DOI:
10.1039/C4RA01214C
(Paper)
RSC Adv., 2014,
4, 14335-14347
Interaction of human serum albumin with liposomes of saturated and unsaturated lipids with different phase transition temperatures: a spectroscopic investigation by membrane probe PRODAN†
Received
20th November 2013
, Accepted 4th March 2014
First published on 4th March 2014
Abstract
The interaction of human serum albumin (HSA) with liposomes made of saturated and unsaturated phosphocholines having distinctly different phase transition temperatures has been studied using circular dichroism (CD), steady state and time resolved fluorescence spectroscopic techniques. We used 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) as the saturated lipid and 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 2-oleoyl-1-palmitoyl-sn-glycero-3-phosphocholine (POPC) as the unsaturated lipid to prepare liposomes. The CD measurement reveals that the liposomes induce some kind of stabilization in HSA. The steady state and time resolved fluorescence spectra of PRODAN (6-propionyl 1,2-dimethylaminonaphthalene) were monitored to unravel the interaction between liposome and HSA. We observed that HSA partially penetrates the liposomes due to hydrophobic interaction and destabilizes the packing order of the lipid bilayer leading to leakage of the probe molecules from the liposome. It was found that HSA preferably penetrates into the liposomes, which are less prehydrated at room temperature. Thus penetration is higher in DPPC and DMPC liposomes as these liposomes are less prehydrated due to higher phase temperature (43 °C and 23 °C respectively). On the other hand HSA has less penetration in DOPC and POPC liposomes because these liposomes are more hydrated owing to lower phase transition temperature (−20 °C and −2 °C respectively). The time resolved fluorescence measurements revealed that penetration of HSA into liposomes brings about release of PRODAN molecules. Incorporation of HSA in all the liposomes results in a significant increase in the rotational relaxation time of PRODAN. This fact confirms that HSA penetrates into the liposome and forms a bigger complex.
1. Introduction
Plasma membrane is a complicated assembly of lipids and proteins, organized into various specialized microdomains with versatile diversity.1 To overcome the problems associated with this diversity it is worthwhile to use synthetic liposomes or vesicles which mimic the geometry and topology of cell membranes.2 Phospholipids form the fundamental matrix of natural membranes and represent the environment in which many proteins and various macro molecules display their activity.3 Therefore characterization of lipid membranes with sufficient selectivity will help to study the variation of its bulk properties.4 Owing to their small size, amphiphilic character, and biocompatibility, liposomes or vesicles are promising systems for drug delivery through the blood stream.5 Therefore it is necessary to visualize the stability of liposomes in presence of serum proteins.6 Human Serum Albumin (HSA) is the most prominent component of blood plasma. It serves as transport protein for several endogenous and exogenous ligands as well as for various drug molecules.7,8 HSA also binds well with fatty acids.9 It is reported that proteins partially penetrate and deform the lipid bi-layer.10 HSA penetrates into the vesicle and gets adsorbed on the surface of vesicles to some extent. Packing of hydrophobic tails of the lipid is also disturbed in presence of HSA.11 Charbonneau et al. suggested that both hydrophobic and hydrophilic interactions occur for liposome–HSA systems.12 Various groups suggested that for prolonged circulation of liposomes in blood stream and to provide stability, cholesterol should be incorporated in the vesicle.13
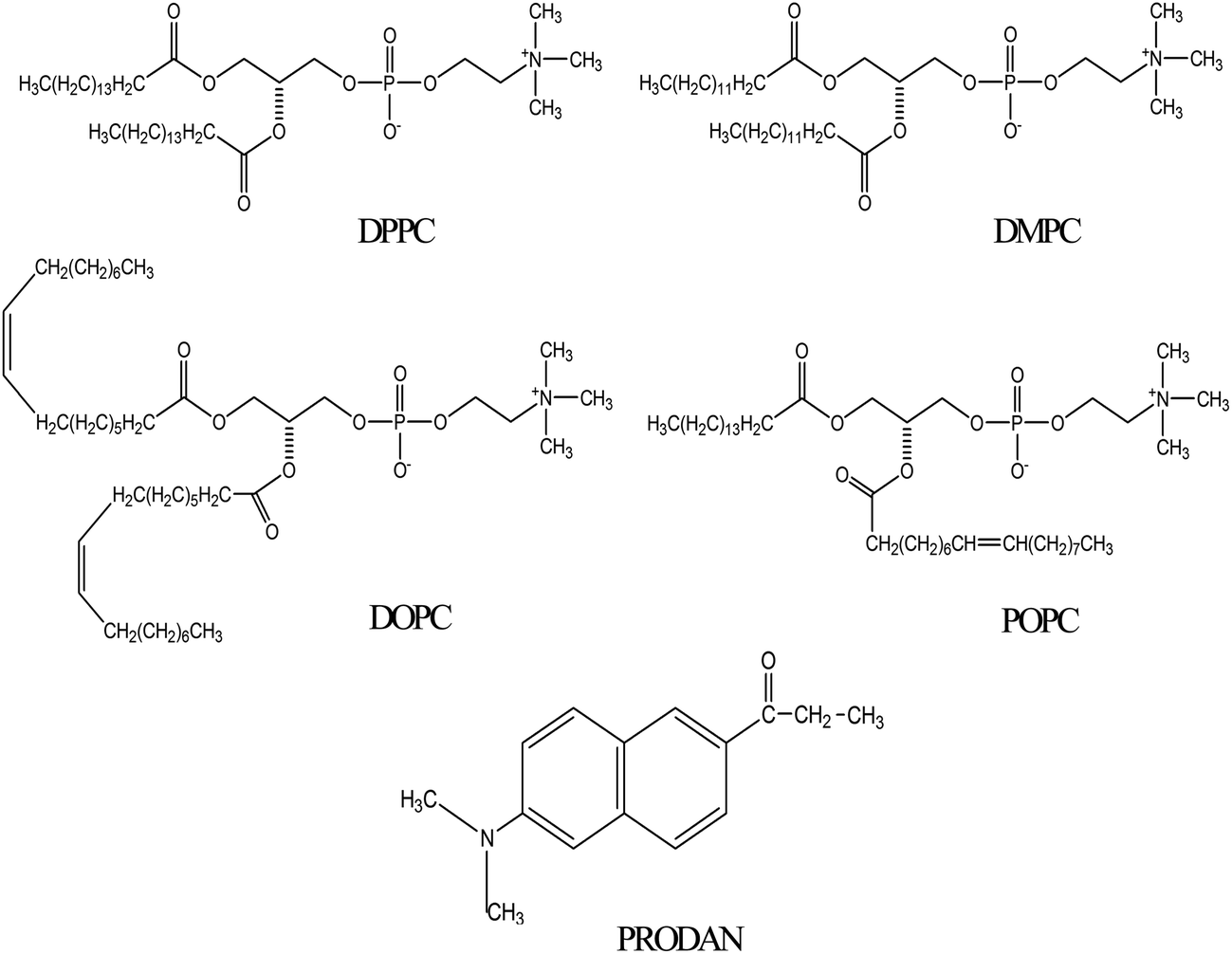
Although there are a few reports regarding liposome–HSA system,10–12 however, none of the studies addressed the nature of interaction between liposome and HSA by fluorescence spectroscopy using a polarity sensitive membrane probe. Moreover, it was not answered what will be fate of encapsulated molecules inside the liposome upon interaction with HSA. Therefore, it is desirable to undertake a study which involves different kind of liposomes. The present work has the novelty because it involves four different phosphatidylcholines lipids with zwitterionic head groups. These lipids are widely different in terms of their phase transition temperature and nature of their acyl chain. 1,2-Dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) are saturated phospholipids while 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 2-oleoyl-1-palmitoyl-sn-glycero-3-phosphocholine (POPC) contain unsaturation in their acyl chain (Scheme 1). Phosphatidylcholines are dominant in eukaryotic membranes.14 The lipids of more metabolically active membranes are considerably more unsaturated. POPC bilayers provide relevant models for the matrix of the endoplasmic reticulum.15 DPPC exhibits properties very similar to those of sphingomyelin which is the most abundant lipid in plasma membrane.16 In our previous studies, we encapsulated anticancer drug ellipticine in DPPC vesicles and studied its release by various bile salts.17 The present study is done to reveal protein–liposome interaction and the transport of various drugs through lipid bilayers via Human Serum Albumin (HSA) to the target site with the help of fluorescence spectroscopy. Fluorescence spectroscopy has several advantages including a high sensitivity, a noninvasive nature, an intrinsic time scale and an excellent response to the physical properties of membrane.18 For this purpose PRODAN (Scheme 1) has been chosen as a probe molecule primarily to study the environment inside the liposomes and to reveal the liposome–HSA interaction. PRODAN is very sensitive towards environmental polarity and the origin of its solvatochromatic nature have been debated.19 It shows large spectral shifts when attached to membranes.20 The sensitivity of emission properties of PRODAN towards polarity is attributed to a large difference between the dipole moments in its ground (S0) and excited (S1) states.21 According to various calculations, difference in dipole moment of PRODAN from 5.50 to 10.20 D causes a shift in the S0 → S1 transition.22a Recently Samanta and co-workers22b have suggested this value to be 4.40 to 5.0 D based on transient dielectric loss measurements. In PRODAN, both locally excited (LE) and twisted internal charge transfer (TICT) states simultaneously exist.23 This amazing feature of PRODAN makes it a useful probe to study structure, function and dynamics of proteins and membranes.20,24 The probe has widely been used to study the dynamics inside a reverse micelles.25 It was reported that in aqueous reverse micelle PRODAN molecules are distributed in three regions according to the polarity of that particular region.25 PRODAN, having higher water solubility is loosely anchored to the bilayer.26 Hof and co-workers studied solvent relaxation of various probes including PRODAN in phosphocholine vesicles.27 The emission maximum of PRODAN depends upon the phase state of phospholipids. It usually emits at 440 nm in gel and at 490 nm in liquid crystalline phase. The shift in emission band from gel to liquid crystalline phase takes place due to dipolar relaxation in liquid crystalline phase of phospholipid but not in gel phase. This dipolar relaxation originates due to a few water molecules present in bilayer at the glycerol backbone where fluorescence moiety of PRODAN actually resides.28 The complex character of emission peak of PRODAN in the lipid bilayer can be explained assuming that both twisted and planar configuration emit in the bilayer.27c As emission of PRODAN is highly sensitive, it has been used to study different protein molecules. Chattopadhyay et al. have reported the red edge excitation spectra (REES) of PRODAN in different proteins like spectrins.29 Hydration dynamics studies of HSA using PRODAN as probe reveals that hydration level of different domains is different and they have different time scales for hydration.30 HSA contains only one tryptophan residue at position 214 (Trp214) in domain II and one free cystine residue at position 34 in domain I, moreover it has 17 disulphide bonds.31 The free thiol group allows site specific labeling of protein with chromophoric or fluorescent probes.32 PRODAN binds with HSA within Sudlow site I i.e. on warfarin binding site.33 So far, interaction of PRODAN with liposome and HSA is reported individually. But interaction of HSA and liposome is still unexplored using a membrane probe. Therefore, photophysics of PRODAN by steady state and time resolved spectroscopy will not only be able to reveal the environment inside the HSA, liposome and liposome–HSA complex but will also give a new insight regarding the interaction between liposomes and HSA. This study may further help in designing a novel drug delivery system for various drugs exhibiting properties very similar to PRODAN.
