“Nail” and “comb” effects of cholesterol modified NIPAm oligomers on cancer targeting liposomes†
Wengang
Li‡
a,
Lin
Deng‡
a,
Basem
Moosa
a,
Guangchao
Wang
b,
Afnan
Mashat
a and
Niveen
M. Khashab
*a
aControlled Release and Delivery Lab (CRD), Physical Science and Engineering Division, Advanced Membranes and Porous Materials Center, King Abdullah University of Science and Technology, Thuwal, 23955-6900, Kingdom of Saudi Arabia. E-mail: niveen.khashab@kaust.edu.sa; Fax: +966128021172; Tel: +966128082410
bAdvanced Nanofabrication, Imaging and Characterization Core Facilities, King Abdullah University of Science and Technology (KAUST), Thuwal, 23955-6900, Kingdom of Saudi Arabia
First published on 7th February 2014
Abstract
Thermosensitive liposomes are a promising approach to controlled release and reduced drug cytotoxicity. Low molecular weight N-isopropylacrylamide (NIPAm) oligomers (NOs) with different architectures (main chain NOs (MCNOs) and side chain NOs (SCNOs)) were synthesized by reversible addition–fragmentation chain transfer (RAFT) polymerization and radical polymerization and then separately used to prepare thermosensitive liposomes. A more controlled and enhanced release was observed for both NO liposomes compared to pristine ones. Two release mechanisms depending on the oligomer architecture, namely “nail” for MCNOs and “comb” for SCNOs, are proposed. In addition to thermosensitivity, the cancer targeting property of NO liposomes was achieved by further biotinylation of the delivery system.
Introduction
Liposomes, first described in the 1960s,1,2 are self-assembled vesicles constructed with lipid bilayers. Due to their biocompatibility, high drug loading and easy preparation process, liposomes have attracted attention for drug delivery ever since their discovery.3–7 Liposomes encapsulated doxorubicin (Doxil®) has been approved to largely reduce the cardiac toxicity associated with free doxorubicin. In the past two decades, stimuli-sensitive liposomes8–11 have been studied for both controllable and enhanced release. Thermosensitive liposomes12–18 were approved to be a promising approach, especially combined with local hyperthermia. When the liposomes reach the heated tumor tissue area, the drug will be released due to changes in lipid membrane permeability.Poly(N-isopropylacrylamide) modified liposomes are one of the most typical thermosensitive liposomes.19–21 Due to the sharp coil-to-globule transition at the lower critical solution temperature (LCST) of the polymer chain, PNIPAm anchored into liposomes can lead to lateral phase separation of lipid bilayers that consequently allows cargo release through membrane defects. It was reported as early as 2005 that PNIPAm with higher molecular weight (Mw) causes faster release than low Mw PNIPAm.22–26 Although proven non-cytotoxic, high Mw PNIPAm is hard to biodegrade and has significant barriers to medical applications such as chronic bioaccumulation.23 On the other hand, NIPAm oligomers (Mw < 2500) can be directly broken down and can have a slower release rate both of which are more desirable in the area of controlled and sustainable release.
Cholesterol (chol), which takes up 30% of cell membranes, is able to decrease the fluidity of cell membranes due to its hydrogen bonding with lipid and flake structure. Many cholesterol derivatives were reported in the modification of liposome surfaces. Owing to the biocompatibility and “anchor-ability” of cholesterol, it is feasible to functionalize liposomes with either cholesterol derivatives or cholesterol modified polymers. Modifying liposomes with targeting moieties such as folic acid,24 biotin,25–27 saccharide,28 peptides,29,30etc. have been heavily studied during the past few years. Biotin, known as vitamin B7, which is highly needed for most cancer cell activities, has been proved to be a promising cancer targeting ligand for many anti-cancer drugs. Biotin-conjugated micelles25,27 and dendrimers26 have been reported to be effective cancer-targeting drug delivery systems;31–34 however, until now, uptake of modified biotin carriers has been improved without targeting a specific cell compartment.
In this report, cholesterol modified NIPAm oligomers with two polymer architectures (main chain, MCNOs, and side chain, SCNOs) were prepared by reversible addition–fragmentation chain transfer (RAFT) polymerization and radical polymerization for a thermosensitive DPPC/Chol liposomes system. Cholesterol molecules were used as the “anchor” groups for the oligomers. Biotinylated cholesterol was also synthesized to enhance cellular uptake. Two mechanisms are proposed to understand the influence of the architecture of the thermosensitive component on the overall release process (MCNOs “nail”, and SCNOs, “comb”) (Scheme 1). Both MCNOs and SCNOs proved biocompatible and showed thermosensitive driven release. Moreover, biotinylation of MCNOs and SCNOs allowed for excellent nuclei targetability in HeLa cells.
