The influence of ionic liquids on the Knoevenagel condensation of 1H-pyrrole-2-carbaldehyde with phenyl acetonitriles – cytotoxic 3-substituted-(1H-pyrrol-2-yl)acrylonitriles†
Ahmed Al Otaibia,
Christopher P. Gordona,
Jayne Gilbertb,
Jennette A. Sakoffb and
Adam McCluskey*a
aCentre for Chemical Biology, Chemistry, School of Environmental and Life Science, The University of Newcastle, University Drive, Callaghan, New South Wales 2308, Australia. E-mail: Adam.McCluskey@newcastle.edu.au; Fax: +61 (0)249 215472; Tel: +61 (0)249 216486
bDepartment of Medical Oncology, Calvary Mater Hospital, Newcastle, New South Wales 2308, Australia
First published on 28th March 2014
Abstract
The Knoevenagel condensation of a series of substituted phenyl acetonitriles with 1H-pyrrole-2-carbaldehyde was examined in seven 1-butyl-3-methylimidazolium based ionic liquids and three protic ionic liquids. Of these [BMIM][Br] and [BMIM][OH], with catalytic piperidine, proved most efficient affording 3-substituted-(1H-pyrrol-2-yl)acrylonitriles 3–17 in good to excellent yields (98%) whilst utilisation of the protic ionic liquid propyl ammonium nitrate resulted in reduced yields (0–66%). Screening of the 3-substituted-(1H-pyrrol-2-yl)acrylonitriles analogues 3–17 against a panel of 11 cancer cell lines and one normal cell line allowed the identification of a series of compounds with broad spectrum cytotoxicity, but more interestingly a significant degree of MCF-7 breast cancer cell line specificity was evident with 6 (7 to >25 fold) and 13 (5.7 to >80 fold). Other analogues show high level of efficacy against specific cell lines with 10 showing excellent activity against MCF-7 (GI50 = 1.7 μM) and A431 (GI50 = 2.8 μM) cell lines. The most promising of the compounds identified herein were the 4-CF3 substituted 10 and the 3,4-dichloro substituted 13 with excellent activities against MCF-7 and A431 cell lines. The 3,4-dichloro-13 was a 0.56 μM potent inhibitor of MCF-7 cell growth.
Introduction
Often significant enhancements in a compound's biological activity can be accessed through a surprisingly small number of robust chemical transformations.1 Arguably this small number of highly utilized transformations should facilitate the greening of drug and lead development pathways. However, in medicinal chemistry access to pure compounds is of paramount importance and is often pursued at the expense of green reaction credentials.2–5 Within our own research efforts we have routinely tolerated low yields and difficult purifications to gain access to the desired compounds.6,7 Recently our interest in developing shorter and more environmentally friendly route to focused compound libraries has seen us explore the application of new technologies in this pursuit. Specifically we have developed a considerable interest in emerging flow chemistry technologies and the possible application of room temperature ionic liquids to enhancing the outcome of simple synthetic procedures.8–10In recent years our medicinal chemistry efforts have been targeted in the areas of clathrin mediated endocytosis,11,12 the synthesis of protein phosphatase 1 and 2A inhibitors for the treatment of cancer and in the development of small molecule anti-parasitic agents for the treatment of livestock.13–16 In all instances we apply a focused compound library development – biological screening iterative cycle approach to enhancing compound activity. A cornerstone of these efforts has been the application of the Knoevenagel condensation to the synthesis of key precursors.8,11,17 These approaches are well documented in the literature.18–24 These analogues were synthesized under phase transfer catalysis (PTC) conditions using PhCH2N(CH3)3(OH) as the PTC.9,24 While the reactions proceeded with moderate efficiency and yield, we believed that ease of anion modification with room temperature ionic liquids would allow us to select the most appropriate system for rapid access to this compound series and potentially simplify reaction work up by product precipitation.24–27 We also viewed the Knoevenagel condensation as an ideal opportunity to probe the effects of different ionic liquids on the reaction outcome.
Results and discussion
Our efforts commenced with the examination of one of the archetypal room temperature ionic liquids, [BMIM][PF6] on the Knoevenagel condensation of pyrrole-2-carboxaldehyde (1) with phenyl acetonitrile (2). In the PhCH2N(CH3)3(OH) mediated process required 5 h and 50 °C and gave the expected products in good yields (67–78%) across a range of substituted phenyl acetonitriles (Scheme 1).24,25 | ||
| Scheme 1 Model Knoevenagel condensation of pyrrole 2-carboxaldehyde with phenyl acetonitrile in an ionic liquid. | ||
In a related study Hangarge et al., reported the Knoevenagel condensation of benzaldehyde and 3-methyl-1-phenylpyrazolin-5-(4H)-one in ethyl ammonium nitrate (EAN) which proceeded in 30 min in a 71% yield,28 while Verdía et al. demonstrated the facile condensation of aromatic aldehydes and active methylene containing compounds in 1,3-dimethylimidazolium methyl sulfate [MMIm][MSO4] containing up to 2.16% of water.29 However our initial reaction using [BMIM][PF6] with pyrrole 2-carboxaldhyde (1) and phenyl acetonitrile (2) under catalyst free conditions gave no evidence of the Knoevenagel product (Table 1, entry 1). Both the initial PTC and the [MMIm][MSO4] approaches required added H2O,28,29 thus we examined the effect of added H2O to [BMIM][PF6] at various temperatures. However, again, even with prolonged heating (2 days at 90 °C), no reaction was evident (Table 1, entries 2–6).
