DOI:
10.1039/C4BM00207E
(Paper)
Biomater. Sci., 2014,
2, 1480-1485
DNA strand exchange reaction activated by cationic comb-type copolymers having ureido groups†
Received
9th June 2014
, Accepted 8th July 2014
First published on 22nd July 2014
Abstract
Strand exchange reactions (SERs) of nucleic acids are essential for genetic recombination and often require nucleic acid chaperone activity. A variety of proteins induce strand exchange reactions in vivo. We have reported that cationic comb-type copolymers composed of a polycation backbone and hydrophilic graft chains greatly accelerate the SER in vitro and exhibit nucleic acid chaperone like activity. To elucidate structure/function relationships involved in the SER accelerating activity of the copolymer, we modified the copolymer with ureido groups. The melting temperature of dsDNA in the presence of the copolymers with ureido groups decreased with increasing ureido content, suggesting that the ureido groups have a destabilizing effect on the DNA duplex. Copolymers modified with ureido groups (about 50 mol%) accelerated a SER more strongly than unmodified copolymers. Given our previous observation that modification of the copolymer with guanidino groups resulted in enhanced chaperone-like activity, we propose that incorporation of chaotropic functional groups into the copolymer structure results in an increase in nucleic acid chaperone-like activity.
Introduction
In a strand exchange reaction (SER) a double-strand of DNA or RNA exchanges one of its strands for a homologous strand from a single-stranded (ss) or double-stranded (ds) donor. SER is a key step in genetic homologous recombination. RecA and its homologous protein Rad51 play central roles in SERs in cells.1–3 SER is also a pivotal process in folding of nucleic acids, especially RNA, into a functional conformation. Nucleic acid chaperone proteins catalyze SER from kinetically-trapped structures into the thermodynamically most favored one. HIV-1 nucleocapsid protein 7 (NCp7),4 the major messenger ribonucleoprotein particle protein p50,5 and the nucleoid associated transcriptional regulator of E. coli StpA6 all exhibit nucleic acid chaperone activity.
SER has been utilized in biotechnological applications. There are many examples for DNA nanomachines driven by SER.7–10 Recently, DNA logic gates employing SER have been also built.11,12 Nanomachines or logic gates work properly because of sequence specific SER. However, there is an issue with the response speed of the action when the DNA concentration is lower. Acceleration of SER is required for the rapid response of the nanomachines or the logic gates.
We have previously demonstrated that cationic comb-type copolymers composed of a polycation backbone and abundant hydrophilic graft chains influence kinetics and thermodynamics of nucleic acid hybridization under physiologically relevant conditions. For example, the poly(L-lysine)-graft-dextran (PLL-g-Dex) copolymer significantly accelerates DNA hybridization.13 Moreover, the copolymer markedly accelerates the SER and increases the stability of dsDNA relative to that in buffer alone.14,15 The copolymer was the first example of an artificially synthesized polymer that activated SER. The SER acceleration of the copolymer has been applied for refining the DNA detection method16–18 and DNA nanomachines.19,20 Other artificial molecule activated SERs were reported. Frykholm et al. reported that positively charged liposome enhances SER.21,22 Chitosan, which is a natural polysaccharide with cationic groups, also activates the SER.23 The ionic interactions between the cationic copolymer and anionic DNA appear to be indispensable in the SER activation, but other interactions are also thought to be involved in the activity of the copolymer. We have prepared a variety of the cationic comb-type copolymers to estimate the effect of the chemical structure of the copolymers on DNA hybridization.24–26 We previously showed that a cationic comb-type copolymer with guanidino groups accelerates SER relative to the unmodified copolymer.27 Guanidine modification did not significantly alter cationic properties of the copolymer but did change the hydrogen-bonding capacity. The guanidinium ion is also known to have chaotropic effects that destabilize dsDNA, which should accelerate SER. To further understand the structure/function relationship of the copolymer, we have focused on non-ionic urea groups in this study. Urea, like the guanidinium ion, has a large hydrogen-bonding capacity and is a strong chaotropic reagent. We prepared cationic comb-type copolymers modified with urea (ureido) groups, poly(allylamine-co-allylurea)-graft-dextran (PAU-g-Dex, Fig. 1), and evaluated their effect on SER.