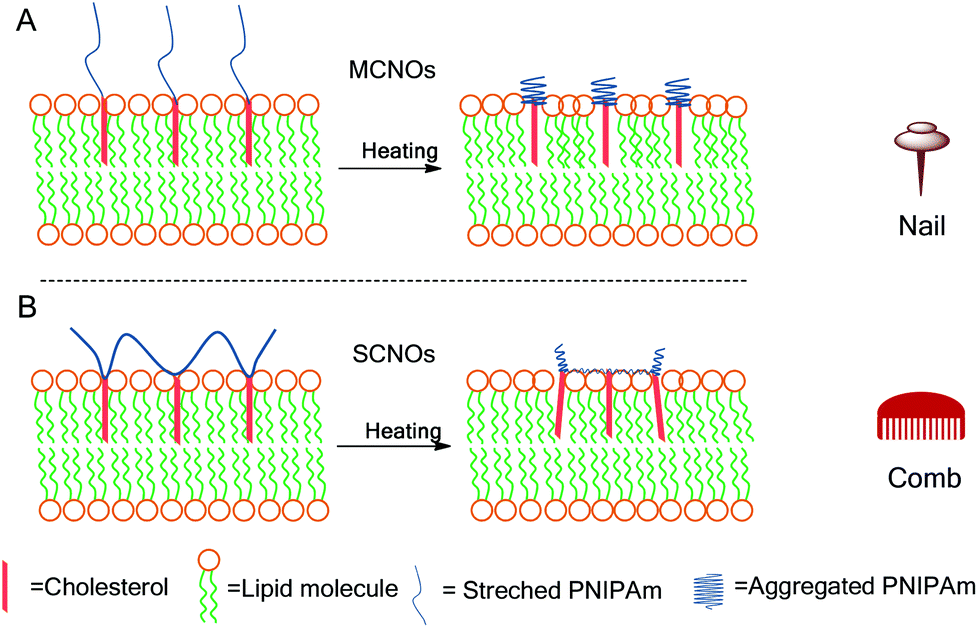 | ||
| Scheme 1 Schematic illustrations for the release of NIPAm oligomer (NO) liposomes at 37 °C: (A) nail effect: main-chain NIPAm oligomers (MCNOs); (B) comb effect: side-chain NIPAm oligomers (SCNOs). | ||
Results and discussion
SCNOs were prepared via radical polymerization while MCNOs were prepared via RAFT polymerization (Fig. 1). Biotinylated cholesterol used for cell targeting was synthesized through a condensation reaction (8, Fig. 1). The chain transfer agent (CTA) was prepared following a literature procedure.35 Cholesterol substituted monomers (3, Fig. 1, for SCNOs) and modified CTA (7, Fig. 1, for MCNOs) were synthesized as described in Fig. 1. Tetraethyleneglycol (TEG) bridges can minimize the steric effect between DPPC molecules and the biotinylated or polymeric cholesterols. 1H NMR spectra are shown in Fig. S1 and S2.† The Mw of SCNOs and MCNOs are determined by GPC (Fig. S3†), and both are around 2200.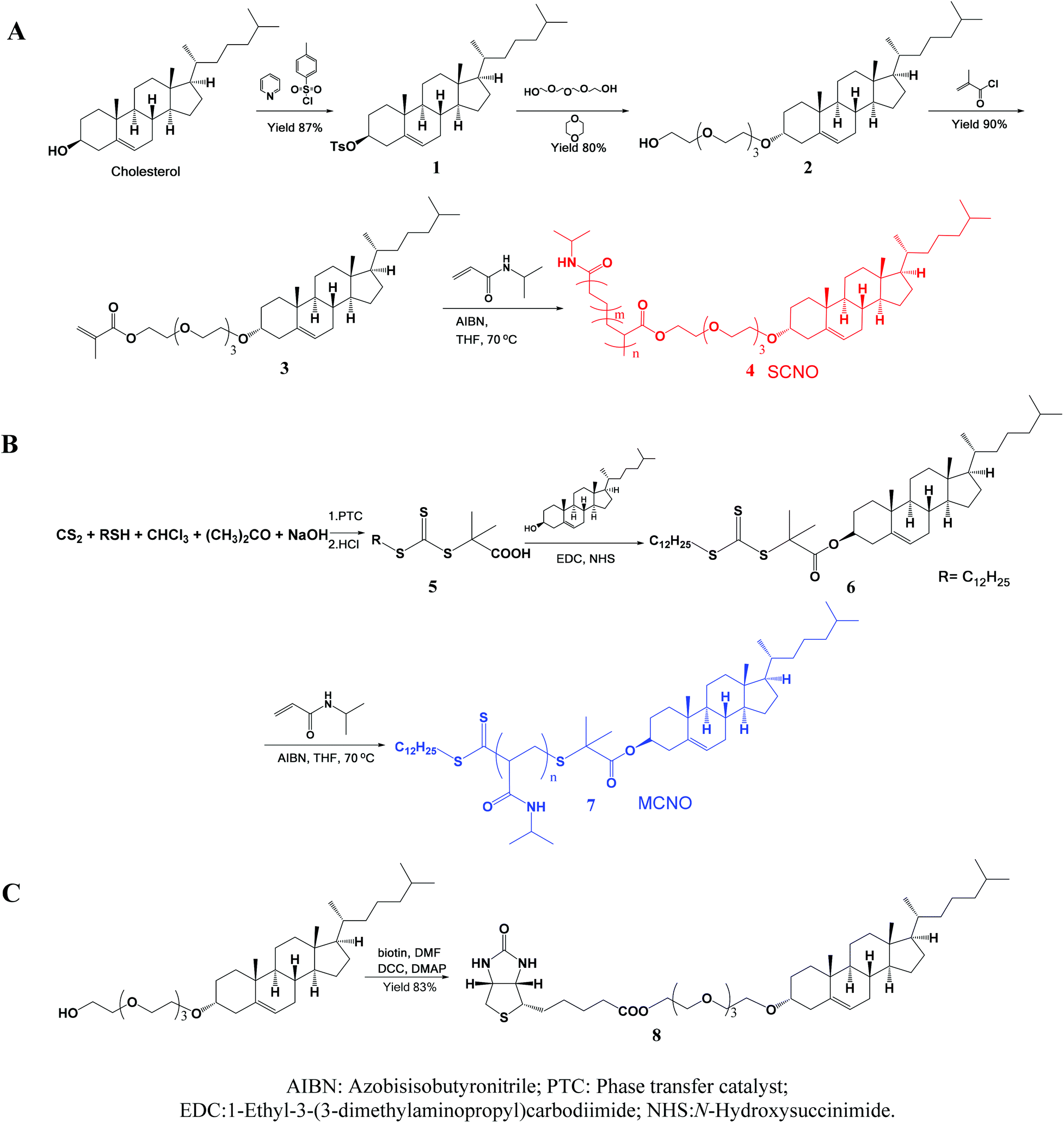 | ||
| Fig. 1 Schematic illustrations of the synthesized process of (A) SCNOs, (B) MCNOs, and (C) biotinylated cholesterol. | ||
In this study, liposomes (DPPC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cholesterol:NOs = 60:40:1, in mol) are prepared by a modified Bangham method. HEPES buffer or drug (calcein: 50 mmol L−1, DOX: 1 mg mL−1) solutions are used as cargo. For the drug loaded liposomes, a Sephadex G50 mini-column was used to remove the extra free drugs. TEM images, negatively stained by uranium acetate, were taken for the morphology of the pristine, SCNO and MCNO liposomes (Fig. 2). Size distribution data were also obtained for the liposomes (Fig. 2G). The results of both TEM and size distribution show that the liposomes were uniform spheres with a diameter of around 150 nm (A. dpristine = 153.6 nm, PDI = 1.279; B. dMCNO = 124.2 nm, PDI = 1.281; C. dSCNO = 140.7 nm, PDI = 1.197).
