The effect of remote substitution on the formation of preferential isomers of cobalt(III)-tetrazolate complexes by microwave assisted cycloaddition†
Received
2nd July 2014
, Accepted 18th July 2014
First published on 24th July 2014
Abstract
The 1,3-dipolar cycloaddition reaction of cis-[Co(N3)2(en)2]NO31 with different organonitriles (NCR) under focussed microwave irradiation produced bis-tetrazolate complexes [Co(N4CR)2(en)2](NO3). Interestingly, in the case of 3-cyano pyridine the reaction produced both cis- and trans-isomers (cis-2 and trans-2), whereas for 4-cyano pyridine the compound obtained was exclusively cis (cis-3) and for 4-bromobenzonitrile it was only the trans- (trans-4) compound which was isolated. This indicates a probable role of remote substitution of the phenyl ring in dictating the formation of the preferential isomer. When starting from the trans-isomer of the diazido complex (trans-[Co(N3)2(en)2]ClO4, 1a), upon reacting with different nitriles a mixture of cis- and trans-isomers of [Co(N4CR)2(en)2]ClO4 was produced in each case, with a greater preference towards cis-geometry [R = 4-NC5H4 (cis-5 and trans-5), 4-BrC6H4 (cis-6 and trans-6) and C6H5 (cis-7 and trans-7)]. The preferential formation of the cis-analogue of compound trans-4 when starting from the trans-precursor was quite curious. A theoretical investigation of compounds trans-4 and cis-6 reveals that the greater stability of the trans-complex 4 may arise from additional van der Waals interactions in the solid state because of the presence of an extra DMF molecule as solvent of crystallization. However, an interacting counter-anion and a probable halogen–halogen interaction may also contribute to the formation of preferential isomers for cycloaddition complexes, even in the solution state.
Introduction
Metal-promoted azidation of nitriles provides an easy and convenient method to synthesize metal tetrazolate complexes. Tetrazole behaves as an interesting ligand as it can act in a unidentate or polydentate fashion, furnishing mono- and polynuclear compounds with interesting topologies.1 Moreover, tetrazole-based complexes have evoked much interest because of their utility in organic synthesis,2 gas generating agents,3 medicinal chemistry4 and anti-corrosion species.5 In recent years many metal tetrazolate complexes have been synthesized using the [2 + 3] cycloadditions of metal ligated azides and nitriles. However, most of them are associated with group 10 transition metals,6 apart from some limited examples of some other transition metal elements.7,8 There are a few examples of cobalt tetrazolate complexes, however in most of these cases the tetrazole itself has been used as the ligand9,10 or it has been generated by in situ [2 + 3] cycloaddition of an azide and a nitrile in a hydrothermal process.11 To the best of our knowledge there are very few examples where the in situ generation of a tetrazolate ligand has been reported using cobalt ions in a controlled and systematic manner.12,13 Moreover, as cobalt(III) ions tend to form different geometrical isomers it was also interesting to investigate if there is any influence of the counter-ions used and the remote substitution of the 5-tetrazole ligands on the overall preferential geometry adopted by the cobalt center.
Herein, we have investigated the reactions of different organonitriles with cobalt(III) coordinated azides and found that cis-[Co(N3)2(en)2]NO3 and trans-[Co(N3)2(en)2]ClO4 react with various RCN compounds to give the corresponding cis- or trans-bis(tetrazolate) complexes or their mixtures depending upon various factors like remote substitution, counter-anion used or solvent of crystallization in the cycloadded tetrazolato ligand.
Experimental section
Materials and instrumentation
All the chemical reagents required were purchased from Sigma and used without further purification. cis-[Co(N3)2(en)2](NO3) 1 and trans-[Co(N3)2(en)2](ClO4) 1a were prepared according to reported methods.14 Infrared spectra (4000–500 cm−1) were recorded with a BRUKER TENSOR 27 instrument in KBr pellets. Mass spectrometric analyses were carried out on a Bruker-Daltonics, microTOF-Q II mass spectrometer. The microwave irradiation experiments were carried out in a focused microwave CEM discover reactor 300W and the reaction tube used had 10 ml capacity and 13 mm internal diameter. Elemental analyses were carried out with a ThermoFlash 2000 elemental analyzer. UV-visible absorption spectra of all the compounds in methanol were recorded on a Carry-100 Bio UV-Visible spectrophotometer. 1H NMR (400 MHz) spectra in DMSO-d6 were measured on an AVANCE III 400 Ascend Bruker BioSpin machine at ambient temperature.
Caution! Azide, tetrazolate and perchlorate compounds are potentially explosive. Only a small amount of material should be prepared and should be handled with care.
Synthesis
cis-[Co{5-(3-pyridyl)-tetrazolato}2(en)2]NO3·DMF (cis-2) and trans-[Co{5-(3-pyridyl)-tetrazolato}2(en)2]NO3 (trans-2).
0.12 g (0.37 mmol) of cobalt diazide complex 1 was dissolved in 5 mL of DMF in a cylindrical pyrex tube and 0.31 g (3 mmol) of 3-cyanopyridine was added to this solution. The system was irradiated for 1 h at 130 °C in a microwave reactor. The solvent was then removed in vacuo and the resulting reddish-brown residue was washed several times with diethyl ether to obtain a brown powder. Both cis- and trans-isomers were detected in the resultant compound by 1H NMR spectroscopy, and are formed tentatively in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio (Fig. S1: ESI†). The mixture was separated by ion-exchange chromatography using SP-Sephadex C-25, which is a strongly acidic (–SO3−Na+) cation exchanger based on cross-linked dextran. 0.1 M NaNO3 solution was used as the elution solvent to run the column. Both isomers were then recrystallized from a DMF–diethyl ether mixture. Although it was possible to grow suitable X-ray diffraction-quality crystals for compound cis-2 during recrystallization, no single crystal was obtained for the corresponding trans isomer.
:4 ratio (Fig. S1: ESI†). The mixture was separated by ion-exchange chromatography using SP-Sephadex C-25, which is a strongly acidic (–SO3−Na+) cation exchanger based on cross-linked dextran. 0.1 M NaNO3 solution was used as the elution solvent to run the column. Both isomers were then recrystallized from a DMF–diethyl ether mixture. Although it was possible to grow suitable X-ray diffraction-quality crystals for compound cis-2 during recrystallization, no single crystal was obtained for the corresponding trans isomer.
cis-[Co{5-(3-pyridyl)-tetrazolato}2(en)2]NO3·DMF (cis-2).
Yield: 11%. IR (KBr, cm−1): νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N 1640, NO3 1384. Anal. Calc. for C19H24N16O4Co (599.43): C 38.03; H 4.00; N 37.36; Found: C 37.92; H 3.89; N 37.20; MS(ESI): m/z = 471.15 [M]+. 1H NMR: δH (400 MHz, DMSO-d6, Me4Si): 2.29–2.90 (m, 8H, CH2), 4.73 (s, 2H, NH2), 5.04 (s, 2H, NH2), 5.81 (s, 2H, NH2), 6.29 (s, 2H, NH2), 6.60 (s, 2H, aromatic), 7.50 (s, 2H, aromatic), 8.30 (s, 2H, aromatic), 8.58 (s, 2H, aromatic) (see Fig. S2: ESI† for 1H NMR spectrum).
N 1640, NO3 1384. Anal. Calc. for C19H24N16O4Co (599.43): C 38.03; H 4.00; N 37.36; Found: C 37.92; H 3.89; N 37.20; MS(ESI): m/z = 471.15 [M]+. 1H NMR: δH (400 MHz, DMSO-d6, Me4Si): 2.29–2.90 (m, 8H, CH2), 4.73 (s, 2H, NH2), 5.04 (s, 2H, NH2), 5.81 (s, 2H, NH2), 6.29 (s, 2H, NH2), 6.60 (s, 2H, aromatic), 7.50 (s, 2H, aromatic), 8.30 (s, 2H, aromatic), 8.58 (s, 2H, aromatic) (see Fig. S2: ESI† for 1H NMR spectrum).
trans-[Co{5-(3-pyridyl)-tetrazolato}2(en)2]NO3·DMF (trans-2).