| Entry | Ionic liquid/H2O | Ratio | Temperature | Time (h) | Yield (%) |
|---|---|---|---|---|---|
| a ‘—’ no product observed, reaction condition: pyrrole-2-carboxaldehyde (165 mg, 1.74 mmol), phenyl acetonitrile (193 μL, 1.65 mmol) and (ionic liquids/H2O). | |||||
| 1 | [BMIM][PF6] | — | 50 | 5 | —a |
| 2 | [BMIM][PF6]/H2O | (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) :10) |
50 | 5 | — |
| 3 | [BMIM][PF6]/H2O | (7:10) |
70 | 5 | — |
| 4 | [BMIM][PF6]/H2O | (7:10) |
90 | 5 | — |
| 5 | [BMIM][PF6]/H2O | (7:10) |
90 | 24 | — |
| 6 | [BMIM][PF6]/H2O | (7:10) |
90 | 48 | — |
| 7 | [BMIM][OH] | — | 50 | 18 | 5 |
| 8 | [BMIM][OH] | — | 50 | 72 | 13 |
| 9 | [BMIM][OH]/H2O | (1:3) |
50 | 18 | 12 |
| 10 | [BMIM][OH]/H2O | (1:15) |
50 | 19 | 14 |
| 11 | [BMIM][OH]/H2O | (2:15) |
50 | 24 | 18 |
| 12 | [BMIM][OH]/H2O | (3:7) |
50 | 5 | 25 |
As we had previously reported on the use of base to catalyse the Knoevenagel condensation,8,11,17 we rationalized that utilisation of a basic ionic liquid would facilitate the desired transformation. We thus examined the use of [BMIM][OH] in our model Knoevenagel condensation of 1 and 2. While the product was clearly observed, the yield was unacceptably low at 5–13% even after 3 days at 50 °C (Table 1, entries 7 and 8). The addition of H2O to [BMIM][OH] had a modest effect on the reaction outcome increasing the observed yield from 12 to 25% (Table 1, entries 9–12).
Given the poor outcomes of the [BMIM][OH] mediated condensation we repeated the experiment with the addition of catalytic quantities of piperidine (see experimental). The effect of added piperidine was stark with [BMIM][OH] subsequently affording 85% of 3 (Table 2, entry 1) which compares very favourably with the 73% yield obtained in the original PTC approach.24 We believe that the additional activation associated with [BMIM][OH] is a direct consequence of the OH-moiety stabilising the imminium intermediate that arises from the catalytic addition of piperidine to the pyrrole-2-carboxaldehyde carbonyl moiety.30
| Entry | ILa | Yield (%) |
|---|---|---|
| a [BMIM][CH3COO], 1-butyl-3-methylimidazolium acetate; [BMIM][HCOO], 1-butyl-3-methylimidazolium formate; [BMIM][OH], 1-methyl-3-butylimidazolium hydroxide; EAN, ethyl ammonium nitrate; PAN, propyl ammonium nitrate; ETA, ethanol ammonium nitrate. Reaction condition: pyrrole-2-carboxaldehyde (165 mg, 1.74 mmol), phenyl acetonitrile (193 μL, 1.65 mmol), H2O (10 mL) and Ionic liquids (7 mL) at 50 °C for 5 h. | ||
| 1 | [BMIM][OH] | 85 |
| 2 | [BMIM][Br] | 56 |
| 3 | [BMIM][BF4] | 31 |
| 4 | [BMIM][PF6] | 37 |
| 5 | [BMIM][NO3] | No reaction |
| 6 | EAN | 24 |
| 7 | PAN | 30 |
| 8 | ETA | 18 |
| 9 | [BMIM][CH3COO] | 29 |
| 10 | [BMIM][HCOO] | 35 |
Neither [BMIM][PF6] nor [BMIM][OH] allowed catalyst free access to the desired 3-substituted-(1H-pyrrol-2-yl)acrylonitriles. We thus examined a broader selection of room temperature ionic liquids previously explored in other aspects of our research endeavours.9,10 These additional room temperature ionic liquids included [BMIM]Br, [BMIM][BF4], [BMIM][NO3], [BMIM][CH3COO], [BMIM][HCOO], and the protic ionic liquids (pILs) EAN, propyl ammonium nitrate (PAN) and ethanol ammonium nitrate (ETA), but in these examples we added piperidine as our catalyst of choice. Interestingly the best yields were noted for the traditional [BMIM]-based ionic liquids (excepting [BMIM][NO3]) with yields ≥31%, while the protic ionic liquids returned yields ≤30%. Of the pILs, PAN returned the highest yield of 30% (Table 2, entry 7). Of these, in the presence of piperidine, only [BMIM][NO3] showed no trace of the desired product (Table 2, entry 5).
The catalytic role of piperidine was investigated by performing the condensation reaction in aqueous medium (Table 3, entries 1 and 2). The results showed that 3% yield for the desired product can be achieved with the use of water alone while a quantitative recovery of the starting materials was observed with the use of a 7:10 mixture of ammonium hydroxide:H2O. This strongly supports a key role for the IL in effecting the Knoevenagel condensation in these instances.
