
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Magnetite-doped polydimethylsiloxane (PDMS) for phosphopeptide enrichment†
Mairi E.
Sandison
a,
K. Tveen
Jensen
b,
F.
Gesellchen
c,
J. M.
Cooper
b and
A. R.
Pitt
*c
aInstitute of Molecular, Cell and Systems Biology, University of Glasgow, Glasgow, G12 8QQ, UK
bSchool of Life and Health Science, Aston University, Birmingham, B4 7ET, UK
cDivision of Biomedical Engineering, University of Glasgow, Glasgow, G12 8LT, UK. E-mail: a.r.pitt@aston.ac.uk
First published on 21st July 2014
Abstract
Reversible phosphorylation plays a key role in numerous biological processes. Mass spectrometry-based approaches are commonly used to analyze protein phosphorylation, but such analysis is challenging, largely due to the low phosphorylation stoichiometry. Hence, a number of phosphopeptide enrichment strategies have been developed, including metal oxide affinity chromatography (MOAC). Here, we describe a new material for performing MOAC that employs a magnetite-doped polydimethylsiloxane (PDMS), that is suitable for the creation of microwell array and microfluidic systems to enable low volume, high throughput analysis. Incubation time and sample loading were explored and optimized and demonstrate that the embedded magnetite is able to enrich phosphopeptides. This substrate-based approach is rapid, straightforward and suitable for simultaneously performing multiple, low volume enrichments.
Introduction
Reversible protein phosphorylation is one of the most common and important protein post-translational modifications.1 Analysis of phosphorylation is commonly performed by mass spectrometry (MS), but this is challenging, primarily due to the low stoichiometry of phosphorylation in biological samples.2 Therefore, samples are typically enriched for phosphopeptides prior to MS analysis.A number of methods for phosphopeptide enrichment have been described in the literature, the most widely used being immobilized metal affinity chromatography (IMAC) and metal oxide affinity chromatography (MOAC).3 In IMAC, phosphorylated species are retained through the formation of metal–ligand complexes, commonly chelated metal ions (Fe3+, Ga3+, Al3+, Zr4+). MOAC exploits the affinity of metal oxide surfaces for phosphate groups4 and appears to have fewer limitations than IMAC.5 MOAC approaches have been growing rapidly, in particular methods employing TiO2 sorbents,6,7 which gained popularity following reports by Pinkse et al. and Kuroda et al. in 2004 (ref. 8 and 9) and by Larsen et al. in 2005.10 Since then a number of TiO2 enrichment strategies have been reported, including the use of TiO2 particles trapped in a polymeric monolith by photopolymerisation,11,12 centrifugation-based protocols using suspensions of TiO2 particles13,14 and the development of capillaries coated with thin TiO2 films by liquid phase deposition.15,16
A number of other metal oxides have also been successfully employed for phosphopeptide enrichment, including ZrO2,17 Fe3O4 (ref. 18 and 19) and Al2O3.20,21 For phosphopeptide analysis by MALDI-MS, several reports have described on-target approaches to enrichment.22–25 The implementation of phosphopeptide enrichment in a lab-on-a-chip (LOC) format26 has many potential advantages including low sample volume requirements, the potential both to multiplex parallel analysis streams and integrate several sample preparation stages in a single system,27–31 decreased analysis times, increased experimental throughput and minimal sample handling.
However, there have been few reports describing LOC phosphopeptide enrichment strategies. A commercial microfluidic HPLC-chip that incorporates a TiO2 bead bed is currently available from Agilent (Phosphochip, Agilent Technologies).32,33 A microfabricated polymeric device whose internal microfluidic channels were coated with TiO2–ZrO2 by liquid phase deposition34 and an acoustophoresis device for efficient on-chip washing of TiO2-coated beads35 have been reported. The former was successfully employed for phosphopeptide enrichment, as demonstrated by the enrichment of the phosphopeptides from a β-casein tryptic digest, but the fabrication processes were complex and time consuming. In the latter, only the washing stages were carried out on-chip. An alternative simple, low-cost, flexible approach that is amenable to straightforward integration with existing LOC platforms would, therefore, be beneficial for phosphoproteomic analysis.
We have developed a new, simple, rapid approach to generating a moldable MOAC sorbent for phosphopeptide enrichment that is compatible with microfluidic technologies. It uses a polydimethylsiloxane (PDMS) substrate doped with magnetite (iron(II/III) oxide) particles, etched to create a highly roughened surface that is primarily composed of magnetite. Magnetite was chosen as the sorbent as it is compatible with the chemical processes necessary for generation of the substrate (polymerisation, curing and etching), and may offer the potential for magnetic patterning of the substrate in future applications. The enrichment protocol is rapid, suitable for low sample concentrations, and has no particle contamination issues. As the magnetite-PDMS material can be formed into a variety of configurations, for example into microwell arrays or microfluidic channels, using standard replica molding techniques, this approach is highly amenable to automated, low-volume, high throughput analysis. Moreover, the sorbent can be reused by re-etching the surface.