:cholesterol:NOs = 60:40:1, in mol) are prepared by a modified Bangham method. HEPES buffer or drug (calcein: 50 mmol L−1, DOX: 1 mg mL−1) solutions are used as cargo. For the drug loaded liposomes, a Sephadex G50 mini-column was used to remove the extra free drugs. TEM images, negatively stained by uranium acetate, were taken for the morphology of the pristine, SCNO and MCNO liposomes (Fig. 2). Size distribution data were also obtained for the liposomes (Fig. 2G). The results of both TEM and size distribution show that the liposomes were uniform spheres with a diameter of around 150 nm (A. dpristine = 153.6 nm, PDI = 1.279; B. dMCNO = 124.2 nm, PDI = 1.281; C. dSCNO = 140.7 nm, PDI = 1.197).
 | ||
| Fig. 2 TEM images (negatively stained) of different liposomal samples both at room temperature and heated at 37 °C for 1 h: (A) pristine liposomes – RT; (B) MCNO liposomes – RT; (C) SCNO liposomes – RT; (D) pristine liposomes – 37 °C; (E) MCNO liposomes – 37 °C; (F) SCNO liposomes – 37 °C; and (G) size distribution results of different liposomes at RT (scale bar is 200 nm (A–C) and 100 nm (D–F)). | ||
The LCST (Fig. S4†) for the two NIPAm oligomers was obtained by the turbidity method with a UV-Vis spectrometer, and both are around 31 °C (TSCNO = 31.8 °C; TMCNO = 30.8 °C). Hence, 37 °C was chosen as the temperature for the release test, because it is the normal body temperature and is above LCST of the NOs. On the other hand, for pure DPPC liposomes, when temperature goes above the main transfer temperature of lipid molecules (Tm-DPPC = 42 °C), lipid bilayers will turn from the gel phase to the liquid crystal phase, and the drug loaded inside liposomes will be able to pass through the membrane easily. However, in our case, as the cholesterol takes 40% of the total lipid, the lipid bilayer is in a fluid phase over a wide range of temperatures36,37 without an exact Tm. One temperature above Tm-DPPC (46 °C) was also chosen for the release test for comparison.
In order to test the thermosensitivity of the NO modified liposomes, a release profile (Fig. 3) was obtained. At 25 °C, pristine, MCNO, and SCNO liposomes are stable during the 120 minutes test period. The liposomes displayed a clear time-dependent calcein release profile at both 37 and 46 °C. A rapid release was observed after 10 min heating for all liposomes at 46 °C and the release of both NO modified liposomes was notably higher than the pristine one.