Yield: 38%. IR (KBr, cm−1): νCN 1617, NO3 1384. Anal. Calc. for C19H24N16O4Co (599.43): C 38.03; H 4.00; N 37.36; Found: C 37.83; H 3.82; N 37.03; MS(ESI): m/z = 471.15 [M]+. 1H NMR: δH (400 MHz, DMSO-d6, Me4Si): 2.06–2.26 (m, 8H, CH2), 4.55 (s, 8H, NH2), 6.78–7.55 (m, 8H, aromatic) (see Fig. S3: ESI† for 1H NMR spectra).
cis-[Co{5-(4-pyridyl)-tetrazolato}2(en)2]NO3·Et2O (cis-3).
This compound was prepared in an identical manner to that of compound cis-2. However, in this case only pure cis-compound has been isolated instead of a mixture and it was recrystallized from a methanol–diethyl ether mixture, which produced some single crystals for X-ray diffraction analysis.
cis-[Co{5-(4-pyridyl)-tetrazolato}2(en)2]NO3·Et2O (cis-3).
Yield: 58%. IR (KBr, cm−1): νCN 1614, NO3 1384. Anal. Calc. for C20H24N15O4Co (597.47): C 40.16; H 4.01; N 35.14; Found: C 39.94; H 3.89; N 35.04; MS(ESI): m/z = 471.20 [M]+. 1H NMR: δH (400 MHz, DMSO-d6, Me4Si): 2.34–2.83 (m, 8H, CH2), 4.60 (s, 2H, NH2), 4.85 (s, 2H, NH2), 5.82 (s, 2H, NH2), 6.31 (s, 2H, NH2), 7.91 (d, 4H, aromatic), 8.87 (d, 4H, aromatic) (see Fig. 1 for 1H NMR spectra).
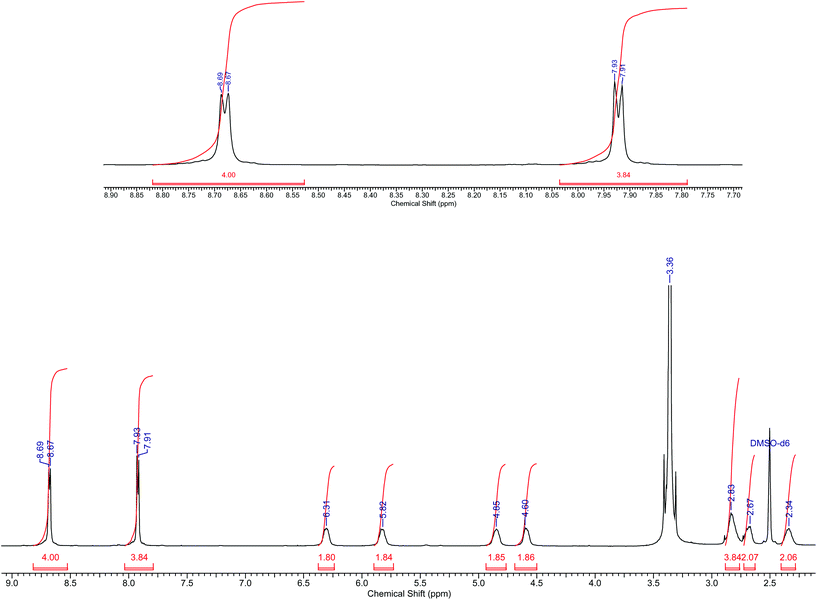 |
| | Fig. 1
1H NMR spectra of cis-3. | |
trans-[Co{5-(4-bromophenyl)-tetrazolato}2(en)2]NO3·DMF (trans-4).
A solution of 0.12 g (0.37 mmol) of cobalt diazide complex 1 and 0.54 g (3 mmol) of 4-bromobenzonitrile in 5 mL of DMF was added to a cylindrical pyrex tube and irradiated for 3 h at 130 °C. The solvent was then removed in vacuo and the resulting reddish-brown residue was washed several times with diethyl ether to obtain a brown powder, which was then recrystallized from a DMF–ether mixture to obtain a reddish-brown crystalline compound along with single crystals suitable for X-ray diffraction studies.
trans-[Co{5-(4-bromophenyl)-tetrazolato}2(en)2]NO3·DMF (trans-4).
Yield: 56%. IR (KBr, cm−1): νCN 1665, NO3 1384. Anal. Calc. for C21H31N14O4Br2Co (762.35): C 33.05; H 4.06; N 25.70; Found: C 32.85; H 3.90; N 25.31; MS(ESI): m/z = 626.99 [M]+. 1H NMR: δH (400 MHz, DMSO-d6, Me4Si): 2.45 (s, 8H, CH2), 5.41 (s, 8H, NH2), 7.73 (d, 4H, aromatic), 8.08 (d, 4H, aromatic) (see Fig. 2 for 1H NMR spectra).
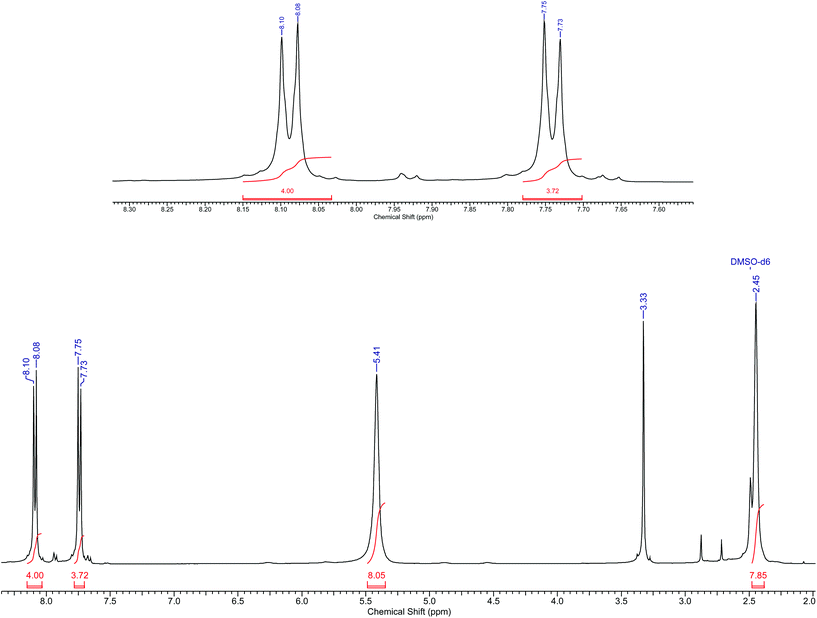 |
| | Fig. 2
1H NMR spectra of trans-4. | |
cis-[Co{5-(4-pyridyl)-tetrazolato}2(en)2]ClO4·H2O (cis-5).
0.12 g (0.33 mmol) of trans-cobalt diazide complex 1a was dissolved in 5 mL of DMF in a cylindrical pyrex tube and 0.31 g (3 mmol) of 4-cyanopyridine was added to this solution. The system was irradiated for 1 h at 130 °C in a microwave reactor. The solvent was then removed in vacuo and the resulting reddish-brown residue was washed several times with diethyl ether to obtain a brown powder. This brown powder was found to be a mixture of cis- and trans-[Co{5-(4-pyridyl)-tetrazolato}2(en)2]ClO4, which was formed tentatively in a 5:2 ratio (see Fig. S4: ESI† for 1H NMR spectrum). Separation of the mixture was attempted by an ion-exchange method with SP-Sephadex C-25. However, only the cis-compound could be isolated upon elution with NaNO3 whereas the trans-compound decomposed in the ion-exchange resin column. The resultant cis-compound was then recrystallized from a methanol–diethyl ether mixture, which furnished some X-ray diffraction-quality crystals.
cis-[Co{5-(4-pyridyl)-tetrazolato}2(en)2]ClO4·H2O (cis-5).
Yield: 56%. IR (KBr, cm−1): νCN 1612, ClO4 1096. Anal. Calc. for C16H26N14O5CoCl (588.85): C 32.60; H 4.41; N 33.28; Found: C 32.06; H 4.10; N 33.07; MS(ESI): m/z = 471.15 [M]+. 1H NMR: δH (400 MHz, DMSO-d6, Me4Si): 1.91–2.82 (m, 8H, CH2), 4.57 (s, 2H, NH2), 4.84 (s, 2H, NH2), 5.82 (s, 2H, NH2), 6.30 (s, 2H, NH2), 7.91 (d, 4H, aromatic), 8.67 (d, 4H, aromatic) (see Fig. S5: ESI† for 1H NMR spectra).
cis-[Co{5-(4-bromophenyl)-tetrazolato}2(en)2]ClO4 (cis-6) and trans-[Co{5-(4-bromophenyl)-tetrazolato}2(en)2]ClO4 (trans-6).
