Synthesis and characterization of isotopically labeled silver nanoparticles for tracing studies†
Adam
Laycock
*ab,
Björn
Stolpe
c,
Isabella
Römer
c,
Agnieszka
Dybowska
b,
Eugenia
Valsami-Jones
bc,
Jamie R.
Lead
cd and
Mark
Rehkämper
ab
aDepartment of Earth Science & Engineering, Imperial College London, South Kensington, London SW7 2AZ, England, UK. E-mail: a.laycock10@imperial.ac.uk; Tel: +44 (0)20 7594 7140
bDepartment of Earth Sciences, Natural History Museum, Cromwell Road, London SW7 5BD, England, UK
cSchool of Geography, Earth and Environmental Sciences, University of Birmingham, Edgbaston, Birmingham, B15 2TT, England, UK
dCenter for Environmental Nanoscience and Risk, Department of Environmental Health Sciences, Arnold School of Public Health, University of South Carolina, Columbia, SC 29208, USA
First published on 27th March 2014
Abstract
Silver nanoparticles (AgNPs) are ever more being used in industrial processes and consumer products, resulting in increasing emissions to the natural environment. To understand the behavior and environmental fate of AgNPs, it is paramount that they can be traced in complex natural samples from exposures at high sensitivity. The technique of stable isotope labeling is ideally suited for this purpose. To support such applications, we present a detailed evaluation of techniques for the preparation of stable isotope labeled AgNPs and demonstrate that isotopically modified particles are only distinguishable from particles with a natural isotope composition by their strong isotopic signature. Monodisperse suspensions of citrate-stabilized AgNPs with target sizes of 17, 20 and 30 nm were synthesized by reduction of silver nitrate solutions with sodium borohydride. The AgNP suspensions were produced using both natural Ag, which is comprised of the two stable isotopes 107Ag (52%) and 109Ag (48%), and Ag enriched to 99.2% in 107Ag. Synthesis was reliably reproduced on three separate occasions in two laboratories. The AgNPs were characterized using dynamic light scattering (DLS) shortly after synthesis and after up to 12 months storage. Some of the batches were also characterized using transmission electron microscopy (TEM) and asymmetric flow field-flow fractionation (FlFFF). The particle size distributions showed good reproducibility between the laboratories and stability over 12 months of storage. Importantly, the 107Ag-enriched particles were indistinguishable in size and shape from particles with a natural isotope composition. The reliability, control on particle size, and high yield of about 80%, demonstrate that the synthesis technique is well suited for small-scale production of isotopically labeled AgNPs. Isotope mass balance calculations furthermore show that the application of labeling enables tracing sensitivities for AgNPs that are at least 40 times, and possibly up to 4000 times, higher than those achievable with bulk Ag concentration measurements and experiments with exposure concentrations that approach predicted environmental levels are possible, if the most precise isotopic measurement techniques are employed.
Nano impactThe application of isotopically labeled nanoparticles enables the accurate and precise tracing of these materials in exposure experiments, even when carried out at low and environmentally relevant concentrations. However, isotopically labeled nanoparticles are not commercially available and published synthesis techniques tend to be unsuitable for use with an isotopically enriched pre-cursor material. This is the first study to provide protocols specifically optimized for the small-scale, high yield synthesis of isotopically labeled silver nanoparticles with defined sizes. The synthesis techniques are rigorously assessed and shown to be fit for purpose. Apart from their isotope composition the silver nanoparticles produced using both a ‘natural’ and an isotopically enriched precursor are shown to be indistinguishable. In addition the nanoparticle suspensions are shown to be stable after 12 months storage. A comprehensive evaluation of the nanoparticles produced here demonstrates the detection capabilities possible when these materials are employed in environmental tracer studies. Detailed equations are also presented to show how the quantification of an enriched isotope label can be achieved from the isotope ratio of a sample. |
1. Introduction
As a consequence of the ubiquitous use of Ag in the photography and imaging industries, anthropogenic emissions of Ag and their environmental fate have been monitored for many decades.1 Whilst the rise of digital photography has significantly reduced such emissions, new concerns have arisen as engineered Ag nanoparticles (AgNPs) are increasingly employed in a broad range of industrial processes and consumer products and emitted into the environment.1,2 Whilst not comprehensive, there are currently 390 products (November 2013) listed on the Woodrow Wilson Database3 that incorporate nano-silver, demonstrating the diverse range of products in which this nanomaterial is found. A focus of current research and debate is whether there are (nano-) specific effects, which increase the bioavailability and toxicity of Ag when present in nanoparticulate form.2 This concern has prompted numerous studies to investigate the environmental transport, behavior, fate and ecotoxicology of engineered AgNPs.Ideally, such studies are carried out at ‘realistic’ environmental conditions, whereby exposures employ NM (nanomaterial) concentrations that are similar to or not far removed from present or predicted values. Recently, Gottschalk, et al.4 compiled and reviewed measured and modeled environmental concentrations of engineered NMs. Based on the reviewed data, predicted environmental levels of engineered nano-Ag are in the range of 0.1 to 100 ng L−1 for surface waters, 1 to 100 ng L−1 for effluents of waste water treatment plants, 1 to 1000 μg kg−1 for biosolids, 1 to 10 μg kg−1 for sediments, and 0.1 to 1000 μg kg−1 for soils. Significantly, these levels are roughly similar to the natural Ag background concentrations of these materials.5
Studies that interrogate the environmental fate of AgNPs typically employ concentration measurements by ICP-MS to quantify the presence of Ag (from NPs) in exposure systems. Such analyses are straightforward but require that exposures employ AgNP concentrations that exceed ‘realistic’ environmental levels by several orders of magnitude. This requirement follows from (i) the comparatively high natural Ag background (see above), and (ii) the condition that unequivocal tracing of AgNPs relies on the detection of anomalies in Ag concentration that clearly exceed these background levels. For example, Shoults-Wilson, et al.6 used AgNP concentrations of 10 to 1000 mg kg−1 in soil exposures of the earthworm Eisenia fetida, whilst Zhao and Wang7 applied AgNPs at 20 to 500 μg L−1 in a waterborne exposure of Daphnia magna. It is conceivable that ecotoxicity studies, which are carried out at such high (and unrealistic) AgNP concentrations, may induce and thus identify distinct patterns of particle behavior and biological interactions that are only relevant for the high particle concentrations that are employed. The challenge is, therefore, to investigate whether the same effects are also observed at low but environmentally relevant NP levels.
To circumvent the limited sensitivity of concentration tracing, alternative NM tracing methods have been explored, particularly fluorescent coatings8,9 and radiolabeling,10–12 but both techniques have associated drawbacks. The application of fluorescent coatings can affect the surface chemistry of the materials and influence their behavior, such as changes to and suppression of dissolution.13 The coatings may furthermore dissociate from the NPs14 and free ions from NP dissolution cannot be traced with this method. Radiolabeled nanomaterials are produced by activation in a nuclear reactor15,16 and the technique is therefore hindered, as specialist equipment, dedicated laboratories and licenses are required for the handling of radioactive materials. A further disadvantage is the often short half-lives of the radiolabels.