Recently Zhao et al. reported the rapid and efficient Knoevenagel condensation catalysed by ultrasonic irradiation in protic ionic liquids,31 however our initial findings presented in Table 2 suggest that there is no requirement for ultrasonic irradiation in the presence of catalytic quantities of piperidine. Obviating the need for ultrasonic approaches brings this synthetic procedure more within the realms of a traditional synthetic or medicinal chemistry laboratory. However IL mediated approaches would only be useful if they showed broad-spectrum applicability across a range of substituted phenyl acetonitrile analogues. We thus set about examining the effect of various substituents and comparing the outcomes of the subsequent Knoevenagel condensation in three ionic liquids: [BMIM][Br], [BMIM][OH] and PAN. This selection allowed comparison of the effects of traditional vs. protic ionic liquids on the reaction outcome. Previous SAR indicated that electronegative moieties were advantageous to cytotoxic activity, thus we initially examined a range of halogenated phenyl acetonitriles (3–14), along with the alkyne (15), nitrile (16) and nitro (17) acetonitriles.28 The outcomes of these studies are shown in Table 4.
:H2O, [BMIM][OH]:H2O, and PAN:H2O (ratio of ionic liquid to H2O is 7 mL:10 mL) at 50 °C, 5 h with catalytic piperidine
| Phenyl acetonitrile | Product | [BMIM][Br]b yield (%) | [BMIM][OH]b yield (%) | PANb yield (%) |
|---|---|---|---|---|
| a S.M. = start material, reaction condition: pyrrole-2-carboxaldehyde (165 mg, 1.74 mmol), with phenyl acetonitriles (193 μL, 1.65 mmol), H2O (10 mL) and ionic liquid (7 mL).b = [BMIM][Br]:H2O, [BMIM][OH]:H2O, and PAN:H2O (ratio of ionic liquid to water is 7 mL:10 mL). |
||||
 |
 |
56 | 85 | 30 |
 |
 |
44 | 34 | S.M.a |
 |
 |
40 | 88 | 59 |
 |
 |
98 | 77 | 53 |
 |
 |
71 | 68 | 51 |
 |
 |
59 | 68 | 61 |
 |
 |
28 | 54 | S.M. |
 |
 |
90 | 81 | 46 |
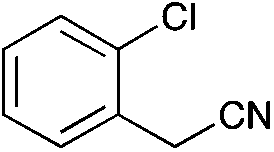 |
 |
42 | 55 | S.M. |
 |
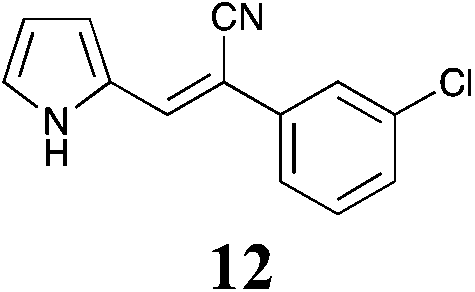 |
79 | 45 | 60 |
 |
 |
89 | 72 | 65 |
 |
 |
93 | 75 | 66 |
 |
 |
77 | 80 | 55 |
 |
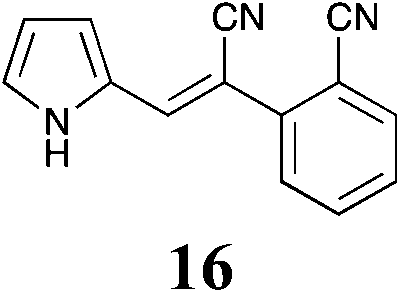 |
60 | 63 | 58 |
 |
 |
58 | 70 | 48 |
Broadly speaking, the Knoevenagel condensation in the selected room temperature ionic liquids detailed in Table 4, leads to the expected 3-substituted-(1H-pyrrol-2-yl)acrylonitriles in moderate to excellent yields across all ionic liquids examined. With [BMIM][Br] the yields of 3-substituted-(1H-pyrrol-2-yl)acrylonitriles 3–17 ranged from 28% (9) to 98% (6); [BMIM][OH] from 34% (4) to 88% (5); and with PAN from 30% (3) to 61% (8). Thus, the protic IL, PAN was the least effective at conducting this transformation. The lower yields (return of unreacted starting material) was most observable in those instances where the starting phenyl acetonitrile possessed an ortho-substituent (Table 4, compounds 4 and 11), and also with the 4-F analogue (9), but this was offset in the disubstituted case (Table 4, compound 13). The lower yields observed with PAN may have been a consequence of the piperidine catalyst interacting with residual free acid that may have been present or free NO3 anions as a result of extraneous H2O. No direct evidence of this was apparent in the 1H NMR of PAN. Overall this IL mediated approach was highly tolerant of simple substituents, and did not require the use of specialist equipment to afford access to the desired products.
As we had proposed that green chemistry approaches were applicable to the development of biologically active compounds, and we have previously reported the cytotoxicity of related compounds,23 we examined compounds 3–17 for their ability to inhibit the cell growth of a panel of cancer cell lines. These cell lines were of colon (HT29, SW480), skin (A431), lung (H460), ovarian (A2780), breast (MCF-7), prostate (Du145), pancreatic (MIA), glioblastoma (SJ-G2, SMA, U87) and neuroblastoma (BE2-C) origin. One normal breast derived cell line, MCF10A, was included as a measure of toxicity to healthy cells. The outcomes of these screening studies are given in Table 5.