Material and methods
Materials
The PDMS employed was Sylgard® 184 Silicon Elastomer Kit from Dow Corning (VWR, Leicestershire, UK). The magnetite particles (iron(II, III) oxide powder, particle size < 5 μm) and all chemicals were purchased from Sigma-Aldrich (Dorset, UK), except for MALDI calibration standards (Peptide Calibration Standards II, 700–4000 Da, Bruker Daltonics) and porcine trypsin (Sequencing Grade Modified Trypsin, Promega).Preparation & characterisation of magnetite-PDMS substrates
Magnetite-PDMS substrates were prepared as follows. The PDMS prepolymer and curing agent were first mixed in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (w/w). Magnetite particles were then added, with a particle:PDMS ratio of either 1:1, 1:2 or 1:4 (w/w), and thoroughly mixed to create a homogenous suspension. To create an array of wells with 96-well plate spacing, the PDMS mixture was poured over an upturned, round-bottomed, 96-well plate (Costar® cell culture plate, Corning®), around which a frame constructed from glass microscope slides had previously been bonded in order to contain the polymer mixture. For microscopy analysis, the PDMS mixture was cast over a polished silicon wafer (Compart Technology, Peterborough). After degassing the PDMS mixture by placing the mold inside a vacuum desiccator and pumping down the chamber, the PDMS was cured in an oven at 50 °C overnight. The cured PDMS was then peeled off the mold and placed in an oven at 95 °C for a further 24 hours to enhance PDMS crosslinking.36
:1 ratio (w/w). Magnetite particles were then added, with a particle:PDMS ratio of either 1:1, 1:2 or 1:4 (w/w), and thoroughly mixed to create a homogenous suspension. To create an array of wells with 96-well plate spacing, the PDMS mixture was poured over an upturned, round-bottomed, 96-well plate (Costar® cell culture plate, Corning®), around which a frame constructed from glass microscope slides had previously been bonded in order to contain the polymer mixture. For microscopy analysis, the PDMS mixture was cast over a polished silicon wafer (Compart Technology, Peterborough). After degassing the PDMS mixture by placing the mold inside a vacuum desiccator and pumping down the chamber, the PDMS was cured in an oven at 50 °C overnight. The cured PDMS was then peeled off the mold and placed in an oven at 95 °C for a further 24 hours to enhance PDMS crosslinking.36
To etch back the surface PDMS matrix and expose the embedded particles, an etchant was prepared from a 75% (w/w) aqueous solution of tetrabutylammonium fluoride (TBAF), which was diluted 1:10 with N-methylpyrrolidinone (NMP) to form a 7.5% TBAF solution. The etchant was prepared immediately prior to use. Substrates were immersed in the etchant and gently agitated for 2 min. They were then rinsed twice with NMP and twice with ethanol, prior to gently blow drying.
The magnetite-PDMS substrates were characterized by both scanning electron (SEM) and atomic force (AFM) microscopies. Prior to SEM analysis, using a Hitachi S4700 SEM with an accelerating voltage of 10 kV and an emission current of 10 μA, the samples were sputter-coated with a thin layer of gold–palladium (approximately 10 nm). AFM characterization was performed using a NanoWizard II Bio AFM (JPK Systems, Berlin).
Tryptic digest of bovine β-casein
Tryptic digestion was carried out overnight at 37 °C in 25 mM ammonium bicarbonate, pH 7.8, using a protein:trypsin ratio of 20:1. The digested casein was aliquoted and frozen at −20 °C. Prior to enrichment, an aliquot of the digest was evaporated to dryness in a SciQuip Christ freeze dryer and resuspended by vortexing in an appropriate volume of loading buffer (80:15:5) acetonitrile (ACN): trifluoroacetic acid (TFA):distilled water (dH2O). The samples were then clarified by centrifugation at 13000 rpm for 30 s.
Preparation of HeLa cell lysate
Human cervical epithelial (HeLa) cells were cultured in T150 flasks in DMEM media supplemented 10% fetal calf serum, 100 μg ml−1 penicillin, 100 μg ml−1 streptomycin at 37 °C in a humidified 5% CO2 atmosphere to 90% confluence. The cells were then washed 3 times in ice-cold PBS and harvested by the addition of 50 mM TRIS, 8 M urea, pH 7.3 including phosphatase inhibitors (cocktail 2 and 3, Sigma) and protease inhibitors (Roche) and detached with a cell scraper. The cells were further lysed by sonication using a microprobe at 100% output for 10 × 10 seconds with 1 minute intervals. The resulting cell lysate was centrifuged at 16900g for 90 minutes at 4 °C. The supernatant was collected and the protein concentration was determined using the Bradford method (Bradford M.M, 1976).
The HeLa cell lysate and a control sample of α-casein were incubated with 15 μl of 0.5 M DTT for 30 minutes at 60 °C and then allowed to cool to room temperature and incubated with 40 μl of 0.5 M iodoacetamide at room temperature for 30 minutes. The proteins were precipitated with trichloroacetic acid (TCA) (final concentration 10%) and washed twice in ice-cold acetone and then dried in a vacuum centrifuge. The dried precipitates were resuspended in 50 μl of 50 mM TRIS, 8 M urea, pH 8.3 and incubated for 30 minutes, then 400 μl of MilliQ water was added to reduce the urea concentration to below 1 M. Sequencing grade trypsin was added in a ratio of 1:40 (w/w, trypsin–protein). The samples were allowed to digest O/N at 37 °C and then acidified by the addition of 5% formic acid and purified on a C18RP column (SepPak, Waters Corporation).
Phosphopeptide enrichment from β-casein
Before performing an enrichment, the PDMS substrates were washed with methanol, 0.1 M ammonium hydroxide, dH2O and then loading buffer. 100 μl of sample containing clarified digest from 5, 20, 50 or 100 ng of β-casein protein was added to each well and left to incubate for 2, 5, 12 or 20 min. After pipetting off the supernatant, the wells were washed twice with loading buffer and once with 10:90 ACN:dH2O, with a 1 min incubation per wash. The substrates were then left to dry in air for 5 min. A 15 min incubation with 150 μl 0.1 M ammonium hydroxide per well was used to elute the phosphopeptides. 4 μl of 20% FA was added to each elution fraction collected to acidify the solution for subsequent analysis and to stabilize the phosphopeptides.
Phosphopeptide enrichment from HeLa lysates
Using the magnetite/PDMS substrate, digested HeLa cell lysate or digested HeLa cell lysate spiked with digested bovine α-casein (1:1 ratio) were added to substrate wells (1 μg protein per well) and incubated and eluted as described above. Enrichment using TiO2 was performed using the Pierce Magnetic Phosphopeptide Enrichment Kit (Thermo Fisher, Rockford, USA), following the manufacturers protocol using 5 μg of lysate per 2.5 μl of beads.