Solid cobalt diazide complex trans-[Co(N3)2(en)2]ClO4 (0.12 g, 0.33 mmol) 1a and 0.54 g (3 mmol) of 4-bromobenzonitrile in 5 mL of DMF were added to a cylindrical pyrex tube and were irradiated for 3 h at 130 °C. After removing the solvent in vacuo, the resulting reddish-brown residue was washed several times with diethyl ether. The resultant brown powder was found to be a mixture of cis- and trans-compounds in a tentative ratio of 4:1 (see Fig. S6: ESI† for 1H NMR spectrum). However the mixtures could not be separated by an ion-exchange method as both compounds decomposed in the column. Recrystallization of the brown powder from a methanol–diethyl ether mixture provides a crystalline compound which has been found to still be a mixture. However, it was possible to grow single crystals of the cis-isomer by this method.
cis- and trans-[Co{5-(4-bromophenyl)-tetrazolato}2(en)2]ClO4 (cis- and trans-6).
Yield: 54%. IR (KBr, cm−1): νCN 1666, ClO4 1126. Anal. Calc. for C18H24N12O4Br2CoCl (726.65): C 29.72; H 3.30; N 23.11; Found: C 30.00; H 3.62; N 22.98; MS(ESI): m/z = 627.18 [M]+. 1H NMR: δH (400 MHz, DMSO-d6, Me4Si): 2.32–2.88 (m, 8H, CH2), 4.52, 4.91, 5.80, & 6.27 (all singlets, 8H, from the cis isomer), 5.41 (s, 8H, from the trans isomer), 7.66–8.10 (m, 8H, aromatic) (see Fig. S7: ESI† for 1H NMR spectra).
cis-[Co(5-phenyltetrazolato)2(en)2]ClO4 (cis-7) and trans-[Co(5-phenyltetrazolato)2(en)2]ClO4 (trans-7).
Solid cobalt diazide complex [Co(N3)2(en)2]ClO4 (0.12 g, 0.33 mmol) 1a and 2 mL of benzonitrile were added to a cylindrical pyrex tube along with 3 mL of DMF. The system was placed in the focused microwave reactor. The reaction mixture was left under irradiation for 1 h at 130 °C. The solvent was then removed in vacuo and the resulting residue was treated with diethyl ether to obtain a reddish-brown powder, which was found to be a mixture of the cis- and trans-isomers, tentatively in a 9:1 ratio (see Fig. S8: ESI† for 1H NMR spectrum). The mixture was separated by an ion-exchange method as previously described for the other compounds and then separately recrystallized from a methanol–ether mixture. However, only the cis-isomer produced some crystals suitable for X-ray analysis.
cis-[Co(5-phenyl-tetrazolato)2(en)2]ClO4·MeOH (cis-7).
Yield: 28%. IR (KBr, cm−1): νCN 1640, ClO4 1048. Anal. Calc. for C19H26N12ClCoO5 (596.9): C 38; H 4.61; N 29.55; Found: C 37.56; H 4.92; N 28.72; MS(ESI): m/z = 469.16 [M]+. 1H NMR: δH (400 MHz, DMSO-d6, Me4Si): 2.27–2.89 (m, 8H, CH2), 4.63 (s, 2H, NH2), 4.99 (s, 2H, NH2), 5.79 (s, 2H, NH2), 6.27 (s, 2H, NH2), 7.42–7.98 (m, 10H, aromatic) (see Fig. S9: ESI† for 1H NMR spectra).
trans-[Co(5-phenyl-tetrazolato)2(en)2]ClO4·MeOH·4C6H5CN (trans-7).
Yield: 6%. IR (KBr, cm−1): νC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N 2230, νCN 1637, ClO4 1046. Anal. Calc. For C47H46N16ClCoO5 (1009.35): C 55.87; H 4.55; N 22.19; Found: C 55.56; H 4.25; N 22.02; MS(ESI): m/z = 469.16 [M]+. 1H NMR: δH (400 MHz, DMSO-d6, Me4Si): 2.01 (s, 4H, CH2), 2.26 (s, 4H, CH2), 4.47 (s, 8H, NH2), 6.49–7.18 (m, 10H + 20H, 5-phenyltetrazole + benzonitrile) (see Fig. S10: ESI† for 1H NMR spectra).
N 2230, νCN 1637, ClO4 1046. Anal. Calc. For C47H46N16ClCoO5 (1009.35): C 55.87; H 4.55; N 22.19; Found: C 55.56; H 4.25; N 22.02; MS(ESI): m/z = 469.16 [M]+. 1H NMR: δH (400 MHz, DMSO-d6, Me4Si): 2.01 (s, 4H, CH2), 2.26 (s, 4H, CH2), 4.47 (s, 8H, NH2), 6.49–7.18 (m, 10H + 20H, 5-phenyltetrazole + benzonitrile) (see Fig. S10: ESI† for 1H NMR spectra).
X-ray crystallography
Single crystal X-ray structural studies of (cis-2), (cis-3), (trans-4), 5 and 7 were performed on a CCD Agilent Technologies (Oxford Diffraction) SUPER NOVA diffractometer. Data for all the complexes were collected at 293(2) K, except for (cis-3), 5 and 7 for which the instrument was at 150(2) K, using graphite-monochromated Mo Kα radiation (λα = 0.71073 Å). The strategy for the data collection was evaluated by using the CrysAlisPro CCD software. The data were collected by standard phi–omega scan techniques and were scaled and reduced using CrysAlisPro RED software. The structures were solved by direct methods using SHELXS-97 and refined using full-matrix least-squares with SHELXL-97, refining on F2.15
The positions of all the atoms were obtained by direct methods. All non-hydrogen atoms were refined anisotropically. The remaining hydrogen atoms were placed in geometrically constrained positions and refined with isotropic temperature factors, generally 1.2Ueq of their parent atoms. The crystal and refinement data are summarized in Table 1, and selected bond distances and bond angles for cis-2, cis-3, trans-4 and (cis-5)–(cis-7) are shown in Tables 2–7 and Table S1 (ESI†).