| Compound | HT29a | U87b | MCF-7c | A2780d | H460e | A431f | Du145g | BE2-Ch | SJ-G2b | MIAi | SMAj | MCF10Ak |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GI50 (μM) | ||||||||||||
| a Colon.b Glioblastoma.c Breast.d Ovarian.e Lung.f Skin.g Prostate.h Neuroblastoma.i Pancreas.j Murine glioblastoma.k Normal breast cell lines.l Not determined. | ||||||||||||
| 3 (ref. 24) | 41 ± 6 | —l | 17 ± 1 | 25 ± 2 | >50 | >50 | >50 | >50 | 46 ± 7 | >50 | —l | —l |
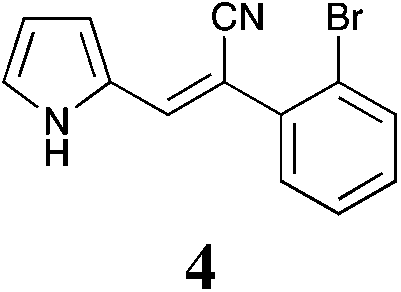
| 4 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
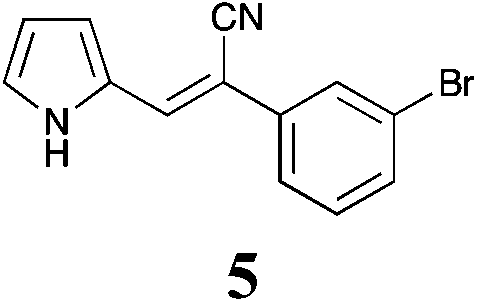
| 5 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
| 6 | 21 ± 1.5 | 40 ± 0.67 | 2.5 ± 0.23 | 18 ± 0.88 | 21 ± 2.8 | 7.2 ± 0.78 | 35 ± 2.2 | —l | 31 ± 0.58 | 40 ± 0.67 | 15 ± 1.9 | 51 ± 5.2 |
| 7 | >50 | >50 | 15 ± 0.67 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
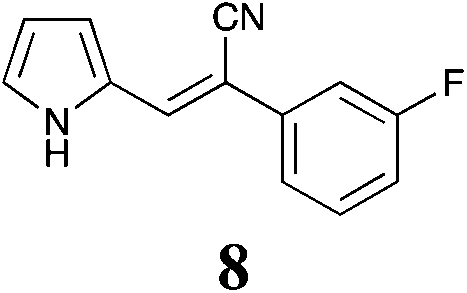
| 8 | 44 ± 1.2 | >50 | 9.8 ± 0.99 | 48 ± 9.0 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
| 9 (ref. 24) | 31 ± 4 | —l | 15 ± 1 | 25 ± 0 | 52 ± 2 | 37 ± 3 | >50 | 43 ± 7 | 30 ± 4 | >50 | —l | —l |
| 10 | 14 ± 0.00 | 32 ± 2.3 | 1.7 ± 0.09 | 21 ± 1.8 | 8.4 ± 2.3 | 2.8 ± 0.12 | 29 ± 1.5 | 31 ± 0.67 | 23 ± 3.2 | 34 ± 1.3 | 24 ± 2.9 | 38 ± 0.58 |
| 11 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
| 12 | 28 ± 1.9 | >50 | 1.9 ± 0.12 | 35 ± 2.3 | 36 ± 1.3 | 4.1 ± 0.35 | >50 | 46 ± 2 | 31 ± 2 | >50 | 49 ± 9 | >50 |
| 13 (ref. 24) | 15 ± 1 | —l | 0.56 ± 0.03 | 16 ± 0 | 5.7 ± 0.7 | 3.2 ± 0.1 | 41 ± 8 | 25 ± 1 | 20 ± 1.0 | 46 ± 7 | —l | —l |
| 14 | 13 ± 0.3 | >50 | 23 ± 2.7 | 15 ± 3.5 | 31 ± 1.0 | 35 ± 1.8 | 35 ± 1.2 | 31 ± 1.2 | 24 ± 4.5 | 40 ± 0.9 | 22 ± 3.8 | >50 |
| 15 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
| 16 (ref. 24) | 25 ± 1 | —l | 18 ± 1 | 18 ± 1 | 32 ± 0 | 27 ± 1 | 26 ± 1 | 22 ± 1 | 24 ± 1 | 34 ± 1 | —l | —l |
| 17 | 32 ± 2.3 | 41 ± 0.67 | 30 ± 0.67 | 32 ± 0.33 | 41 ± 0.3 | 39 ± 0.88 | 39 ± 0.67 | —l | 31 ± 0.33 | 46 ± 1.5 | 26 ± 1.0 | >50 |
The screening data presented in Table 5 shows that analogues 4, 5, 11 and 15 were inactive returning GI50 values >50 μM across all cell lines examined. The parent phenyl analogue, 3, was active against the HT29, MCF-7 and A2780; the 2-F substituted 7 against MCF-7; the 3-F substituted 8 against HT29, MCF-7 and A2780 cell lines. The remaining analogues 9, and 12 displayed modest; and 6, 10, 13, 16 and 17 displayed good activity across cell lines evaluated. Of the brominated analogues only the p-Br (6) was active with GI50 values ranging from 2.5 (MCF-7) to 40 μM (U87 and MIA) and a GI50 value of 51 μM against the normal breast cell line (MCF0A), but the corresponding fluorinated analogues, 7–10, were most active against the breast cancer cell line with GI50 values of 15, 9.8, 1.7 μM, respectively. The chlorinated analogues, 12–14, displayed modest to good levels of broad spectrum activity and retained specificity for the MCF-7 breast cancer cell line with GI50 values of 1.9 and 0.56 for 12 and 13 μM, respectively. Interestingly while the introduction of a second chlorine moiety with 12 and 13 gave rise to sub micromolar potency against the MCF-7 cell line, this activity was reduced significantly in the case of the 3,5-dichloro substituent pattern (14, MCF-7 GI50 23 μM) compared with the 3,4-dichloro substituent pattern of 13.