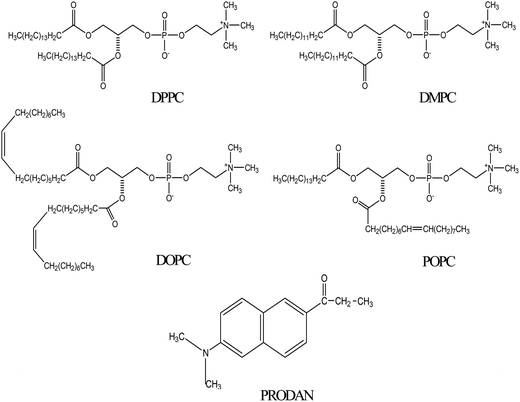 |
| | Scheme 1 | |
2. Experimental section
2.1. Materials
PRODAN, HSA, urea and all the lipids (DPPC, DMPC, POPC and DOPC) were purchased from Sigma-Aldrich. Na2HPO4 and NaH2PO4 were purchased from Merck. All the chemicals were used without further purification. All the experiments were performed in Milli Q water. Stock solution of PRODAN was prepared in methanol. Required amount of methanolic solution was taken in a volumetric flask and dried under vacuum to create a thin film of PRODAN. An appropriate amount of phosphate buffer (25 mM) was added to it and was sonicated for two hours. Small Unilamellar Vesicles (SUV) were prepared by ethanol injection method as described earlier.17 The stock solution of lipid was prepared in ethanol. The desired amount of ethanolic lipid solution was rapidly injected into the aqueous solution of PRODAN (above the phase transition temperature of respective lipids) and was equilibrated for 60 minutes. The concentration of lipid in the solution was 0.4 mM and the percentage of ethanol was less than 1% (v/v). The molar ratio of PRODAN to lipid was around 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200. Required amount of HSA was added to the solution of SUV to prepare stock solution of HSA and Lipid. This solution was incubated for 30 minutes before the measurements.
:200. Required amount of HSA was added to the solution of SUV to prepare stock solution of HSA and Lipid. This solution was incubated for 30 minutes before the measurements.
2.2. Spectroscopic measurements
Steady state absorption spectra were taken in a Varian UV-Vis spectrometer (Model: Cary 100). Emission spectra were taken in a Fluoromax-4p fluorimeter from Horiba Jobin Yovon (Model: FM-100). The samples were excited at 375 nm. The fluorescence spectra were corrected for the spectral sensitivity of the instrument. The excitation and emission slits were 2/2 nm for the emission measurements. All the measurements were done at 25 °C.
For the time resolved studies, we used a picosecond time correlated single photon counting (TCSPC) system from IBH (Model: Fluorocube-01-NL). The experimental setup for TCSPC has been described elsewhere.34 The samples were excited at 375 nm using a picosecond diode laser (Model: PicoBrite-375L). The repetition rate was 5 MHz. The signals were collected at magic angle (54.70°) polarization using a photomultiplier tube (TBX-07C) as detector which has a dark counts less than 20 cps. The instrument response function of our setup is ∼140 ps. The data analysis was done using IBH DAS (version 6) decay analysis software. The fluorescence decay was described as a sum of exponential functions:
| |
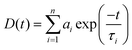 | (1) |
where
D(
t) is the normalized fluorescence decay and
τi are the fluorescence lifetimes of various fluorescent components and
ai are the normalized pre-exponential factors. The amplitude weighted lifetime is given by:
| |
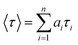 | (2) |
The quality of the fit was judged by reduced chi square (χ2) values and the corresponding residual distribution. To obtain the best fitting in all of the cases, χ2 was kept near to unity. The same setup was used for anisotropy measurements. For the anisotropy decays, we used a motorized polarizer in the emission side. The emission intensities at parallel and perpendicular polarizations were collected alternatively until a certain peak difference between parallel (‖) and perpendicular (⊥) decay was achieved. The same software was also used to analyze the anisotropy data. The time resolved anisotropy was described with the following equation:
| |
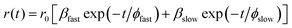 | (3) |
where
r(
t) is the rotational relaxation correlation function and
r0 is the limiting anisotropy and
ϕfast and
ϕslow are the individual rotational relaxation time and
βfast and
βslow are the amplitudes of rotational relaxation time.
2.3. Circular dichroism (CD)
CD is a sensitive technique to monitor the conformational changes in proteins. CD spectra of HSA and its lipid complexes were recorded with a Jasco J-815 spectrometer (Jasco, Tokyo, Japan). For measurements in the far UV region (200–270), a quartz cell with a path length of 0.1 cm (Hellma, Muellheim/Baden, Germany) was used in nitrogen atmosphere. The HSA concentration was kept constant (10 μm) while varying lipid concentration (0.1 mM, 0.3 mM and 0.6 mM). An accumulation of five scans with a scan speed of 20 nm min−1 was performed and data were collected for each sample from 200 to 270 nm. The sample temperature was maintained at 25 °C using Escy temperature controller circulating water bath connected to the water-jacketed quartz cuvettes. Spectra were corrected for buffer signal.
3. Results and discussion
3.1. Interaction of PRODAN with HSA
PRODAN exhibits emission maxima at 520 nm in aqueous buffer solution. Addition of HSA to the buffer solution of PRODAN diminishes the intensity at 520 nm and additionally a band appears at 455 nm. The band at 455 nm is attributed to the LE state of HSA bound PRODAN. The appearance of LE state indicates that PRODAN experiences a less polar environment in HSA. With increase in HSA concentration, the contribution of TICT band decreases while that of LE band increases which implies that more number of PRODAN molecules bind with HSA. Interestingly, we obtained an isoemissive point at around 510 nm which implies that there are two emitting species in the excited state. The emission spectrum of PRODAN in presence of 20 μM of HSA was deconvoluted by a combination of the lognormal functions to show simultaneous existence of CT and LE species (Fig. 1A) by taking the emission spectrum of PRODAN in aqueous buffer solution as reference. We estimated binding constant of PRODAN molecules with HSA using Bensei–Hidlebrand equation for 1:1 complex as following.| |
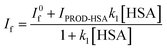 | (4) |
where, I0f is the fluorescence intensity of PRODAN in absence of HSA and I is the fluorescence intensity when all PRODAN molecules form complex with HSA. The nonlinear regression analysis following eqn (4) yields the binding constant (k1) around 7 × 105 M−1 (Fig. 1B). The earlier report29a states that PRODAN binds with HSA at Sudlow site I i.e. on warfarin binding site. This observation is consistent with our experimental result (The data are not shown). The formation of complex between PRODAN and HSA is exothermic with value of ΔH° = −22.82 kJ mol−1.33 It was also reported that PRODAN does not bring about any conformational changes in HSA.29a
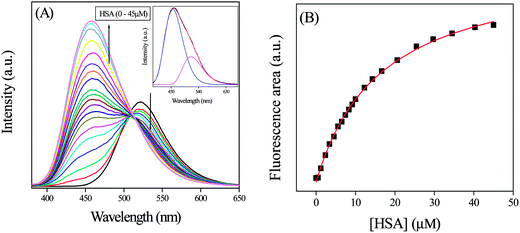 |
| | Fig. 1 (A) The emission spectra of PRODAN at different concentration of HSA. Inset is the emission spectrum of PRODAN in presence of 20 μM HSA that has been deconvoluted in LE and TICT state. (B) The fitted binding curve between PRODAN and HSA following eqn (4). | |
To gain more specific local information about the binding of PRODAN to HSA, we estimated the energy transfer efficiency between PRODAN and HSA by monitoring the emission spectra of Trp214 of HSA (Fig. 2). The distance between Trp214 and PRODAN was estimated from the energy transfer efficiency expression:
| |
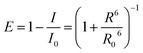 | (5) |
where
I0 and
I are the intensities of Trp214 emission measured for the protein alone and for PRODAN-HSA complex, respectively.