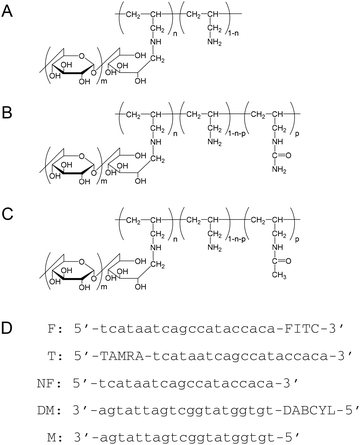 |
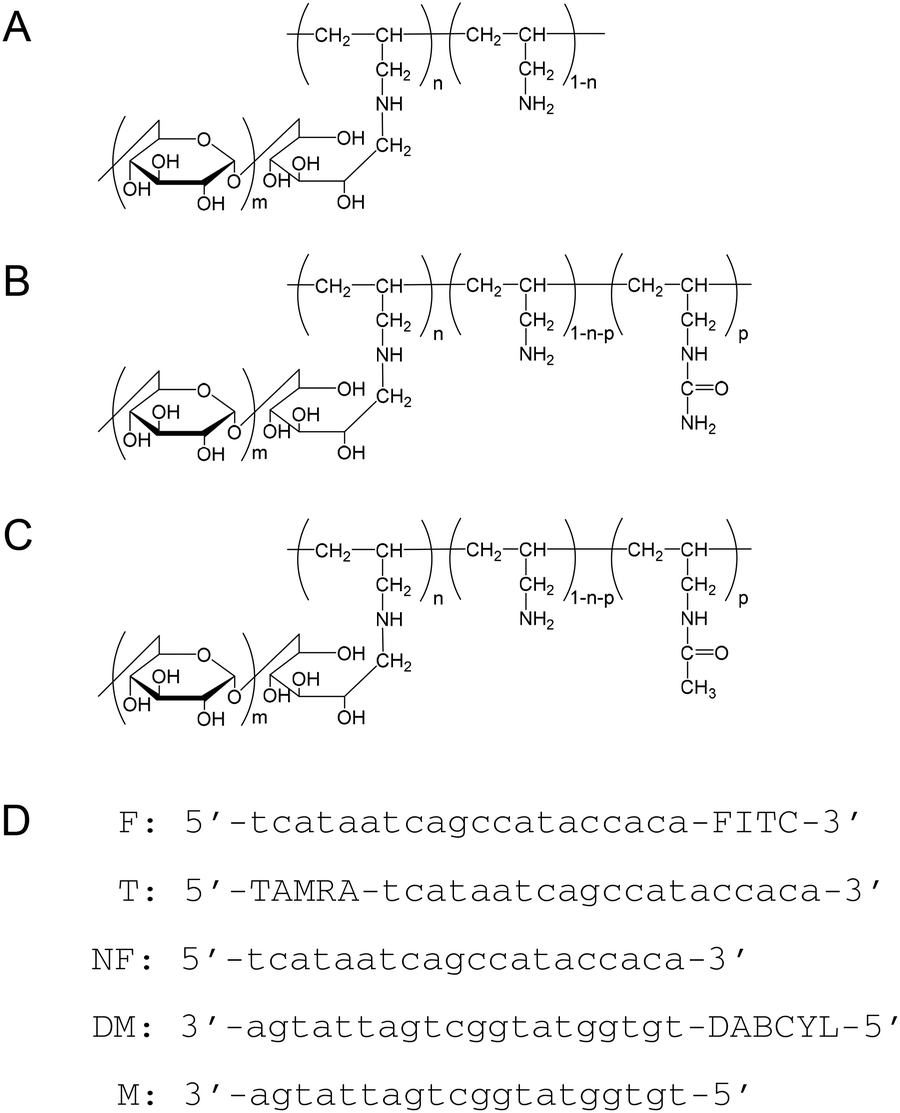
| | Fig. 1 Structural formulas of (A) PA-g-Dex, (B) PAU-g-Dex, (C) PAAc-g-Dex, and (D) DNA oligonucleotide sequences used in this study. | |
Experimental
Materials
Poly(allylamine) hydrochloride (PA·HCl) aqueous solution (Mw: 1.5 × 104), kindly supplied by Nitto Boseki, was purified by precipitation from methanol. Dextran (Mw: 8 × 103) was obtained from Extrasynthese. Sodium cyanoborohydride and poly(vinylsulfonic acid), sodium salt (PVS) were purchased from Sigma-Aldrich. Oligonucleotide DNAs (Fig. 1) were purchased from FASMAC Co. Other reagents were purchased from Wako Pure Chemical Industries.
Preparation of graft copolymers
Preparation of poly(allylamine)-graft-dextran (PA-g-Dex, Fig. 1) was described in a previous report.26 Briefly, the copolymer was prepared by a reductive amination reaction of PA·HCl with dextran using sodium cyanoborohydride as a reductant in sodium borate buffer (pH 8.5).28 The composition of the resulting copolymer was determined by the 1H-NMR spectrum in D2O to be 92 wt% dextran and 8 wt% PA. To prepare PAU-g-Dex copolymers with different ureido contents, various amounts of potassium cyanate were added to PA-g-Dex in water (300 mg per 5 mL), and the mixtures were incubated at 60 °C for 3 days. The polymers were dialyzed against water and then lyophilized. The ureido contents of the copolymers were determined by 1H NMR (Bruker Avance 400) in 0.1% NaOD at 50 °C (Table 1). PAU-g-Dex: δ = 0.6–1.7 (polymer (PA) backbone, –CH2–CH–), 2.5 (–CH2–NH2), 3.0 (–CH2–NH–CONH2), 3.2–4.1 and 4.9 ppm (dextran). Partially acetylated PA-g-Dex (PAAc-g-Dex, Fig. 1) was prepared by acetylation of PA-g-Dex. Acetic anhydride (24 μL) was added to PA-g-Dex in DMSO (300 mg per 5 mL), and the mixture was incubated at 50 °C for 3 days. The degree of acetylation of the polymer dialyzed against water was determined by 1H NMR. PAAc-g-Dex: δ = 1.0–2.0 (polymer (PA) backbone, –CH2–CH–), 1.9 (–CH2–NH–COCH3), 2.5 (–CH2–NH2), 3.0 (–CH2–NH–COCH3), 3.2–4.1 and 4.9 ppm (dextran).
Table 1 Properties of cationic comb-type copolymers used in this study
|
|
Modification degree |
|
| Polymer code |
In feeda |
In polymerb |
Rate constant for SERc (s−1) |
|
Mol ratio of KOCN to amino groups of PA-g-Dex ([KOCN]/[PA-g-Dex]amino groups).
Determined by 1H NMR in D2O containing 0.1% NaOD.