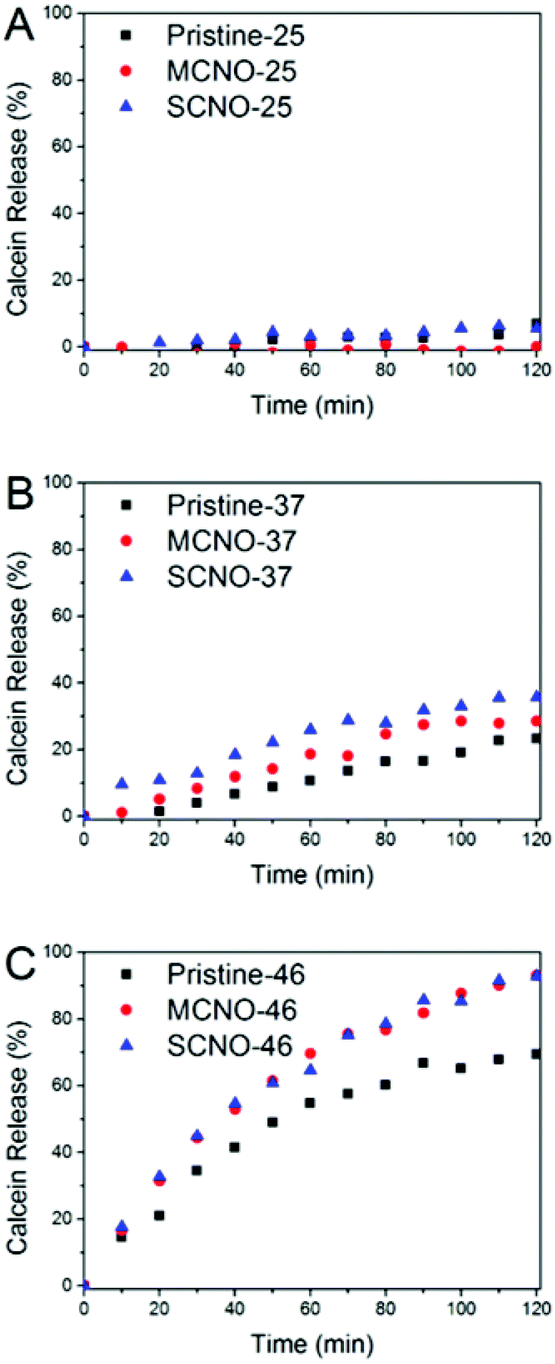 | ||
| Fig. 3 Release profiles of liposomes at different temperatures: (A) 25 °C, (B) 37 °C and (C) 46 °C. | ||
When incubated at 37 °C, pristine liposomes show a comparable release rate, which is due to the thermally-activated barrier density fluctuations in the lipid bilayer. However, both NO modified liposomes tend to release faster than pristine. This is due to the lateral phase separation that occurs when the temperature is raised above the LCST of the NIPAm chains causing their consequent aggregation.
From the release scheme of Fig. 1, the difference between MCNO and SCNO liposomes’ release at 37 °C lies mainly in the different aggregation forms of the oligomers. For MCNO when the NIPAm backbone shrinks, it will form a hydrophobic bolus on top of the cholesterol “anchor”; thus the oligomer will tend to go inside the lipid bilayers where the environment is hydrophobic. Such behaviour is just like pressing a nail into a wall (nail effect). On the other hand for SCNO, more cholesterol molecules are inserted into the lipid bilayer; thus when the side chains get aggregated, it will look like a comb inserted into hair (comb effect). When temperature goes higher than the LCST of NOs, hydrophobic bolus were formed and inserted into lipid bilayers to cause defects for cargo to release in both cases. The main difference between “nail” and “comb” effects is that the backbone shrink of the latter can produce intramolecular lateral forces accelerating the formation of defects so as to promote the drug release, and the “comb” effect will bring in more release than “nail”. Due to the limited chain length used in this study, the difference in release was not excessive. In fact, this is another advantage of NIPAm oligomers as the oligomer chain length can further control the speed of the release, i.e. longer chains will lead to faster release. This was verified by synthesizing SCNO with longer chain length (SCNO′), which showed an increased release at 37 °C (Fig. S5†).
To further verify the thermosensitivity of NO liposomes intuitively, negatively stained TEM images (Fig. 2D–F) of both the pristine and NO liposomes were obtained at 37 °C (incubation for one hour). Liposomes kept at room temperature look almost like isolated spheres, due to the NO chain that prohibits the contact between liposomes (Fig. 2A–C). Highly shrunk vesicles were observed in NO liposomes as a result of heating-induced phase transition (Fig. 2E and F (red arrow)). To better understand the release mechanism, the Tb3+/DPA assay38 (Fig. S6†) and the NBD/RhB assay39 (Fig. S7†) were performed following published procedures. Slight lipid mixing was observed indicating that some aggregation occurred when the liposomes were heated. However, no fusion was obtained from the Tb3+/DPA assays, which indicates that the defects caused by shrunk NOs were the main trigger of the release. The visualized results further confirm the thermosensitivity of both NO systems.
The in vitro cytotoxicity of liposomes was assessed by the MTT assay with the HeLa cell line at different concentrations of liposomes (10−7 ∼ 1 mg mL−1, total lipids). Fig. S8(A–C†) shows that liposomes are safe at low concentrations (Clipid < 10−3 mg mL−1). For all further in vitro tests, the total lipid concentration used was lower than 10−3 mg mL−1 to minimize the cytotoxicity of liposomes themselves. MTT assays for both NO liposomes (DOX loaded) are also available in Fig. S8D,† where no obvious difference between the two systems is noted.