Fig. 2 reveals that the efficiency of energy transfer between Trp214 of HSA and PRODAN is around 43%. Such weak energy transfer indicates that PRODAN molecules bind at a location which is away from tryptophan. In the equality,
R is the distance between Trp214 and PRODAN in Angstrom.
R0 is a characteristic Forster distance for 50% energy transfer efficiency related to the properties of donor and acceptor, and can be calculated using following equation
| | |
R06 = 8.79 × 10−5n−4κ2ϕ0∫ε(λ)f(λ)λ4dλ/∫f(λ)
| (6) |
where,
n is the refractive index of the medium,
κ2 is a geometric factor related to the relative orientation of the transition dipole moments of the donor and acceptor,
ε(
λ) is the molar absorptivity of PRODAN, and
f(
λ) is the normalized fluorescence intensity of Trp214. Therefore,
R0 is calculated from
eqn (6) using geometrical parameter
κ2 as 2/3. These parameters yielded a value for
R0 of 26 Å, leading to an estimate for
R, the apparent distance between Trp214 and PRODAN being 24 Å. We fitted the quenching data with a modified Stern–Volmer equation as follows:
| |
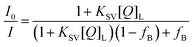 | (7) |
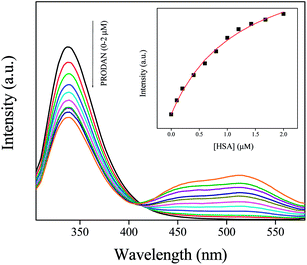 |
| | Fig. 2 Emission spectra of HSA (10 μM) in presence of different concentration of PRODAN (0–2 μM). | |
In this equation I0 is the intensity of HSA in absence of PRODAN. KSV is the Stern–Volmer quenching constant and
| |
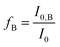 | (8) |
where
I0,B is fluorescence intensity of the tryptophan accessible to quencher. Thus the estimated
KSV and
fB were around 1.77 × 10
6 M
−1 and 0.54 respectively.
We measured the fluorescence lifetime of PRODAN at 457 and 520 nm at different concentration of HSA (Table S1 in the ESI†). In presence of 1 μM HSA, PRODAN exhibits the lifetime components around 0.73 ns (55%) and 3.60 ns (45%) at 457 nm. The decay at 50 μM concentration of HSA at the same wavelength is comprised of 0.90 ns (33%) and 4.00 ns (67%) components with an average lifetime of 3.00 ns. We assign the species with time component of 4.00 ns to HSA bound PRODAN and the species with time component of 0.90 ns to the free PRODAN species in aqueous medium. Our result is consistent with the measurement made by Basak and co-workers.29b The increase in fluorescent quantum yield and lifetime of PRODAN and its derivative when bound to protein is due to reduced conformational freedom of the amine and carbonyl groups because of the close packing of surrounding protein.30 The significant increase in longer component from 45% to 67% upon addition of 50 μm HSA clearly indicates that PRODAN molecules are entrapped inside the hydrophobic pocket of HSA. Table S1† reveals a similar component at 520 nm when PRODAN binds with HSA. The lifetime at 520 nm was fitted with a bi-exponential function. The increase in nanosecond component which represents HSA bound PRODAN species from 26% to 45% confirms the binding of PRODAN molecules with HSA.
3.2. Interaction of PRODAN with liposomes
In this section we first encapsulated PRODAN in different liposomes. Addition of liposomes to aqueous solution of PRODAN causes a blue shift in emission spectra followed by a new band at 435 nm. This band is assigned as LE state of PRODAN. The appearance of LE band indicates that PRODAN molecules are encapsulated inside the liposomes. Interestingly, we observe an isoemissive point in DPPC and DMPC liposomes which indicates the existence of two emissive species in these two liposomes (Fig. 3).
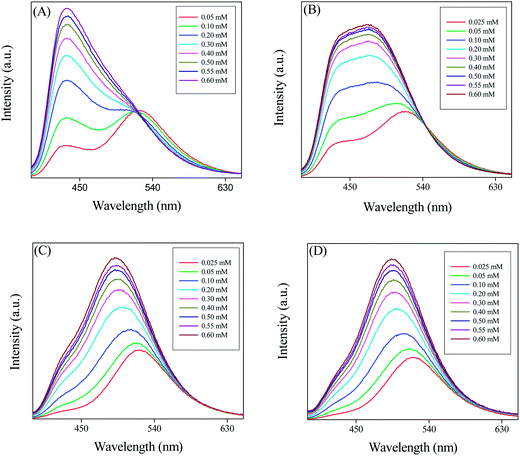 |
| | Fig. 3 The emission spectra of PRODAN at different concentration of liposomes (A) DPPC (B) DMPC (C) DOPC and (D) POPC liposomes. | |
On the other hand isoemissive point was not observed when PRODAN is incorporated into DOPC and POPC liposomes. A similar observation was reported by Correa and co-workers in case of DOPC liposomes.35 They explained this observation with the model proposed by Chong and co-workers.26a The lack of isoemissive point in POPC and DOPC liposomes may be due to absence of a prominent LE state in DOPC and POPC liposomes. To explain this observation we consider the differences in the hydration level of liposomes which further depends on their phase transition temperature. DPPC and DMPC have phase transition temperatures around 43 °C and 23 °C respectively while POPC and DOPC have phase transition temperature around – 2 °C and – 20 °C respectively. It is reported by Horta and co-workers36 that the liposomes having lower phase transition temperature is more hydrated than liposomes having higher phase transition temperature. Again because of much lower phase transition temperature, DOPC and POPC are significantly much more hydrated and are much softer than DPPC and DMPC at room temperature. PRODAN molecules are mostly encapsulated in the interfacial region of DOPC and POPC. Since DOPC and POPC exist in the liquid crystalline phase at room temperature, the non polar region is less motionally restricted compared to that in interfacial region. Thus lack of a prominent LE state may be responsible for absence of an isoemissive point.
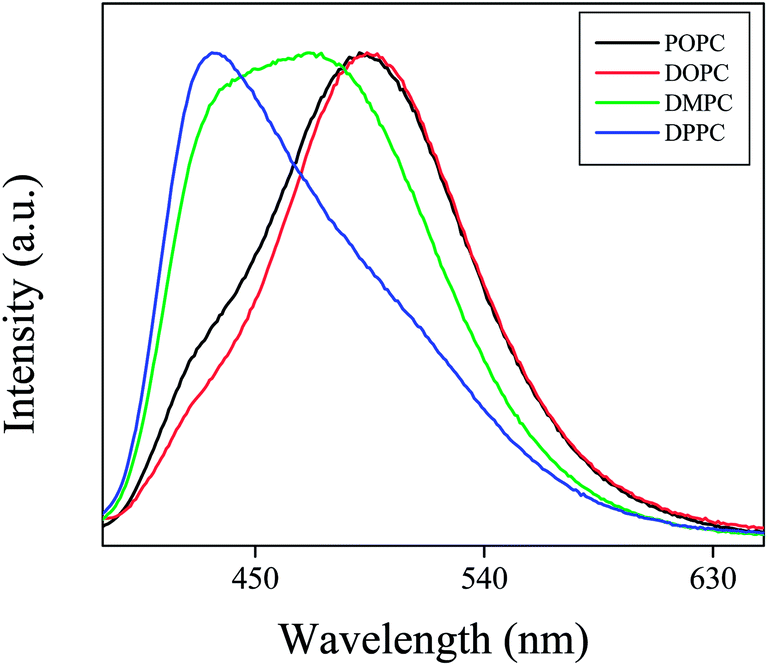
The normalized emission spectra of PRODAN (Fig. 4) in different liposomes reveal that maximum blue shift takes place in DPPC liposomes. This fact also indicates that DPPC and DMPC are more hydrophobic as compared to DOPC and POPC due to their higher phase transition temperature and due to difference in their prehydration levels. Correa and co-workers correlated the maximum emission band energy of PRODAN (Eem-PRODAN) with ET(30) polarity scale for different solvents by the following equation.35
| | |
ET(30) = 147 ± 5 − (1.62 ± 0.02)Eem-PRODAN n = 23, r = 0.98
| (9) |
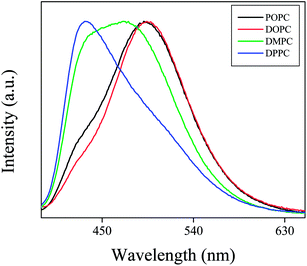 |
| | Fig. 4 Normalized emission spectra of PRODAN in different liposomes. | |
Following this equation E values as obtained for DPPC, DMPC, POPC and DOPC liposomes are 41.7, 47, 52 and 54 kcal mol−1 respectively. The different micropolarity as experienced by PRODAN in different liposomes could be attributed to the difference in their prehydration levels which further depends on phase transition temperatures.