The fluorine substitution pattern also had a marked effect on cytotoxicity with the 2-F (7) active against only the breast cancer cell line MCF-7 with a GI50 = 15 ± 0.67 μM. Both the 3-F (8) and 4-F (9) analogues displayed a broad spectrum of cytotoxicity with 8 active against two cell lines (HT29, GI50 44 ± 1.2 μM and MCF-7, 9.8 ± 0.99 μM) and 9 active against seven cell lines displaying a modest level of selectivity for the MCF-7 cell line. In keeping with the trend observed with the fluorinated analogues, the 2-Cl analogue (11) was inactive; the 3-Cl analogue (12) displays excellent activity across all cell lines excepting U87, Du145, MIA and MCF10A (normal breast cells) with GI50 values for the active cell lines ranging from 1.9 ± 0.12 to 49 ± 9 μM. The activity of 12 was most notable with MCF-7 (1.9 ± 0.12 μM) and A431 (4.1 ± 0.35 μM) both of which are oestrogen positive cell lines. Introduction of a second chlorine substituent with 13 saw a significant enhancement in cytotoxicity across essentially all of the cell lines examined. In some cases there was a six fold potency enhancement relative to the mono-Cl analogue 12 with the H460 cell line (12 GI50 = 36 ± 1.3 vs. 13 GI50 = 5.7 ± 0.7 μM), and this extra –Cl substituent resulted in sub micromolar potency against MCF-7 cell lines (0.56 ± 0.03 μM). Both the ethynyl (15) and nitro (17) display moderate levels of cytotoxicity across most cell lines examined.
Conclusion
The Knoevenagel condensation of 1H-pyrrole-2-carbaldehyde with a range of phenyl acetonitriles proceeded smoothly and in excellent yields (up to 98%) in [BMIM] based ionic liquids ([BMIM][OH] and [BMIM][Br]) but only with added catalytic piperidine. The corresponding condensations in the pIL, PAN gave consistently lower isolated yields, typically between 30–66% although in a number of cases no product was observed. It is conceivable that this was a function of the catalytic piperidine interacting with either residual HNO3 or free NO3 in PAN.Screening of the 3-substituted-(1H-pyrrol-2-yl)acrylonitriles analogues 3–17 against a panel of 11 cancer cell lines and one normal cell line allowed the identification of a series of compounds with broad spectrum cytotoxicity, but more interestingly that a significant degree of MCF-7 breast cancer cell line specificity was evident with 6 (7–>25 fold) and 13 (5.7–82 fold). Other analogues show high level of efficacy against specific cell lines with 10 showing excellent activity against MCF-7 (GI50 = 1.7 μM) and A431 (GI50 = 2.8 μM) cell lines.
These data suggest that ionic liquids do facilitate ease of access to a range of cytotoxic 3-substituted-(1H-pyrrol-2-yl)acrylonitriles and that these analogues display an interesting breadth of cytotoxic activity and that with further development may produce significantly more potent potential development candidates. The most promising of the compounds identified herein are the 4-CF3 substituted 10 and the 3,4-dichloro substituted 13 with excellent activities against MCF-7 and A431 cell lines. The 3,4-dichloro-13 was a 0.56 μM potent inhibitor of MCF-7 cell growth.
Experimental
All reagents were purchased from Sigma-Aldrich, Matrix Scientific or Lancaster Synthesis and were used without purification. With the exception of THF (anhydrous >99%) obtained from Sigma-Aldrich, all solvents were re-distilled from glass prior to use.1H and 13C NMR spectra were recorded on a Brüker Avance™ AMX 400 MHz spectrometer at 400 and 101 MHz, respectively. Chemical shifts (δ) are reported in parts per million (ppm) measured relative to the internal standards, and coupling constants (J) are expressed in Hertz (Hz). Mass spectra were recorded on a Shimadzu LCMS 2010 EV using a mobile phase of 1:1 acetonitrile:H2O with 0.1% formic acid.
General method for synthesis of (Z)-2-phenyl-3-(1H-pyrrol-2-yl)acrylonitrile (3)
1H-Pyrrole-2-carbaldehyde (1) (165 mg, 1.74 mmol) was added to vigorously stirred H2O (10 mL) and heated to 50 °C upon which it dissolved. Phenyl acetonitrile (2) (193 mg, 1.65 mmol) was then slowly added forming a suspension and then piperidine (2 drops, catalytic) was added. Heating was continued at 50 °C and once a clear solution was evident, typically 5–10 min, the ionic liquid (7 mL) was added dropwise. The reaction vessel was then sealed and the mixture stirred at 50 °C for 5 h. The reaction solution was filtered hot, washed with warm H2O and dried under vacuum to yield a solid. The crude solid was recrystallised from EtOH to afford compound (Z)-2-phenyl-3-(1H-pyrrol-2-yl)acrylonitrile (3) as a brown solid.MP 93–94 °C; IR ν(cm−1): 3396, 2206, 1600, 1588, 1129, 753, 729, 681, 588, 484; 1H NMR (CDCl3): δ 9.80 (brs, 1H), 7.61–7.55 (m, 2H), 7.44–7.38 (m, 3H), 7.36–7.29 (m, 2H), 7.10–7.04 (m, 1H), 6.74–6.66 (m, 1H), 6.37–6.32 (m, 1H); 13C NMR (101 MHz, CDCl3): δ 134.0, 131.4, 129.2, 128.3, 127.9, 125.1, 124.1, 120.8, 119.3, 110.9, 101.5.