MALDI mass spectrometry analysis
Prior to mass spectrometry analysis, collected fractions were dried to completion by freezer drying, before resuspending in 3 μl of a 20% dilution of saturated dihydroxybenzoic acid (DHB) in 50% ACN, 0.1% TFA. 1.5 μl of each sample was spotted onto a stainless steel MALDI target (MTP 384 target plate, Bruker Daltonics), alongside peptide standard calibration spots.Fractions were analysed using a Bruker Daltonics Ultraflex III MALDI TOF/TOF tandem time-of-flight mass spectrometer in positive ion reflectron mode. The laser spot size was set to minimum (10 μm), the matrix suppression deflection to m/z 690 and the detection range to m/z 700–3600. The laser power intensity was optimized to give maximum sensitivity without saturation for the fractions with the strongest signals and this power intensity was then used for all spectra acquired. For each spot, data was collected from 5000 shots fired at numerous points across the entire area. Biotools software version 3.1 (Bruker Daltonics) was used to deconvolute the spectra obtained using the SNAP algorithm (S/N threshold of 4, quality factor threshold of 20) to produce a list of monoisotopic masses with normalized intensities. Phosphopeptide enrichment was quantified by comparing the measured intensities of five phosphopeptide peaks (m/z: 2061.83, 2432.05, 2962.42, 3042.39, 3122.35 Da) to the seven most common non-phosphopeptide peaks (m/z: 742.45, 780.50, 830.45, 873.49, 1013.52, 2186.17, 2909.60 Da). A script was written in Matlab (Mathworks) to extract the intensities of each of these peaks and to return a value corresponding to the percentage of the summed phosphopeptide signal intensity relative to the total intensity of all 12 peaks. For each experimental condition, mean values from a minimum of three replicates are reported, with error bars corresponding to one standard error.
Nano LC-MSMS analysis
The dried phosphopeptide enriched samples and an unenriched sample were resuspended in 2% acetonitrile (0.1% formic acid) and analysed by LC-MSMS. Peptides were separated and analysed using an Ultimate 3000 system (Dionex, Camberley) and a 5600 TripleTOF (ABSciex, Warrington, UK) controlled by Chromeleon Xpress and Analyst software (1.5.1 or TF, ABSciex, Warrington). The peptides were purified on a C18RP pre-column (C18 PepMapTM, 5 μm, 5 mm × 0.3 mm i.d. Dionex, Bellefonte, PA, USA) by washing for 4 min with 2% aqueous acetonitrile (0.1% formic acid) at 30 μL min−1. The peptides were then separated on a C18 nano-HPLC column (C18 PepMapTM, 5 μm, 75 μm i.d. × 150 mm, Dionex, Bellefonte, PA, USA) using a gradient elution running from 2% to 45% aqueous acetonitrile (0.1% formic acid) in 3 hours and a final washing step running from 45% to 90% aqueous acetonitrile (0.1% formic acid) in 1 min. The system was then washed with 90% aqueous acetonitrile (0.1% formic acid) for 5 min and the equilibrated with 2% aqueous acetonitrile (0.1% formic acid).High resolution TOF MS mode was used to collect scans in positive mode from 350 to 1200 Da for 250 ms. MS/MS data was collected using information-dependent acquisition (IDA) with the following criteria: the 10 most intense ions with +2 to +5 charge states and a minimum of intensity of 200 counts-per-second (cps) were chosen for analysis, using dynamic exclusion for 20 s, 250 ms acquisition and a fixed collision energy setting of 50 ± 5 V.
Mascot analysis and extracted ion chromatogram (XIC) generation
Peptides were identified using Mascot version 2.4.1. (Matrix Science, London), selecting the fixed modification of carbomidomethyl (C) and variable modifications; deamidation, phosphoserine, phosphotyrosine, phosphotheonine and oxidized methioine. In all cases the data was searched against the Swissprot data base (v. SwissProt_2013_08) using Mammalia taxonomy. Both the peptide and the fragment tolerance were set at 0.1 Da, choosing +2, +3 and +4 charge states and allowing for one missed cleave by trypsin. Phosphopeptides were manually validated from raw data files and manual de novo sequencing and XIC were generated using PeakView (ABSciex). using a 0.05 Da window, and where necessary Gaussian smoothed with a 5 point window and baseline subtracted with a 3 min window.Results and discussion
Characterization of Magnetite-PDMS substrates
The aim of this work was to develop a simple, moldable, flexible, rapidly produced MOAC sorbent for phosphopeptide enrichment that is compatible with microfluidic technologies and that could also be employed within a conventional laboratory to create microwell arrays. PDMS is the most ubiquitously used material in the LOC field, with a number of advantageous characteristics,37 and was employed to create a composite metal oxide–polymer for surface. A series of substrates was produced using various particle:PDMS doping ratios to determine the optimum doping level. Prepolymer mixtures with particle loadings greater than 1:1 were too viscous for reliable casting and so substrates with particle:PDMS ratios of 1:1, 1:2 and 1:4 were characterized. These mixtures can be used as normal PDMS for replica molding, including casting over microfabricated structures. The fabrication process and example of such structures, which demonstrates that this composite PDMS material can easily be incorporated into microfluidic systems, are shown in Fig. 1.