Table 1 Crystal data and structure refinement of complexes cis-2, cis-3, trans-4, cis-5, cis-6 and cis-7
| |
cis-
2
|
cis-
3
|
trans-
4
|
| Empirical formula |
C38H48N32O8Co2 |
C20H24N15O4Co |
C21H31N14O4CoBr2 |
| Formula weight |
1198.94 |
597.47 |
762.35 |
| Wavelength (Å) |
0.71073 |
0.71073 |
0.71073 |
| Temperature (K) |
293(2) |
150(2) |
293(2) |
| Crystal system |
Monoclinic |
Monoclinic |
Triclinic |
| Colour and shape |
Yellow needle |
Yellow needle |
Yellow needle |
| Space group |
P21/c |
P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a (Å) |
18.5537(5) |
11.6957(6) |
8.5104(7) |
| b (Å) |
20.4481(7) |
18.9480(6) |
12.417(2) |
| c (Å) |
14.4208(5) |
12.5618(3) |
16.104(2) |
| α (°) |
90 |
90 |
109.442(14) |
| β (°) |
91.001(3) |
92.388(4) |
100.647(9) |
| γ (°) |
90 |
90 |
95.584(10) |
| Volume (Å3) |
5470.2(3) |
2781.40(18) |
1553.7(4) |
| Z |
4 |
4 |
2 |
|
D
calcd (mg m−3) |
1.456 |
1.427 |
1.672 |
|
μ(Mo Kα) (mm−1) |
0.685 |
0.673 |
3.193 |
|
F(000) |
2472 |
1232 |
788 |
| Crystal size (mm) |
0.32 × 0.28 × 0.24 |
0.33 × 0.26 × 0.21 |
0.38 × 0.34 × 0.29 |
|
θ range (°) |
3.00 to 25.00 |
3.17 to 25.00 |
3.05 to 25.00 |
| Limiting indices |
−22 ≤ h ≤ 22 |
−13 ≤ h ≤ 11 |
−10 ≤ h ≤ 10 |
| −24 ≤ k ≤ 24 |
−22 ≤ k ≤ 22 |
−14 ≤ k ≤ 12 |
| −17 ≤ l ≤ 17 |
−14 ≤ l ≤ 14 |
−19 ≤ l ≤ 19 |
| Total/unique no. of reflns. |
31738/9616 |
15822/4888 |
10996/5454 |
|
R
int
|
0.0629 |
0.0387 |
0.0545 |
| Data/restr./params. |
9616/0/721 |
4888/25/361 |
5454/0/384 |
| GOF(F2) |
1.026 |
1.034 |
1.057 |
|
R
1, wR2 |
0.0589, 0.1390 |
0.0502, 0.1328 |
0.0751, 0.2081 |
|
R
1, wR2 (all data) |
0.1020, 0.1721 |
0.0676, 0.1467 |
0.1078, 0.2381 |
| Peak and hole (e Å–3n) |
0.729 and −0.449 |
0.587 and −0.316 |
0.907 and −0.719 |
| |
cis-
5
|
cis-
6
|
cis-
7
|
| Empirical formula |
C32H50N28O9Co2Cl2 |
C18H24N12O4Br2CoCl |
C19H26N12O5CoCl |
| Formula weight |
1159.76 |
726.65 |
596.9 |
| Wavelength (Å) |
0.71073 |
0.71073 |
0.71073 |
| Temperature (K) |
150(2) |
293(2) |
150(2) |
| Crystal system |
Monoclinic |
Trigonal |
Orthorhombic |
| Color and shape |
Yellow needle |
Yellow needle |
Yellow needle |
| Space group |
P21/c |
P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c1 c1 |
P212121 |
| a (Å) |
11.6499(7) |
12.545(3) |
13.0541(4) |
| b (Å) |
16.1363(13) |
12.545(3) |
13.3594(7) |
| c (Å) |
13.5916(11) |
32.893(16) |
14.8320(4) |
| α (°) |
90.00 |
90.00 |
90 |
| β (°) |
95.163(6) |
90.00 |
90 |
| γ (°) |
90.00 |
120.00 |
90 |
| Volume (Å3) |
2544.7(3) |
4483(3) |
2586.63(17) |
| Z |
4 |
6 |
10 |
|
D
calcd (mg m−3) |
1.511 |
1.585 |
1.533 |
|
μ(Mo Kα) (mm−1) |
0.834 |
3.381 |
0.823 |
|
F(000) |
1192 |
2108 |
1232 |
| Crystal size (mm) |
0.26 × 0.21 × 0.18 |
0.33 × 0.25 × 0.08 |
0.23 × 0.18 × 0.13 |
|
θ range (°) |
2.94 to 25.00 |
1.24 to 20.92 |
3.14 to 24.99 |
| Limiting indices |
−13 ≤ h ≤ 13 |
−12 ≤ h ≤ 12 |
−15 ≤ h ≤ 15 |
| −19 ≤ k ≤ 19 |
−12 ≤ k ≤ 12 |
−12 ≤ k ≤ 15 |
| −16 ≤ l ≤ 14 |
−33 ≤ l ≤ 32 |
−17 ≤ l ≤ 17 |
| Total/unique no. of reflns. |
21371/4471 |
33225/1589 |
18243/4554 |
|
R
int
|
0.0740 |
0.1818 |
0.0367 |
| Data/restr./params. |
4471/0/334 |
1589/0/164 |
4554/0/343 |
| GOF(F2) |
1.067 |
1.156 |
1.104 |
|
R
1, wR2 |
0.0678, 0.1706 |
0.0896, 0.2471 |
0.0418, 0.1103 |
|
R
1, wR2 (all data) |
0.0908, 0.1951 |
0.1374, 0.3084 |
0.0451, 0.1131 |
| Peak and hole (e Å–3n) |
0.820 and −0.715 |
1.762 and −0.819 |
0.524 and −0.474 |
Table 2 Selected bond lengths (Å) and bond angles (°) for cis-2
| Co(1)–N(5) |
1.904(4) |
N(5)–Co(1)–N(10) |
91.90(16) |
| Co(1)–N(10) |
1.907(4) |
N(1)–Co(1)–N(4) |
178.46(16) |
| Co(1)–N(1) |
1.955(4) |
N(5)–Co(1)–N(3) |
174.28(16) |
| Co(1)–N(3) |
1.958(4) |
N(5)–Co(1)–N(2) |
87.56(15) |
| Co(1)–N(2) |
1.958(3) |
N(10)–Co(1)–N(1) |
90.31(16) |
| Co(1)–N(4) |
1.952(4) |
|
|
| N(5)–N(7) |
1.305(5) |
|
|
| N(7)–N(8) |
1.341(5) |
|
|
Table 3 Selected bond lengths (Å) and bond angles (°) for cis-3
| Co(01)–N(10) |
1.919(2) |
N(10)–Co(01)–N(5) |
87.98(12) |
| Co(01)–N(5) |
1.919(3) |
N(10)–Co(01)–N(2) |
174.76(13) |
| Co(01)–N(4) |
1.953(3) |
N(10)–Co(01)–N(4) |
91.38(11) |
| Co(01)–N(3) |
1.953(3) |
N(3)–Co(01)–N(1) |
176.90(11) |
| Co(01)–N(1) |
1.956(3) |
N(4)–Co(01)–N(1) |
92.42(12) |
| Co(01)–N(2) |
1.956(3) |
|
|
| N(5)–N(6) |
1.314(4) |
|
|
| N(6)–N(7) |
1.340(4) |
|
|
Table 4 Selected bond lengths (Å) and bond angles (°) for trans-4
| Co(1)–N(3) |
1.915(6) |
N(3)#1–Co(1)–N(3) |
180.0(3) |
| Co(1)–N(5) |
1.956(5) |
N(3)–Co(1)–N(6) |
89.5(2) |
| Co(1)–N(6) |
1.951(5) |
N(6)–Co(1)–N(6)#1 |
180.0(3) |
| N(3)–N(2) |
1.297(7) |
|
|
| N(2)–N(1) |
1.350(8) |
|
|
Table 5 Selected bond lengths (Å) and bond angles (°) for cis-5
| Co(1)–N(9) |
1.902(4) |
N(9)–Co(1)–N(5) |
92.38(17) |
| Co(1)–N(4) |
1.960(4) |
N(9)–Co(1)–N(3) |
90.79(17) |
| Co(1)–N(5) |
1.913(4) |
N(5)–Co(1)–N(3) |
89.99(17) |
| Co(1)–N(3) |
1.942(4) |
N(9)–Co(1)–N(1) |
90.19(17) |
| Co(1)–N(1) |
1.949(4) |
N(5)–Co(1)–N(1) |
90.12(18) |
| Co(1)–N(2) |
1.955(4) |
N(3)–Co(1)–N(1) |
179.01(17) |
| N(5)–N(6) |
1.311(6) |
N(9)–Co(1)–N(2) |
175.50(17) |
| N(5)–N(8) |
1.341(6) |
N(5)–Co(1)–N(2) |
89.40(17) |
| N(6)–N(7) |
1.345(6) |
N(3)–Co(1)–N(2) |
93.34(17) |
| N(9)–N(10) |
1.321(6) |
N(1)–Co(1)–N(2) |
85.67(17) |
| N(9)–N(12) |
1.339(5) |
N(9)–Co(1)–N(4) |
86.82(18) |
| N(10)–N(11) |
1.344(6) |
N(5)–Co(1)–N(4) |
175.79(18) |
|
|
|
N(3)–Co(1)–N(4) |
85.89(18) |
|
|
|
N(1)–Co(1)–N(4) |
94.02(19) |
|
|
|
N(2)–Co(1)–N(4) |
91.70(18) |
Table 6 Selected bond lengths (Å) and bond angles (°) for cis-6
| Co(1)–N(4) |
1.936(11) |
N(4)–Co(1)–N(4)#3 |
90.9(6) |
| Co(1)–N(4)#3 |
1.936(11) |
N(4)–Co(1)–N(1) |
91.6(4) |
| Co(1)–N(1) |
1.935(9) |
N(4)#3–Co(1)–N(1) |
91.1(4) |
| Co(1)–N(1)#3 |
1.935(9) |
N(4)–Co(1)–N(1)#3 |
91.1(4) |
| Co(1)–N(2)#3 |
1.943(10) |
N(4)#3–Co(1)–N(1)#3 |
91.6(4) |
| Co(1)–N(2) |
1.943(10) |
N(1)–Co(1)–N(1)#3 |
176.1(6) |
| N(4)–N(5) |
1.312(13) |
N(4)–Co(1)–N(2)#3 |
175.7(4) |
| N(4)–N(3) |
1.328(13) |
N(4)#3–Co(1)–N(2)#3 |
89.2(4) |
| N(6)–N(5) |
1.321(14) |
N(1)–Co(1)–N(2)#3 |
92.8(4) |
|
|
|
N(1)#3–Co(1)–N(2)#3 |
84.5(4) |
|
|
|
N(4)–Co(1)–N(2) |
89.2(4) |
|
|
|
N(4)#3–Co(1)–N(2) |
175.7(4) |
|
|
|
N(1)–Co(1)–N(2) |
84.5(4) |
|
|
|