MP 122–123 °C; IR ν(cm−1): 3309, 2205, 1596, 1139, 731, 596, 444. 1H NMR (CDCl3): δ 9.99 (brs, 1H), 7.79 (dd, J = 8.0, 1.2 Hz, 1H), 7.57–7.45 (m, 2H), 7.40 (s, 1H), 7.41–7.32 (m, 1H), 7.27–7.20 (m, 1H), 6.84–6.77 (m, 1H), 6.53–6.46 (m, 1H). 13C NMR (CDCl3): δ 137.5, 135.7, 133.8, 131.0, 130.1, 128.1, 127.4, 124.5, 123.0, 120.1, 119.6, 110.9, 100.7.
MP 12–22 °C; IR ν(cm−1): 3309, 2205, 1597, 1430, 1139, 731, 596, 444; 1H NMR (CDCl3): δ 9.79 (brs, 1H), 7.71 (t, J = 1.9 Hz, 1H), 7.52–7.47 (m, 1H), 7.47–7.42 (m, 1H), 7.39 (s, 1H), 7.31–7.27 (m, 1H), 7.09 (q, J = 2.6 Hz, 1H), 6.73 (t, J = 3.7 Hz, 1H), 6.40–6.33 (m, 1H); 13C NMR (CDCl3): δ 136.2, 132.2, 131.2, 130.7, 127.9, 127.6, 124.8, 123.8, 123.4, 120.4, 120.1, 111.2, 99.8.
MP 123–124 °C; IR ν(cm−1): 3374, 2213, 1487, 1131, 817, 739, 594, 489; 1H NMR (CDCl3): δ 9.77 (brs, 1H), 7.53 (d, J = 8.6 Hz, 2H), 7.44 (d, J = 8.6 Hz, 2H), 7.38 (s, 1H), 7.11–7.06 (m, 1H), 6.74–6.66 (m, 1H), 6.40–6.32 (m, 1H); 13C NMR (101 MHz, CDCl3): δ 133.1, 132.3, 131.6, 127.7, 126.6, 124.6, 122.2, 120.4, 119.8, 111.2, 100.3.
MP 100–102 °C; IR ν(cm−1): 3409, 2205, 1603, 1125, 740, 677; 1H NMR (CDCl3): δ 9.83 (brs, 1H), 7.57–7.47 (m, 2H), 7.35–7.06 (m, 4H), 6.75–6.65 (m, 1H), 6.40–6.31 (m, 1H); 13C NMR (CDCl3): δ 159.7 (d, J = 248.7 Hz), 135.9 (d, J = 9.4 Hz), 129.7 (d, J = 8.6 Hz), 129.1 (d, J = 2.5 Hz), 127.8, 124.8 (d, J = 3.7 Hz), 124.6, 122.4 (d, J = 10.7 Hz), 120.6, 119.9, 116.6 (d, J = 22.2 Hz), 111.0, 95.9 (d, J = 2.2 Hz).
MP 104–107 °C; IR ν(cm−1): 3396, 2208, 1583, 1407, 1121, 745, 594, 522; 1H NMR (CDCl3): δ 9.79 (s, 1H), 7.43–7.34 (m, 3H), 7.30–7.27 (m, 0H), 7.09 (td, J = 2.8, 1.4 Hz, 1H), 6.73 (dd, J = 3.7, 1.6 Hz, 1H), 6.37 (dt, J = 3.7, 2.4 Hz, 1H); 13C NMR (101 MHz, CDCl3): δ 163.3 (d, J = 245.2 Hz), 136.3 (d, J = 8.1 Hz), 132.1, 130.8 (d, J = 8.5 Hz), 127.6, 124.7, 120.9 (d, J = 2.9 Hz), 120.4, 120.0, 115.1 (d, J = 21.2 Hz), 111.9 (d, J = 23.4 Hz), 111.2, 100.2 (d, J = 2.9 Hz).
MP 10–103 °C; IR ν(cm−1): 3393, 2208, 1593, 1122, 857, 745, 687; 1H NMR (CDCl3): δ 9.76 (s, 1H), 7.54 (dd, J = 8.7, 5.2 Hz, 2H), 7.32 (s, 1H), 7.16–7.04 (m, 3H), 6.69 (s, 1H), 6.35 (q, J = 2.8 Hz, 1H); 13C NMR (101 MHz, CDCl3): δ 162.7 (d, J = 246.7 Hz), 131.3 (d, J = 22.1 Hz), 130.3 (d, J = 3.4 Hz), 127.7, 126.9 (d, J = 8.2 Hz), 124.2, 120.6, 119.3, 116.2 (d, J = 21.9 Hz), 111.0, 100.4.
MP 149–150 °C; IR ν(cm−1): 3389, 2205, 1590, 1167, 1111, 833, 752, 583; 1H NMR (CDCl3): δ 9.83 (brs, 1H), 7.72–7.62 (m, 4H), 7.48 (s, 1H), 7.16–7.09 (m, 1H), 6.80–6.73 (m, 1H), 6.42–6.35 (m, 1H); 13C NMR (CDCl3): δ 137.6 (q, J = 1.4 Hz), 132.8, 130.0 (q, J = 32.6 Hz), 127.6, 126.2 (q, J = 3.8 Hz), 125.2, 124.0 (q, J = 270.3 Hz), 120.6, 120.3, 111.4, 99.7.