An attractive alternative method of labeling, which does not suffer from the above drawbacks, involves the application of enriched stable isotopes. The basic approach of stable isotope labeling relies on the detection of changes in a diagnostic isotope ratio, which are produced in an experimental system by the introduction of a ‘contaminant’ that is prepared from a highly enriched single isotope of the target element. In the case of Ag, the natural form of the element consists of nearly equal amounts of the isotopes 107Ag and 109Ag but highly enriched (to >99%) 107Ag can be purchased from a number of sources. The introduction of essentially pure 107Ag into an exposure will not only increase the total Ag concentration of the system but also change the 107Ag/109Ag isotope ratio, such that this exceeds the natural value of ~1. The magnitude of the isotopic perturbation hereby depends on the relative contributions of enriched 107Ag to natural Ag (Fig. 1). As (i) natural variations in isotope compositions are typically small for most elements and (ii) isotope ratios can be measured much more precisely than absolute concentrations, the use of enriched stable isotopes provides an extremely selective and sensitive means of elemental tracing, even in the presence of high natural background levels.17,18 Consequently, the stable isotope approach has been used successfully for several decades in the fields of medical, life and environmental sciences, when highly sensitive and precise tracing methods were required.19,20
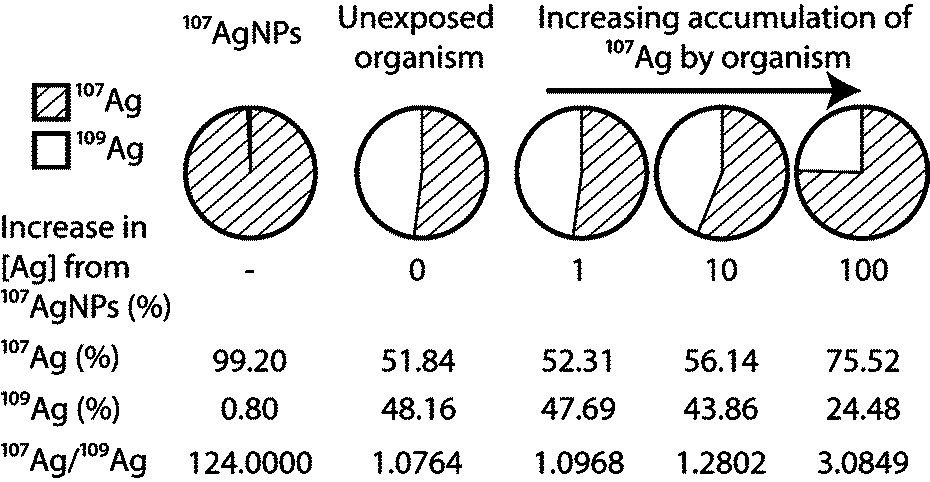 | ||
| Fig. 1 Demonstrates how the contribution from isotopically enriched 107Ag to a natural Ag background will result in a deviation in the 107Ag/109Ag that can be directly related to the degree of accumulation. | ||
More recently, it has been shown that stable isotope tracing can also be applied to nanomaterials, with pioneering studies of ZnO and CuO NPs.17,18,21–23 A unique requirement of such investigations is that the labeled NPs must be specifically prepared, from a single, highly enriched stable isotope form of the element. Particular advantages of this approach are (i) high versatility, as it can, in principle, be applied to any NP with a multi-isotopic element as the main constituent; (ii) the label cannot be lost by dissociation or degradation; and (iii) the unique isotopic signature of the element remains traceable even if the speciation of the original material changes, for example by dissolution. The methodology, however, also requires that a suitable protocol for the preparation of labeled NPs is available. Whilst recipes for the synthesis of various NMs with defined sizes and properties are abundant in the literature, most of these methods are either unsuitable for the synthesis of stable isotope labeled NPs or they provide insufficient documentation for this purpose. This conclusion follows from the relatively high price of commercially available enriched stable isotopes, which typically cost about $2 to $20 per mg of metal or metal oxide. Given this non-negligible cost, methods for the preparation of isotopically labeled NMs must be optimized with a focus on small-scale synthesis, reliability and high yields. These factors, however, are generally not considered in conventional synthesis protocols, where particle size and shape are typically of higher priority.
In this study, we have examined existing protocols to develop techniques for the optimized preparation of isotopically labeled AgNPs with distinct sizes. Rigorous evaluations demonstrate that the methods are suitable for routine and reliable small-scale synthesis of stable isotope labeled AgNPs at yields of ~80%. The characterization of both labeled AgNPs and control samples produced from natural Ag furthermore unequivocally establishes that the labeling process does not generate distinct or unusual physico-chemical particle properties. This is essential in order to demonstrate that isotopically modified AgNPs are equivalent to AgNPs with a natural isotope composition and to validate the findings of any studies carried out using isotopically modified AgNPs, such as the recent investigation of Gigault and Hackley.24 These workers24 used asymmetric-flow field flow fractionation system coupled with on-line ICP-MS detection to show how a stable isotope label may be used to confirm the presence of Ag from an isotopically enriched source within a natural sample. While this technique offers rapid and definitive results it does not allow accurate quantification of the enriched material in a sample. Our study assesses the utility of the isotope tracing approach beyond the level of simply detecting the isotope label in a sample. To this end, we present the results of simple model calculations, which reveal the lowest levels at which isotopically labeled AgNPs can be detected and accurately quantified in relevant natural samples by stable isotope tracing, when different common mass spectrometric methods are employed for analysis.
2. Experimental
The AgNPs were synthesized by appropriately modified standard reduction methods.25,26 In particular, batches of particles with 3 target sizes were produced from both natural Ag and enriched 107Ag. Two operators carried out the AgNP synthesis on three occasions in two laboratories and the reproducibility of the methods was assessed by comprehensive characterization of all NP batches produced.2.1 Choice of isotope label
When selecting an enriched stable isotope for labeling there are several key considerations, including degree of isotopic enrichment, cost of the material and the diagnostic isotope ratio that will be monitored for the purpose of tracing. A detailed discussion of these factors is provided by Larner and Rehkämper.27 Silver has two naturally occurring stable isotopes, 107Ag and 109Ag, that have near equal natural abundances of 51.8% and 48.2%, respectively, and both are available at an enrichment of about 99% (Isoflex U.S.A., February 2012). As both isotopes are equally suitable for tracing, 107Ag was chosen here for labeling because it was marginally cheaper than 109Ag (Isoflex U.S.A., February 2012).2.2 Chemicals and reagents
Powdered Ag metal enriched to 99.2% in 107Ag (Isoflex U.S.A.) was converted to 107AgNO3 by dissolution in 15.4 M HNO3 and subsequent evaporation to dryness. Natural AgNO3 (>99% purity), trisodium citrate dihydrate (>99% purity) and sodium borohydride (99.99% trace metals basis) were purchased from Sigma-Aldrich. Water of >18.2 MΩ cm quality (Millipore, UK) and quartz-distilled mineral acids were used throughout. Dilute artificial seawater (dilute-ASW) with a salinity of about 16 PSU was produced by dissolution of 32 g of Tropic Marin Salt in 2 L purified water and used as medium for a dialysis study to assess dissolution.2.3 Synthesis
The synthesis protocols used here were adapted from the methods outlined by Römer, et al.26 Three protocols, denoted AgNP1, AgNP2, and AgNP3 (Table 1), were applied to produce particles with target sizes of 30, 20 and 17 nm, both with enriched 107Ag (denoted as 107-AgNP1, 107-AgNP2 and 107-AgNP3, respectively) and natural Ag (denoted as Nat-AgNP1, Nat-AgNP2 and Nat-AgNP3). The NP batches are further designated to indicate (i) the date of synthesis, either May or October 2012 (May12 or Oct12), and (ii) the laboratory in which synthesis was conducted, either at the Facility for Environmental Nanoscience Analysis and Characterization (FENAC) at the University of Birmingham or the MAGIC (Mass Spectrometry and Isotope Geochemistry at Imperial College) Laboratories at Imperial College London, (F and IC respectively). For example, the non-labeled AgNPs with a 20 nm target size prepared at FENAC in October 2012 are identified as Nat-AgNP2-Oct12-F (Table 1).| Freshly prepared particles | Particles after storage | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Particle batcha | DLS | TEM | FIFFF | DLS | ||||||||
| Yield (%) | Diameterb (nm) | PDIc | Diameterd (nm) | CVe | d n (nm) | d w (nm) | PDh | FWHMi (nm) | Storagej (months) | Diameterb (nm) | PDIc | |
