black line) at 532 nm generated an excited state which absorbed between 400–500 nm and decayed over ca. 400 ps to form a new intermediate with narrower absorption bands at 425 and 650 nm which decayed much more slowly (τ425 ≈ 300 (±10) ns in air, τ425 ≈ 860 ns, τ650 ≈ 730 ns in Ar) to the ground state. We have assigned this long-lived transient to a triplet bodipy (31*) excited state (see ESI† for further details).20,21
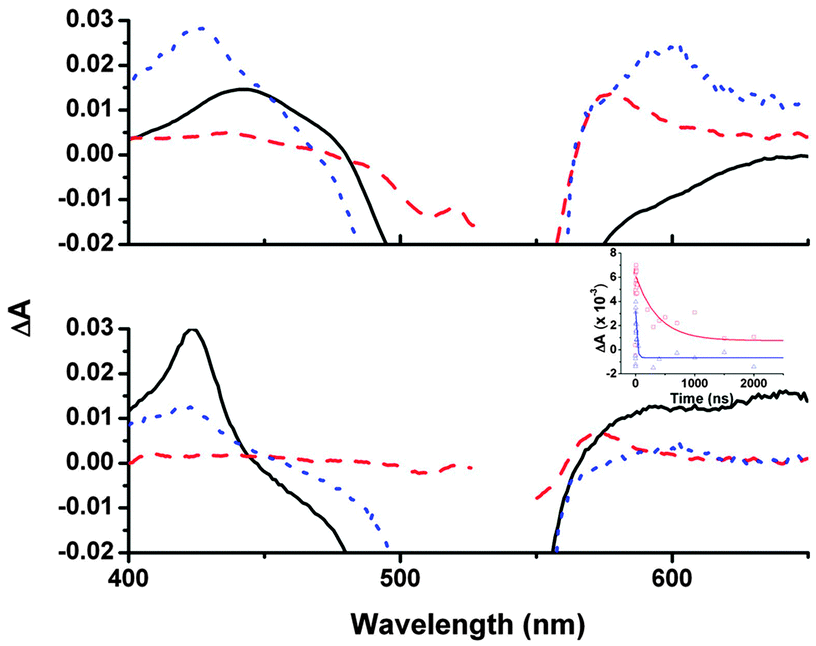 | ||
| Fig. 3 Transient absorption spectra of 1 top: 10 ps after excitation (1.2 μJ), bottom: 2 ns after excitation (1.6 μJ); black solid line = 1 in CH2Cl2, red dashed line = 1/NiO, blue dotted line = 1/NiO and I3−/I−. Inset: kinetic traces of 1/NiO (red) at 575 nm and 1/NiO and I3−/I− at 600 nm (blue). Symbols represent data, lines represent fit of single exponential decay. | ||
The transient absorption results were different for 1 adsorbed on NiO (Fig. 3 red dashed line). On the ps timescale, the broad features between 400–470 nm corresponding to 11* that were observed in CH2Cl2 were less intense. Instead a peak at 575 nm formed rapidly (τrise ≈ 1 ps) which was characteristic of 1− by comparison with our spectroelectro-chemistry experiments (Fig. S26, ESI†). A broad featureless absorption persisted throughout the ps- and ns-transient spectra. This is attributed to oxidised NiO and indicates that the charge-separated state (NiO+/1−) had formed. The dynamics for each band were characteristically heterogeneous and we have fitted the rise and decay in each experiment to first order kinetics giving a short (τ1 ps) and long (τ2 ns) time constant (Table S4, ESI†) to enable us to compare the general behaviour of the intermediates. The rapid time constant for the formation of NiO+/1− can be compared to the lifetime of 11* recorded in solution to give an estimate of the injection efficiency.‡ Using this approach gives a very high efficiency of 99% but it should be noted that any slower injection processes were masked by the subsequent decay of this peak. To try and provide a more realistic value for this process we have compared the relative amplitudes after excitation of the signals at 425 nm (which contains contributions from both 11* and 1−) and 575 nm (NiO+/1− only) at early (2 ps) and late (2 ns) times. This analysis suggests that the charge-separation efficiency is closer to 50% and this result is more in agreement with the maximum IPCE for the solar cells. The peak attributed to NiO+/1− at 575 nm (τ2 ≈ 180 ns) lived three orders of magnitude longer than the charge-separated state of the widely studied P1 dye.16 This substantial increase in the charge-separated state lifetime prompted us to investigate the effect of substituting the I3−/I− for the tris(4,4′-di-tert-butyl-2,2′-dipyridyl)cobalt(III/II) redox shuttle in our DSCs in an attempt to increase the photovoltage.7 Unfortunately the photocurrent was negligible in this case, suggesting that the lifetime was still not sufficient for re-oxidation of 1− by the cobalt electrolyte.§