MP 110–112 °C; IR ν(cm−1): 3309, 2207, 1595, 1141, 730, 595; 1H NMR (CDCl3): δ 9.87 (brs, 1H), 7.47–7.43 (m, 1H), 7.42–7.38 (m, 1H), 7.34–7.29 (m, 2H), 7.18 (s, 1H), 7.12–7.06 (m, 1H), 6.72–6.65 (m, 1H), 6.40–6.32 (m, 1H); 13C NMR (101 MHz, CDCl3): δ 137.5, 133.8, 133.1, 130.6, 130.5, 129.9, 127.5, 124.5, 120.2, 119.6, 110.9, 98.9.
MP 111–112 °C; IR ν(cm−1): 3386, 2212, 1605, 1528, 1398, 1132, 1039, 732, 681, 590; 1H NMR (CDCl3): δ 9.79 (brs, 1H), 7.62–7.51 (m, 1H), 7.50–7.42 (m, 2H), 7.40 (s, 1H), 7.34 (t, J = 7.8 Hz, 1H), 7.13–7.06 (m, 1H), 6.76–6.69 (m, 1H), 6.40–6.33 (m, 1H); 13C NMR (CDCl3): δ 135.9, 135.3, 132.1, 130.4, 128.2, 127.6, 125.0, 124.8, 123.3, 120.4, 120.1, 111.2, 100.0.
MP 140–141 °C; IR ν(cm−1): 3416, 2200, 1604, 1125, 748, 590, 492; 1H NMR (CDCl3): δ 9.78 (brs, 1H), 7.65 (d, J = 2.2 Hz, 1H), 7.47 (d, J = 8.5 Hz, 1H), 7.41 (d, J = 2.3 Hz, 1H), 7.38 (s, 1H), 7.15–7.07 (m, 1H), 6.77–6.72 (m, 1H), 6.40–6.34 (m, 1H); 13C NMR (CDCl3): δ 134.2, 133.6, 132.2, 131.1, 130.1, 127.5, 126.7, 125.1, 124.2, 120.4, 120.1, 111.4, 98.9.
MP 148–156.6 °C; IR ν(cm−1): 3381, 2208, 1593, 1141, 742, 594; 1H NMR (CDCl3): δ 9.84 (brs, 1H), 7.47 (d, J = 2.0 Hz, 1H), 7.36–7.27 (m, 2H), 7.16 (s, 1H), 7.12–7.08 (m, 1H), 6.71–6.66 (m, 1H), 6.39–6.33 (m, 1H). 13C NMR (CDCl3): δ 137.65, 135.20, 133.81, 132.38, 131.34, 130.40, 127.86, 127.34, 124.86, 120.15, 119.98, 111.10, 97.68.
MP 100–102 °C; IR ν(cm−1): 3446, 3268, 2194, 1588, 1037, 831, 720, 520, 435; 1H NMR (CDCl3): δ 9.80 (brs, 1H), 7.60–7.47 (m, 4H), 7.41 (s, 1H), 7.12–7.05 (m, 1H), 6.75–6.68 (m, 1H), 6.40–6.32 (m, 1H), 3.16 (s, 1H); 13C NMR (101 MHz, CDCl3): δ 134.4, 132.9, 131.7, 127.8, 124.8, 124.7, 121.9, 120.4, 119.9, 111.2, 100.6, 83.2, 78.8.
MP 148–149 °C; IR ν(cm−1): 3420, 2218, 2199, 1605, 1403, 1333, 740, 572; 1H NMR (CDCl3): δ 9.81 (brs, 1H), 7.75 (d, J = 7.7 Hz, 1H), 7.68–7.61 (m, 2H), 7.56 (s, 1H), 7.43 (ddd, J = 7.7, 5.9, 2.7 Hz, 1H), 7.14 (q, J = 2.7 Hz, 1H), 6.85–6.81 (m, 1H), 6.41–6.37 (m, 1H); 13C NMR (CDCl3): δ 138.2, 137.3, 134.7, 133.4, 129.2, 128.5, 127.2, 125.6, 121.1, 120.0, 117.9, 111.6, 110.0, 97.2.
MP 130–133 °C; IR ν(cm−1): 3368, 2208, 1507, 1576, 1327, 1034, 757, 683, 482; 1H NMR (CDCl3): δ 9.87 (brs, 1H), 8.27 (d, J = 8.9 Hz, 2H), 7.72 (d, J = 8.9 Hz, 2H), 7.55 (s, 1H), 7.22–7.10 (m, 1H), 6.88–6.73 (m, 1H), 6.47–6.29 (m, 1H); 13C NMR (CDCl3): δ 147.1, 140.5, 133.8, 127.6, 126.2, 125.4, 124.6, 121.8, 120.0, 111.8, 98.8.
Acknowledgements
AAO thanks the Saudi Arabian government for the provision of a PhD scholarship. AM acknowledges project funding from the Australian Research Council, and CPG is the recipient of an Australian Research Council DECRA fellowship.References
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
- M. C. Bryan, B. Dillon, L. G. Hamann, G. J. Hughes, M. E. Kopach, E. A. Peterson, M. Pourashraf, I. Raheem, P. Richardson, D. Richter and H. F. Sneddon, J. Med. Chem., 2013, 56, 6007–6021 CrossRef CAS PubMed.
- D. J. C. Constable, C. Jimenez-Gonzalez and R. K. Henderson, Org. Process Res. Dev., 2007, 11, 133–137 CrossRef CAS.