Pseudo-first-order rate constant (k′) in HEPES buffer containing 10 mM NaCl.
|
|
PA-g-Dex
|
|
|
|
| U0 |
— |
0 |
18.9 × 10−5 |
|
PAU-g-Dex
|
|
|
|
| U18 |
0.1 |
0.18 |
43.4 × 10−4 |
| U29 |
0.2 |
0.29 |
53.1 × 10−5 |
| U53 |
0.5 |
0.53 |
15.6 × 10−4 |
| U75 |
0.75 |
0.75 |
27.6 × 10−5 |
| U80 |
0.8 |
0.80 |
12.5 × 10−5 |
| U88 |
0.9 |
0.88 |
9.2 × 10−6 |
|
PAAc-g-Dex
|
|
|
|
| A54 |
0.6 |
0.54 |
33.6 × 10−5 |
UV-melting temperature measurements
dsDNA was obtained by mixing complementary strands, NF and M (Fig. 1), in equimolar amounts and annealing at 95 °C for 5 min in the presence of cationic copolymers if indicated, followed by slow cooling to 25 °C. Melting temperature (Tm) measurements were carried out at a final dsDNA concentration of 1 μM in 10 mM HEPES (pH 7.5) containing 10 mM, 50 mM, or 150 mM NaCl. The cationic copolymers were added to dsDNA solution at an [amino groups]copolymer/[phosphate groups]DNA (N/P) ratio of 5. UV melting curves at 260 nm were recorded with a V-630 spectrophotometer (JASCO) equipped with a temperature controller at a heating rate of 1 °C min−1. The differential absorbance (ΔA = hyperchromicity at 260 nm) was calculated to correct for baseline shift.
PAGE assay to quantify DNA strand exchange reaction
3′-Fluorescein isothiocyanate (FITC)-labeled duplexes (F/M) was used in a gel electrophoresis assay to quantify DNA strand exchange. The duplex was prepared by mixing FITC-labeled ssDNA (F) and complementary ssDNA (M) in equimolar amounts and annealing at 95 °C for 5 min, followed by slow cooling to 25 °C. FITC-labeled dsDNA (F/M, 0.5 μM) was incubated at 30 °C with complementary ssDNA (NF, 15 μM) in 10 mM HEPES (pH 7.5) containing 10, 50, or 150 mM NaCl in the absence or presence of the copolymer (N/P = 5) for various time periods. After incubation, PVS (final 0.3 wt%) was added to the reaction mixture to dissociate the copolymer from DNA before electrophoresis. The mixtures were analyzed on a 13% polyacrylamide gel electrophoresed at 100 V and 3 °C for 2 h. The DNA was visualized with a Pharos FX™ Molecular Imager (BIO-RAD).
Fluorescence resonance energy transfer (FRET) assay for real-time monitoring of DNA strand exchange reaction
FITC/dabcyl-labeled dsDNA (F/DM, 0.06 μM) was incubated at 30 °C with a ssDNA target (NF, 0.6 μM) in 10 mM HEPES (pH 7.5) containing 10, 50, or 150 mM NaCl in the absence or presence of cationic copolymers (N/P = 5). The change in fluorescence intensity of the mixture (total volume = 2 mL) in a plastic cell was recorded on a Jasco FP-6500 spectrofluorometer equipped with a Peltier thermostated cell holder at excitation and emission wavelengths of 490 and 520 nm, respectively, with excitation and emission slits at 5 nm. The value of relative fluorescence intensity was calculated using the following equation:
| Relative fluorescence intensity = ([FI]t − [FI]0)/([FI]∞ − [FI]0) |
where [FI]0 is the initial fluorescence intensity and [FI]t and [FI]∞ are fluorescence intensities at time t and after the reaction reached equilibrium, respectively. The value of [FI]∞ was obtained by measuring fluorescence intensity after re-annealing the DNA mixture (heating at 95 °C for 5 min followed by slow cooling at 30 °C).
DNA-copolymer binding assay by fluorescence polarity measurement
DNA-copolymer binding was evaluated by fluorescence polarity measurements with an Olympus MF20. Various concentrations of the cationic copolymer were added to 5′-tetramethylrhodamine (TAMRA)-labeled ssDNA (T) or TAMRA-labeled dsDNA (T/M) solution (final concentration 5 nM) in 10 mM HEPES (pH 7.5) containing 300 mM NaCl plus 5 μg mL−1 NewPol PE64 surfactant. After the mixtures were incubated for 30 min at room temperature, fluorescence polarities of the mixtures were measured at room temperature.