The cellular localization of DOX-loaded liposomes was tested using a Zeiss LSM 710 inverted confocal microscope (Fig. 4). All the tests were done with the same setup. The NBD labelled liposomes (NBD-DPPE:DPPC:Chol:NOs = 1:60:40:1, in mol) with DOX loaded were prepared and then diluted with cell culture medium, and after incubation with HeLa cells at 37 °C, the cellular uptake for DOX loaded liposomes was monitored. After one hour incubation, the liposome uptake was observed due to NBD fluorescence spreading inside the cytoplasm, and the nuclear uptake of DOX was also visualized in all samples. According to fluorescence intensity, almost all the DOX released was taken up by the nuclei, with SCNO liposomes releasing the most DOX which is in agreement with the release profiles. The same test was repeated with HepG2 cells (Fig. S9†), and similar results were obtained.
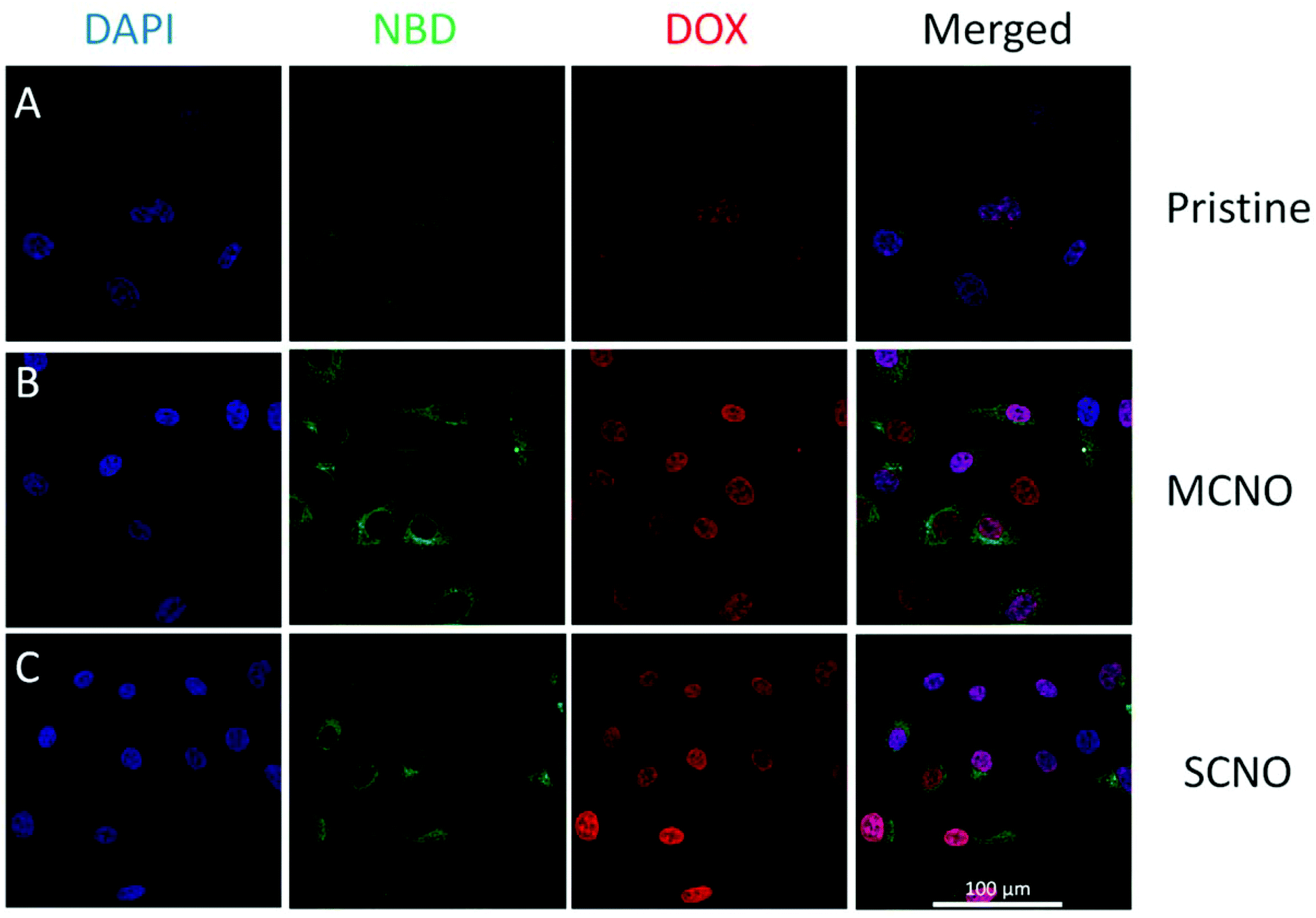 | ||
| Fig. 4 CLSM images of DOX loaded liposomes incubated with HeLa cells at 37 °C for 1 hour (from left to right, DAPI, NBD, DOX, merged): (A) pristine liposomes, (B) MCNO liposomes, (C) SCNO liposomes. | ||
To achieve better targeting, biotinylated cholesterol was used to enhance the cell uptake and a time-dependent liposome uptake test was evaluated by CLSM. Biotinylated and pristine liposomes with NBD labelled (biotin-cholesterol: total lipid = 5% in mole; no doxorubicin loaded) were prepared, and were then incubated with HeLa cells (10−3 mg mL−15, 30 min) to see the localization of liposomes. The cellular uptake could be observed after 5 min for both samples (Fig. 5). An enhancement was detected for biotinylated liposomes after 30 min treatment, while for the pristine liposomes no increase was observed. Moreover, a Z-stack confocal microscopic image (Fig. S10,† biotinylated liposomes, 30 min) shows an intense fluorescence both in the cytoplasm and around the nuclei area, which indicates that the biotinylated liposomes got aggregated around the nuclei membrane. From the in vitro studies, biotinylation could significantly enhance the cellular uptake of liposomes, which indicated that the biotinylated liposomes would largely enhance the drug efficiency.