N(1)#3–Co(1)–N(2) |
92.8(4) |
|
|
|
N(2)#3–Co(1)–N(2) |
91.1(6) |
Table 7 Selected bond lengths (Å) and bond angles (°) for cis-7
| Co(01)–N(1) |
1.935(4) |
N(1)–Co(01)–N(2) |
86.0(1) |
| Co(01)–N(2) |
1.972(3) |
N(1)–Co(01)–N(3) |
91.5(1) |
| Co(01)–N(3) |
1.947(3) |
N(1)–Co(01)–N(4) |
177.5(1) |
| Co(01)–N(4) |
1.962(4) |
N(1)–Co(01)–N(5) |
90.8(1) |
| Co(01)–N(5) |
1.920(3) |
N(1)–Co(01)–N(9) |
92.1(1) |
| Co(01)–N(9) |
1.906(3) |
N(2)–Co(01)–N(3) |
92.7(1) |
| N(5)–N(6) |
1.343(4) |
N(2)–Co(01)–N(4) |
94.0(1) |
| N(5)–N(7) |
1.310(6) |
N(2)–Co(01)–N(5) |
175.7(1) |
| N(7)–N(8) |
1.339(5) |
N(2)–Co(01)–N(9) |
86.9(1) |
| N(9)–N(10) |
1.327(5) |
N(3)–Co(01)–N(4) |
85.9(1) |
| N(9)–N(12) |
1.345(4) |
N(3)–Co(01)–N(5) |
90.2(1) |
| N(10)–N(11) |
1.345(4) |
N(3)–Co(01)–N(9) |
176.3(1) |
|
|
|
N(4)–Co(01)–N(5) |
89.4(1) |
|
|
|
N(4)–Co(01)–N(9) |
90.4(1) |
|
|
|
N(5)–Co(01)–N(9) |
90.4(1) |
However, the single crystal X-ray diffraction data collection for compound cis-6 was performed using a Bruker-APEX-II CCD diffractometer with graphite monochromated MoKα radiation (λ = 0.71073 Å). The structures were solved by SIR-9216, available in WinGX, which successfully located most of the non-hydrogen atoms. Subsequently, least square refinements were carried out on F2 using SHELXL-97 (WinGX version)15 to locate the remaining non-hydrogen atoms. All non-hydrogen atoms were refined anisotropically and most of the hydrogen atoms were refined isotropically on calculated positions using a riding model.
Liberation of 5-substituted tetrazoles from cobalt-complex
The following method has been adopted for the release of the pure tetrazoles from the corresponding cobalt-tetrazolato complexes. In a typical example, 0.1 g (0.164 mmol) of complex cis-2 was dissolved in 10 mL of methanol and about 0.069 g (1.064 mmol) of NaN3 dissolved in 10 mL of methanol was added to it. The reaction mixture was refluxed for 2 h. No precipitate was formed. The solution was filtered and the filtrate was evaporated to dryness under reduced pressure. The solid obtained was washed several times with diethyl ether and the washings were concentrated to obtain a colorless solid which was identified as 5-phenyl-1H-tetrazole. The different tetrazoles were isolated in moderate to good yields (40 to 60%) and characterized by ESI mass spectrometry, furnishing similar data to that reported elsewhere.17–25 All the tetrazoles were identified in their molecular protonated form [MH]+ in the ESI-MS spectra.
Results and discussion
Synthesis and isolation of the metal complexes and 5-substituted tetrazoles
Treatment of the diazidocobalt(III) complex cis-[Co(N3)2(en)2]NO31 under microwave irradiation with the respective organonitriles RCN (R = 3-NC5H4, 4-NC5H4 and 4-BrC6H4) gives the corresponding bis(tetrazolato) complexes [Co(N4CR)2(en)2]NO3 (Scheme 1). Use of the microwave technique in metal-assisted cycloaddition reactions is well known as it can drive the reaction much faster than the conventional refluxing method.7,8 The reaction with 3-cyanopyridine furnished a mixture of cis- and trans-isomers of the tetrazolato complex (cis-2 & trans-2), tentatively in a 1:4 ratio (in the absence of any other clear cut demarcation between both isomers, calculations are based on the integration of the 1H NMR peak observed due to the nitrogen protons in ethylenediamine for the cis- and trans-isomers). However, the reaction with 4-cyanopyridine yielded purely the cis-isomer (cis-3) as a reddish-brown product. A similar reaction with 4-bromobenzonitrile furnished exclusively the trans-isomer of the bis(tetrazolato) complex (trans-4), indicating a possible role of remote substitution at the 5-position of the phenyl ring in the preferential geometry of the cycloaddition complex. In all the reactions, DMF was used as the solvent because of the poor solubility of the precursor complexes in other common organic solvents. In view of the above findings, we were interested in checking the outcome when the starting material is in the trans-configuration. As there is no report, to the best of our knowledge, of the existence of any compound like trans-[Co(N3)2(en)2]NO3, we have therefore synthesized a known compound trans-[Co(N3)2(en)2]ClO41a as per the literature procedure.14 Upon reaction of 1a with 4-cyanopyridine, 4-bromobenzonitrile and benzonitrile we have obtained a mixture of cis- and trans-isomers of the bis(tetrazolate)complexes [Co(N4CR)2(en)2]ClO4 in tentatively 5:2, 4:1 and 9:1 ratios, respectively (Scheme 2). This result indicates a possible role of the counter-anion in dictating the preferential isomer of the tetrazolate complexes. However, only the cis-isomer of the tetrazolate complex synthesized from 4-cyanopyridine has been separated by the ion-exchange chromatographic method (cis-5). A similar effort to separate the isomers of the bromo-substituted tetrazolato complexes (cis-6 and trans-6) was futile as both compounds decomposed in the ion-exchange column. However, both geometrical isomers (cis-7 & trans-7) of [Co(5-phenyltetrazolato)2(en)2]ClO4 can be separated by ion-exchange column chromatography.
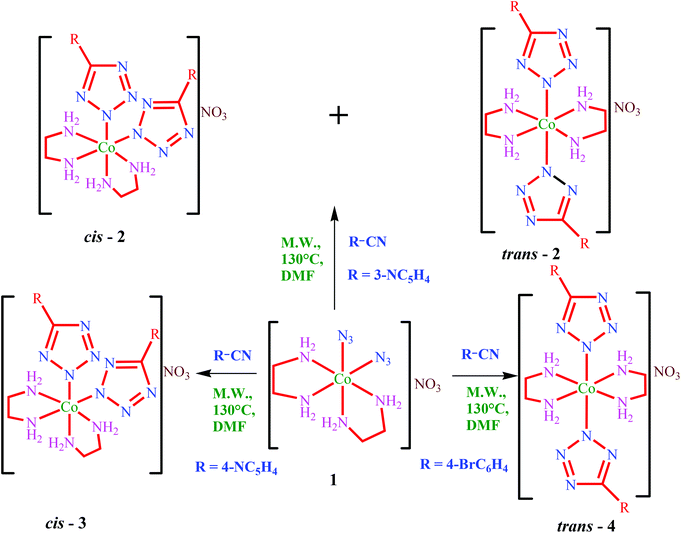 |
| | Scheme 1 | |
 |
| | Scheme 2 | |
All the reactions were carried out using focussed microwave radiation at 130 °C for 1–3 h. Most of the complexes are soluble in methanol, dichloromethane and DMF.
All the complexes (except for trans-5, cis-6 & trans-6) have been characterized by elemental analyses, IR spectroscopy, ESI-MS and 1H NMR spectroscopy. As cis-6 and trans-6 could not be separated from the mixture, it was characterized as a whole by IR spectroscopy, elemental analysis, ESI-MS and 1H NMR spectroscopy. None of the IR spectra of the compounds show the typical azide band that was present at ca. 2078 and 2019 cm−1 for complex 1 and 2016 cm−1 for complex 1a, and display a new strong band within the 1612–1665 cm−1 range due to the tetrazole ring, in agreement with the literature.7,8,26,27 In the ESI-MS of all the bis(tetrazolate) complexes the molecular ion peaks have been observed, confirming the presence of the bistetrazolate ligands in the metal complexes.