τ 1 for the NiO+/1− signal differed little in the presence of I3−/I− (Fig. 3 blue dotted line) and absence of I3−/I− (Fig. 3 red dashed line, shifted to λmax = 600 nm), hence there was no measurable effect of the electrolyte on a short timescale. However, on a ns timescale the signal for NiO+/1− decayed more rapidly (τ2 ≈ 23 ns) in the presence of the electrolyte, indicating that interception of the electron by the redox shuttle efficiently competes with recombination. This re-oxidation is very fast compared to the μs timescale reported for re-reduction of dyes on TiO2.22 The mechanism for the dye regeneration pathway in n-DSCs is still under debate and it is currently believed that two equivalents of I− per dye molecule are required.22–24 For p-DSCs, re-oxidation of the dye requires only one molecule of I3− per dye and this may be why we observe this process proceeding more quickly. Reported timescales for the regeneration reaction range from ps to μs.9,25 Reasons for the differing kinetics could be the different driving force for the reaction (reported ΔGreg < 200 meV to 1 eV; ΔGreg = 640 meV for 1) and the overlapping signals for reduced dye and I2− produced from the reduction of I3− (λ = 420–475 nm).
In our TA experiments on 1/NiO in the presence of electrolyte we also observed an additional peak centred at 425 nm which decayed with τ = 500 ns. At first we were concerned that the presence of the heavy atoms in the electrolyte had driven the S1 → T1 conversion as observed by Morandeira et al. for Coumarin 343/NiO25 but the band at 650 nm that accompanied the higher energy peak for 31* in CH2Cl2 was absent in this sample. Instead, we have assigned this transient as I2−. The extinction coefficient of I2− should be three times greater than that of the reduced bodipy. The different amplitudes at 420 nm vs. 600 nm in the TA spectrum 2 ns after excitation indicates that both reduced dye and reduced bodipy are present in similar quantities. Therefore we have compelling evidence that electron transfer from the photoreduced dye to I3− generating I2− contributes to the photocurrent in p-DSCs.
In conclusion, the three orders of magnitude increase in charge-separated state lifetime is a significant breakthrough in our efforts to improve the efficiency of dye-sensitized photocathodes. We anticipate that tuning the electronic coupling by modifying the substituents on the bodipy will increase the charge-separated state yield and lifetime further. This will enable higher photocurrents to be obtained and alternative electrolytes to be used which increase the photovoltage. These results have wider implications to the field of “solar fuels” since photocatalysis requires long-lived charge-separated intermediates for the desired chemical reactions to take place.
We thank the University of Nottingham and The Royal Society for funding and MWG gratefully acknowledges receipt of a Wolfson Merit Award.
Notes and references
- F. Odobel, Y. Pellegrin, E. A. Gibson, A. Hagfeldt, A. L. Smeigh and L. Hammarström, Coord. Chem. Rev., 2012, 256, 2414 CrossRef CAS PubMed.
- J. He, H. Lindström, A. Hagfeldt and S.-E. Lindquist, Sol. Energy Mater. Sol. Cells, 2000, 62, 265 CrossRef CAS.
- A. L. Smeigh, L. Le Pleux, J. Fortage, Y. Pellegrin, E. Blart, F. Odobel and L. Hammarström, Chem. Commun., 2012, 48, 678 RSC.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed.
- A. Nattestad, A. J. Mozer, M. K. R. Fischer, Y.-B. Cheng, A. Mishra, P. Bäuerle and U. Bach, Nat. Mater., 2010, 9, 31 CrossRef CAS PubMed.
- L. Li, E. A. Gibson, P. Qin, G. Boschloo, M. Gorlov, A. Hagfeldt and L. Sun, Adv. Mater., 2010, 22, 1759 CrossRef CAS PubMed.
- E. A. Gibson, A. L. Smeigh, L. Le Pleux, J. Fortage, G. Boschloo, E. Blart, Y. Pellegrin, F. Odobel, A. Hagfeldt and L. Hammarström, Angew. Chem., 2009, 48, 4402 CAS.