Results
Effect of PAU-g-Dex on melting temperature of DNA
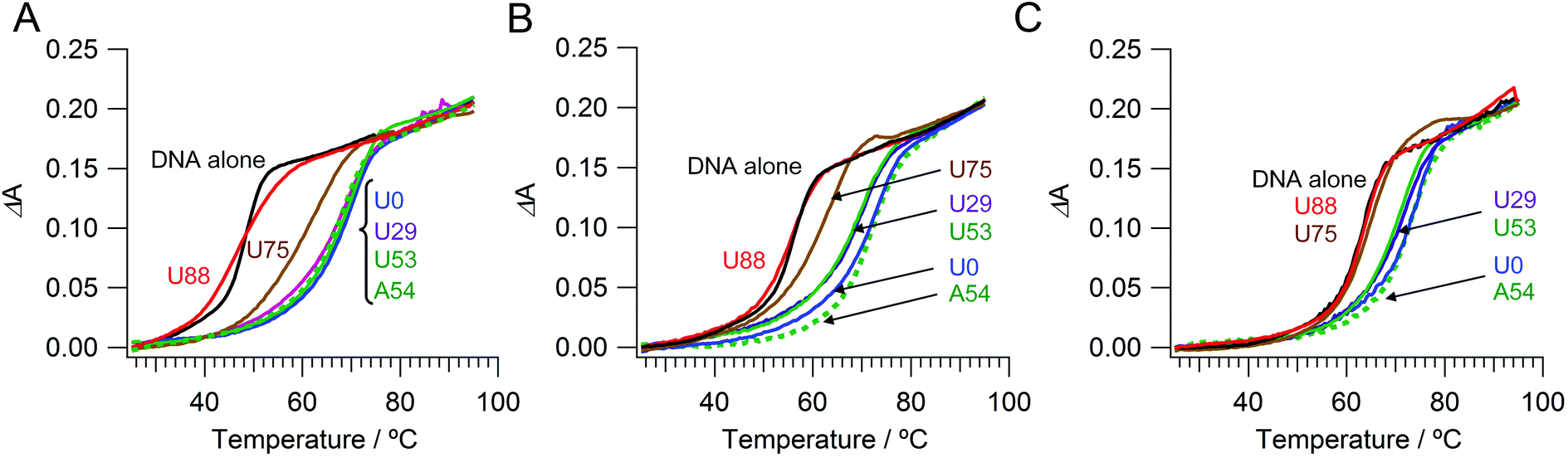
We prepared a series of PAU-g-Dex copolymers with different ureido contents to estimate the effect of ureido groups on the stability of dsDNA. Fig. 2A shows UV-melting curves of 20-base pair dsDNA in the presence of copolymers in 10 mM HEPES buffer (pH 7.5) containing 10 mM NaCl. As previously reported,26,28,29 the Tm value of the dsDNA in the presence of PA-g-Dex (67 °C) was 20 °C higher than that of dsDNA alone (47 °C). PAU-g-Dex copolymers having moderate ureido contents (U29 and U53) stabilized dsDNA to a similar extent to that observed in the presence of PA-g-Dex.
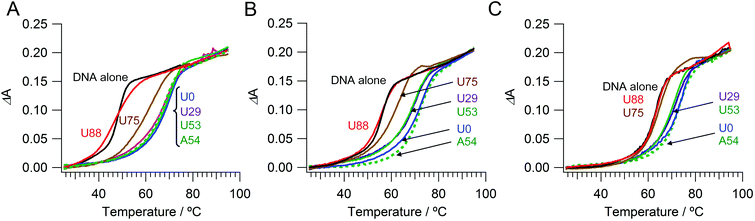 |
| | Fig. 2 UV melting curves of dsDNA with cationic comb-type copolymers in 10 mM HEPES (pH 7.5) containing (A) 10 mM, (B) 50 mM, or (C) 150 mM NaCl. | |
PAU-g-Dex with higher ureido contents (U75 and U88) was less stabilizing. The Tm value in the presence of U88 was 47 °C, the same as the Tm of dsDNA in the absence of the copolymer. Similar ureido content dependences on Tm were observed when the NaCl concentration was increased to 50 mM and to 150 mM (Fig. 2B, C). The loss of the stabilization effect of the copolymers at high ureido content was primarily caused by decrease in positive charge density. However, other contributions from the ureido modification are likely because the Tm curve in the presence of U88 had a much broader transition than that of dsDNA alone. To further assess the effect of ureido modification, we modified PA-g-Dex with acetyl groups. PAU-g-Dex having 53 mol% ureido groups (U53; Fig. 2, solid green lines) was less stabilizing than acetylated PA-g-Dex (PAAc-g-Dex, A54; Fig. 2, dashed green lines) although both copolymers have the same cationic density.
Effect of ureido modification on strand exchange reaction
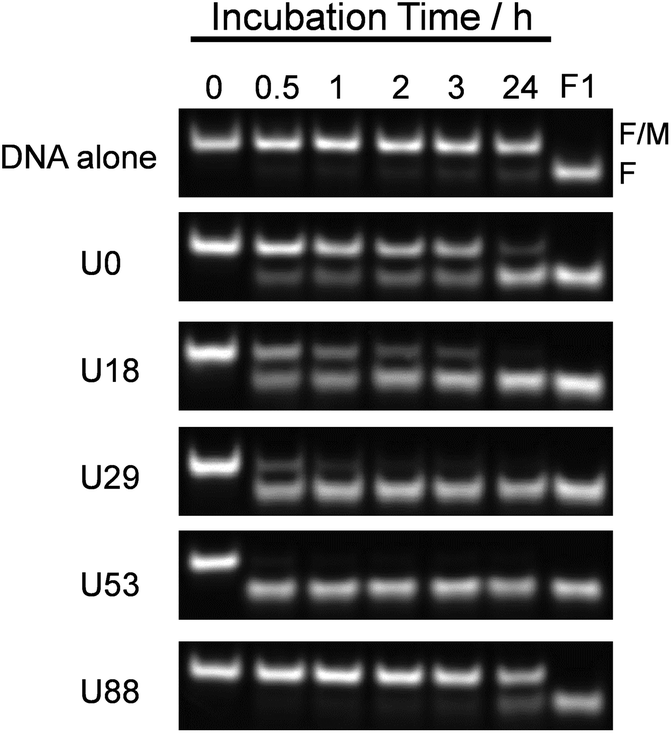
To estimate the effect of ureido modification of the cationic comb-type copolymer on a DNA SER, SERs in the presence of PAU-g-Dex copolymers with varying ureido contents were explored. A PAGE assay was used to follow the SER between FITC-labeled dsDNA (F/M) and its homologous non-labeled ssDNA (NF, 30 times molar excess to the labelled dsDNA). A band corresponding to dsDNA disappeared and a new band corresponding to ssDNA appeared as a function of time (Fig. 3). The intensity of the dsDNA band slightly decreased in the absence of the copolymer after 24 hours. In the presence of PA-g-Dex (U0), a band corresponding to ssDNA was visible after 30 min incubation and increased in intensity as a function of time. This result indicated that the cationic comb-type copolymer (U0) accelerated the SER. The SER was observed in the presence of all ureido-modified PA-g-Dex copolymers tested with the exception of U88. The reaction in the presence of the copolymer U53, which is modified with 53 mol% ureido groups, was complete within 30 min. The presence of the ureido modification significantly enhanced SER relative to the unmodified copolymer; however, when the polymer was highly modified with ureido groups (U88, 88% modification), the SER was not accelerated significantly relative to the reaction in the absence of the copolymer. This is likely due to a lack of ionic interactions between U88 and DNA.