1H NMR spectroscopy was used to determine the relevant ratios of the geometrical isomers that have been obtained as a result of the cycloaddition reactions. In the absence of any other clear cut distinguishable protons between the cis- and trans-isomers, integration of the NH2 protons has been used to calculate a tentative ratio of the geometrical isomers generated in the solution state. Each individual isomer has been further characterized with the help of 1H NMR spectroscopy. All the cis-isomers show four signals within the range of 4.51–6.37 ppm, typical for nitrogen protons of ethylenediamine in the cis-[Co(en)2] group,28 while the CH2 groups of ethylenediamine show a more complex multiplet within the range of 1.91–2.90 ppm as reported elsewhere.29 The results obtained are in good agreement with similar types of (ethylenediamine)cobalt complexes reported earlier.30–32 All the trans-isomers show a single peak in the range of 4.47–5.41 ppm, attributed to the nitrogen protons of ethylenediamine, while the CH2 protons are observed in the range of 2.01–2.45 ppm. Some additional protons in the aromatic region observed for compound trans-7 may be attributed to the presence of free benzonitrile, which might have been entrapped during the crystallization of the compound. This fact is also further supported by the presence of a small nitrile peak at 2130 cm−1 in the IR spectrum of the compound and by elemental analysis result.
Liberation of 5-substituted tetrazole from the bis(tetrazolato) complexes was carried out by treating the precursor tetrazolato-metal complexes with an excess of sodium azide in methanol. After refluxing the mixture for 2 hours the filtrate was evaporated to dryness and the residue was washed with diethyl ether, which upon concentration provides the respective 5-substituted tetrazole in its neutral form rather than the sodium salt, as is evident by the presence of the protonated form of the molecular ion peak [MH]+ in ESI-MS. In all probability, the tetrazolate ion picks up one proton from the water present in the solvent. Apart from using ESI-MS, all these tetrazoles have been identified using 1H and 13C NMR spectroscopy, which correspond well with earlier reports.17–25
The Co3+ ion with d6 configuration in an octahedral crystal field in the solution state gives mostly one absorption band due to the d–d transitions in the octahedral CoN6 crystal field. The strong band which is observed in the range of 443.7–450.9 nm is assigned to the 1A1g→1T1g transition while the absorptions due to the transition 1A1g→1T2g are mostly masked (correlated to a very weak shoulder) due to the presence of a long tail of the charge-transfer peaks.33,34 The presence of other electronic transitions which appear in the UV region can be correlated to the π→π* transition in the ligand (Table 8). One thing to be noted here is that although not much difference has been observed in the absorption spectra of the different isomers, it should be mentioned that they are not totally identical (Fig. S11: ESI†).
Table 8 Absorption spectra peaks of compounds cis-2, trans-2, cis-3, trans-4, cis-5, cis-6–trans-6, cis-7 and trans-7 in methanol
| Compound no. |
λ, nm (ε, M−1 cm−1) |
|
cis-
2
|
446.8 (180) |
|
trans-
2
|
447.5 (171) |
|
cis-
3
|
445.1 (147), 249.8 (22400), 202.5 (42200) |
|
trans-
4
|
443.7 (174), 252.9 (47000), 203.5 (71300) |
|
cis-
5
|
446.7 (165) |
|
trans-
5
|
Cannot be separated |
| Mixture of cis-6 and trans-6 |
447.6 (72), 242 (137000), 204.6 (120900) |
|
cis-
7
|
450.86 (72) |
|
trans-
7
|
446.27 (91) |
Crystal structures of cis-[Co{5-(3-pyridyl)-tetrazolato}2(en)2]NO3·DMF (cis-2), cis-[Co{5-(4-pyridyl)-tetrazolato}2(en)2]NO3·Et2O (cis-3), cis-[Co{5-(4-pyridyl)-tetrazolato}2(en)2]ClO4·H2O (cis-5), cis-[Co{5-(4-bromophenyl)-tetrazolato}2(en)2]ClO4 (cis-6) and cis-[Co(5-phenyl-tetrazolato)2(en)2]·ClO4 (cis-7)
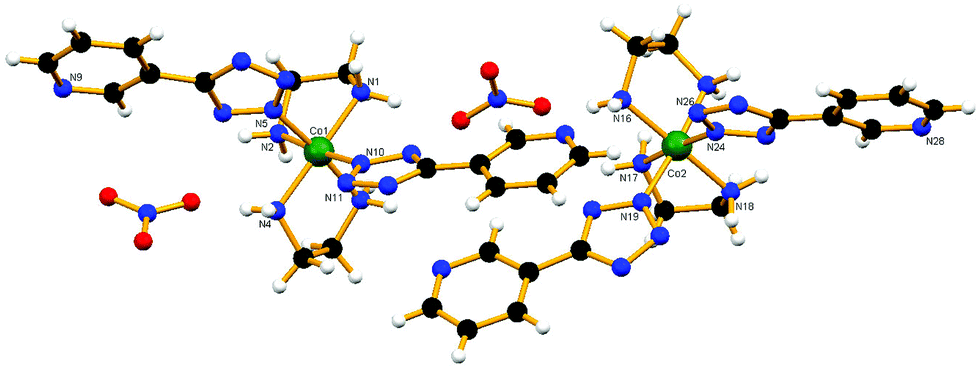
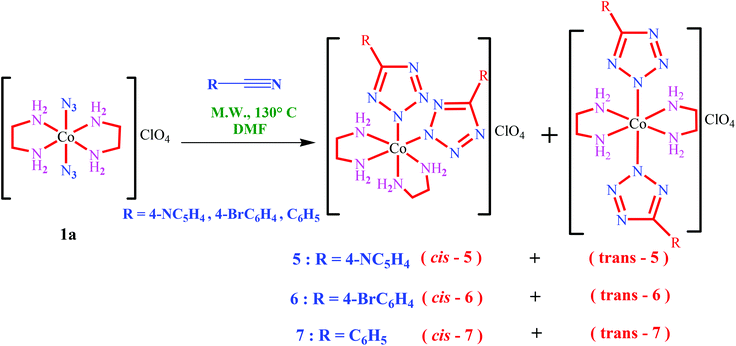
The nature of all the above complexes has been established by single-crystal X-ray crystallography. The corresponding crystallographic data are given in Table 1 and selected bond distances and angles in Tables 2–7. The corresponding hydrogen bonding interactions are summarized in Table S1 (ESI†). X-ray structure determination of the cis-2 compound revealed that there are two crystallographically independent complex molecules with very similar geometries in the unit cell (Fig. 3). The central ion is situated in a distorted octahedral environment. Both tetrazolate anions coordinate in a monodentate fashion and their disposition around the metal center is cis. The dihedral angles between the coordinated tetrazoles and substituted pyridyl rings are found to be 7.96° and 10.44°, respectively, for the first cobalt center. However, for the second cobalt center they are 3.40° and 3.73°, respectively. The complex cation and non-coordinated anion are associated in ionic pairs through very close hydrogen bonding interactions from both ethylenediamine molecules towards the oxygen atoms of the nitrate ion.
 |
| | Fig. 3 View of the two crystallographically independent complex molecules of compound cis-2. | |
The first type of molecule is coordinated to different surrounding nitrate ions through N2–H2B⋯O555, N4–H4B⋯O555, C1–H1C⋯O111, N1–H1B⋯O111, N1–H1B⋯O222 and C16–H16⋯O222 interactions (Fig. S12a†). The second independent molecule is also connected to the counter-ions by several similar hydrogen bonding interactions. In addition, these molecules are interconnected through the pyridyl nitrogens [N3–H3B⋯N23 and N4–H4A⋯N28] to form two strands of a one-dimensional chain structure interconnected through hydrogen bonding via nitrate anions along the a-axis (Fig. S12b†) (N2–H2B⋯O555, N4–H4B⋯O555, N1–H1B⋯O111, C1–H1C⋯O111, N16–H16⋯O011, N17–H17A⋯O011, N15–H15B⋯O666 and N17–H17B⋯O666). One of the independent molecules is further stabilized by hydrogen bond formation between DMF and one of the en molecules attached to the metal centre (N1–H1A–O888). There are two types of nitrate ion. One is forming a hydrogen bonded 1D polymer by forming bridges between the two crystallographically independent molecules and the other is helping to join the two strands together by hydrogen bond formation (Fig. S12c†).