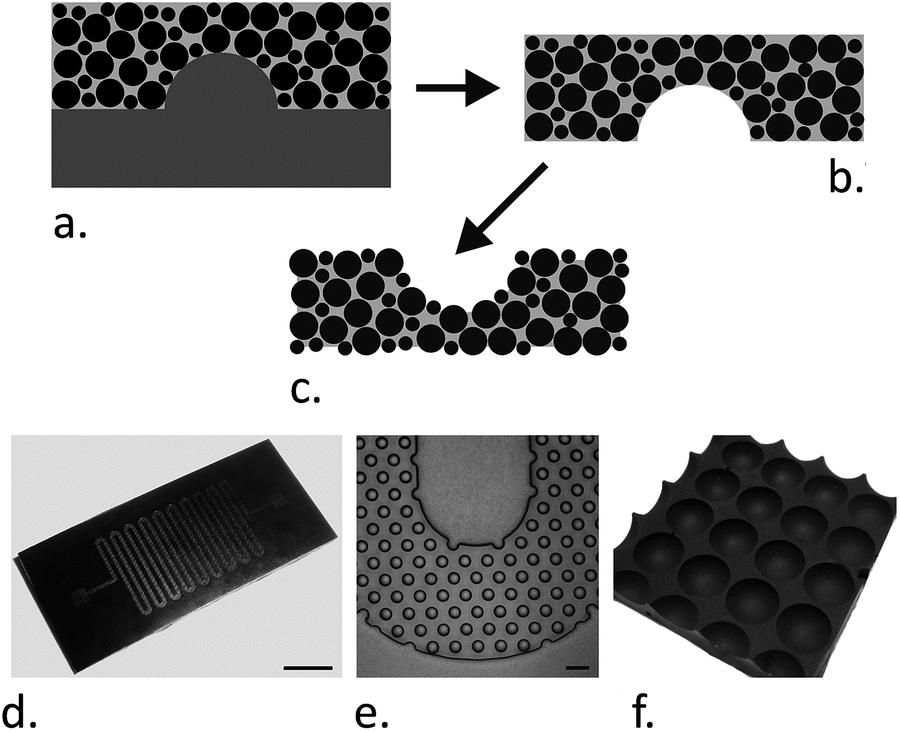 | ||
| Fig. 1 Fabrication of magnetite-PDMS structures. (a–c) The fabrication process. PDMS prepolymer is mixed with magnetite particles and cast over a suitable mold (a). After curing, the magnetite-PDMS is peeled off (b) and the surface PDMS etched back (c) to expose the embedded magnetite particles. (d and e) show an example of a microfluidic channel (50 μm deep) fabricated from magnetite-PDMS (1:1 particle:PDMS ratio). The mold used for casting this channel was fabricated as previously described.29 The scale bar in photograph (d) is 3 mm. Micrograph (e) shows a magnified region of the serpentine channel packed with 50 μm diameter internal pillars, the scale bar is 100 μm. (f) A photograph of a 4 × 4 well array with 96-well plate spacing (the centre–centre distance is 9 mm). | ||
For characterization of the surface by SEM and AFM, the magnetite-PDMS was cast over a polished silicon wafer to produce a substrate with an initially smooth, level surface. Following curing, the substrates were etched using a TBAF solution,38 resulting in the removal of the surface PDMS to expose the embedded magnetite particles (Fig. 2a). Increasing the particle doping level increased the concentration of particles at the surface (Fig. 2a), with a 1:1 particle:PDMS ratio producing a highly roughened surface that is predominantly composed of magnetite particles. The rms surface roughness of this substrate was measured by AFM to be 264 ± 74 nm, with a peak-valley height of 1.25 ± 0.22 μm (both mean ± standard error, taken from the measurement of seven 5 × 5 μm regions across the substrate). For all results reported below, a magnetite:PDMS ratio of 1:1 was employed.
 | ||
| Fig. 2 SEM and AFM characterization of magnetite-PDMS substrates. (a) SEM images taken at 400× and 10000× magnification of three substrates with different magnetite:PDMS ratios. (b) A typical AFM image of 1:1 magnetite-PDMS substrate. | ||
Enrichment of phosphopeptides
To demonstrate that the fabrication procedure did not affect the phosphopeptide binding properties of the magnetite, and to optimize the adsorption and elution conditions, enrichment of a β-casein digest was used, which allowed comparison with previous work.18,19 To enable rapid optimization of multiple enrichment parameters, magnetite-PDMS microwell arrays with a volume of approximately 100 μl were created using the optimum 1:1 particle:PDMS doping ratio by casting the magnetite-PDMS over the back of a 96-well cell culture plate (Fig. 1f). After etching back the surface, phosphopeptide enrichment of a tryptic digest of β-casein was carried out. The enriched and unenriched fractions were analysed by MALDI-TOF mass spectrometry and the results compared to those obtained previously. Samples were loaded into the wells in an acidic buffer to minimize the binding of non-phosphorylated peptides and incubated for the required time. The wells were then briefly washed with buffers containing ACN, which lowers the surface tension of these solutions allowing them to better wet the surface, before bound phosphopeptides were eluted in 0.1 M ammonium hydroxide.
Eluted fractions were analyzed with MALDI-MS (sample spectra in ESI Fig. S-1, peptide data in ESI Table 1†). Following magnetite-PDMS enrichment, the strong signals from non-phosphopeptide peaks are very significantly reduced, and clear signals from both phosphopeptides can be seen. The identity of the phosphopeptides was confirmed by MSMS (data not shown). This high level of enrichment is very similar to that reported before for magnetite enrichment,18,19 demonstrating that the procedure to generate the sorbent does not affect its binding properties. Higher ammonium hydroxide concentrations in the elution buffer (0.4 M or 1 M) did not improve the data. Adding 10% ACN to the eluent improved its surface wetting properties but the enrichment results obtained were poorer, with no phosphopeptides being clearly detected at the same laser power (ESI Fig. S-2†). Magnetite is a fairly well characterized substrate for phosphopeptide enrichment.18,19 As with other iron-based enrichments substrates, it shows a slightly different bias in physiochemical properties of the enriched peptides to the more commonly used TiO2 substrates,39 with a stronger enrichment of more acidic phosphopeptides, although it is still able to enrich a broad range of phosphopeptides with high selectivity.
Optimisation of adsorption and elution conditions
The effect of both analyte concentration and incubation time was assessed using β-casein peptides. To quantify the enrichment levels, the normalized MALDI peak intensities were extracted for seven non-phosphopeptide peaks and five phosphopeptide peaks. The enrichment level was then expressed as the percentage of the summed phosphopeptide intensities with respect to the sum of the intensities of both phosphopeptide and non-phosphopeptide peaks (%phospho, where 100% signifies the detection of only phosphopeptides). The %phospho value obtained when analyzing a series of 50 ng samples of the unenriched β-casein digest was 0.84% ± 0.21% (mean ± SEM, n=17).Sample loadings from 5 to 100 ng (corresponding to low pmol to fmol quantities of sample) and incubation times from 2 to 20 min were tested. The mean %phospho values obtained are reported in Fig. 3 (n ≥ 3 for each experimental condition). These demonstrate that enrichment of the phosphopeptides is maintained across these conditions, and is good even at lower sample loadings and short incubation times. A 5 min sample incubation produced high enrichment levels for all loading conditions with good reproducibility (mean over all loadings was 96.3 ± 3.1%, n = 13). Lower sample loadings down to 5 ng did not significantly compromise enrichment, but higher sample loadings (i.e. 100 ng) or extended incubation times (i.e. 20 min) reduced enrichment and increased sample to sample variation. The use of the microwell array format was particularly beneficial for rapidly optimizing the enrichment protocol.