| DLS – Dynamic light scattering, TEM – Transmission electron microscopy, FIFFF – Asymmetric Flow Field Flow Fractionation.a Each particle batch was designated depending on (i) whether the batch was labeled or non-labeled (107-AgNP or Nat-AgNP), (ii) which synthesis protocol was used for the production (1, 2 or 3; see Table 2), (iii) whether the particles were made in May or October 2012 (May12 or Oct12) and (iv) the laboratory in which the synthesis was carried out, either at FENAC, University of Birmingham (F) or the MAGIC Laboratory, Imperial College London (IC).b The hydrodynamic diameter was calculated as the average of individual DLS measurements.c Polydispersity index.d The average TEM diameter ±1sd is calculated from analyses of 234–337 individual particles.e Coefficient of variance = Standard deviation/TEM diameter.f The number average hydrodynamic diameter.g The weighted average hydrodynamic diameter.h Polydispersity (dn/df).i Full width at half maximum.j Particle storage was in the dark at 4 °C. | ||||||||||||
| AgNP1 protocol – target particle size 30 nm | ||||||||||||
| Nat-AgNP1-May12-F | 91 | 41.6 | 0.25 | 36 | 49 | 1.3 | 24 | 12 | 41.5 | 0.25 | ||
| 107-AgNP1-May12-F | 64 | 29.3 | 0.22 | 27 | 31 | 1.2 | 26 | 12 | 29.7 | 0.20 | ||
| Nat-AgNP1-May12-IC | 14 | 35.5 | 0.26 | 12 | 35.3 | 0.23 | ||||||
| 107-AgNP1-May12-IC | 31.2 | 0.16 | ||||||||||
| Nat-AgNP1-Oct12-F | 79 | 26.5 | 0.13 | 22.3 ± 6.7 | 0.30 | 6 | 26.8 | 0.11 | ||||
| 107-AgNP1-Oct12-F | 98 | 31.2 | 0.18 | 28.2 ± 8.3 | 0.29 | 6 | 33.7 | 0.22 | ||||
| AgNP2 protocol – target particle size 20 nm | ||||||||||||
| Nat-AgNP2-May12-F | 87 | 20.7 | 0.33 | 18 | 21 | 1.2 | 18 | 12 | 19.7 | 0.25 | ||
| 107-AgNP2-May12-F | 83 | 16.7 | 0.25 | 17 | 22 | 1.3 | 17 | 12 | 14.6 | 0.34 | ||
| Nat-AgNP2-May12-IC | 61 | 17.2 | 0.28 | 8 | 16.8 | 0.35 | ||||||
| 107-AgNP2-May12-IC | 61 | 16.3 | 0.27 | 12 | 17.5 | 0.18 | ||||||
| Nat-AgNP2-Oct12-F | 63 | 22.6 | 0.15 | 19.5 ± 5.7 | 0.29 | 6 | 22.9 | 0.10 | ||||
| 107-AgNP2-Oct12-F | 78 | 23.9 | 0.20 | 20.1 ± 6.3 | 0.31 | 6 | 24.1 | 0.18 | ||||
| AgNP3 protocol – target particle size 17 nm | ||||||||||||
| Nat-AgNP3-May12-F | 17.1 | 0.20 | 16 | 18 | 1.1 | 11 | ||||||
| 107-AgNP3-May12-F | 24.3 | 0.24 | ||||||||||
| Nat-AgNP3-May12-IC | 77 | 18.2 | 0.23 | 12 | 16.8 | 0.31 | ||||||
| 107-AgNP3-May12-IC | 31 | 20.6 | 0.29 | 12 | 24.1 | 0.21 | ||||||
| Nat-AgNP3-Oct12-F | 80 | 17.2 | 0.25 | 18.0 ± 5.2 | 0.29 | 6 | 32.5 | 0.52 | ||||
| 107-AgNP3-Oct12-F | 76 | 28.1 | 0.44 | 16.3 ± 4.8 | 0.29 | 6 | 26.1 | 0.39 | ||||
For synthesis, 100 mL aqueous solutions of 0.25 mM sodium citrate and 0.30 mM silver nitrate were cooled to 4 °C, combined in a conical flask and vigorously stirred. To the AgNP1 and AgNP2 batches, 6 mL aqueous NaBH4 was added at a concentration of 10 and 1 mM, respectively, whilst nothing was added to the AgNP3 batch at this stage. For all protocols, the mixtures were then boiled for 90 minutes, after which the AgNP1 and AgNP2 mixtures were removed from the heat. For the AgNP3 batches, 6 mL of 10 mM aqueous NaBH4 solution were added to the mixture after 90 minutes of boiling, followed by boiling for a further 10 minutes. All solutions were thereafter allowed to cool overnight in the dark at room temperature. A summary of the synthesis protocols is given in Table 2.
| Step | Synthesis protocol | ||
|---|---|---|---|
| AgNP1 | AgNP2 | AgNP3 | |
| a 100 mL of 0.30 mM Na-citrate solution in >18.2 MΩ cm water. b 100 mL of 0.25 mM Ag-nitrate solution in >18.2 MΩ cm water. c Aliquot, with appropriate dilution, of 100 mL of 10 mM NaBH4 stock solution in >18.2 MΩ cm water. d RT = room temperature. | |||
| Mix Na-citratea & Ag-nitrateb solutions then stir for | 10 min | 10 min | 10 min |
| Add 6 mL NaBH4c solution | 10 mM | 1 mM | — |
| Boil mixture | 90 min | 90 min | 90 min |
| Add 6 mL NaBH4c solution | — | — | 10 mM |
| Continue to boil | — | — | 10 min |
| Allow to cool in the dark | RTd | RTd | RTd |
The particle suspensions were filtered through 0.1 μm pore-size cellulose acetate filters to remove any larger (non-nano) particulates. Non-reacted reagents were thereafter removed by either diafiltration or dialysis. In the diafiltration method, the volume of the suspensions was reduced to ~20 mL without drying, by forcing through a 1 kDa (~1 nm) molecular weight cut-off (MWCO) regenerated cellulose ultrafiltration membrane (Amicon, Millipore) using a nitrogen gas pressurized and stirred filtration cell. The suspension volume was then re-adjusted back to ~200 mL using 0.15 mM sodium citrate solution and this process was repeated three times. For the dialysis method, the suspensions were transferred to 1 kDa MWCO dialysis bags (Spectra/Por) followed by dialysis against 5 L of 0.15 mM sodium citrate solution for 72 hours. The AgNP suspensions in aqueous Na-citrate from all protocols and filtration methods were thereafter stored in the dark at 4 °C.
2.4 Characterization of particle size
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 60 minutes. Grids were then rinsed by immersing in 18 MΩ water for a few seconds and dried overnight. The TEM micrographs were obtained from 3 to 5 different areas on each grid, at a magnification of 30000×, 100000× and 300000×. The diameters of at least 200 particles in each sample were then determined on the micrographs with 300000× magnification using the computer software Digital Micrograph (Gatan Inc.).
000 rpm for 60 minutes. Grids were then rinsed by immersing in 18 MΩ water for a few seconds and dried overnight. The TEM micrographs were obtained from 3 to 5 different areas on each grid, at a magnification of 30000×, 100000× and 300000×. The diameters of at least 200 particles in each sample were then determined on the micrographs with 300000× magnification using the computer software Digital Micrograph (Gatan Inc.).
For quantification, the samples were analyzed relative to a standard solution with Ag and Pd abundances of 20 ng mL−1 in the same acid matrix, using a Nu Plasma multiple collector ICP-MS (MC-ICP-MS) instrument at the Imperial College MAGIC Laboratories. In these analyses the total Ag (107Ag + 109Ag) signal intensities obtained for samples were compared to the standard solution and 105Pd was monitored to correct for any drifts in instrumental sensitivity. The measurements provided the total Ag concentrations of the solutions and the yields of the synthesis methods were then calculated based on (i) the dilutions factors used for the preparation of the measuring solutions and (ii) the total mass of Ag used for particle preparation.
3. Results & discussion
All information gathered from the characterization of the 18 batches of AgNPs that were prepared during the study is summarized in Table 1 and discussed in the following.3.1 Size characterization of nanoparticles
In the following, we present and discuss the results of a statistical evaluation that interrogates whether isotopic labeling or other factors (e.g., date or laboratory of synthesis) had a significant impact on the size of the prepared AgNPs. In order to incorporate all particle suspensions in this evaluation, including batches with different target sizes, we adopted the following procedure:
(i) All particle suspensions were split into one of three groups depending on their target particle size (17, 20 and 30 nm). For each individual batch, the proportional difference from the mean size (PDMS) of the group was calculated. This was achieved by finding the mean particle size (MS), as determined by DLS, for each group. The particle size determined for an individual suspension (SamS) was then subtracted from the mean value, to calculate the absolute difference between the individual and mean particle sizes. The proportional difference was then determined by dividing the absolute difference by the mean particle size (eqn (1), see ESI†).
| PDMS = (MS − SamS)/MS | (1) |
This normalization procedure was validated by t-tests that, where possible, were carried out using both measured absolute size data and the calculated relative size differences. Specifically, this was possible when the DLS sizes of Nat-AgNPs were compared with 107-AgNPs for each of the three target sizes (30 nm, 20 nm, 17 nm; see ESI†) Notably, these three t-tests yielded exactly identical results when they were conducted with both the absolute and the normalized size data (see ESI†). This result validates the application of the normalized particles size data (obtained as detailed above) in further t-tests.