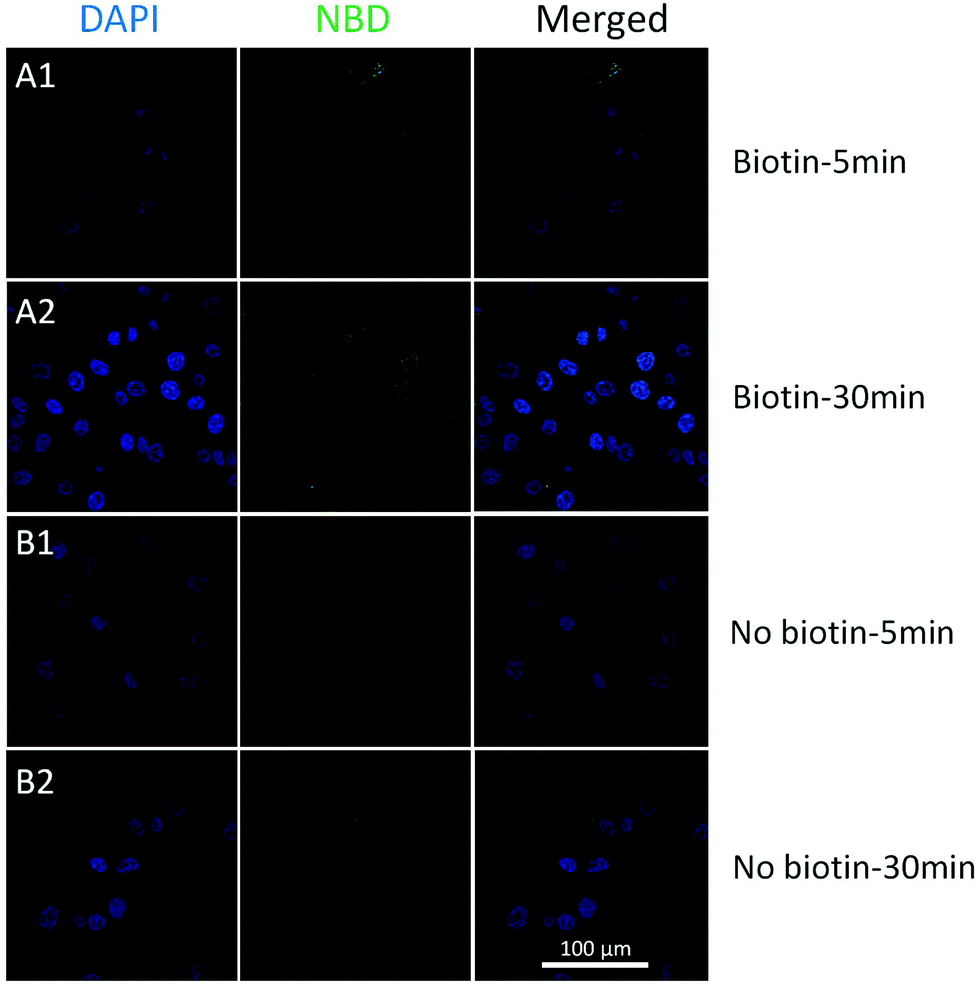 | ||
| Fig. 5 CLSM images of the cellular uptake of biotinylated and non-biotinylated liposomes in HeLa (from left to right, DAPI, NBD, merged): (A1) biotinylated liposomes incubated with HeLa for 5 min; (A2) biotinylated liposomes incubated with HeLa for 30 min; (B1) pristine liposomes incubated with HeLa for 5 min; (B2) pristine liposomes incubated with HeLa for 30 min. | ||
Conclusion
Liposomes modified with two types of low Mw cholesterol-NIPAm oligomers instead of high Mw industrial PNIPAm copolymers have been prepared and proved to be efficient in thermosensitive release. “Nail” and “comb” effects on the release of MCNO and SCNO liposomes are proposed with the “comb” effect leading to a faster release. Biotinylation is further verified to be effective in cancer targeting leading to an enhancement of cellular uptake efficiency. Our liposomal systems modified with low Mw NOs could serve as promising carriers for either drug or gene delivery with potential high transfer efficiency.Experiments and materials
Materials
Pyridine, p-toluenesulfonylchloride, dichloromethane (DCM), chloroform, n-hexanes, ethyl acetate (EtOAc), tetraethyleneglycol (TEG), dioxane, methanol (MeOH), triethylamine (TEA), methacryloyl chloride, N-isopropylacrylamide (NIPAm, 2200), N,N′-dimethylformide (DMF), azobisisobutyronitrile (AIBN), doxorubicin (DOX), tetrahydrofuran (THF), isopropanol, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), N-hydroxysuccinimide (NHS), 1-dodecanethiol, acetone, tricaprylmethylammonium chloride, sodium hydroxide, hydrochloride (HCl), carbon disulfide and 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) were bought from Sigma-Aldrich; biotin was bought from Fluka; 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), cholesterol (chol), and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (ammonium salt) (NBD-DPPE) were bought from Avantipolar Lipid. All solutions were prepared by deionized water (Milli-Q, 18.2 MΩ cm, 25 °C).Characterization
All 1H NMR were obtained on a Bruker AV-III 600 MHz NMR spectrometer. GPC data were obtained on an Agilent 1200 GPC system. TEM images were acquired on an FEI Tecnai T12 transmission electron microscope at 120 keV. FTIR spectra were acquired on a Thermo Nicolet 6700 FT-IR system. Particle size distribution data were taken on a Malvern NanoZS. DSC data were obtained from a Netzsch DSC 204 HP Phoenix differential scanning calorimeter. TGA data were acquired from Netzsch TG 209 F1 Iris FTIR coupling. A fluorescence spectrum was acquired on a Varian Eclipse fluorescence spectrometer. CLSM images were obtained from a Zeiss LSM 710 inverted microscope.:1) to give cholesterol-OTs as a white powder (87% yield).