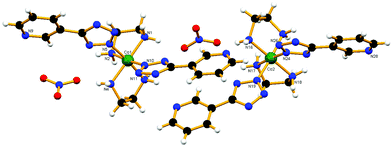
Compound cis-3, which crystallizes in the monoclinic space group P21/c, crystallographically shows one independent structure. The cis configuration of the compound has been enforced by weak H-bonding interactions between one nitrate ion and two en molecules attached to the same metal center (N1–H1A⋯O111, N4–H4B⋯O111 and N1–H1A⋯O333) (Fig. 4). Two such molecules are hydrogen bonded to each other via a mutual interaction through N4–H4A⋯N14, involving a pyridyl nitrogen atom and one of the hydrogens of the en ligand (Fig. S13a†). These dimers are further mutually interconnected via C16–H16⋯N9, forming a ladder type of structure along the c-axis (Fig. S13b†). These strand type ladder structures are interconnected by a hydrogen bonded nitrate structure (N3–H3B⋯O222, N2–H2B⋯O222 and N4–H4B⋯O111) in a three dimensional way to form a hydrogen bonded framework (Fig. S13c†). A diethylether molecule has also been observed as the solvent of crystallization, which is also associated with one of the ethylene diamine hydrogens via hydrogen bonding (N1–H1B⋯O101).
 |
| | Fig. 4 View of weak H-bonding interactions between the nitrate ion and two ethylenediamine molecules attached to the same metal center in cis-3. | |
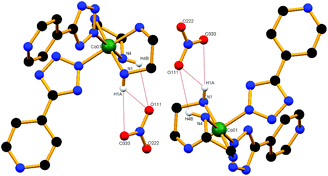
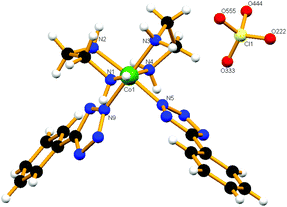
The single crystal X-ray structure of cis-5 confirms the formation of the cis configuration of the compound. It crystallizes in the monoclinic space group P21/c and exhibits 3-D supramolecular arrays formed by hydrogen bond interactions. In 5, the Co(III) resides in an octahedral co-ordination geometry (Fig. 5). Each cationic unit is connected to three neighbouring oxygen atoms of the perchlorate anion via N1–H1B⋯O333, N1–H1B⋯O444, C2–H2A⋯O333, N4–H4A⋯O333, N2–H2A⋯O111, C3–H3A⋯O111 and N3–H3A⋯O444 interactions (Fig. S14a†). Furthermore, in this network each unit is connected to two adjacent units through the pyridyl nitrogen atom and hydrogens of the en ligand via N3–H3B⋯N14, C2–H2B⋯N13 and N2–H2B⋯N13 interactions along the b-axis (Fig. S14b†). All of these H-bonds together form a 3D supramolecular framework structure.
 |
| | Fig. 5 View of local co-ordination geometry at the Co(III) centre in cis-5 where the tetrazolato ligands are cis to each other. | |
Although the refinement factor for the crystal structure of compound cis-6 is on the high side, it unequivocally indicates that the two tetrazolate ions coordinate the metal ion in a cis-fashion through the N2-atoms (Fig. 6). The dihedral angle between the tetrazole and the phenyl ring is found to be 22.73° for both ligands. Each perchlorate ion is found to be connected with three molecules through three equivalent oxygen atoms (C1–H1A⋯O1 and C9–H9⋯O1). This is further supported by N1–H1B⋯O2 hydrogen bonding (Fig. S15a†) and may also be helping to enforce the cis-geometry within the molecule. The independent molecules are packed along the a-axis and b-axis through H-bonding between different molecules and the perchlorate counter ions help in forming a hydrogen bonded 2D polymer in the ab-plane. All these networks, which are running in the direction of the ab-plane, orient in such a way that due to the complexes being in the cis conformation the para-substituted bromine atoms get stacked one upon another along the b-axis as well as the a-axis (Fig. S15b†). However, long distance Br⋯Br interactions between the bromine atom of one molecule and two other bromine atoms of neighbouring molecules [Br–Br 4.969 Å] may also contribute to the preferential cis-geometry of complex cis-6.
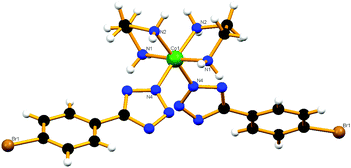 |
| | Fig. 6 Molecular structure of compound cis-6. | |
Compound cis-7 crystallizes in space group P212121 and the metal–nitrogen distances are found to be in the range of 1.906–1.972 Å, typical for the Co3+ ion. The tetrazolato ligands are cis to each other (Fig. 7). The perchlorate counter ions are found to be connecting different molecules via hydrogen bonding interactions, viz. N1–H1A⋯O222, C3–H3D⋯O222, N3–H3B⋯O333, N3–H3B⋯O555, N4–H4B⋯O555, C3–H3C⋯O444, N4–H4B⋯O444 and N2–H2B⋯O444 (Fig. S16a†), leading to the formation of a hydrogen bonded 3D network (Fig. S16b†).
 |
| | Fig. 7 View of local co-ordination geometry at the Co(III) centre in cis-7. | |
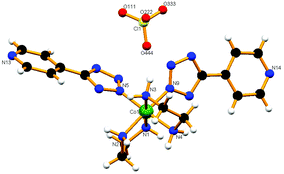
Crystal structure of trans-[Co{5-(4-bromophenyl)-tetrazolato}2(en)2]NO3 (trans-4)
Compound trans-4 has crystallized in space group P having two crystallographically independent complex molecules with very similar geometries (Fig. 8). Two tetrazolato ligands coordinate in a trans fashion and the twist of the phenyl ring with respect to the coordinated tetrazole rings is different for the two independent molecules. The dihedral angle has been found to be 4.74° for the first type, whereas for the second type this is about 25.02° for both 5-substituted tetrazole ligands. The twist in the ring can be attributed to the H-bond formation between an ethylenic hydrogen of one molecule and tetrazolyl nitrogen of the adjacent complex (N5–H5B⋯N11) (Fig. S17a†). These molecules are further interconnected by different ethylenic hydrogens and tetrazolyl nitrogens (N5–H5B⋯N11 and N8–H8A⋯N1) to form a 1D-chain along the b-axis (Fig. S17b†). This chain structure is further supported by nitrate ions which connect the four different molecules surrounding each one by hydrogen bonds (C7–H7⋯O111, N8–H8B⋯O111, N8–H8B⋯O333, N6–H6B⋯O333 and C9–H9A⋯O222), which altogether forms a 2D-network in the ab-plane (Fig. S17c†). Furthermore, one DMF molecule has been found hydrogen bonded to the first type of cobalt center via a N5–H5A⋯O101 hydrogen bond. The consequence is that all 5-substituting bromine atoms are found to be aligned in a single line when viewed along the a- or b-axis (Fig. S17d†). The nearest Br⋯Br [Br(1)⋯Br(2)] contact has been found to be 4.082 Å, slightly larger than the sum of the van der Waals radii. However, the formation of a long distance Br⋯Br interaction could be one of the driving forces for the crystallization of the molecule in the trans-geometry.
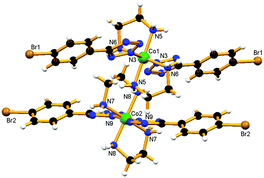 |
| | Fig. 8 View of compound trans-4 having two crystallographically independent units. | |
Computational studies
Density functional theory (DFT) calculations are carried out to understand the driving force behind the crystallization of complex trans-4 in the preferred geometry. Interestingly, the reaction of 4-bromobenzonitrile with trans-[Co(N3)2(en)2]ClO4 predominantly produced the cis-isomer (cis-6). These two complexes (trans-4 and cis-6) are considered for our calculations as both complexes have the same set of ligands (R = 4-BrC6H4) though have different counter anions. Therefore to understand the factors which may be playing a role in the preferred geometry, we have modelled these two complexes (trans-4 and cis-6) for our comparative study.