At room temperature DPPC vesicles remain in sol gel phase (SG) and DMPC vesicles remain in nearly liquid crystalline (LC) phase. As, torsion of the –N(CH3)2 are more restricted in SG phase of the phospholipid bilayer than that in LC phase, LE band has less contribution in LC phase compared to that in sol gel phase. DOPC and POPC liposomes exist completely in LC phase at room temperature. Thus they have similar emission spectra for PRODAN at room temperature which is clear from Fig. 4. We estimated the partition coefficient of prodan in different liposomes using the following equation37
| |
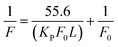 | (10) |
where
F0 and
F are fluorescence intensities of PRODAN molecules in aqueous and in lipid phase, respectively,
L is the lipid concentration and the molar concentration of water was considered to be 55.6 M. Thus using
eqn (10) and the slopes from
Fig. 5, the calculated
KP values are 1.0 × 10
5, 5.8 × 10
5, 2.8 × 10
5, 2.6 × 10
5 for DPPC, DMPC, POPC and DOPC liposomes respectively. Notably the lower partition coefficient in DPPC liposomes compared to that in other liposomes stems from the fact that the interfacial region of DPPC is much more rigid due to its sol gel phase and this rigidity hinders the encapsulation of more number of PRODAN molecules. The liposomes like DMPC, DOPC and POPC remain in liquid crystalline phase at room temperature and they allow PRODAN to penetrate in the interfacial region.
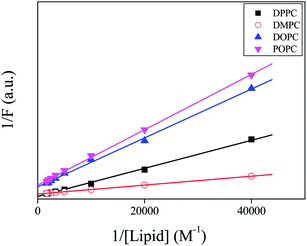 |
| | Fig. 5 Double reciprocal plot of the intensity of PRODAN with respect to concentration different liposomes. | |
We estimated the lifetime of PRODAN at various concentrations of liposomes at 440 and 520 nm. The values of lifetime for different vesicles are summarized in Tables S2A and S2B (in ESI†) and the representative decays are shown in Fig. 6. PRODAN exhibits a bi-exponential decay in aqueous buffer solution at 520 nm with the lifetime components 0.62 ns (τ1) and 1.8 ns (τ2) with a population of 74% and 26% respectively. Thus the average lifetime of PRODAN at 520 nm is around 0.93 ns. In DPPC liposome, at 520 nm, where the emission spectra is predominantly from TICT state of PRODAN has conspicuously dependence on the concentration of lipid and is well described by a tri-exponential function. The picosecond component i.e. τ1 remains same throughout the concentration of DPPC and the nanosecond component i.e. τ2 increased up to around 2.17 ns. A third component of around 5.19 ns (τ3) with a population of 31% appeared at higher concentration of DPPC (Table S2A†). We, therefore, assign τ1 component to the PRODAN molecules remaining in the aqueous phase which drops from 74% to 53% upon increasing the concentration of DPPC from 0 to 0.6 mM. The 2.17 ns component i.e. τ2 may be ascribed to the PRODAN molecules in aqueous phase or loosely bound in the interfacial region and the longest component i.e. 5.20 ns (τ3) component may come from the PRODAN molecules strongly held inside the liposome. Interestingly, PRODAN at 435 nm in 0.6 mM DPPC liposome where emission mainly comes from LE state exhibits a tri-exponential decay. The components are 0.62 ns (17%), 2.73 ns (31%) and 6.57 ns (52%). We already assigned these components to different locations in liposomes.
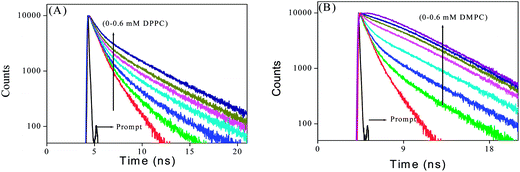 |
| | Fig. 6 Decay of PRODAN at different concentration of liposomes (A) DPPC liposome (B) DMPC liposome. | |
In DMPC liposome, the decays at 520 nm were fitted to a bi-exponential function with a picosecond and a nanosecond component. As the liposome concentration increases and incorporate more number of PRODAN molecules, the picosecond component disappeared leading to a rise component around 1.65 ns (31%) and a nanosecond component around 4.72 ns (69%). The decay proceeds with a rise component indicates that solvation takes place. Surprisingly, in case of DPPC liposomes, we did not observe any rise component which could be because of the fact that the aliphatic tail region of DPPC is more dehydrated than that of DMPC. Table S2B† reveals that the decay of PRODAN in DMPC liposomes at 435 nm is tri-exponential with picosecond component (τ1) and two nanosecond components (τ2 and τ3). The components are 0.74 ns (14%), 1.77 ns (31%) and 4.25 ns (55%). It is noteworthy that τ3 is significantly less in DMPC liposomes compared to that in DPPC liposomes. The probable reason is that DMPC remains in nearly liquid crystalline phase at room temperature while DPPC remains in sol–gel phase which brings in additional rigidity in DPPC compared to that in DMPC. This may be responsible for higher time component in DPPC compared to that in DMPC.
In DOPC and POPC liposomes initially decay at 520 nm was bi-exponential with a picosecond component (∼670 ps) originating from the PRODAN molecules in the aqueous phase and a nanosecond component around 2.65 to 2.69 ns. Due to low concentration of lipid two components are observed picosecond component in aqueous phase and the other nanosecond component in lipid phase. However, at higher concentration the decay becomes single exponential with time constant around 3.52 ns and 3.38 ns for DOPC and POPC liposomes respectively. These results are in accordance with that reported by Correa and co-workers.35 At 435 nm the decays in DOPC and POPC were fitted to a biexponential function having a picosecond component around 0.840 ns and a nanosecond component around 2.60 to 2.70 ns. Notably, the longer components in POPC and DOPC liposomes at 520 and 435 nm are significantly smaller compared to longer component of DPPC and DMPC liposomes at the same wavelength. This observation may be explained by considering the fact that at room temperature both DOPC and POPC remain in liquid crystalline phase due to significant lower phase transition temperature compared to DPPC and DMPC. Therefore, PRODAN experiences a less constrained environment in DOPC and POPC liposomes giving rise to a shorter lifetime component as compared to DPPC and DMPC liposomes.