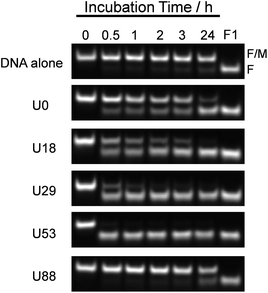 |
| | Fig. 3 DNA strand exchange reaction analyzed by 13% PAGE. ssDNA (NF) was added to dsDNA (F/M) in 10 mM HEPES (pH 7.5) containing 10 mM NaCl in the absence or presence of copolymers (N/P = 5) for indicated time at 30 °C. [NF] = 15 μM, [F/M] = 0.5 μM. | |
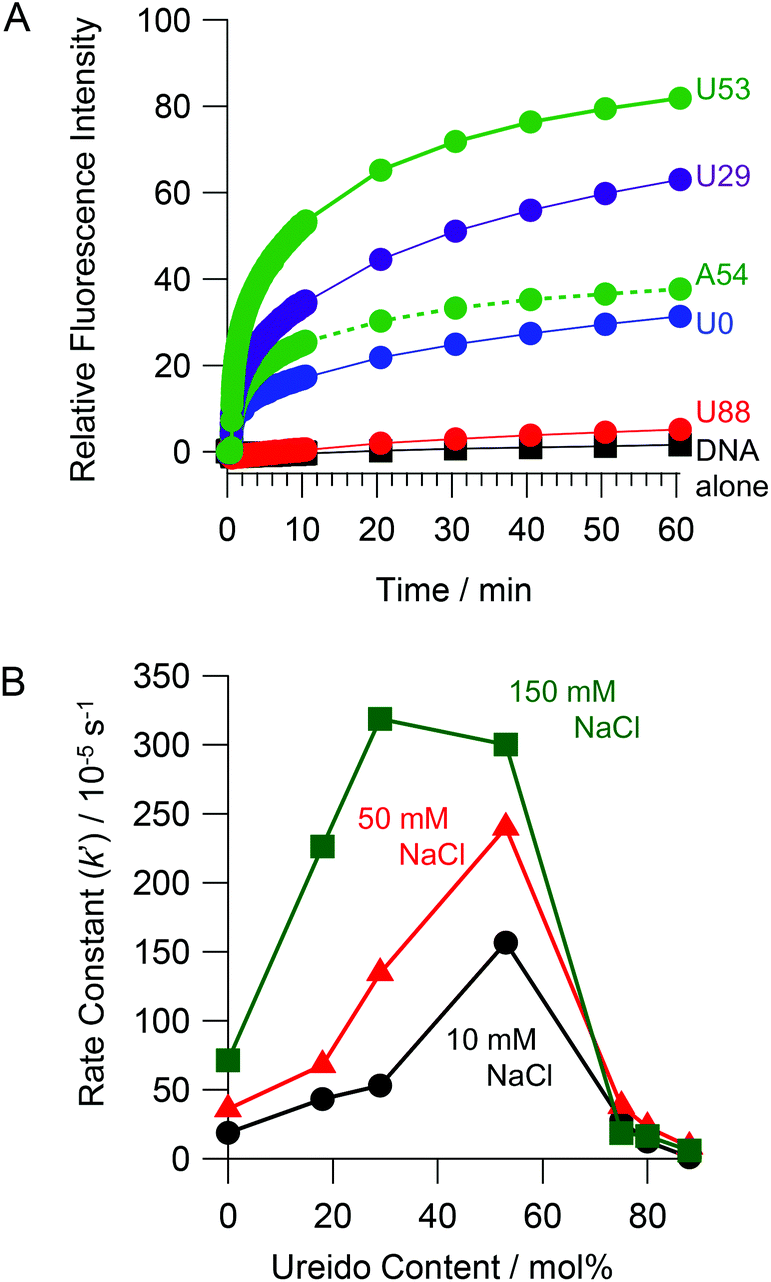
In order to evaluate the kinetics of SER more quantitatively, we employed a FRET assay. The extent of strand exchange over time in the absence and presence of ureido-modified copolymers is shown in Fig. 4A. No significant increase in the fluorescence intensity (which is a measure of SER) was observed in the absence of the copolymer. An increase in fluorescence intensity over time was observed in the presence of all copolymers except U88. The copolymer with moderate ureido modification (U53) accelerated the SER to the greatest extent. The pseudo first order rate constants (k′) for the SER determined by FRET are summarized in Table 1. In the absence of the copolymer, the SER was very slow (k′ < 1 × 10−6 s−1). The presence of U0 (k′ = 18.9 × 10−5 s−1) accelerated SER at least 100 times relative to the reaction in the absence of the copolymer. U53 (k′ = 15.6 × 10−4 s−1) accelerated SER about 8 fold relative to U0. The rate of the SER in the presence of the acetylated copolymer PAAc-g-Dex (A54, k′ = 33.6 × 10−5 s−1) was slower than that in the presence of ureido-modified PA-g-Dex U53, even though both copolymers have the same modification degree and cationic charge density. This result strongly suggests that ureido groups facilitate SER. Fig. 4B shows the rate constants as a function of ureido content of the copolymers. In 10 mM NaCl, the rate constant increased with increasing ureido content, peaking at 53 mol%, and then decreased with further increase in the ureido content. Peaks were also observed in 50 and 150 mM NaCl; the peak shifted to lower ureido content as the NaCl concentration increased. This is reasonable because the ionic interaction between the copolymer and DNA is weakened with increasing NaCl concentration. Higher cationic density (i.e., lower modification degree) of the copolymer is required to maintain ionic interactions required for SER acceleration at higher NaCl concentration. The observed SER enhancement at higher NaCl concentration in the presence of copolymers was also observed in a PAGE experiment (Fig. S1†).