:1:1) to end up with a colourless syrup (80% yield).
Negatively stained TEM
A drop of liposomes at room temperature was dropped on the grid, and then the grid was washed with deionized water 3 times. The grid was thereafter merged into U(OAc)2 solution (2%) for 3 min for staining. The pre-heated liposomes (37 °C) were stained in the same way.where Fi and Ft mean the initial and intermediary fluorescence intensities of the liposome suspension, respectively. Ff is the fluorescent intensity of the liposomes after the addition of Triton X-100 (final concentration of 1%).
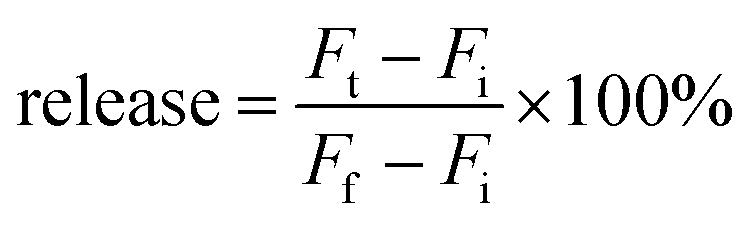
In order to test the release of NO liposomes, the calcein loaded liposomes were diluted 1000 times with HEPES buffer and then time-dependent release profiles were acquired using a fluorescence spectrometer in 120 min at 25, 37 and 46 °C. The loading efficiency of calcein/DOX is 3–5% w/w.
The biological tests were finished with DOX loaded liposomes. The fluorescence setup for DOX is λex. = 490 nm, λem. = 580 nm.
The cytotoxicity of DOX loaded liposomes was also evaluated by the MTT assay. The same procedure as cell viability was followed, but after washing with DPBS buffer, the cells were incubated in the culture media with liposomes for 2 h at 37 °C.
The difference between biotinylated liposomes and non-biotinylated liposomes were investigated following the same procedure with HeLa cells. And the incubation time by liposome containing medium was 5 min and 30 min.
Acknowledgements
This research is fully sponsored by King Abdullah University of Science and Technology (KAUST).References
- A. D. Bangham and R. W. Horne, Nature, 1962, 196, 952–953 CAS.
- R. W. Horne, A. D. Bangham and V. P. Whittaker, Nature, 1963, 200, 1340–1340 CAS.
- E. A. Forssen and Z. A. Tökès, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 1873–1877 CrossRef CAS.
- A. Gabizon, R. Catane, B. Uziely, B. Kaufman, T. Safra, R. Cohen, F. Martin, A. Huang and Y. Barenholz, Cancer Res., 1994, 54, 987–992 CAS.
- A. H. Negussie, J. L. Miller, G. Reddy, S. K. Drake, B. J. Wood and M. R. Dreher, J. Controlled Release, 2010, 143, 265–273 CrossRef CAS PubMed.
- E. Yuba, C. Kojima, A. Harada, T. Tana, S. Watarai and K. Kono, Biomaterials, 2010, 31, 943–951 CAS.
- A. D. Tagalakis, L. He, L. Saraiva, K. T. Gustafsson and S. L. Hart, Biomaterials, 2011, 32, 6302–6315 CAS.
- N. Bertrand, J. G. Fleischer, K. M. Wasan and J.-C. Leroux, Biomaterials, 2009, 30, 2598–2605 CAS.
- K. Kono, T. Ozawa, T. Yoshida, F. Ozaki, Y. Ishizaka, K. Maruyama, C. Kojima, A. Harada and S. Aoshima, Biomaterials, 2010, 31, 7096–7105 CAS.
- D. Pornpattananangkul, S. Olson, S. Aryal, M. Sartor, C.-M. Huang, K. Vecchio and L. Zhang, ACS Nano, 2010, 4, 1935–1942 CAS.
- M. de Smet, E. Heijman, S. Langereis, N. M. Hijnen and H. Grüll, J. Controlled Release, 2011, 150, 102–110 CAS.