(ii) The PDMSs calculated for each individual particle suspension were pooled in different manners to produce populations that were evaluated using a student's t-test. These tests evaluated whether there are significant and systematic differences in size that can be assigned to a specific factor (see ESI†).
Based on two-tailed t-tests for populations of unequal variance, there was no significant difference at the 95% confidence level (p = 0.05) when the PDMSs were grouped into the following populations; (i) Nat-AgNPs and labeled 107-AgNPs (p = 0.57), (ii) synthesis at the FENAC and Imperial College laboratories (p = 0.18), (iii) synthesis date (e.g., May12-F, May12-IC and Oct12-F; p = 0.27 to 0.73), (iv) NP preparation by operators AL and BS (particle synthesis in May and October 2012 respectively; p = 0.45), and (v) freshly prepared and aged particles after 6–12 months storage (p = 0.98). These results were obtained with the omission of batches 107-AgNP1-May12-F and 107-AgNP3-Oct12-F, which have particularly anomalous sizes of 41.6 nm (target size 30 nm) and 28.1 nm (target size 17 nm), respectively. These unusually large deviations from the target size may reflect experimental problems during synthesis. However, very similar results are produced when these data are included in the above t-test evaluations. A detailed summary of the t-test data and calculated p-values is presented in the ESI.†
Notably, the average particle diameters of the suspensions after storage deviated by less than 3.5 nm from the average diameters that were measured directly after preparation. The only exception is sample Nat-AgNP3-Oct-F, where the average diameter increased by about 90% (15.3 nm; Table 1). The PDIs determined by DLS were less than 0.45 for all AgNP suspensions and less than 0.3 for 16 of the 18 batches, indicating that the particle size distributions were fairly monodisperse (Table 1). A t-test demonstrated that there was no significant difference between the PDIs of (i) Nat-AgNPs and 107-AgNPs (p = 0.59) and (ii) pristine particles and particles after storage (p = 0.65) (see ESI†).
Whilst the t-tests discussed above indicate that there are no statistically significant differences in particles size and PDI that can be assigned to a single factor, the data of Table 1 nonetheless demonstrate small but resolvable differences in particle size for NP batches prepared with the same protocol. Given the results of the t-tests, these disparities most likely reflect ‘normal’ batch-to-batch variations that occur, if syntheses are carried out on different days, in different laboratories and by different operators. These results hence provide an indication of the particle size variability that can be expected if different scientists apply the protocols of Table 2 in their own laboratories. In contrast, Römer, et al.26 previously demonstrated that, using similar protocols for the preparation of AgNPs, repeated synthesis by the same operator, in the same laboratory with the same equipment and reagents, can routinely provide particle batches with highly reproducible sizes. In summary, these results thus demonstrate that despite small batch-to-batch variations in particles size that reflect slight differences in experimental conditions, the protocols summarized in Table 2 are suitable for the robust and reliable synthesis of isotopically labelled AgNPs in different laboratories.
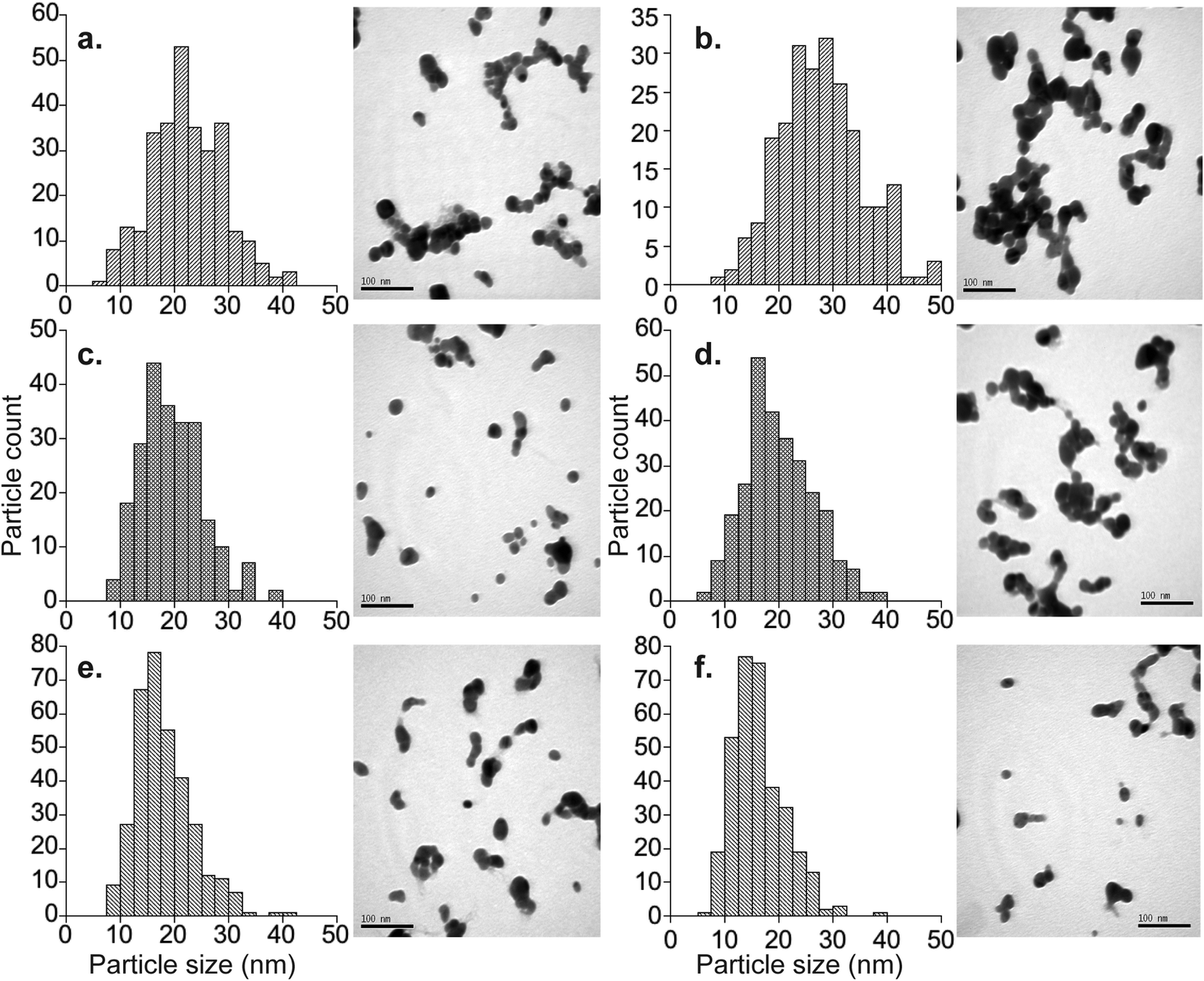 | ||
| Fig. 2 TEM images and measured particle size distributions for particles batches synthesized in October 2012. all images are at a magnification of 300000x; scale bars are 0.1 μm. (a) Nat-AgNP1-Oct12-F average particle size = 22.3 ± 6.7 nm (1sd) n = 290; (b) 107-AgNP1-Oct12-F average particle size = 28.2 ± 8.3 nm (1sd) n = 236; (c) Nat-AgNP2-Oct12-F average particle size = 19.5 ± 5.7 nm (1sd) n = 234; (d) 107-AgNP2-Oct12-F average particle size = 20.1 ± 6.3 nm (1sd) n = 283; (e) Nat-AgNP3-Oct12-F average particle size = 18.0 ± 5.3 nm (1sd) n = 337; (f) 107-AgNP3-Oct12-F average particle size = 16.3 ± 4.8 nm (1sd) n = 333. | ||
The average diameters determined from the TEM micrographs were typically 10 to 15% (about 3–4 nm) smaller than the average hydrodynamic diameters determined by DLS. This is a well-known phenomenon, which reflects that larger particles scatter proportionally more light, such that the average DLS particle diameters are biased towards larger values.28 The exceptions to this are samples Nat-AgNP3-Oct12-F and 107-AgNP3-Oct12-F. The Nat-AgNP3-Oct12-F particle batch shows an average TEM particle size that is very close to that determined by DLS, whereas there is a large discrepancy between the DLS (28.1 nm) and TEM (16.3 nm) size data for 107-AgNP3-Oct12-F. These observations are likely a consequence of the polydispersity of the respective samples. In particular, the particle size distribution histogram of Nat-AgNP3-Oct12-F shows the smallest size range (Fig. 2), suggesting it is highly monodisperse and therefore the DLS analysis shows very little size bias. In contrast, 107-AgNP3-Oct12-F is the most polydisperse sample (PDI = 0.44), and this may reflect a small number of larger particles in the suspension that heavily bias the DLS measurement.