- J. C. Freys, J. M. Gardner, L. D'Amario, A. M. Brown and L. Hammarström, Dalton Trans., 2012, 41, 13105 RSC.
- L. Le Pleux, A. L. Smeigh, E. A. Gibson, Y. Pellegrin, E. Blart, G. Boschloo, A. Hagfeldt, L. Hammarström and F. Odobel, Energy Environ. Sci., 2011, 4, 2075 CAS.
- S. Erten-Ela, M. D. Yilmaz, B. Icli, Y. Dede, S. Icli and E. U. Akkaya, Org. Lett., 2008, 10, 3299 CrossRef CAS PubMed.
- S. Kolemen, O. A. Bozdemir, Y. Cakmak, G. Barin, S. Erten-Ela, M. Marszalek, J.-H. Yum, S. M. Zakeeruddin, M. K. Nazeeruddin, M. Grätzel and E. U. Akkaya, Chem. Sci., 2011, 2, 949 RSC.
- Y. Ooyama, Y. Hagiwara, T. Mizumo, Y. Harima and J. Ohshita, New J. Chem., 2013, 37, 2479 RSC.
- S. Hattori, K. Ohkubo, Y. Urano, H. Sunahara, T. Nagano, Y. Wada, N. V. Tkachenko, H. Lemmetyinen and S. Fukuzumi, J. Phys. Chem. B, 2005, 109, 15368 CrossRef CAS PubMed.
- P. Qin, M. Linder, T. Brinck, G. Boschloo, A. Hagfeldt and L. Sun, Adv. Mater., 2009, 21, 2993 CrossRef CAS.
- E. A. Gibson, L. Le Pleux, J. Fortage, Y. Pellegrin, E. Blart, F. Odobel, A. Hagfeldt and G. Boschloo, Langmuir, 2012, 28, 6485 CrossRef CAS PubMed.
- P. Qin, J. Wiberg, E. A. Gibson, M. Linder, L. Li, T. Brinck, A. Hagfeldt, B. Albinsson and L. Sun, J. Phys. Chem. C, 2010, 114, 4738 CAS.
- Y.-S. Yen, W.-T. Chen, C.-Y. Hsu, H.-H. Chou, J. T. Lin and M.-C. P. Yeh, Org. Lett., 2011, 13, 4930 CrossRef CAS PubMed.
- M. Mao, J.-B. Wang, Z.-F. Xiao, S.-Y. Dai and Q.-H. Song, Dyes Pigm., 2012, 94, 224 CrossRef CAS PubMed.
- C. Y. Lee and J. T. Hupp, Langmuir, 2010, 26, 3760 CrossRef CAS PubMed.
- A. Harriman, J. P. Rostron, M. Cesario, G. Ulrich and R. Ziessel, J. Phys. Chem. A, 2006, 110, 7994 CrossRef CAS PubMed.
- M. Galletta, S. Campagna, M. Quesada, G. Ulrich and R. Ziessel, Chem. Commun., 2005, 4222 RSC.
- J. N. Clifford, E. Palomares, M. K. Nazeeruddin, M. Gratzel and J. R. Durrant, J. Phys. Chem. C, 2007, 111, 6561 CAS.
- B. H. Farnum, J. J. Jou and G. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15628 CrossRef CAS PubMed.
- I. Montanari, J. Nelson and J. R. Durrant, J. Phys. Chem. B, 2002, 2203 Search PubMed.
- A. Morandeira, G. Boschloo, A. Hagfeldt and L. Hammarström, J. Phys. Chem. C, 2008, 112, 9530 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, electronic spectra, electrochemical characterisation, DFT calculations, J–V curves for 1vs. P1, transient absorption spectra, spectroelectrochemistry of 1/NiO. See DOI: 10.1039/c3cc46133e |
| ‡ ΔGinj = e[EVB(NiO) − ED*/D−]; ΔGreg = e[E(I3−/I2˙−) – ED/D−]; E(D*/D)− = E(D/D)− + E0-0; EVB(NiO) ≈ −0.12 V vs. Fe(Cp)2+/0;15E0′(I3−/I2˙−) = −0.82 V vs. Fe(Cp)2+/0 in acetonitrile;15ηinj = 1 − (τ1/NiO/τ1*). |
| § This is consistent with the work of Le Pleux et al. who reported that the oxidation of the PMI-NDI sensitiser with Co(III/II) occurred on the μs timescale (τ = 3.5 μs) and was competitive with charge recombination. |
| This journal is © The Royal Society of Chemistry 2014 |