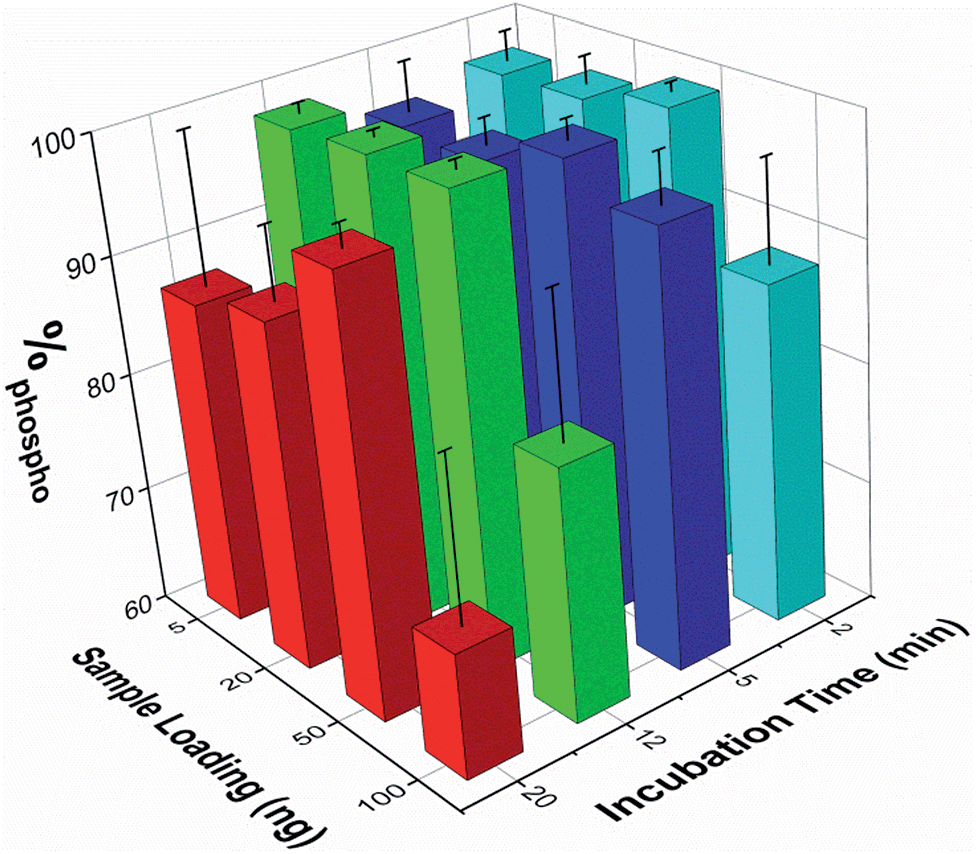 | ||
| Fig. 3 Variation in phosphopeptide enrichment levels with sample loading and incubation time. A summary of the mean %phospho values obtained for different sample loadings (the mass of β-casein protein used to produce the clarified digest samples) and sample incubation times. A script was written in Matlab (Mathworks) to extract the intensities of each of these peaks and to return a value corresponding to the percentage of the summed phosphopeptide signal intensity relative to the total intensity of all 12 peaks. The error bars correspond to +/− one standard error (n ≥ 3). | ||
The optimum loading level and incubation time is likely to vary for different samples since it is important to get the correct balance between phosphopeptide enrichment and non-specific adsorption, which appears to increase with longer incubation times or loading highly concentrated samples, possibly due to differential kinetics of binding. The microwell array format reported here is well suited to rapidly optimizing the enrichment protocol, as several different sample loadings or other experimental conditions, each requiring only a low volume sample, can be performed in parallel.
Reuse of substrate
One distinct advantage of this system is that following enrichment the magnetite-PDMS substrates can readily be re-etched to reveal a fresh surface, enabling a substrate to be re-used many times. Four etching-enrichment cycles have been tested, each fresh surface having successfully enriched phosphopeptides with no discernible loss of affinity, and many more cycles could be performed as the magnetite particle concentration is uniform throughout the substrate. One previously identified drawback of PDMS is the potential for leaching of low molecular weight (LMW) siloxanes, which could result in sample contamination.40–43 Therefore, spectra obtained from four separate elution fractions were extensively searched for signs of LMW siloxane contamination (details in ESI Table 2†), over an increased mass range (100–3600 m/z). However, no evidence of contamination attributable to PDMS oligomers was found, probably due to a combination of the extended 95 °C bake (which should enhance PDMS crosslinking), the high level of magnetite particle doping (which results in a reduced PDMS volume fraction) and the etching of the surface PDMS (which creates a surface that is primarily composed of magnetite).Enrichment from complex samples
