
Effect of support and solvent on the activity and stability of NiCoB amorphous alloy in cinnamic acid hydrogenation
Abstract
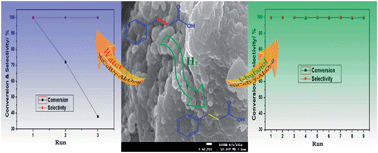
Selective hydrogenation of cinnamic acid was studied over different supported NiCoB amorphous alloys; a γ-Al2O3 supported NiCoB catalyst showed particularly good activity. The application of ultrasound during catalyst preparation was found to make the Ni active sites more dispersed, thus enhancing the catalyst activity. The NiCoB/γ-Al2O3-u catalyst so obtained could be recycled effectively for nine runs in tert-butanol, in contrast it deactivated after only three runs in water. XPS, SEM and XRD characterizations indicated that loss of Ni and hydration of the γ-Al2O3 support were the main reasons for catalyst deactivation in water. Thus, an efficient and stable catalytic system involving NiCoB/γ-Al2O3-u and tert-butanol was established for cinnamic acid hydrogenation in this study.
 Please wait while we load your content...
Please wait while we load your content...