3.3. Interaction between liposomes and HSA
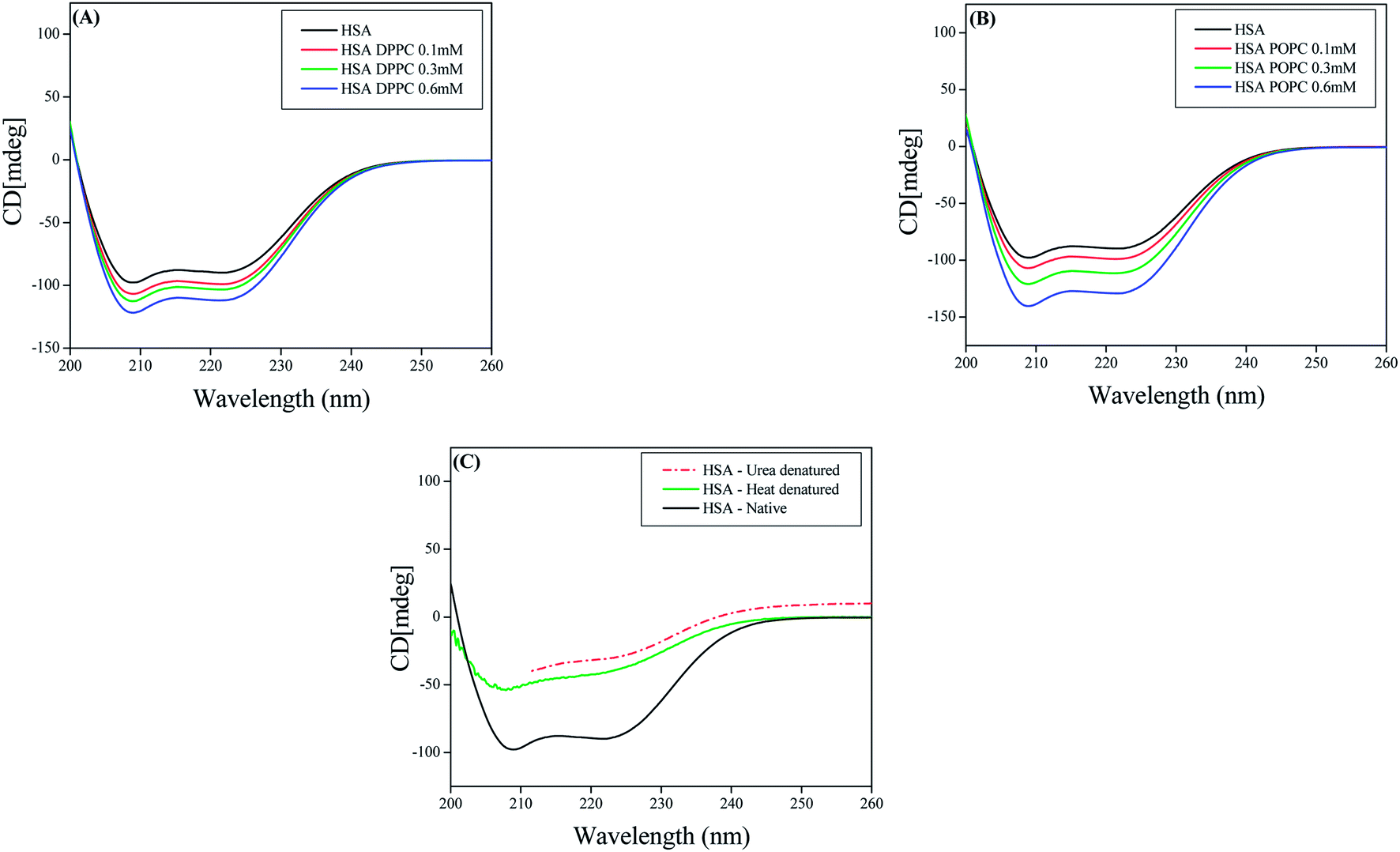
3.3.1 CD Measurements. To gain a better insight on interaction of HSA with various liposomes CD measurements were performed using HSA, DPPC liposomes and POPC liposomes at various concentrations of these lipids. The CD spectra of HSA exhibit two negative minima at 208 and 217 nm, which is typical characterization of α-helix structure of proteins.38a Interaction between DPPC–HSA and POPC–HSA caused an increase in band intensity at all wavelengths of the far UV CD without any significant shift of the peaks (Fig. 7). This indicates that both DPPC liposomes and POPC liposomes causes a slight increase in the α-helical structure of HSA. While heating HSA till 90 °C and addition of 8 M urea causes decrease in the band intensity at all the wavelengths (Fig. 7C). This signifies decrease in α-helical content upon denaturation of HSA.38b Thus we conclude that both the lipid upon interaction with HSA cause perturbation in the secondary structure of HSA but increase in α-helical content suggest that HSA is not denatured or unfolded during the interaction.12
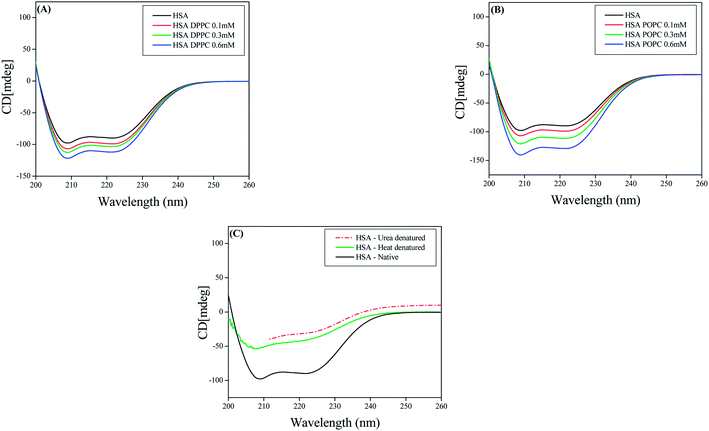 |
| | Fig. 7 CD spectra of (A) HSA and DPPC liposomes (B) HSA and POPC liposomes (C) native HSA and denatured HSA. | |
3.3.2 Steady state and time resolved measurement. Addition of HSA to PRODAN impregnated liposomes causes a quenching in the fluorescence intensity of PRODAN. The continuous decrease in the intensity with addition of HSA to PRODAN impregnated liposomes indicates that HSA interacts with the liposomes. Interestingly, we observe a red shift in the emission spectra of PRODAN in DPPC liposomes (from 435 to 460 nm) while a blue shift is observed in DMPC (from 460 to 455 nm), DOPC (from 497 to 471 nm), and POPC (from 490 to 467 nm) liposomes (Fig. 8).
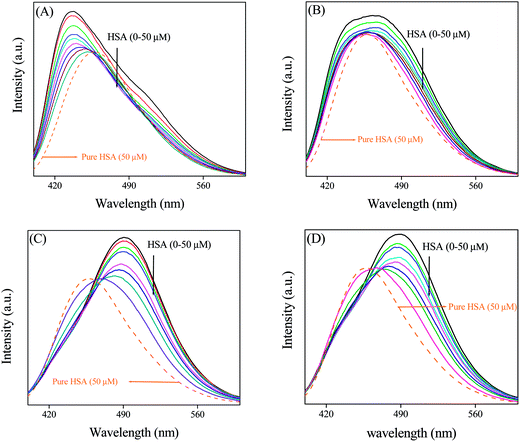 |
| | Fig. 8 The steady state emission spectra of PRODAN in different liposomes as a function of HSA concentration. (A) DPPC, (B) DMPC, (C) DOPC and (D) POPC. The dashed graph represents emission spectra of PRODAN in native HSA which is normalized with respect to highest concentration of HSA in liposomes. | |
There are two reasons that may be accounted for the observed quenching in liposomes. The first one is the penetration of HSA into liposomes and the second is the release of PRODAN molecules from liposome and subsequent migration to the hydrophobic core of HSA. Sabin and co-workers11a reported that the forces which are involved in the interaction between liposomes and HSA are electrostatic and hydrophobic in nature. Primarily HSA interacts with the liposome through electrostatic interaction to form HSA liposome complex and destabilize the packing of lipid within bilayer and the order of acyl chain is reduced. The zeta potential (ξ) was used to monitor the electrostatic interaction between liposomes and HSA.11a It was found that ξ decreases exponentially with the protein concentration. The strong dependence of ξ was reported as a patent evidence that the attractive electrostatic contribution has a major role in the formation of liposome–HSA complex. A similar type of electrostatic interaction has been invoked by Charbonneau and co-workers.12 Sabin and co-workers11a also reported the protein penetration inside the liposome. DSC measurement by them reveal that pretransition temperature of DMPC liposomes decreases by more than one degree at the same time the enthalpy change (ΔH) increases. A similar type of results were reported by Gatlantai and co-workers.11b,c The effect of HSA over DMPC and DPPC liposomes indicates that protein penetrates into hydrophobic bilayer affecting the packing of the hydrocarbon tails of lipids. Therefore, there is contribution of hydrophobic forces in formation of liposome–HSA-complexes. The contribution comes from interaction between the lipid tails and parts of HSA that penetrate into the lipid bilayer. The decrease in the interfacial tension indicates that protein molecules intercalate between the hydrophobic tails of the lipid. This intercalation causes the leakage in the interfacial region which facilitates the migration of the probe molecules from liposome to either aqueous phase or hydrophobic pocket of HSA. Notably, upon addition of HSA to PRODAN impregnated liposomes, the emission maxima are shifted towards the emission maximum of native HSA (Fig. 8). This observation led to the conclusion that PRODAN molecules being released from the liposome are trapped in the hydrophobic pocket of HSA.
We compared the extent of quenching from Fig. 9 in different liposomes by plotting ϕ0/ϕ as a function of concentration of HSA. Since we cannot calculate the local concentration of HSA, so; we did not estimate Stern–Volmer quenching constant from this plot. It is observed from Fig. 9 that among the un-conjugated lipids, quenching is higher in DPPC liposome compared to that in DMPC liposomes. On the other hand in case of conjugated lipids, the quenching is little higher in POPC liposomes compared to that in DOPC liposomes. The extent of quenching depends upon the extent of perturbation of lipid bilayer by HSA. The significant difference in quenching in DPPC and DMPC liposomes and little difference in DOPC and POPC liposomes may be explained by considering the structural differences of different lipids, phase transition temperature and prehydration level.
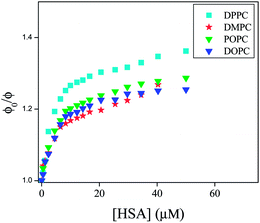 |
| | Fig. 9 ϕ0/ϕ plot as a function of concentration of HSA (0–50 μM) in different liposomes. | |
In the present study, all the four lipids are zwitterionic and they possess similar head groups but differ in their acyl chains. While DPPC and DMPC contain saturated acyl chain with different chain length, POPC and DOPC contain unsaturated acyl chain with different number of carbon atoms. As the length of hydrophobic acyl chain is the measure of hydrophobicity and it is already reported that saturated fatty acids bind with greater affinity to albumins due to increase in hydrophobic interaction,39 in that sense the order of quenching follows the right trend. DPPC has higher phase transition temperature than DMPC. The liposomes with lower phase transition temperature remains more hydrated as compared to liposome with higher phase transition temperature. Therefore DPPC is less hydrated as compared to DMPC. So; higher quenching is observed in DPPC as compared to DMPC. Among POPC and DOPC bilayers, POPC is monounsaturated while DOPC is bi-unsaturated with CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH in cis position. The unsaturated fatty acids with a cis double bond, faces little steric restriction on binding to various sites on protein.40 Therefore perturbation of lipid bilayer by HSA will be more pronounced in POPC bilayers as compared to DOPC bilayers. Along with this phase transition temperature of POPC is higher than DOPC. The DOPC bilayers will remain in a more prehydrated state as compared to POPC. Thus lower quenching is observed in DOPC as compared to POPC.