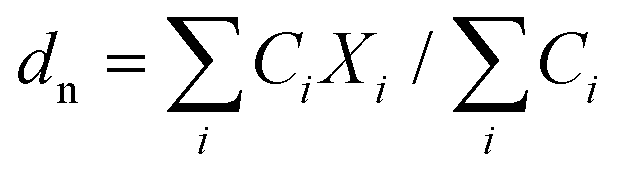 | (2) |
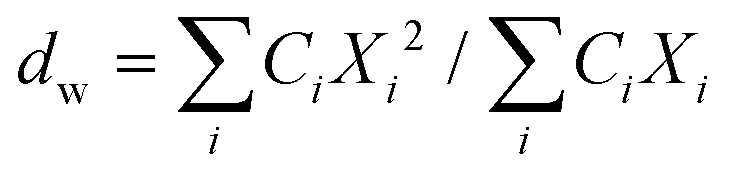 | (3) |
| PD = dw/dn | (4) |
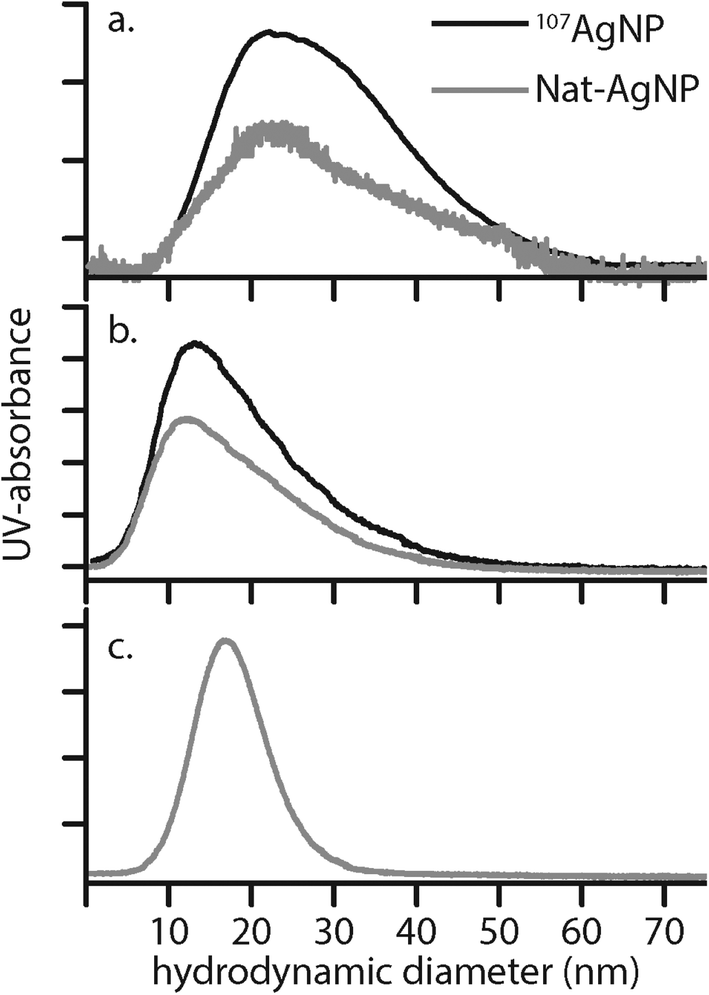 | ||
| Fig. 3 Hydrodynamic diameter distributions determined by FIFFF for selected AgNP batches. (a): 107-AgNP1-May12-F and Nat-AgNP1-May12-F, (b) 107-AgNP2-May12-F, Nat-AgNP2-May12-F and (c) Nat-AgNP3-May12-F. | ||
Here, Ci is the UV-absorbance signal at a given hydrodynamic diameter, Xi. The dn values were typically slightly smaller (up to 13%) whilst dw was slightly larger (up to 32%) compared to the particle size determined by DLS. The dn values for 107-AgNP2-May12-F (17 nm) and Nat-AgNP2-May12-F (18 nm) were only slightly larger than for Nat-AgNP3-May12-F (16 nm). The two former samples also showed a higher PD value than Nat-AgNP3-May12-F, with size distributions tailing towards larger diameters (Fig. 3). As a consequence, there is a slightly lager difference in dw values between 107-AgNP2-May12-F (22 nm) and Nat-AgNP2-May12-F (21 nm) versus Nat-AgNP3-May12-F (18 nm).
3.2 Synthesis yields
Reliable small-scale synthesis techniques with high yields are required for the production of isotopically labeled NPs, due to the expense of using enriched stable isotopes (e.g., about $3.50 per mg for 107Ag purchased from Isoflex USA in February 2012). The total Ag concentrations of 24 different particle suspensions, encompassing 16 batches of labeled 107AgNPs and 8 batches of non-labeled AgNPs, were determined to assess the yield of the synthesis protocols (Table 1). Typically the yields were between 60% and 98%, with an average of 78% ± 23% (1sd). Considering that only small amounts of 107Ag are required for a single synthesis (~2.7 mg) and due to the excellent yields achieved here, the preparation of 107AgNPs is quite affordable, with a cost of approximately $4 to $5 per milligram.3.3 Dissolution behavior
The results of the dialysis experiments are shown in Fig. 4. The particle stock suspension was added directly to the dilute-ASW without prior treatment or washing to remove excess sodium citrate. Although this effect was not investigated, the presence of sodium citrate at the expected concentration of ~4 mg L−1 is not anticipated to have a significant influence on the dissolution behavior. The Ag contents measured on the outside of the dialysis bags represent the total Ag concentration of the dilute-ASW, encompassing both particulate and dissolved forms of Ag. In contrast, the Ag measured inside the bags has crossed a 1 kDa MWCO dialysis membrane and therefore reflects the dissolved component only. The experimental setup therefore ensures that identical Ag concentrations for the dialysis bags and the container are only achieved, when all Ag in the latter reservoir is in dissolved form.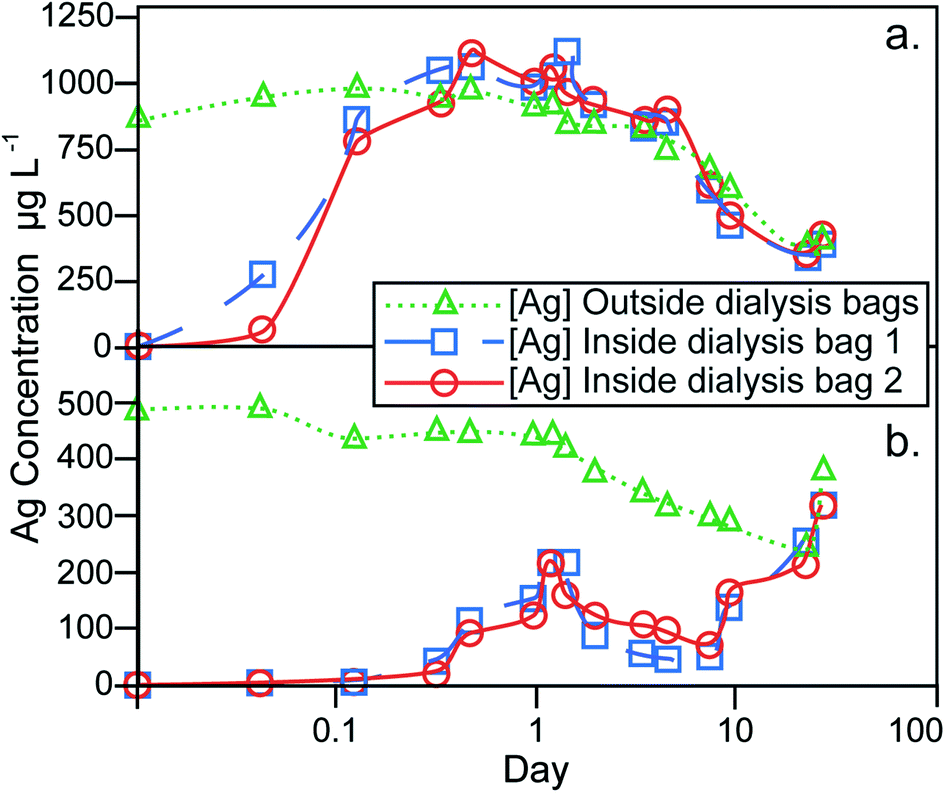 | ||
| Fig. 4 Results for AgNP dissolution behavior in dilute-ASW, assessed by dialysis. (a) Control experiment, whereby aqueous AgNO3 solution is added to the dilute asw outside of the dialysis bags to a concentration of 1 mg L−1. (b) Experiment where 107AgNPs, from particle batch 107-AgNP2-Oct12-F, were added to the dilute-asw outside of the dialysis bags to a concentration of 0.9 mg L−1. | ||
The dialysis control experiment, where 1.00 mg L−1 of aqueous Ag was introduced into the containers, can be used to assess the behavior of dissolved Ag in the absence of AgNPs. At the beginning of the control experiment, the Ag concentrations inside the dialysis bags increased rapidly and reached the same level (of about 0.8–1.1 mg L−1) as on the outside after 8 hours, demonstrating the time required for equilibrium to establish over the dialysis membrane (Fig. 4a). During the remaining 27 days of the experiment, the Ag concentrations inside and outside the dialysis bags were always essentially identical, showing that the setup functioned well to provide a reliable monitor of dissolved Ag contents on the outside of the dialysis bags. However, the Ag concentration outside the bags decreased during most of the experiment to a value of about 0.4 mg L−1 after 22 days. This indicates that dissolved Ag was lost, presumably as a result of adsorption onto the container walls and the dialysis membrane. The small final increase in dissolved Ag concentration that is seen at day 27, is likely due to desorption of the Ag from the surfaces of the container and dialysis bags.