To demonstrate the use of the substrate for a more complex sample, phosphopeptides were enriched from an α-casein spiked HeLa lysate to show the degree of enrichment, and from a normal HeLa lysate for general phosphopeptide enrichment, using the optimized procedure described above. It was found that for the complex samples, the optimal sample loading per well was higher, with 1 μg per well giving good results, but phosphopeptides could be detected down to 100 ng of sample. Samples were analyzed by LC-ESI-MSMS followed by generation of extracted ion chromatograms (XIC) for relative quantification for a number of phospho and non-phosphopeptides. As described for the pure β-casein digest, the magnetite doped substrate showed a high degree of enrichment of α-casein phosphopeptides from the spiked lysate (details of peptides in ESI Table S-3†), with only traces of the non-phosphopeptides remaining (Fig. 4).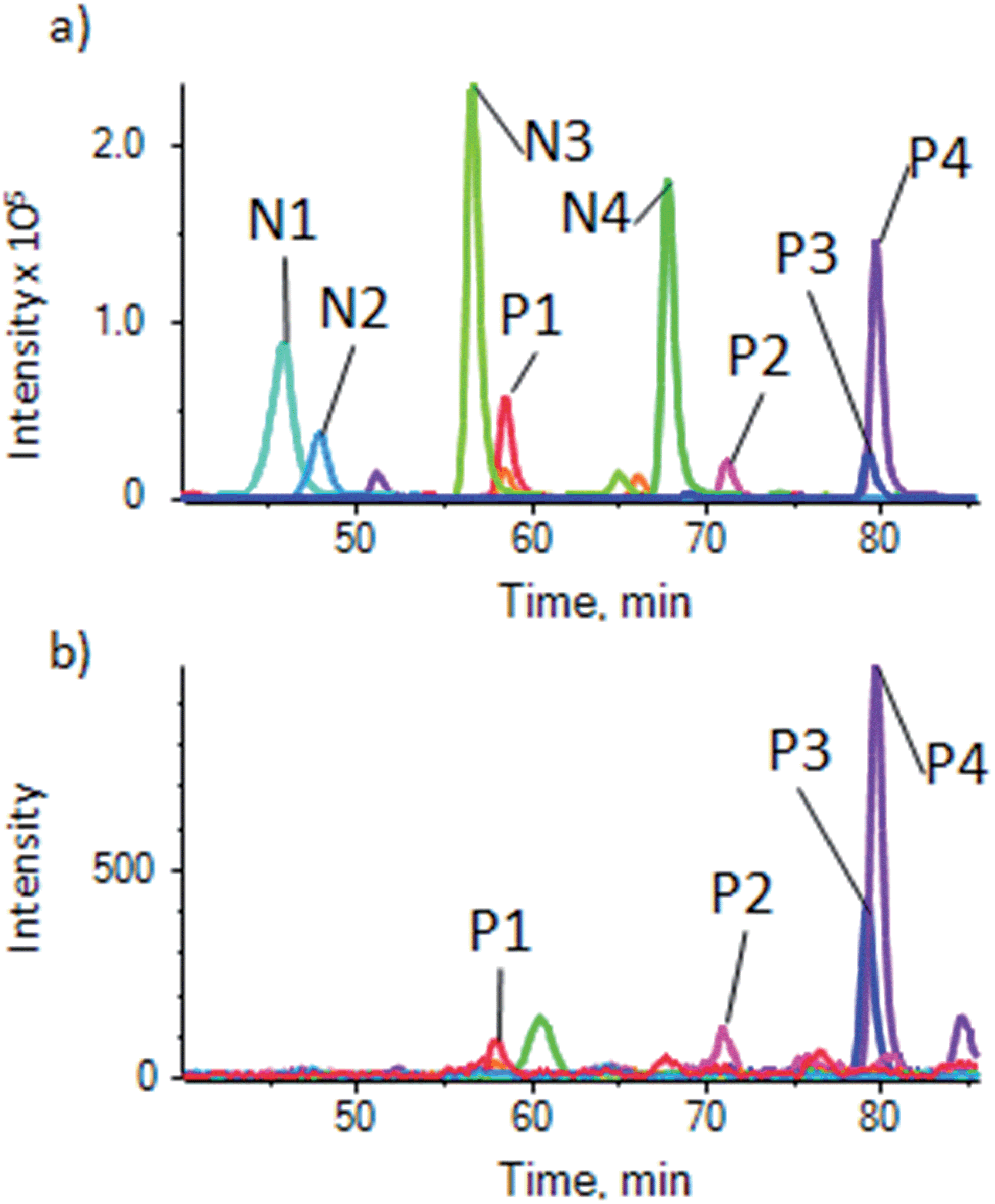 | ||
| Fig. 4 Enrichment of α-casein phosphopeptides from a casein spiked HeLa cell lysate. (a) XIC traces for unenriched control showing 4 phosphopeptides (P1–P4) and 4 non-phosphopeptides (N1–N4) (details of peptides in ESI table S-3a†) and (b) sample enriched using the magnetite-PDMS substrate showing XIC of the same peptides. Peaks for non-phosphopeptides are very much reduced or absent. | ||
To demonstrate the use of the substrate for enrichment of phosphopeptides from a more general sample, and to make a comparison with enrichment using the most commonly used metal oxide, TiO2, phosphopeptides were enriched from a HeLa lysate using a commercial TiO2 based kit (Pierce Magnetic Phosphopeptide Enrichment Kit) and our substrate. Enrichment of phosphopeptides was seen in both cases. XIC were generated for a number of phosphopeptides with a range of physicochemical properties (ESI Table S-3b†) that were identified from a MASCOT search of the data, to demonstrate enrichment and compare the two substrates. Fig. 5 shows the XIC for 10 phosphopeptides using the magnetite doped PDMS (Fig. 5a) and the commercial TiO2 kit (Fig. 5b). For comparison, Fig. 5c shows the XIC generated at the same masses as the phosphopeptides used to generate Fig. 5a and b from an unenriched HeLa lysate. A number of isobaric, non-phosphorylated peptides are picked up, and peaks corresponding to the phosphopeptides cannot be seen, clearly demonstrating the enrichment in Fig. 5a and b. There is, therefore, significant enrichment using both substrates, but as has been noted before, there are some differences in enrichment efficiencies for peptides with different physicochemical properties between the two different sorbants. Differences in intensities between Fig. 5a and b relate to the significant difference in sorbent surface areas between the two methods, resulting in lower capacity for the magnetite embedded system than the large volume of particulate TiO2 used in the commercial kit. However, the magnetite doped PDMS appears to perform well, and the processing of the substrate does not appear to have affected its ability to enrich phosphopeptides.