CH in cis position. The unsaturated fatty acids with a cis double bond, faces little steric restriction on binding to various sites on protein.40 Therefore perturbation of lipid bilayer by HSA will be more pronounced in POPC bilayers as compared to DOPC bilayers. Along with this phase transition temperature of POPC is higher than DOPC. The DOPC bilayers will remain in a more prehydrated state as compared to POPC. Thus lower quenching is observed in DOPC as compared to POPC.
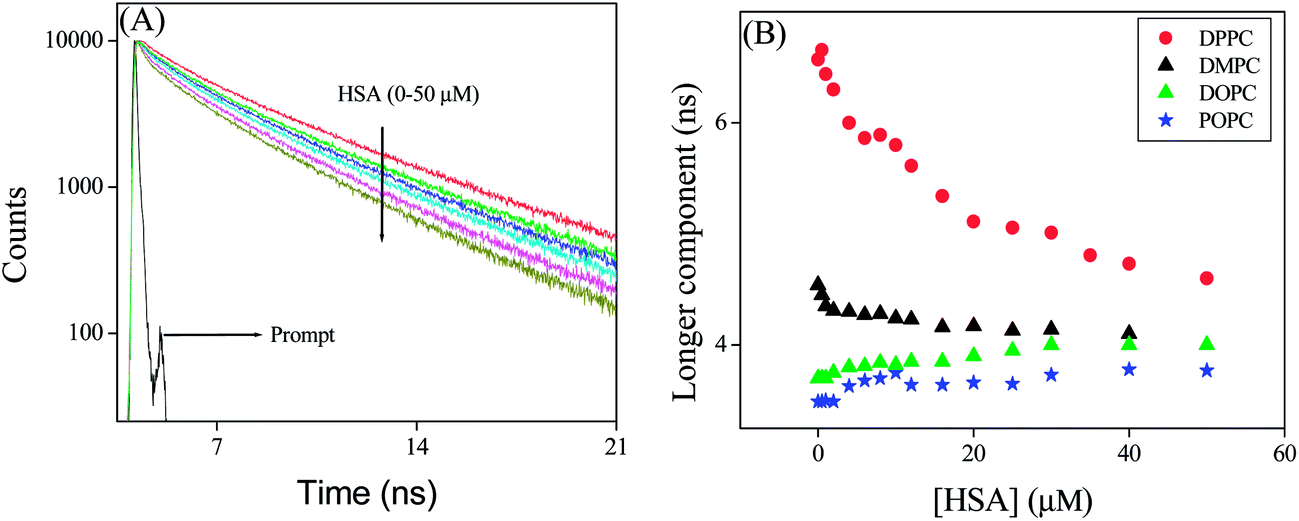
In this context the lifetime data may be helpful to unravel the dynamics of PRODAN inside the liposome. We already mentioned that the fluorescence decay of PRODAN in aqueous buffer solution is adequately fitted to a bi-exponential function with time constant 0.60 ns (74%) and 1.80 ns (26%). The lifetime of PRODAN is significantly enhanced when encapsulated in liposomes. This is already discussed in the previous section. Table S3A (in the ESI†) reveals that PRODAN exhibits a tri-exponential decay with time components of 0.62 ns (15%), 2.73 ns (31%) and 6.57 ns (54%) in DPPC liposomes. We already assigned that the picosecond component corresponds to the PRODAN molecules in the aqueous phase, 2.73 ns component is attributed to the PRODAN molecules loosely bound in the interfacial region and third component i.e. 6.57 ns component perhaps comes from those PRODAN molecules which are strongly held inside the liposome. Addition of HSA to PRODAN impregnated DPPC liposomes causes quenching in the lifetime components of PRODAN (Fig. 10). After addition of 2 μM HSA, the decays became bi-exponential and at 50 μM HSA the decay is comprised of the components of 1.48 (40%) and 4.60 ns (60%). The significant quenching in the longer component (from 6.48 ns to 4.60 ns) implies the penetration of HSA into liposome. The striking observation is that the lifetime components of PRODAN in presence of 50 μM HSA in DPPC liposomes (1.48 and 4.60 ns) are very similar to that in pure HSA (1.49 ns and 4.0 ns, Table S1†) which indicates that PRODAN molecules upon interaction with HSA are released from liposome and migrate to the hydrophobic pocket of HSA. Had the PRODAN molecules migrated to aqueous phase, we would have obtained a picosecond component. We already mentioned that a shift is observed in the steady state emission spectra of PRODAN upon addition of HSA which indicates that PRODAN is migrating to the hydrophobic pocket of HSA. Thus this fact is supported by the lifetime data.
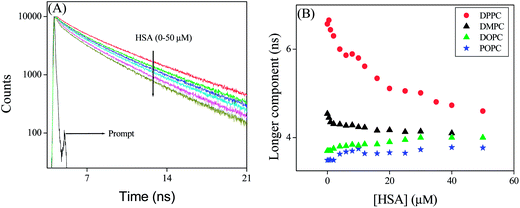 |
| | Fig. 10 (A) Time resolved decays of PRODAN at different concentration of HSA in DPPC liposome. (B) Longer component of PRODAN in liposomes at different concentration of HSA. | |
However, a different result is obtained in DMPC–HSA system. It is revealed that unlike in DPPC system, DMPC offers only little decrement in lifetime components in presence of HSA. Addition of HSA causes a little quenching in the longer component from 4.55 to 4.10 ns while the shorter component decreases from 2.2 ns to 1 ns. The little quenching in the longer component indicates that, HSA has a less penetration in DMPC liposomes. The less penetration stems from the fact that DMPC because of its low phase transition temperature (23 °C) remains nearly in liquid crystalline phase at room temperature and is much more hydrated than DPPC. As the hydrophobic interaction is responsible for the penetration of HSA into liposomes, therefore, HSA prefers DPPC over DMPC as former is more dehydrated hence is more hydrophobic compared to the latter. Although one should expect that DMPC is loosely packed and thus eases HSA to penetrate inside the liposome. However, higher quenching of the longer component in DPPC liposomes made it clear that hydrophobic interaction dominates over the other factors. On the other hand significant decrement in the shorter time component in DMPC liposomes indicates that HSA destabilize the interfacial region leaving the liposome core intact. Therefore, from the above results, we may conclude that the changes in the shorter component takes place when HSA destabilizes the interfacial region and the change in longer component in liposomes takes place when HSA affects the core of the liposomes due to penetration by hydrophobic interaction.11a The latter process depends on prehydration level of liposomes.
The above finding is again supported by the time resolved data in conjugated liposomes (Table S3B in the ESI†). In case of DOPC and POPC liposomes, we observe that the lifetime of PRODAN is single exponential with a component around 3.49 ns and 3.77 ns respectively. Addition of HSA results in quenching and time resolved decay becomes bi-exponential. Surprisingly we observe a marginal increment in the longer component in POPC (from 3.77 to 3.90 ns) and DOPC liposomes (3.40 to 3.77 ns). The observation in these two liposomes clearly indicates the HSA does not penetrate in these two liposomes. On the other hand appearance of picosecond component (0.86 to 0.95 ns) and increment in its amplitude up to 35–40% in both the liposomes indicates the leakage of PRODAN molecules and confirms the fact that HSA destabilize the interfacial region of these liposome and core of the interfacial region remains intact. It is noteworthy that in case of DPPC liposome similar kind of changes in the population of shorter and longer components was observed. Therefore, it may unambiguously be concluded that the leakage of PRODAN molecules takes place due to destabilization of interfacial region of liposomes.
We carried out time resolved anisotropy measurements to probe interaction of liposomes with HSA. The anisotropy decays are shown in Fig. 11 and the results are summarized in Table 1. PRODAN exhibits a single exponential decay with a time constant of 0.170 ns at 520 nm in aqueous buffer solution at pH 7.40. In liposomes and liposomes–HSA complex the anisotropy was measured at 450 nm. PRODAN exhibits bi-exponential anisotropy decays consisting of a picosecond and a nanosecond component in all liposomes. The fast components (ϕfast) are around 0.47 (44%), 0.50 (45%), 0.40 (32%) and 0.416 ns (37%) in DPPC, DMPC, DOPC and POPC liposomes respectively. On the other hand the slow components (ϕslow) are 2.77 (56%), 2.93 (55%), 2.25 (68%), and 2.20 ns (63%) in DPPC, DMPC, DOPC and POPC liposomes respectively. It is revealed that addition of HSA to PRODAN loaded liposomes causes a significant increment in ϕslow. Thus in presence 15 μM HSA, ϕslow were found to be 3.5 (60%), 3.90 ns (56%), 3.75 (40%), 3.60 (53%) in DPPC, DMPC, DOPC and POPC liposomes respectively. Since liposomes and liposomes–HSA complex are big in the size, the motion of liposome and liposome–HSA is too slow to impact on the overall rotational relaxation of PRODAN. The increment in ϕslow may be due to the fact that the interfacial region of liposome becomes compact due to electrostatic interaction between liposomes and HSA. It is revealed from above mentioned result that the increment in ϕslow in DPPC and DMPC liposomes is less compared to that in DOPC and POPC liposomes. The higher increment in ϕslow in DOPC and POPC liposomes may be attributed to the fact that the interfacial region of DOPC and POPC becomes more compact due to adsorption of HSA. The other explanation is that possibly lipid packing order of DPPC and DMPC are much more affected by higher penetration of HSA in these liposomes as compared to that in DOPC and POPC liposomes. This prevents the increment in ϕslow in DPPC and DMPC liposomes. A similar conclusion was drawn in the discussion of quenching in the time resolved data. Interestingly, we observed a decrement in the population of ϕslow in case of DOPC and POPC liposomes. However, we do not observe any increment in the population of slower component (βslow). Since these two liposomes are soft in the interfacial region, there is possibility that these two liposomes can accommodate more number of HSA molecules which causes a significant leakage. Moreover, HSA penetrates deeper in DPPC and DMPC liposomes, so; for these liposomes PRODAN can migrate to hydrophobic pocket of HSA.