In the dialysis experiment of sample 107-AgNP2-Oct12-F, the Ag concentration outside the bags immediately after NP addition was 0.5 mg L−1, a recovery of only 56% (Fig. 4b). The container was furthermore black in color after the addition of the 107AgNPs, which indicates that AgNP adsorption to container surfaces was responsible for the loss of Ag. During the experiment, the Ag concentration outside of the bags generally decreased until day 22. For the same time period, the Ag contents inside both dialysis bags initially showed an increase over the first 28 hours, followed by a decrease until day 8, with increasing concentrations observed thereafter (Fig. 4b). On day 22, the Ag concentrations outside and within both dialysis bags were identical and all three concentrations rose continuously during the final time period to day 27. This final increase in concentration is similar to that seen in the control experiment, which suggests that it is also most likely caused by desorption of Ag from surfaces that are in contact with the dilute-ASW.
Due to the adsorptive losses of both dissolved and nanoparticulate Ag during dialysis, it is difficult to quantify the dissolution behavior of the AgNPs. It may be assumed, however, that the dissolution of AgNPs will be similar, regardless of whether they are in suspension or adsorbed to container walls. With this assumption, our data can be applied to derive some general conclusions about the dissolution behavior of AgNPs. In particular, after an initial 8 hour equilibration time, the control experiment with dissolved Ag exhibited nearly identical Ag concentrations inside and outside of the dialysis bags (Fig. 4a). During the same time period, the dialysis experiment with NP sample 107-AgNP2-Oct12-F showed low Ag concentrations inside the dialysis bags (0.02–0.04 mg L−1), which correspond to only 2–5% of the 107AgNP concentration that was initially added to the container (Fig. 4b). These observations indicate that the 107AgNP suspension in dilute-ASW produced only a very small fraction of dissolved Ag during the first few hours of the experiment. Over the following 27 days of the 107AgNP dialysis experiment, the Ag concentrations inside the dialysis bags displayed an overall increase, demonstrating extensive 107AgNP dissolution (Fig. 4b). This conclusion is supported by the observation that very similar Ag concentrations were observed for the inside and outside of the dialysis bags on day 27, with a final Ag concentrations of 0.32 mg L−1 for both bags (Fig. 4b). As some dissolved Ag was probably lost to adsorption onto the container walls, the value of 0.32 mg L−1 can be taken to represent a tentative minimum estimate for the solubility of the 107AgNPs in dilute-ASW. Given the adsorption problems encountered here with the dialysis technique, future AgNP dissolution studies should consider using alternative methods, such as ultrafiltration, to obtain improved constraints on the dissolution behavior of AgNPs.
3.4 Reproducibility and reliability of synthesis method
The three synthesis protocols produced fairly monodisperse AgNP suspensions throughout, but suspensions prepared with the same protocol display small but significant batch-to-batch variations in size, regardless of the isotopic composition of the AgNPs (Table 1). A detailed evaluation of the results (Table 1) demonstrates, however, that the protocols for particle target sizes of 30 nm (AgNP1), 20 nm (AgNP2) and 17 nm (AgNP3) still offer acceptable control on particle size. Protocol AgNP1 produced the largest particles (26.5–31.2 nm, average = 30.7 ± 2.9 nm; 1sd, n = 5), whilst the particles from protocols AgNP2 (16.3–23.9 nm, average = 19.6 nm ± 3.0 nm; 1sd, n = 6) and AgNP3 (17.1–24.3 nm, average = 19.5 ± 2.7 nm; 1sd, n = 5) were smaller but similar. These quoted averages were calculated from all DLS data obtained for freshly prepared particles (Table 1), excluding the two most obvious outliers (Nat-AgNP1-May12-F and 107-AgNP3-Oct12-F), but very similar results are obtained when the complete dataset is considered (see ESI†).These findings demonstrate that the methods provide a robust means of obtaining small batches of isotopically labeled AgNPs with diameters that closely match desired target sizes. However, due to the observed batch-to-batch variations in size, particle characterization will need to be carried out for applications, which require that particles diameters are known to a high degree of accuracy. Although only three synthesis protocols were utilized here, it is likely that the experimental conditions can be adjusted to target larger and smaller particle sizes. For example, slightly modified versions of the synthesis methods applied here were used in previous studies to produce AgNPs with diameters as small as 11.5 nm33 and 7.2 nm26 when measured by TEM.
3.5 Stability of particles
The particle suspensions in aqueous Na-citrate solutions were stored in the dark at 4 °C to help prevent ageing. To assess the stability of the particle suspensions under these conditions, a selection of particle batches were characterized by DLS periodically during 12 months of storage (Table 1). Batch Nat-AgNP3-Oct12-F showed the most significant signs of ageing, as after 6 months storage the average particle size and PDI increased from 17.2 to 32.5 nm and from 0.25 to 0.52, respectively. Whilst this is the type of change that would be expected as a result of particle aggregation, it was shown by Römer, et al.26 that small citrate-capped Ag NPs generally have a greater stability and exhibit less agglomeration than larger particles. This indicates that the aggregation determined for sample Nat-AgNP3-Oct12-F may be an atypical result. All other particle batches exhibited no significant signs of ageing with all but one sample showing less than a 10% change in the average DLS particle size (Table 1). In addition, the PDI data also display only minor changes, indicating that the particle suspensions remained fairly monodisperse. The size and PDI results, furthermore, reveal no difference in aging behavior between natural and isotopically labeled AgNPs (Table 1). Any changes that might occur to the stock suspensions, such as agglomeration, may influence the particle behavior, but would have no effect on the isotope label or tracing capabilities.3.6 Isotope label & detection capabilities
The motivation for synthesizing AgNPs from enriched 107Ag is to produce a material with a distinct non-natural isotope signature, which can be used in experiments and exposures, so that changes induced in the 107Ag/109Ag isotope ratio of relevant samples (e.g. biological materials, soil or water) can be used to trace and quantify the presence of the labeled material17,18,22,23,27 (Fig. 1). The contribution of enriched 107Ag to a sample is only detectable when the deviation induced in the 107Ag/109Ag ratio of the sample clearly exceeds (i) the precision of the isotopic measurement or (ii) any natural variations in the isotope composition of Ag. These factors are discussed below.Isotopic analyses of Ag are generally carried out using ICP-MS instrumentation, as measurements by thermal ionization mass spectrometry (TIMS) are significantly more cumbersome.34 Using standard quadrupole ICP-MS (Q-ICP-MS) instruments, 107Ag/109Ag ratios can typically be measured with a precision of about 1–5% (2sd). Improved data with a precision of ±0.2 to 1% (2sd) can be obtained with Q-ICP-MS at optimal analytical conditions (including separation of Ag from matrix elements) or by the application of single collector sector field ICP-MS (SF-ICP-MS).35 Even more precise isotopic measurements are possible by multiple collector ICP-MS (MC-ICP-MS), which can achieve uncertainties of better than ±0.02% (2sd) for 107Ag/109Ag on purified solutions of Ag.36,37
Studies to investigate natural stable isotope fractionation of Ag are extremely limited in number and scope, with a primary focus on Ag ores (which are straightforward to analyze) and a few igneous rocks.37–39 Data are also available from an archeological study on the provenance of coins40 and an investigation into natural isotope fractionation based on a limited number of environmental samples.41 These analyses revealed only minor differences of about ±0.05% in the 107Ag/109Ag ratios of the samples, relative to the NIST SRM 978a Ag isotope standard. Such small variations are only resolvable using MC-ICP-MS and hence have no impact on the sensitivity that can be achieved for the detection of isotopically labeled AgNPs by Q-ICP-MS and SF-ICP-MS. Given the published results, it is furthermore unlikely that samples, such as soils or sediments, from a single location will exhibit geological Ag isotope fractionations that exceed ±0.02% and hence impact the AgNP detection sensitivity of MC-ICP-MS. However, there are currently no studies that investigate whether changes in Ag isotope composition are generated by biological uptake and processing of this element. Based on data acquired for other elements, such Zn42 and Cd,43 it is conceivable that biotic reactions may induce Ag isotope effects in the range of 0.02% to 0.05% and such fractionations may be preserved in biological tissue and fluids, water samples or soils and sediments. It is unlikely, however, that biological isotope effects will normally exceed 0.1% to 0.2% and hence no serious impact on the detection sensitivity of MC-ICP-MS for isotopically labeled AgNPs is expected. Nonetheless, it will be important for future work to carefully characterize possible biological Ag isotope fractionations. If natural Ag isotope variations of ≥0.05% are found to be common and deemed to be obstructive, this problem can be readily circumvented by analyses of appropriate control samples. Such measurements can characterize the natural Ag isotope composition and variability of the background, and this permits appropriate corrections and/or uncertainties to be applied to the isotopic data collected for exposed samples.