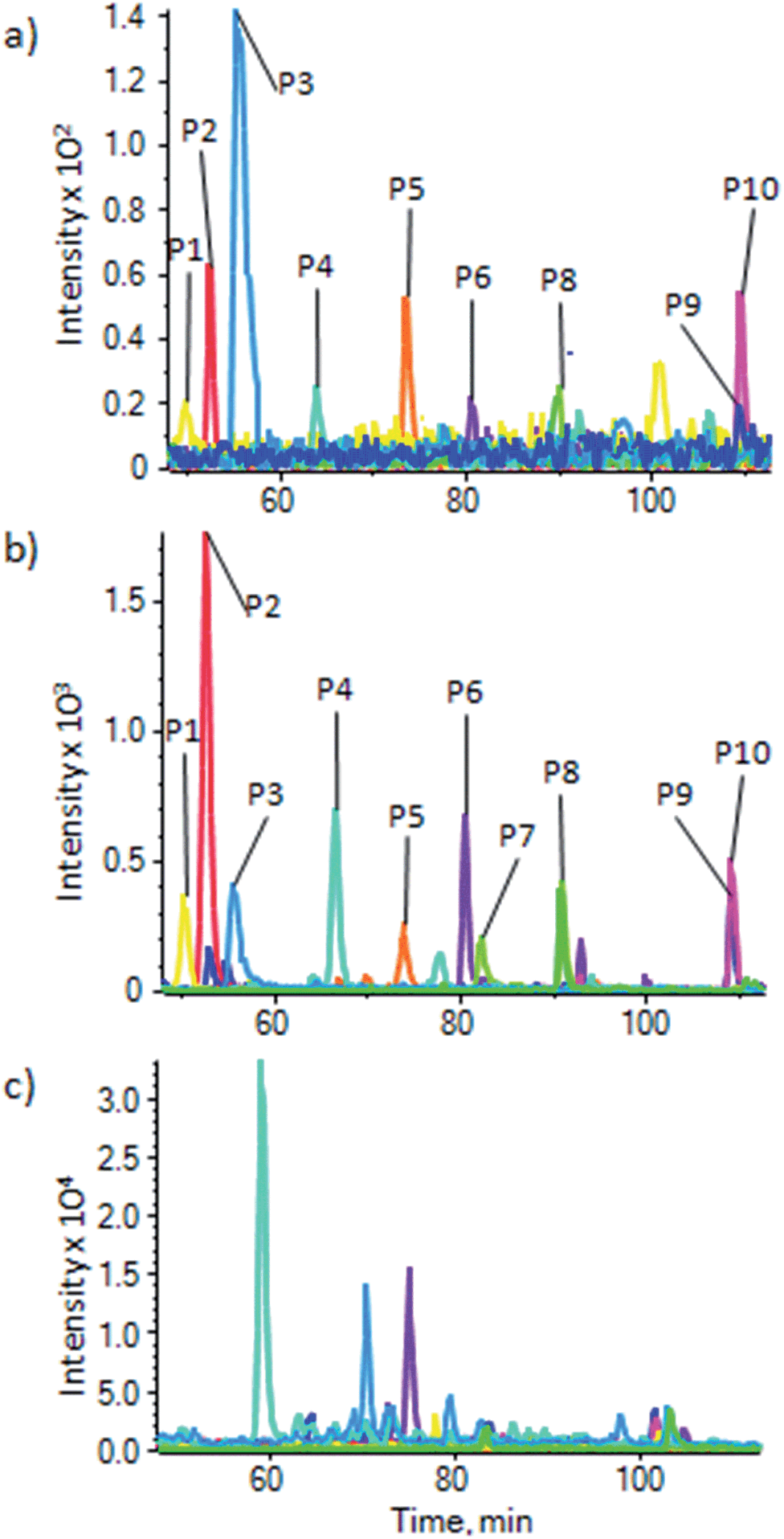 | ||
| Fig. 5 Enrichment of phosphopeptides from a HeLa cell lysate. XIC for 10 phosphopeptides from a HeLa cell lysate (P1–P10, details in ESI Table S-3b†) using (a) our substrate and (b) a commercial TiO2 based kit (Pierce), showing a similar enrichment, but with some differing affinities between the two methods. (c) XIC for the same masses as (a) and (b) but in an unenriched HeLa lysate; this shows peptides isobaric to the phosphopeptides eluting at different times to the phosphopeptides, which are not seen in (a) and (b), and no traces of the phosphopeptides, demonstrating the high levels of enrichment for both methods. | ||
Conclusions
A new magnetite-PDMS based material for performing phosphopeptide enrichment is reported. This uses a rapid and straightforward protocol, with key enrichment parameters having been optimised. Levels of phosphopeptide enrichment similar to those seen previously for magnetite were obtained. The principal benefits of this composite polymeric material, whose surface can be simply refreshed to enable reuse, are its compatibility with LOC systems and the ability to simply produce large-scale, low-volume microwell arrays for high throughput analysis. The simple enrichment protocol, which involves no particle suspensions (thereby minimizing the risk of particle contamination) or centrifugation steps, and the benefits of being able to re-etch the substrate, means that this magnetite-PDMS provides a cheap, rapid and straightforward approach for phosphopeptide enrichment that is amenable to LOC methods, high throughput analysis and automation.Acknowledgements
Funding for this work was from the RASOR (“Radical Solutions for Researching the proteome”) BBSRC IRC programme (BB/C511572/1) and the Proxomics EPSRC Cross-Disciplinary Research Landscape Award (EP/I017887/1). The authors thank Dr Michele Zagnoni for help in writing the Matlab script used for data analysis and Dr Huabing Yin for assistance in acquiring the AFM images.Notes and references
- K. A. Resing and N. G. Ahn, FEBS Lett., 2005, 579, 885–889 CrossRef CAS PubMed.
- H. Steen, J. A. Jebanathirajah, J. Rush, N. Morrice and M. W. Kirschner, Mol. Cell. Proteomics, 2006, 5, 172–181 CAS.
- A. Tichy, B. Salovska, P. Rehulka, J. Klimentova, J. Vavrova, J. Stulik and L. J. Hernychova, Proteomics, 2011, 74, 2786–2797 CAS.
- A. Leitner, TrAC, Trends Anal. Chem., 2010, 29, 177–185 CrossRef CAS PubMed.
- J. D. Dunn, G. E. Reid and M. L. Bruening, Mass Spectrom. Rev., 2010, 29, 29–54 CAS.
- K. Engholm-Keller and M. R. Larsen, J. Proteomics, 2011, 75, 317–328 CrossRef CAS PubMed.
- H. Matsuda, H. Nakamura and T. Nakajima, Anal. Sci., 1990, 6, 911–912 CrossRef CAS.
- I. Kuroda, Y. Shintani, M. Motokawa, S. Abe and M. Furuno, Anal. Sci., 1990, 20, 1313–1319 CrossRef.
- M. W. H. Pinkse, P. M. Uitto, M. J. Hilhorst, B. Ooms and A. R. J. Heck, Anal. Chem., 2004, 76, 3935–3943 CrossRef CAS PubMed.
- M. R. Larsen, T. E. Thingholm, O. N. Jensen, P. Roepstorff and T. J. Jorgensen, Mol. Cell. Proteomics, 2005, 5, 873–886 Search PubMed.
- H. C. Hsieh, C. Sheu, F. K. Shi and D. T. Li, J. Chromatogr. A, 2007, 1165, 128–135 CrossRef CAS PubMed.