- R. Gani, C. Jimenez-Gonzalez and D. J. C. Constable, Comput. Chem. Eng., 2005, 29, 1661–1676 CrossRef CAS PubMed.
- K. Grodowska and A. Parczewski, Acta Pol. Pharm., 2010, 67, 3–12 CAS.
- A. B. McGeachie, L. R. Odell, A. Quan, N. Chau, T. A. Hill, D. J. Keating, M. A. Cousin, E. M. van Dam, J. Daniel, A. Mariana, A. Whiting, S. Perera, A. Novelle, J. Gilbert, J. A. Sakoff, M. Chircop, A. McCluskey and P. J. Robinson, ACS Chem. Biol., 2013, 8, 1507–1518 CrossRef CAS PubMed.
- L. R. Odell, D. Howan, C. P. Gordon, M. J. Robertson, N. Chau, A. Mariana, A. E. Whiting, R. Abagyan, J. A. Daniel, N. N. Gorgani, P. J. Robinson and A. McCluskey, J. Med. Chem., 2010, 53, 5267–5280 CrossRef CAS PubMed.
- C. P. Gordon, B. Venn-Brown, M. J. Robertson, K. A. Young, N. Chau, A. Quan, P. J. Robinson and A. McCluskey, J. Med. Chem., 2013, 56, 46–59 CrossRef CAS PubMed.
- M. Tarleton and A. McCluskey, Tetrahedron Lett., 2011, 52, 1583–1586 CrossRef CAS PubMed.
- C. P. Gordon, N. Byrne and A. McCluskey, Green Chem., 2010, 12, 1000–1006 RSC.
- L. von Kleist, W. Stahlschmidt, H. Bulut, K. Gromova, D. Puchkov, M. J. Robertson, K. A. MacGregor, N. Tomlin, A. Pechstein, N. Chau, M. Chircop, J. A. Sakoff, J. von Kries, W. Saenger, H.-G. Kräusslich, O. Shupliakov, P. J. Robinson, A. McCluskey and V. Haucke, Cell, 2011, 146, 471–484 CrossRef CAS PubMed.
- C. B. Harper, M. R. Popoff, A. McCluskey, P. J. Robinson and F. A. Meunier, Trends Cell Biol., 2013, 23, 90–101 CrossRef CAS PubMed.
- B. E. Campbell, M. Tarleton, C. P. Gordon, A. McCluskey, J. A. Sakoff, J. Gilbert and R. B. Gasser, Bioorg. Med. Chem. Lett., 2011, 21, 3277–3281 CrossRef CAS PubMed.
- M. J. Robertson, S. G. Stewart, C. P. Gordon, J. Gilbert, J. A. Sakoff and A. McCluskey, Bioorg. Med. Chem., 2011, 19, 5734–5741 CrossRef CAS PubMed.
- B. E. Campbell, A. Hoffman, A. McCluskey and R. B. Gasser, Biotechnol. Adv., 2011, 29, 28–39 CrossRef CAS PubMed.
- A. McCluskey, A. T. R. Sim and J. A. Sakoff, J. Med. Chem., 2002, 45, 1151–1175 CrossRef CAS.
- W. H. Correa, J. K. Edwards, A. McCluskey, I. McKinnon and J. L. Scott, Green Chem., 2003, 5, 30–33 RSC.
- F. Freeman, Chem. Rev., 1980, 80, 329 CrossRef CAS.
- L. F. Tietze, Chem. Rev., 1996, 96, 115 CrossRef CAS PubMed.
- Y. O. Aharma and M. S. Degani, Green Chem., 2009, 11, 526 RSC.
- K. Ebitani, K. Motokura, K. Mori, T. Mizugaki and K. Kaneda, J. Org. Chem., 2006, 71, 5440 CrossRef CAS PubMed.
- E. Knoevenagel, Ber. Dtsch. Chem. Ges., 1896, 29, 172 CrossRef CAS.
- L. F. Tietze, Pure Appl. Chem., 2004, 76, 1967 CrossRef CAS.
- M. Tarleton, J. Gilbert, M. J. Robertson, A. McCluskey and J. A. Sakoff, MedChemComm, 2011, 2, 31–37 RSC.
- M. Ali, J.-A. Bliese, M. Rasmussen, R. M. Sargent, S. Saubern, D. G. Sawutz, J. S. Wilkie, D. A. Winkler, K. N. Winzenberg and R. C. J. Woodgate, Bioorg. Med. Chem. Lett., 2007, 17, 993–997 CrossRef PubMed.
- G. Alberghina, M. E. Amato, F. A. Bottino, A. Corsaro and S. Fisichella, J. Heterocycl. Chem., 1986, 23, 1747–1752 CrossRef CAS.
- W. Herz and J. Brasch, J. Org. Chem., 1958, 23, 711–714 CrossRef CAS.
- R. V. Hangarge, D. V. Jarikote and M. S. Shingare, Green Chem., 2002, 4, 266–268 RSC.
- P. Verdía, F. Santamarta and E. Tojo, Molecules, 2011, 16, 4379–4388 CrossRef PubMed.
- W. H. Correa, J. K. Edwards, A. McCluskey, I. McKinnon and J. L. Scott, Green Chem., 2003, 5, 30–33 RSC.
- S. Zhao, X. Wang and L. Zhang, RSC Adv., 2013, 3, 11691–11696 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra. See DOI: 10.1039/c3ra47418f |
| This journal is © The Royal Society of Chemistry 2014 |