Ecotoxicology studies typically generate and require analyses for a large number of samples (more than 100), making it important to consider the balance between optimal data quality and high sample throughput. Whilst MC-ICP-MS can provide unrivalled precision for the determination of 107Ag/109Ag, such analyses are also particularly laborious, because Ag must be separated from any sample matrix prior to analysis. In contrast, quadrupole and sector field ICP-MS can be employed for direct 107Ag/109Ag measurements without chemical separation. As sample preparation is much more straightforward, the latter instruments can achieve significantly higher sample throughput but at the cost of somewhat poorer precision for the data, which ultimately limits the tracing capabilities.
In the following, we discuss the tracing sensitivities that can be achieved using labeled 107Ag (and thus 107AgNPs) for two hypothetical cases with relevance to ecotoxicology, and where the endmember methods of Q-ICP-MS and MC-ICP-MS are evaluated for detection (Table 3). Case 1 involves analyses of 5 mg dry weight samples of biological tissue, as might be obtained from invertebrates following exposures, with a realistic Ag background of 100 ng g−1.5 Case 2 is for a 5 mL water sample (a realistic volume for removal and analysis from a laboratory exposure) that features a Ag background concentration of 200 ng L−1, as would be appropriate for river, lake and estuarine waters.5 Based on the concentrations quoted above, the case 1 and 2 samples would contain only 0.5 ng and 1 ng of Ag, respectively (Table 3), and these small amounts will limit the precision of the isotopic analyses by ICP-MS. Based on literature data and test measurements conducted at Imperial College (for MC-ICP-MS), it is estimated that the diagnostic 107Ag/109Ag isotope ratio can be determined in these samples with a precision of about ±5% for Q-ICP-MS and ±0.05% for MC-ICP-MS (an intermediate precision is expected for SF-ICP-MS).
| Case 1 | Case 2 | |
|---|---|---|
| a Smallest increase in the Ag concentration from the addition of highly enriched 107Ag, which is resolvable by measurement of the 107Ag/109Ag isotope ratio using a given technique. | ||
| Sample | 5 mg biological tissue | 5 mL water |
| Background Ag concentration | 100 ng g−1 | 200 ng L−1 |
| Total natural Ag content of sample | 0.5 ng | 1 ng |
| Analysis by Q-ICP-MS, precision ±5% (2sd) | ||
| Minimum concentration change detectable by 107Ag/109Ag analysisa | 2.6 ng g−1 | 5.2 ng L−1 |
| Uncertainty in quantification of label at: | ||
| 10× minimum concentration | 126 ± 3.12 ng g−1 | 252 ± 6.24 ng L−1 |
| 100× minimum concentration | 360 ± 4.37 ng g−1 | 720 ± 8.76 ng L−1 |
| Analysis by MC-ICP-MS, precision ±0.05% (2sd) | ||
| Minimum concentration change detectable by 107Ag/109Ag analysisa | 0.026 ng g−1 | 0.052 ng L−1 |
| Uncertainty in quantification of label at: | ||
| 10× minimum concentration | 100.26 ± 0.026 ng g−1 | 200.52 ± 0.053 ng L−1 |
| 100× minimum concentration | 102.6 ± 0.027 ng g−1 | 205.2 ± 0.055 ng L−1 |
The precision of mass spectrometric measurements is defined here as the 2sd calculated from replicate isotope analyses of samples or a standard reference material. The analytical precision is a key factor because it determines the smallest change in the 107Ag/109Ag isotope ratio (ΔRres), as induced by addition of the 107Ag tracer, which is analytically resolvable as a deviation from the natural 107Ag/109Ag ratio (Rnat), such that ΔRres = 2sd.
The addition of an enriched 107Ag tracer to a natural sample will increase the 107Ag/109Ag ratio of the system. In the following, we define Rres, as the 107Ag/109Ag ratio, which is associated with the smallest addition of 107Ag tracer that is resolvable by measurement. Rres is therefore given by:
| Rres = Rnat + ΔRres | (5) |
Isotope mass balance can then be used to calculate the minimum molar amount of enriched 107Ag (Nres-Agen), from labeled 107AgNPs, that must be added to a given molar amount of natural Ag (N-Agnat) in a system, to increase the 107Ag/109Ag ratio from Rnat to Rres:
| Nres-Agen = N-Agnat × ((Rres × 109Abnat) − 107Abnat)/(107Aben − (Rres × 109Aben)) | (6) |
The final 107Ag/109Ag isotope ratio of an exposed sample (Rsam), as measured by ICP-MS, is used to quantify the amount of enriched 107Ag label present. This calculation is outlined in the following. The molar fraction of enriched 107Ag to total Ag (fren) present in the sample is given by:
| fren = N-Agen/(N-Agnat + N-Agen) | (7) |
| fren = [(107Aben − (Rsam × 109Aben))/((Rsam × 109Abnat) − 107Abnat) + 1]−1 | (8) |
The fractional molar abundance of 109Ag in the exposed sample (109Absam) is then:
| 109Absam = (fren × 109Aben) + ((1 − fren) × 109Abnat) | (9) |
The molar abundance of total Ag (N-Ag) and enriched Ag (N-Agen) in the sample can be determined by quantifying N-109Ag, the molar amount of 109Ag present. This can be readily achieved using standard ICP-MS techniques, where a solution prepared from a known aliquot of the sample is analyzed, such that N-109Ag can be determined relative to a suitable calibration curve. Following this, N-Ag and N-Agen can be calculated from:
| N-Ag = (N-109Ag/109Absam) | (10) |
| N-Agen = (N-109Ag/109Absam) × fren | (11) |
The mass M-Agen and mass concentration [M-Agen] of enriched Ag in the sample can then be obtained as:
| M-Agen = N-Agen × AtWen | (12) |
| [M-Agen] = (N-Agen × AtWen)/M | (13) |
| AtWen = (107Aben × 107AtW) + (109Aben × 109AtW) | (14) |
The results of these calculations for cases 1 and 2 are given in Table 3. In detail, they indicate that isotopic tracing with Q-ICP-MS requires that the addition of enriched 107Ag raises the total Ag concentration by at least 2.6%, to 102.6 ng g−1 for the tissue (case 1) and 205.2 ng L−1 for the water sample (case 2; Table 3). With MC-ICP-MS, measurements of the 107Ag/109Ag ratio are about 100-fold more precise and consequently the tracing sensitivity is also improved by two orders of magnitude. In this scenario the Ag sample concentration only needs to increase by 0.026% from the addition of 107Ag, to generate analytically resolvable differences in the 107Ag/109Ag ratio. For the tissue and water samples, this means that increases in the Ag concentration to 100.026 ng g−1 (from 100 ng g−1) and 200.052 g L−1 (from 200 g L−1), respectively, are already traceable by isotopic analyses. Evaluation of the detection sensitivity for a given isotope label, analytical approach and natural background concentration in this manner, enables appropriate experimental plans and dosing levels to be prepared.