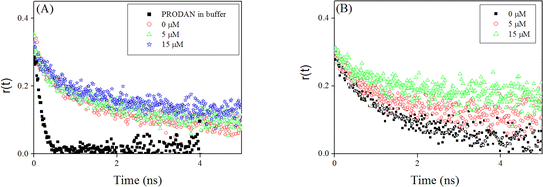 |
| | Fig. 11 Fluorescence anisotropy decays of PRODAN at different concentration of HSA (A) DMPC liposomes (B) DOPC liposomes. | |
Table 1 Rotational relaxation parameters of PRODAN in different liposomes and liposome–HSA complex ata λem = 450 nm
| System |
βfast (%) |
βslow (%) |
ϕfast (ns) |
ϕslow (ns) |
r0 |
| The estimated error in the measurement is around 5%. |
| PRODAN in buffer solution, pH 7.4 |
1 |
|
0.17 |
— |
0.29 |
| DPPC |
0.44 |
0.56 |
0.47 |
2.77 |
0.32 |
| DPPC + 5 μM HSA |
0.40 |
0.60 |
0.60 |
3.20 |
0.32 |
| DPPC + 15 μM HSA |
0.40 |
0.60 |
0.57 |
3.50 |
0.33 |
| DMPC |
0.45 |
0.55 |
0.50 |
2.93 |
0.35 |
| DMPC + 5 μM HSA |
0.42 |
0.58 |
0.39 |
3.46 |
0.35 |
| DMPC + 15 μM HSA |
0.45 |
0.55 |
0.37 |
3.90 |
0.33 |
| DOPC |
0.32 |
0.68 |
0.40 |
2.25 |
0.31 |
| DOPC + 5 μM HSA |
0.42 |
0.58 |
0.62 |
3.51 |
0.32 |
| DOPC + 15 μM HSA |
0.60 |
0.40 |
0.57 |
3.75 |
0.31 |
| POPC |
0.37 |
0.63 |
0.42 |
2.20 |
0.32 |
| POPC + 5 μM HSA |
0.39 |
0.61 |
0.45 |
2.99 |
0.33 |
| POPC + 15 μM HSA |
0.46 |
0.54 |
0.40 |
3.60 |
0.33 |
Conclusion
The present study reveals a clear understanding of how HSA interacts with liposomes of saturated and unsaturated lipids having different phase transition temperature. The CD measurement indicates that HSA is stabilized upon interaction with liposomes. Steady state and time resolved fluorescence analysis reveal that HSA alters the packing order of liposome through penetration and releases the encapsulated probe molecules from liposome, which simultaneously migrates in the hydrophobic pocket of HSA. The penetration is apparently caused by hydrophobic interaction between liposomes and HSA. The extent of penetration depends on the prehydration level of liposomes. The liposomes of saturated lipids (DPPC and DMPC) having higher phase transition temperature are less prehydrated at room temperature and hence have stronger affinity towards HSA than that of liposomes of unsaturated lipids. The penetration caused by hydrophobic interaction is also revealed in anisotropy measurement.
Acknowledgements
The authors would like to thank SIC, IIT Indore for providing the facility. The work was supported by the fast track project funded by Department of Science and Technology, New Delhi, India.
References
- L. J. Pike, J. Lipid Res., 2003, 44, 655 CrossRef CAS PubMed
 .
. -
(a) N. Ashgarian and Z. A. Schelly, Biochim. Biophys. Acta, 1999, 1418, 295 CrossRef ;
(b) N. M. Correa, Z. A. Schelly and H. Zhang, J. Am. Chem. Soc., 2000, 122, 6432 CrossRef CAS .
- T. Parassasi, G. D. Satasio, A. Ubaldo and E. Gratton, Biophys. J., 1990, 57, 1179 CrossRef .
- A. S. Klymchenko and A. P. Demchenko, Langmuir, 2002, 18, 5637 CrossRef CAS .
-
(a) A. Sytnik and I. Litvinyuk, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 12959 CrossRef CAS ;
(b) S. K. Sahoo and V. Labhasetwar, Drug Discovery Today, 2003, 8, 1112 CrossRef CAS .
- A. Chonn, S. C. Semple and P. R. Cullis, J. Biol. Chem., 1992, 267, 18759 CAS .
- A. O. Pedersen, K. L. Mesenberg and U. Kragh-Hansen, Eur. J. Biochem., 1995, 233, 395 CAS .
-
(a) S. Sugio, A. Kashima, S. Mochizuki, M. Noda and K. Kobayashi, Protein Eng., 1999, 12, 439 CrossRef CAS PubMed ;
(b) D. C. Carte and J. X. Ho, Adv. Protein Chem., 1994, 45, 153 CrossRef ;
(c) H. M. He and D. C. Carter, Nature, 1992, 358, 209 CrossRef PubMed ;
(d) T. Peters, Adv. Protein Chem., 1985, 37, 161 CrossRef CAS ;
(e) S. Curry, P. Brick and N. P. Frank, Biochim. Biophys. Acta, 1999, 1441, 131 CrossRef CAS ;
(f) I. Petitpas, T. Grune, A. A. Battacharya and S. Curry, J. Mol. Biol., 2001, 314, 955 CrossRef CAS PubMed ;
(g) E. L. Gelamo, C. H. T. P. Silva, H. Imasato and M. Tabak, Biochim. Biophys. Acta, 2002, 1594, 84 CrossRef CAS ;
(h) V. T. G. Chuang and M. Otagiri, Biochim. Biophys. Acta, 2001, 1546, 337 CrossRef CAS .
- A. A. Bhattacharya, T. Grune and S. Curry, J. Mol. Biol., 2000, 303, 721 CrossRef CAS PubMed .
-
(a) L. J. Lis, J. W. Kauffman and D. F. Shriver, Biochim. Biophys. Acta, 1976, 436, 513 CrossRef CAS ;
(b) D. Hoekstra and G. Scherphoft, Biochim. Biophys. Acta, 1979, 551, 109 CrossRef CAS .
-
(a) J. Sabin, G. Prieto, J. M. Ruso, P. V. Messina, F. J. Salgado, M. Nogueira, M. Costas and F. I. Sarmiento, J. Phys. Chem. B, 2009, 113, 1655 CrossRef CAS PubMed ;
(b) R. Galantai and I. Bárdos-Nagy, Int. J. Pharm., 2000, 195, 207 CrossRef CAS ;
(c) I. Bárdos-Nagy, R. Galantai, M. Laberg and J. Fidy, Langmuir, 2013, 19, 146 CrossRef .
- D. Charbonneau, M. Beauregard and H. A. Tajmir-Riahi, J. Phys. Chem. B, 2009, 113, 1777 CrossRef CAS PubMed .
-
(a) L. P. Tseng, H. J. Liang, T. W. Chung, Y. Y. Huang and D. Z. Liu, J. Med. Biol. Eng., 2007, 27, 29 Search PubMed ;
(b) C. K. Kim and D. K. Park, Arch. Pharmacal Res., 1987, 10, 75 CrossRef ;
(c) J. Hernandez, J. Estelrich, M. T. Montero and O. Valls, Int. J. Pharm., 1989, 57, 211 CrossRef CAS ;
(d) T. P. W. McMullen and R. N. McElhaney, Curr. Opin. Colloid Interface Sci., 1996, 1, 83 CrossRef CAS .
- D. E. Vance, and J. E. Vance, Biochemistry of lipids, Lipoproteins and membranes, Elsevier Science, Vancouver, B. C. Canada, 1995 Search PubMed .
- A. L. Arbuzova and R. E. Koeppe, Annu. Rev. Biophys. Biomol. Struct., 2007, 37, 107 Search PubMed .
- D. L. Nelson and M. M. Cox, Principles of Biochemistry, W. H. Freeman and company, U. S. A, New York, 4th edn, 2008 Search PubMed .
- R. Thakur, A. Das and A. Chakraborty, Phys. Chem. Chem. Phys., 2012, 14, 15369 RSC .