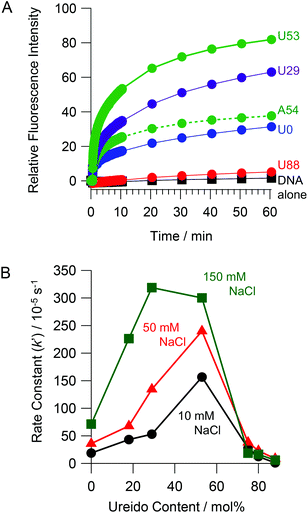 |
| | Fig. 4 (A) Real time monitoring of SER with copolymers (N/P = 5) in 10 mM HEPES (pH 7.5) containing 10 mM NaCl at 30 °C. (B) Rate constants of SER in the presence of PAU-g-Dex with various ureido contents in 10 mM HEPES (pH 7.5) containing 10 mM (black circles), 50 mM (red triangles) or 150 mM NaCl (green squares) at 30 °C. [NF] = 0.6 μM, [F/DM] = 0.06 μM. | |
Binding affinity of PAU-g-Dex for dsDNA and ssDNA
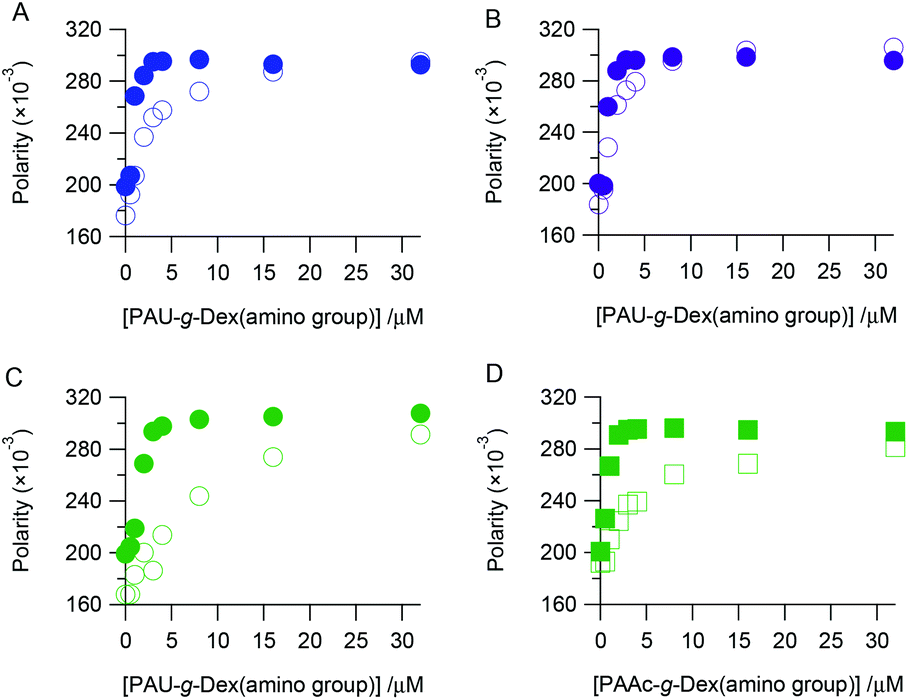
We assessed the binding affinity of DNA to PAU-g-Dex to understand the role of the ureido group in the SER acceleration. Fluorescently labeled ssDNA or dsDNA was mixed with the copolymer, and the change in fluorescence polarity was observed. Fig. 5 plots the fluorescence polarity as a function of concentration of the copolymer. U0 interacted with dsDNA more strongly than with ssDNA (Fig. 5A). This was expected because in cationic polymers the 20-bp dsDNA has more anionic charges and a higher anionic charge density than the 20-nt ssDNA. PAU-g-Dex (U29 and U53) also bound more strongly to dsDNA than to ssDNA (Fig. 5B, C). Binding profiles of A54 to dsDNA and ssDNA were not significantly different from those of U53 (Fig. 5D). Furthermore, affinities of these two copolymers for both dsDNA and ssDNA were weaker than that of U0. These results suggest that binding affinity of PAU-g-Dex copolymers to DNAs is primarily determined by electrostatic interactions.