- S. S. Liang and S. H. Chen, J. Chromatogr. A, 2009, 1216, 2282–2287 CrossRef CAS PubMed.
- M. P. Alcolea and P. R. Cutillas, Methods Mol. Biol., 2010, 658, 111–126 CAS.
- G. Pocsfalvi, M. Cuccurullo, G. Schlosser, S. Scacco, S. Papa and A. Malorni, Mol. Cell. Proteomics, 2007, 6, 231–237 CAS.
- B. Lin, T. Li, Y. Zhao, F. K. Huang, L. Guo and Y. Q. Feng, J. Chromatogr. A, 2008, 1192, 95–102 CrossRef CAS PubMed.
- S. Lü, Q. Luo, X. Li, J. Wu, J. Liu, S. Xiong, Y. Q. Feng and F. Wang, Analyst, 2010, 135, 2858–2863 RSC.
- M. Rainer, H. Sonderegger, R. Bakry, C. W. Huck, S. Morandell, L. A. Huber, D. T. Gjerde and G. K. Bonn, Proteomics, 2008, 8, 4593–4602 CrossRef CAS PubMed.
- Y. Huang, Q. Shi, C. K. Tsung, H. P. Gunawardena, L. Xie, Y. Yu, H. Liang, P. Yang, G. D. Stucky and X. Chen, Anal. Biochem., 2011, 408, 19–31 CrossRef CAS PubMed.
- A. Lee, H. J. Yang, E. S. Lim, J. Kim and Y. Kim, Rapid Commun. Mass Spectrom., 2008, 22, 2561–2564 CrossRef CAS PubMed.
- Y. Li, Y. Liu, J. Tang, H. Lin, N. Yao, X. Shen, C. Deng, P. Yang and X. Zhang, J. Chromatogr. A, 2007, 1172, 57–71 CrossRef CAS PubMed.
- C. T. Chen, W. Y. Chen, P. J. Tsai, K. Y. Chien, J. S. Yu and Y. C. Chen, J. Proteome Res., 2007, 6, 316–325 CrossRef CAS PubMed.
- A. Eriksson, J. Bergquist, K. Edwards, A. Hagfeldt, D. Malmström and V. A. Hernández, Anal. Chem., 2010, 82, 4577–4583 CrossRef CAS PubMed.
- G. R. Blacken, M. Volný, T. Vaisar, M. Sadílek and F. Tureček, Anal. Chem., 2007, 79, 5449–5456 CrossRef CAS PubMed.
- M. L. Niklew, U. Hochkirch, A. Melikyan, T. Moritz, S. Kurzawski, H. Schlüter, L. Ebner and M. W. Linscheid, Anal. Chem., 2010, 82, 1047–1053 CrossRef CAS PubMed.
- H. Wang, J. Duan and Q. Cheng, Anal. Chem., 2011, 83, 1624–1631 CrossRef CAS PubMed.
- P. S. Dittrich and A. Manz, Nat. Rev. Drug Discovery, 2006, 5, 210–218 CrossRef CAS PubMed.
- H. Lu, M. A. Schmidt and K. F. Jensen, Lab Chip, 2005, 5, 23–29 RSC.
- C. Martino, M. Zagnoni, M. E. Sandison, M. Chanasakulniyom, A. R. Pitt and J. M. Cooper, Anal. Chem., 2011, 83, 5361–5368 CrossRef CAS PubMed.
- M. E. Sandison, S. A. Cumming, W. Kolch and A. R. Pitt, Lab Chip, 2010, 10, 2805–2813 RSC.
- J. Wen, E. W. Wilker, M. B. Yaffe and K. F. Jensen, Anal. Chem., 2010, 82, 1253–1260 CrossRef CAS PubMed.
- D. Wu, J. Qin and B. Lin, J. Chromatogr. A, 2008, 1184, 542–559 CrossRef CAS PubMed.
- S. Mohammed, K. Kraiczek, M. W. Pinkse, S. Lemeer, J. J. Benschop and A. J. Heck, J. Proteome Res., 2008, 7, 1565–1571 CrossRef CAS PubMed.
- R. Raijmakers, K. Kraiczek, A. P. de Jong, S. Mohammed and A. J. Heck, Anal. Chem., 2010, 82, 824–832 CrossRef CAS PubMed.
- K. Tsougeni, P. Zerefos, A. Tserepi, A. Vlahou, S. D. Garbis and E. Gogolides, Lab Chip, 2011, 11, 3113–3120 RSC.
- P. Augustsson, J. Persson, S. Ekström, M. Ohlin and T. Laurell, Lab Chip, 2009, 9, 810–818 RSC.
- D. T. Eddington, J. P. Puccinelli and D. J. Beebe, Sens. Actuators, B, 2006, 114, 170–172 CrossRef CAS PubMed.
- J. Zhou, A. V. Ellis and N. H. Voelcker, Electrophoresis, 2010, 31, 2–16 CrossRef CAS PubMed.
- S. Takayama, E. Ostuni, X. Qian, J. C. McDonald, X. Jiang, P. LeDuc, M. H. Wu, D. E. Ingber and G. M. Whitesides, Adv. Mater., 2001, 13, 570–574 CrossRef CAS.
- B. Bodenmiller, L. N. Mueller, M. Mueller, B. Domon and R. Aebersold, Nat. Methods, 2007, 4, 231–237 CrossRef CAS PubMed.
- K. J. Regehr, M. Domenech, J. T. Koepsel, K. C. Carver, S. J. Ellison-Zelski, W. L. Murphy, L. A. Schuler, E. T. Alarid and D. J. Beebe, Lab Chip, 2009, 9, 2132–2139 RSC.
- P. Kaali, D. Momcilovic, A. Markström, R. Aune, G. Czel and S. Karlsson, J. Appl. Polym. Sci., 2010, 115, 802–810 CrossRef CAS PubMed.
- S. M. Hunt and G. A. George, Polym. Int., 2000, 49, 633–635 CrossRef CAS.
- X. Sun, R. T. Kelly, K. Tang and R. D. Smith, Analyst, 2010, 135, 2296–2302 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4an00750f |
| This journal is © The Royal Society of Chemistry 2014 |