- L. Li, H. T. L. M. Ten, D. Schipper, T. M. Wijnberg, R. G. C. van, A. M. M. Eggermont, L. H. Lindner and G. A. Koning, J. Controlled Release, 2010, 143, 274–279 CAS.
- M. C. Sandstrom, L. M. Ickenstein, L. D. Mayer and K. Edwards, J. Controlled Release, 2005, 107, 131–142 Search PubMed.
- M. Hossann, T. Wang, M. Wiggenhorn, R. Schmidt, A. Zengerle, G. Winter, H. Eibl, M. Peller, M. Reiser, R. D. Issels and L. H. Lindner, J. Controlled Release, 2010, 147, 436–443 CAS.
- L. Li, H. T. L. M. Ten, D. Schipper, T. M. Wijnberg, R. G. C. van, A. M. M. Eggermont, L. H. Lindner and G. A. Koning, J. Controlled Release, 2010, 143, 274–279 CAS.
- M. Hossann, Z. Syunyaeva, R. Schmidt, A. Zengerle, H. Eibl, R. D. Issels and L. H. Lindner, J. Controlled Release, 2012, 162, 400–406 CAS.
- B. M. Dicheva, T. L. M. T. Hagen, L. Li, D. Schipper, A. L. B. Seynhaeve, G. C. V. Rhoon, A. M. M. Eggermont, L. H. Lindner and G. A. Koning, Nano Lett., 2013, 13, 2324–2331 CAS.
- L. Li, H. T. L. M. Ten, M. Hossann, R. Suss, R. G. C. van, A. M. M. Eggermont, D. Haemmerich and G. A. Koning, J. Controlled Release, 2013, 168, 142–150 CAS.
- A. Polozova and F. M. Winnik, Langmuir, 1999, 15, 4222–4229 CAS.
- H. D. Han, B. C. Shin and H. S. Choi, Eur. J. Pharm. Biopharm., 2006, 62, 110–116 CAS.
- A. Polozova, A. Yamazaki, J. L. Brash and F. M. Winnik, Colloids Surf., A, 1999, 147, 17–25 CAS.
- K. Kono, T. Murakami, T. Yoshida, Y. Haba, S. Kanaoka, T. Takagishi and S. Aoshima, Bioconjugate Chem., 2005, 16, 1367–1374 CAS.
- M. Patenaude and T. Hoare, ACS Macro Lett., 2012, 1, 409–413 CrossRef CAS.
- A. Gabizon, H. Shmeeda, A. T. Horowitz and S. Zalipsky, Adv. Drug Delivery Rev., 2004, 56, 1177–1192 CAS.
- C. Cheng, H. Wei, B.-X. Shi, H. Cheng, C. Li, Z.-W. Gu, S.-X. Cheng, X.-Z. Zhang and R.-X. Zhuo, Biomaterials, 2008, 29, 497–505 CAS.
- W. Yang, Y. Cheng, T. Xu, X. Wang and L.-P. Wen, Eur. J. Med. Chem., 2009, 44, 862–868 CrossRef CAS PubMed.
- J. H. Kim, Y. Li, M. S. Kim, S. W. Kang, J. H. Jeong and D. S. Lee, Int. J. Pharm., 2012, 427, 435–442 CAS.
- K. Y. Choi, H. Chung, K. H. Min, H. Y. Yoon, K. Kim, J. H. Park, I. C. Kwon and S. Y. Jeong, Biomaterials, 2010, 31, 106–114 CAS.
- S. Sakuma, T. Yano, Y. Masaoka, M. Kataoka, H. Ken-ichiro, H. Tachikawa, Y. Shoji, R. Kimura, H. Ma, Z. Yang, L. Tang, R. M. Hoffman and S. Yamashita, J. Controlled Release, 2009, 134, 2–10 CAS.
- W. J. M. Mulder, R. Koole, R. J. Brandwijk, G. Storm, P. T. K. Chin, G. J. Strijkers, C. de Mello Donegá, K. Nicolay and A. W. Griffioen, Nano Lett., 2005, 6, 1–6 Search PubMed.
- T. T. Nguyen, K. L. Sly and J. C. Conboy, Anal. Chem., 2011, 84, 201–208 Search PubMed.
- T. R. Ward, Acc. Chem. Res., 2010, 44, 47–57 Search PubMed.
- C. L. Zavaleta, W. T. Phillips, A. Soundararajan and B. A. Goins, Int. J. Pharm., 2007, 337, 316–328 CAS.
- J. Pierron, C. Malan, M. Creus, J. Gradinaru, I. Hafner, A. Ivanova, A. Sardo and T. R. Ward, Angew. Chem., 2008, 120, 713–717 Search PubMed.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CAS.
- K. Sanat, V. A. Raghunathan and M. Satyajit, J. Phys.: Condens. Matter, 2005, 17, S1177 CrossRef.
- S. Karmakar, B. R. Sarangi and V. A. Raghunathan, Solid State Commun., 2006, 139, 630–634 CAS.
- R. D. Morero, A. L. Vinals, B. Bloj and R. N. Farias, Biochemistry, 1985, 24, 1904–1909 CAS.
- D. K. Struck, D. Hoekstra and R. E. Pagano, Biochemistry, 1981, 20, 4093–4099 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3bm60222b |
| ‡ The authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2014 |