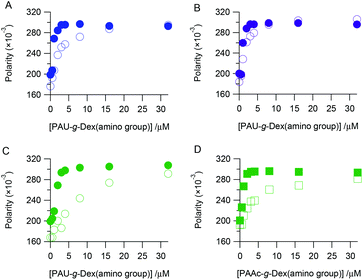 |
| | Fig. 5 Fluorescence polarity of fluorescence labeled dsDNA (filled symbols) or ssDNA (opened symbols) with various concentrations of U0 (A), U29 (B), U53 (C) or A54 (D) in 10 mM HEPES (pH 7.5) containing 300 mM NaCl. | |
Discussion
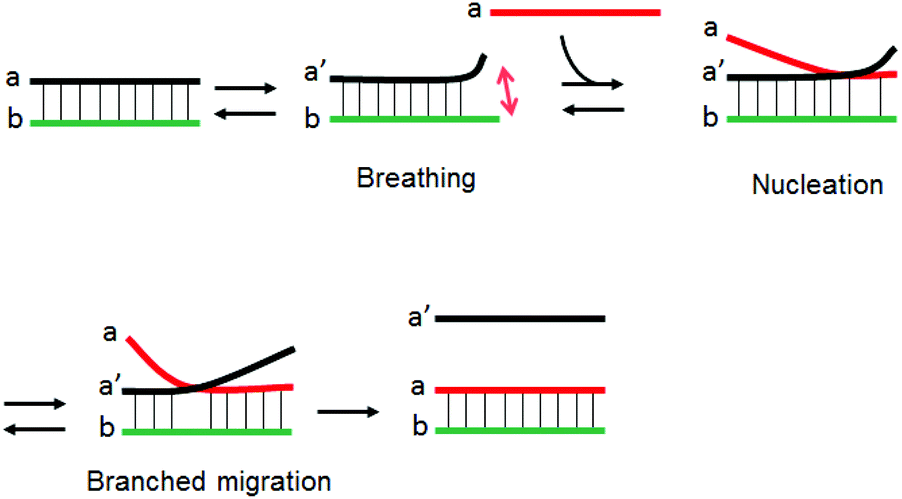
The strand exchange reaction between short dsDNA and a homologous ssDNA likely proceeds through nucleation and branch migration as depicted in Fig. 6. The terminal ends of dsDNA have breathing dynamics as base pairs associate and dissociate. The SER is initiated by a nucleation step where the ssDNA partially binds to the breathing dsDNA terminus to form a three-stranded intermediate.30 The nucleation step is followed by a rapid branched migration step. The three-stranded intermediate must be thermodynamically unstable because of limited number of base-pairs and local accumulation of anionic charges due to the phosphate ions. This nucleation step is considered to be the rate determining step. We speculate that cationic comb-type copolymers accelerate SER by stabilizing the three-stranded intermediate by reducing electrostatic repulsion.15 When the three-stranded intermediate is stabilized by the presence of the copolymer, the rate-determining step of SER should be the breathing step. Since the cationic comb-type copolymers increased the thermal stability of dsDNA, breathing frequency must be decreased in the presence of the copolymer, and this may cause a bottleneck in SER. Hence the SER accelerating activity of the copolymers might be enhanced by reducing the stabilizing effect of the copolymer for dsDNA.
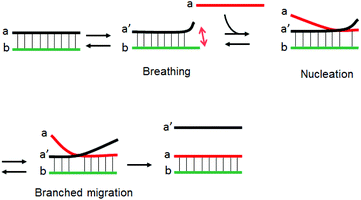 |
| | Fig. 6 Schematic illustration of a mechanism of DNA SER. | |
In this paper, we focused on modification of the copolymer with ureido groups. Due to chaotropic activity, urea denatures highly ordered structures of proteins and nucleic acids, and, therefore, we assumed that incorporation of the chaotropic activity in the copolymers would result in less stabilization of dsDNA. As expected, in the presence of PA-g-Dex modified with ureido groups the thermal stability of dsDNA was decreased relative to the stability in the presence of unmodified copolymers (Fig. 2). Since the ureido modification of the copolymer resulted in decrease in cationic charge density of the copolymer, both decrease in cationic charge density and incorporation of the ureido activity are seemingly involved in the destabilizing effect. Comparison of ureido and acetyl modified copolymers, however, strongly indicated a substantial role of the ureido groups in the destabilization activity (U53 vs. A54, Fig. 2B, C). A 54 mol% level of acetyl modification of PA-g-Dex did not alter the Tm of dsDNA relative to that in the presence of unmodified copolymer; however, the same level of ureido modification decreased the Tm significantly.
As summarized in Fig. 4, the SER acceleration effect due to the copolymer was enhanced by ureido modification. The ureido modification likely facilitates nucleation frequency by increasing dsDNA breathing. Although there were no significant differences in Tm of dsDNA in the presence of U53 or U0 under 10 mM NaCl conditions (Fig. 2A), U53 showed higher acceleration activity than U0 (Fig. 3 and 4). This may be due to local destabilization by U53. As clearly seen in the data shown in Fig. 4, there is an optimum degree of modification. This suggests that ionic interactions are prerequisite for copolymer facilitation of SER. A shift in the peak position to lower ureido content with increasing NaCl concentration supports this idea. We previously analyzed the effect of a copolymer modified with guanidinium groups,27 another chaotropic agent. The guanidino modification also resulted in a loss in the stabilizing effect and an increase in SER acceleration relative to the unmodified copolymer. Taking into account these observations, DNA denaturing activity of these functional groups was maintained when these groups were conjugated with polymer chains. This destabilization effect on dsDNA may enhance breathing dynamics of base pair association/dissociation, leading to the enhancement of the strand exchange reaction.
Conclusions
In this study, we explored the effect of cationic copolymers modified with chaotropic ureido groups on a DNA strand exchange reaction. The copolymers modified to about 50 mol% ureido content accelerated the SER 8 fold relative to the unmodified copolymer. Ureido groups in the copolymer likely increased breathing dynamics of dsDNA to accelerate SER. We propose that incorporation of chaotropic functional groups such as ureido and guanidinium groups into the copolymer structure can be used in design of artificial nucleic acid chaperones.