We have studied these complexes in their solid state structures retaining their space group geometries. Periodic boundary conditions are used for the three dimensional structure modelling. First-principles calculations are used, using the projected augmented wave (PAW) method as implemented in the Vienna ab initio simulation package (VASP) for their structural relaxation.35–37 The exchange–correlation interaction is treated in the level of the GGA using the Perdew–Burke–Ernzerhof model (GGA-PBE).38 We have calculated the van der Waals corrected interaction energies using the semi-empirical correction of Grimme,39 as available with VASP for the accurate treatment of the weak intermolecular interactions between the metal complexes. Our calculations show that both complexes (trans-4 and cis-6) are minima in their potential energy surfaces. The calculated van der Waals interaction energies for the complexes trans-4 and cis-6 are 4.90 and 4.30 eV (per molecular unit), respectively. Therefore, the van der Waals interaction energy is more for the trans geometry. Interestingly, in the trans (trans-4) geometry, we found that one solvent molecule (DMF) is trapped for each molecular unit. To understand the possible role of the solvent in the preferred geometry, we have removed the solvent from the crystal structure of the trans-4 complex and relaxed the structure, retaining the same space group geometry. We find that the calculated van der Waals interaction energies are reduced from 4.90 eV to 3.99 eV per molecular unit. Therefore, each solvent molecule is contributing around 20.98 kcal (0.91 eV) towards the total van der Waals interaction energy. Therefore the weak interaction directed by the solvent molecule possibly plays a major role in crystallizing the complex trans-4 in the trans geometry.
Conclusions
In this work we have investigated the interaction of the diazido complexes cis-[Co(N3)2(en)2](NO3) 1 and trans-[Co(N3)2(en)2](ClO4) 1a with various organonitriles under focussed microwave irradiation. Interaction of 1 with 3-cyanopyridine forms cis- and trans-isomers of the cycloaddition bis-tetrazolato complexes, tentatively in a 1:4 ratio. However, the interaction with 4-cyanopyridine produced exclusively the cis-isomer of the bis(tetrazolate) complex and reaction with 4-bromobenzonitrile produced the bis(tetrazolate) complex in the trans disposition, indicating the role of remote substitution of the phenyl ring in the formation of the preferential isomer. The interaction of trans-[Co(N3)2(en)2]ClO4 under similar conditions with 4-cyanopyridine, 4-bromobenzonitrile and benzonitrile furnished mixtures of cis- and trans-isomers of the bis(tetrazolate) complexes where the cis-geometry predominates. It has been assumed that it is possibly the counter-anion which is helping the compound to crystallize and is also interacting with the complex in the solution state to contribute to the preferential geometrical isomer formation upon cycloaddition. Further, a DFT calculation study indicates that the trans isomer trans-4 is being further stabilized by means of van der Waals interactions through an extra DMF molecule (compared to cis-6), which is present as solvent of crystallization, along with some Br⋯Br halogen interactions. Furthermore, 5-substituted tetrazole can be isolated from the bis(tetrazolate) complexes by treating them with excess sodium azide.
Acknowledgements
We are grateful for the financial support received from the Council of Scientific and Industrial Research, New Delhi. One of us (M. S.) thanks CSIR for the award of JRF in a CSIR sponsored project. We are also thankful to Sophisticated Instrument Center, IIT Indore for the structure elucidation.
References
- G. Aromi, L. A. Barrios, O. Roubeau and P. Gamez, Coord. Chem. Rev., 2011, 255, 485–546 CrossRef CAS PubMed.
-
Other Five-membered Rings with Three or more Heteroatoms, and their Fused Carbocyclic Derivatives, in Comprehensive Heterocyclic Chemistry III, ed. V. A. Ostrovskii, G. I. Koldobskii, R. E. Trifonov, A. R. Katritzky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, Oxford, 2008, vol. 6, pp. 257–423 Search PubMed.
- P. N. Gaponik, S. V. Voitekhovich and O. A. Ivashkevich, Russ. Chem. Rev., 2006, 75, 507–539 CrossRef CAS PubMed.
- J. R. Anacona and P. Alvarez, Transition Met. Chem., 2002, 27, 856–860 CrossRef CAS.
- Z. H. Chohan, C. T. Supuran and A. Scozzafava, J. Enzyme Inhib. Med. Chem., 2004, 19, 79–84 CrossRef CAS PubMed.
- E. A. Popova, R. E. Trifonov and V. A. Ostrovskii, ARKIVOC, 2012, 1, 45–65 Search PubMed.
- S. Mukhopadhyay, J. Lasri, M. A. J. Charmier, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2007, 5297–5304 RSC.
- P. Smoleński, S. Mukhopadhyay, M. F. C. Guedes da Silva, M. A. J. Charmier and A. J. L. Pombeiro, Dalton Trans., 2008, 6546–6555 RSC.
- Y. Li, C.-Q. Wang, H.-D. Bian, F.-P. Huang, H. Liang and Q. Yu, J. Coord. Chem., 2012, 65, 3665–3673 CrossRef CAS.
- W.-W. Dong, J. Zhao and L. Xu, Cryst. Growth Des., 2008, 8, 2882–2886 CAS.
- Y. Chen, Y. Song, Y. Zhang and J.-P. Lang, Inorg. Chem. Commun., 2008, 11, 572–575 CrossRef CAS PubMed.
- M. Saha, R. Nasani, S. M. Mobin, B. Pathak and S. Mukhopadhyay, Inorg. Chem. Commun., 2013, 34, 62–67 CrossRef CAS PubMed.
- P. Paul and K. Nag, Inorg. Chem., 1987, 26, 2969–2974 CrossRef CAS.
- P. J. Staples and M. L. Tobe, J. Chem. Soc., 1960, 4812–4820 RSC.
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343–350 CrossRef.
- T. Jin, S. Kamijo and Y. Yamamoto, Tetrahedron Lett., 2004, 45, 9435–9437 CrossRef CAS PubMed.
- M. Nasrollahzadeh, Y. Bayat, D. Habibi and S. Moshaee, Tetrahedron Lett., 2009, 50, 4435–4438 CrossRef CAS PubMed.
- J. Bonnamour and C. Bolm, Chem. – Eur. J., 2009, 15, 4543–4545 CrossRef CAS PubMed.
- J. He, B. Li, F. Chen and Z. X. G. Yin, J. Mol. Catal. A: Chem., 2009, 304, 135–138 CrossRef CAS PubMed.
- S. Rostamizadeh, H. Ghaieni and R. Aryan, Chin Chem. Lett., 2009, 20, 1311–1314 CrossRef CAS PubMed.
- P. Z. Demko and K. B. Sharpless, J. Org. Chem., 2001, 66, 7945–7950 CrossRef PubMed.
- D. Amantini, R. Beleggia, F. Fringuelli, F. Pizzo and L. Vaccoro, J. Org. Chem., 2004, 69, 2896–2898 CrossRef CAS PubMed.
- M. L. Kantam, K. B. S. Kumar and P. K. Raja, J. Mol. Catal. A: Chem., 2006, 247, 186–188 CrossRef CAS PubMed.
- M. L. Kantam, K. B. Shiva Kumar and C. Sridhar, Adv. Synth. Catal., 2005, 347, 1212–1214 CrossRef CAS PubMed.
- R. Nasani, M. Saha, S. M. Mobin and S. Mukhopadhyay, Polyhedron, 2013, 55, 24–36 CrossRef CAS PubMed.
- M. Saha, R. Nasani, M. Das, A. Mahata, B. Pathak, S. M. Mobin, L. M. Carella, E. Renschler and S. Mukhopadhyay, Dalton Trans., 2014, 43, 8083–8093 RSC.
- R. Sharma, R. P. Sharma, K. Karaghiosoff and T. M. Klapoetke, Inorg. Chem. Commun., 2007, 10, 139–142 CrossRef CAS PubMed.
- K. Nagaraj and S. Arunachalam, Transition Met. Chem., 2013, 38, 649–657 CrossRef CAS.
- R. P. Sharma, R. Sharma, R. Bala, J. M. Salas and M. Quiros, J. Mol. Struct., 2006, 794, 341–347 CrossRef CAS PubMed.
- D. A. House and J. W. Blunt, Inorg. Nucl. Chem. Lett., 1975, 11, 219–223 CrossRef CAS.
- P. Kofod, P. Harris and S. Larsen, Inorg. Chem., 1997, 36, 2258–2266 CrossRef CAS PubMed.
- A. Wojciechowska, D. Dobrzyńska and J. Janczak, Polyhedron, 2012, 47, 118–125 CrossRef CAS PubMed.
- R. P. Sharma, R. Sharma, R. Bala, K. N. Singh, L. Pretto and V. Ferretti, J. Mol. Struct., 2006, 784, 109–116 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter, 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter, 1994, 49, 14251–14269 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter, 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, J. A. Chevary and C. Fiolhais, Phys. Rev. B: Condens. Matter, 1992, 46, 6671–6687 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 933043–933047 and 935043. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qi00089g |
|
| This journal is © the Partner Organisations 2014 |