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Kluwer Academic Plenum Publisher, New York, 3rd edn, 2006 Search PubMed .
-
(a) A. B. J. Parusel, J. Chem. Soc., Faraday Trans., 1998, 94, 2923 RSC ;
(b) A. B. J. Parusel, W. Nowak and S. Grimme, J. Phys. Chem. A, 1998, 102, 7149 CrossRef CAS ;
(c) W. Nowak, P. Adamczak, A. Balter and A. Sygula, J. Mol. Struct.: THEOCHEM, 1986, 32, 13 CrossRef CAS ;
(d) A. I. Harianawala and R. H. Bogner, J. Lumin., 1998, 79, 97 CrossRef CAS ;
(e) D. Marks, P. Prosposito, H. Zhang and M. Glasbeek, Chem. Phys. Lett., 1998, 289, 535 CrossRef CAS .
-
(a) S. Guha, K. Sahu, D. Roy, S. K. Mondal, S. Roy and K. Bhattacharyya, Biochemistry, 2005, 44, 8940 CrossRef CAS PubMed ;
(b) E. K. Krasnowska, L. A. Bagatolli, E. Gratton and T. Parasassi, Biochim. Biophys. Acta, 2001, 1151, 330 CrossRef ;
(c) N. A. Pasdar and E. C. Y. L. Chan, Food Hydrocolloids, 2001, 15, 285 CrossRef ;
(d) O. P. Bondar and E. S. Rowe, Biophys. J., 1999, 76, 956 CrossRef CAS .
-
(a) R. B. Macgregor and G. Weber, Nature, 1986, 319, 70 CrossRef CAS PubMed ;
(b) A. Balter, W. Nowak and W. Pawelkiewicz, Chem. Phys. Lett., 1988, 143, 565 CrossRef CAS .
-
(a) C. E. Bunker, T. L. Bowen and Y. P. Sun, Photochem. Photobiol., 1993, 58, 499 CrossRef CAS ;
(b) A. Samanta and R. W. Fessenden, J. Phys. Chem. A, 2000, 104, 8972 CrossRef CAS .
-
(a) S. K. Pal, D. Mandal and K. Bhattacharyya, J. Phys. Chem. B, 1998, 102, 11017 CrossRef CAS ;
(b) B. C. Lobo and C. J. Abelt, J. Phys. Chem. A, 2003, 107, 10938 CrossRef CAS ;
(c) B. Mennucci, M. Caricato, F. Ingrosso, C. Cappelli, R. Cammi, J. Tomasi, G. Scalmani and M. J. Frisch, J. Phys. Chem. B, 2008, 112, 414 CrossRef CAS PubMed ;
(d) M. Brozis, V. I. Tomin and J. Heldt, Zh Prikl Spectrosk, 2002, 69, 589 Search PubMed .
-
(a) H. Rottenberg, Biochemistry, 1992, 31, 9473 CrossRef CAS ;
(b) O. P. Bondar and E. S. Rowe, Biophys. J., 1999, 71, 1440 CrossRef ;
(c) A. Sommer, F. Paltauf and A. Hermetter, Biochemistry, 1990, 29, 11134 CrossRef CAS ;
(d) E. K. Krasnowska, E. Gratton and T. Parasassi, Biophys. J., 1998, 74, 1984 CrossRef CAS ;
(e) J. Zeng and P. L. G. Chong, Biochemistry, 1991, 30, 9485 CrossRef CAS .
-
(a) B. Sengupta, J. Guharay and P. K. Sengupta, Spectrochim. Acta, Part A, 2000, 56, 1433 CrossRef ;
(b) F. M. Agazzi, J. Rodriguez, R. D. Falcone, J. J. Silber and N. M. Correa, Langmuir, 2013, 29, 3556 CrossRef CAS PubMed ;
(c) M. Novaira, F. Moyano, M. A Biasutti, J. J Silber and N. M. Correa, Langmuir, 2008, 24, 4637 CrossRef CAS PubMed ;
(d) M. Novaira, M. A. Biasutti, J. J. Silber and N. M. Correa, J. Phys. Chem. B, 2007, 111, 748 CrossRef CAS PubMed ;
(e) S. S. Quintana, R. D. Falcone, J. J. Silber and N. M. Correa, ChemPhysChem, 2012, 13, 115 CrossRef CAS PubMed ;
(f) A. M. Durantini, R. D. Falcone, J. J. Silber and N. M. Correa, ChemPhysChem, 2009, 10, 2034 CrossRef CAS PubMed ;
(g) R. Adhikary, C. A. Barnes and J. W. Petrich, J. Phys. Chem. B, 2009, 113, 11999 CrossRef CAS PubMed .
-
(a) P. L. G. Chong, Biochemistry, 1988, 27, 399 CrossRef CAS ;
(b) C. C. de Vequi-Suplicy, C. R. Benatti and M. T. Lamy, J. Fluoresc., 2006, 16, 365 Search PubMed .
-
(a) J. Sýkora, P. Kapusta, V. Fidler and M. Hof, Langmuir, 2002, 18, 571 CrossRef ;
(b) P. Jurkiewicz, A. Olzùyńska, M. Langner and M. Hof, Langmuir, 2006, 22, 8741 CrossRef CAS PubMed ;
(c) L. Cwiklik, A. J. A. Aquino, M. Vazdar, P. Jurkiewicz, J. Pittner, M. Hof and H. Lischka, J. Phys. Chem. A, 2011, 115, 11428 CrossRef CAS PubMed .
-
(a) T. Parasassi, G. De Stasio, A. Ubaldo and E. Gratton, Biophys. J., 1990, 57, 179 CrossRef ;
(b) T. Parasassi, E. K. Krasnowska, L. Bagatolli and E. Gratton, J. Fluoresc., 1998, 8, 365 CrossRef CAS .
-
(a) S. S. Krishnakumar and D. Panda, Biochemistry, 2002, 41, 7443 CrossRef CAS PubMed ;
(b) S. Basak, D. Debnath, H. Haque, S. Ray and A. Chakrabarti, Indian J. Biochem. Biophys., 2001, 38, 84 CAS ;
(c) P. Abbyad, X. Schi, W. Childs, T. B. McAnaney, B. E. Cohen and S. G. Boxer, J. Phys. Chem. B, 2007, 111, 8269 CrossRef CAS PubMed ;
(d) A. Chakrabarti, Biochem. Biophys. Res. Commun., 1996, 226, 495 CrossRef CAS PubMed ;
(e) A. Chakrabarti, D. A. Kelkar and A. Chattopadhyay, Biosci. Rep., 2006, 26, 386 CrossRef PubMed ;
(f) A. Chattopadhyay, S. S. Rawat, D. A. Kelkar, S. Ray and A. Chakrabarti, Protein Sci., 2003, 12, 2403 Search PubMed .
-
(a) D. Zhong, A. Douhal and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 14056 CrossRef CAS PubMed ;
(b) J. K. A. Kamal, L. Zhao and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13411 CrossRef CAS PubMed .
-
(a) D. C. Carter and J. X. Ho, Adv. Protein Chem., 1994, 45, 152 CrossRef ;
(b) T. Peters Jr, All about Albumins: Biochemistry, Genetics, and Medical Applications, Academic Press, San Diego, 1996 Search PubMed .
-
(a) B. Farruggia, F. Garcia and G. Picó, Biochim. Biophys. Acta, 1995, 1252, 59 CrossRef ;
(b) B. Nerli, D. Romanini and G. Picó, Chem.–Biol. Interact., 1997, 104, 179 CrossRef CAS .
- F. Moreno, M. Cortijo and J. G. Jimenez, Photochem. Photobiol., 1999, 69, 8 CrossRef CAS .
- R. Thakur, A. Mallik and A. Chakraborty, Photochem. Photobiol., 2012, 88, 1248 CrossRef CAS PubMed .
- F. Moyano, M. A. Biasutti, J. J. Silber and N. M. Correa, J. Phys. Chem. B, 2006, 110, 11838 CrossRef CAS PubMed .
- B. A. C. Horta, A. H. de Vries and P. H. Heunenberger, J. Chem. Theory Comput., 2010, 6, 2488 CrossRef CAS .
- Z. Huang and R. P. Haugland, Biochem. Biophys. Res. Commun., 1991, 181, 166 CrossRef CAS .
-
(a) A. Gerbanowski, C. Malabat, C. Rabiller and J. Gueguen, J. Agric. Food Chem., 1999, 47, 5218 CrossRef CAS PubMed ;
(b) Y. Moriyama and K. Takeda, Langmuir, 1999, 15, 2003 CrossRef CAS .
- A. A. Spector, J. Lipid Res., 1975, 16, 165 CAS .
-
(a) V. G. Richieri, A. Anel and M. K. Alan, Biochemistry, 1993, 32, 7574 CrossRef ;
(b) D. P. Cistola, D. M. Small and J. A. Hamilton, J. Biol. Chem., 1987, 262, 10971 CAS ;
(c) J. S. Parks, D. P. Cistola, D. M. Small and J. A. Hamilton, J. Biol. Chem., 1983, 258, 9262 CAS .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra01214c |
|
| This journal is © The Royal Society of Chemistry 2014 |