Acknowledgements
Parts of this work were supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Molecular Robotics” (no. 24104003), “Nanomedicine Molecular Science” (no. 2306) and the Cooperative Research Program of “Network Joint Research Center for Materials and Devices” from the Ministry of Education, Culture, Sports, Science and Technology, by Center of Innovation (COI) Program, Japan Science and Technology Agency (JST), and by KAKENHI (no. 23240074, 25350552) from Japan Society for the Promotion of Science.
Notes and references
- C. DasGupta, A. M. Wu, R. Kahn, R. P. Cunningham and C. M. Radding, Cell, 1981, 25, 507–516 CrossRef CAS.
- P. Baumann, F. E. Benson and S. C. West, Cell, 1996, 87, 757–766 CrossRef CAS.
- C. M. Radding, J. Biol. Chem., 1991, 266, 5355–5358 CAS.
- Z. Tsuchihashi and P. O. Brown, J. Virol., 1994, 68, 5863–5870 CAS.
- M. A. Skabkin, V. Evdokimova, A. A. M. Thomas and L. P. Ovchinnikov, J. Biol. Chem., 2001, 276, 44841–44847 CrossRef CAS PubMed.
- A. Zhang, V. Derbyshire, J. L. Salvo and M. Belfort, RNA, 1995, 1, 783–793 CAS.
- P. Alberti and J.-L. Mergny, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1569–1573 CrossRef CAS PubMed.
- H. Yan, X. Zhang, Z. Shen and N. C. Seeman, Nature, 2002, 415, 62–65 CrossRef CAS PubMed.
- Y. Tian and C. Mao, J. Am. Chem. Soc., 2004, 126, 11410–11411 CrossRef CAS PubMed.
- A. J. Turberfield, J. C. Mitchell, B. Yurke, A. P. Mills Jr., M. I. Blakey and F. C. Simmel, Phys. Rev. Lett., 2003, 90, 118102 CrossRef CAS.
- W. Li, Y. Yang, H. Yan and Y. Liu, Nano Lett., 2013, 13, 2980–2988 CrossRef CAS PubMed.
- L. Qian and E. Winfree, Science, 2011, 332, 1196–1201 CrossRef CAS PubMed.
- L. Wu, N. Shimada, A. Kano and A. Maruyama, Soft Matter, 2008, 4, 744–747 RSC.
- W. J. Kim, Y. Sato, T. Akaike and A. Maruyama, Nat. Mater., 2003, 2, 815–820 CrossRef CAS PubMed.
- W. J. Kim, T. Akaike and A. Maruyama, J. Am. Chem. Soc., 2002, 124, 12676–12677 CrossRef CAS PubMed.
- K. Yamana, Y. Fukunaga, Y. Ohtani, S. Sato, M. Nakamura, W. J. Kim, T. Akaike and A. Maruyama, Chem. Commun., 2005, 2509–2511 RSC.
- H. Asanuma, T. Osawa, H. Kashida, T. Fujii, X. Liang, K. Niwa, Y. Yoshida, N. Shimada and A. Maruyama, Chem. Commun., 2012, 48, 1760–1762 RSC.
- J. Michaelis, A. Maruyama and O. Seitz, Chem. Commun., 2013, 49, 618–620 RSC.
- S. W. Choi, N. Makita, S. Inoue, C. Lesoil, A. Yamayoshi, A. Kano, T. Akaike and A. Maruyama, Nano Lett., 2006, 7, 172–178 CrossRef PubMed.
- J. Du, L. Wu, N. Shimada, A. Kano and A. Maruyama, Chem. Commun., 2013, 49, 475–477 RSC.
- K. Frykholm, B. Nordén and F. Westerlund, Langmuir, 2009, 25, 1606–1611 CrossRef CAS PubMed.
- K. Frykholm, F. Baldelli Bombelli, B. Norden and F. Westerlund, Soft Matter, 2008, 4, 2500–2506 CAS.
- D. Lee, K. Singha, M.-K. Jang, J.-W. Nah, I.-K. Park and W. J. Kim, Mol. BioSyst., 2009, 5, 391–396 RSC.
- R. Moriyama, J. Mochida, A. Yamayoshi, N. Shimada, A. Kano and A. Maruyama, Curr. Nanosci., 2011, 7, 979–983 CrossRef CAS.
- N. Shimada, M. Yamamoto, A. Kano and A. Maruyama, Biomacromolecules, 2010, 11, 3043–3048 CrossRef CAS PubMed.
- Y.-i. Sato, Y. Kobayashi, T. Kamiya, H. Watanabe, T. Akaike, K. Yoshikawa and A. Maruyama, Biomaterials, 2005, 26, 703–711 CrossRef CAS PubMed.
- S. W. Choi, A. Kano and A. Maruyama, Nucleic Acids Res., 2008, 36, 342–351 CrossRef CAS PubMed.
- A. Maruyama, M. Katoh, T. Ishihara and T. Akaike, Bioconjugate Chem., 1997, 8, 3–6 CrossRef CAS PubMed.
- Y. Sato, R. Moriyama, S. W. Choi, A. Kano and A. Maruyama, Langmuir, 2006, 23, 65–69 CrossRef PubMed.
- L. P. Reynaldo, A. V. Vologodskii, B. P. Neri and V. I. Lyamichev, J. Mol. Biol., 2000, 297, 511–520 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: PAGE experiment of SER at higher NaCl concentration in the presence of copolymers. See DOI: 10.1039/c4bm00207e |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.