The uncertainties of the isotope ratio analyses can be propagated through the above equations to assess the impact of the measurement precision on the calculated mass of enriched tracer material present in a sample or its mass concentration. Importantly, the uncertainties do not scale in a linear manner and depend on the isotopic enrichment of the tracer and the proportion of enriched to natural Ag in the sample. To demonstrate this, the uncertainties for [M-Agen] of cases 1 and 2 (Table 3) were calculated for the addition of 107Ag-enriched tracer at levels, which exceed the smallest detectable molar concentration Nres-Agen by a factor of 10× and 100×.
The results of these calculations are promising because they indicate that despite of the unfavorable natural isotope composition of Ag, stable isotope labeling of Ag can provide detection sensitivities and uncertainties of quantification that are far better than those achievable by concentration tracing. Considering that the latter methodology can only provide unequivocal results if the concentration anomalies generated by elemental uptake exceed the natural background levels by at least a factor of 2 (but preferably more), the application of stable isotope labeling enhances the tracing sensitivity by a least a factor of 40 (using Q-ICP-MS for detection) and possibly by up to a factor of ~4000 (with use of MC-ICP-MS). Furthermore, with the superior precision of MC-ICP-MS for isotopic analyses, the tracing sensitivities are likely to be sufficiently high, to enable experiments to be carried out at low and environmentally relevant exposure concentrations.
4. Conclusions
In this study, published methods were modified for small-scale synthesis of citrate-capped AgNPs from both natural Ag and enriched 107Ag. Three different synthesis protocols were used on three occasions and in two laboratories to obtain AgNPs with target sizes of 17, 20 and 30 nm. All particles batches produced were thoroughly characterized and the results demonstrate that the new procedures provide reproducible, monodisperse particle suspensions at high yields of ~80% and with a long-term stability of up to 12 months. Whilst the measurements revealed small but significant batch-to-batch variations in size, the particles were generally found to have diameters that closely matched the desired target sizes. Detailed statistical tests furthermore argue against systematic differences in size between AgNPs prepared from enriched 107Ag versus natural Ag. In summary, these results show that the new methods are ideally suited for the small-scale preparation of isotopically labeled AgNPs. Simple isotope mass balance calculations furthermore demonstrate that application of stable isotope labeling can increase the detection sensitivity of AgNPs in environmental samples by at least a factor of 40 and possibly by up to 4000×, in comparison to commonly employed bulk Ag concentration measurements. Use of the most precise isotopic measurement techniques should therefore, permit environmentally realistic exposure scenarios.Acknowledgements
Katharina Kreissig and Barry Coles of the Imperial College MAGIC Laboratories are thanked for technical assistance. The research received funding from the NERC Facility for Environmental Nanoscience Analysis and Characterization (FENAC) at the University of Birmingham and was carried out as part of PROSPEcT, a public–private partnership between DEFRA, EPSRC, TSB and the Nanotechnology Industries Association (NIA Ltd.) and its members. Further funding was provided by a UK Research Council studentship to A.L. and the joint US-EPA and UK-NERC Research Program grant RD-834557501-0. The authors also thank three anonymous reviewers and editor Greg Lowry, for providing constructive comments and helpful suggestions.References
- T. W. Purcell and J. J. Peters, Environ. Toxicol. Chem., 1998, 17, 539–546 CrossRef CAS.
- S. W. P. Wijnhoven, W. J. G. M. Peijnenburg, C. A. Herberts, W. I. Hagens, A. G. Oomen, E. H. W. Heugens, B. Roszek, J. Bisschops, I. Gosens, D. Van de Meent, S. Dekkers, W. H. De Jong, M. Van Zijverden, A. J. A. M. Sips and R. E. Geertsma, Nanotoxicology, 2009, 3, 109–138 CrossRef CAS.
- Woodrow Wilson consumer products inventory for products containing nano-silver, http://www.nanotechproject.org/cpi/browse/nanomaterials/silver-nanoparticle/, Accessed December 2013.
- F. Gottschalk, T. Sun and B. Nowack, Environ. Pollut., 2013, 181, 287–300 CrossRef CAS PubMed.
- R. Eisler, Silver hazards to fish, wildlife and invertebrates: A synoptic review, U.S. Department of the Interior, 1996 Search PubMed.
- W. A. Shoults-Wilson, B. C. Reinsch, O. V. Tsyusko, P. M. Bertsch, G. V. Lowry and J. M. Unrine, Soil Sci. Soc. Am. J., 2011, 75, 365–377 CrossRef CAS.
- C. M. Zhao and W. X. Wang, Environ. Toxicol. Chem., 2011, 30, 885–892 CrossRef CAS PubMed.
- L. Maretti, P. S. Billone, Y. Liu and J. C. Scaiano, J. Am. Chem. Soc., 2009, 131, 13972–13980 CrossRef CAS PubMed.
- T. Xia, M. Kovochich, M. Liong, L. Madler, B. Gilbert, H. B. Shi, J. I. Yeh, J. I. Zink and A. E. Nel, ACS Nano, 2008, 2, 2121–2134 CrossRef CAS PubMed.
- A. Chrastina and J. E. Schnitzer, Int. J. Nanomed., 2010, 5, 653–659 Search PubMed.
- C. Coutris, E. J. Joner and D. H. Oughton, Sci. Total Environ., 2012, 420, 327–333 CrossRef CAS PubMed.
- M. Zuykov, E. Pelletier and S. Demers, Mar. Environ. Res., 2011, 71, 17–21 CrossRef CAS PubMed.
- N. Lewinski, V. Colvin and R. Drezek, Small, 2008, 4, 26–49 CrossRef CAS PubMed.
- T. Tenuta, M. P. Monopoli, J. Kim, A. Salvati, K. A. Dawson, P. Sandin and I. Lynch, PLoS One, 2011, 6 Search PubMed.
- N. Gibson, U. Holzwarth, K. Abbas, F. Simonelli, J. Kozempel, I. Cydzik, G. Cotogno, A. Bulgheroni, D. Gilliland, J. Ponti, F. Franchini, P. Marmorato, H. Stamm, W. Kreyling, A. Wenk, M. Semmler-Behnke, S. Buono, L. Maciocco and N. Burgio, Arch. Toxicol., 2011, 85, 751–773 CrossRef CAS PubMed.
- D. H. Oughton, T. Hertel-Aas, E. Pellicer, E. Mendoza and E. J. Joner, Environ. Toxicol. Chem., 2008, 27, 1883–1887 CrossRef CAS.
- F. R. Khan, A. Laycock, A. Dybowska, F. Larner, B. D. Smith, P. Rainbow, S. N. Luoma, M. Rehkämper and E. Valsami-Jones, Environ. Sci. Technol., 2013, 47, 8532–8439 CAS.
- F. Larner, Y. Dogra, A. Dybowska, J. Fabrega, B. Stolpe, L. J. Bridgestock, R. Goodhead, D. J. Weiss, J. Moger, J. R. Lead, E. Valsami-Jones, C. R. Tyler, T. S. Galloway and M. Rehkämper, Environ. Sci. Technol., 2012, 46, 12137–12145 CrossRef CAS PubMed.
- N. A. Matwiyoff and D. G. Ott, Science, 1973, 181, 1125–1133 CAS.
- C. Sridar, I. Hanna and P. F. Hollenberg, Xenobiotica, 2013, 43, 336–345 CrossRef CAS PubMed.
- S. K. Misra, A. Dybowska, D. Berhanu, M. N. Croteau, S. N. Luoma, A. R. Boccaccini and E. Valsami-Jones, Environ. Sci. Technol., 2012, 46, 1216–1222 CrossRef CAS PubMed.
- M. N. Croteau, A. D. Dybowska, S. N. Luoma and E. Valsami-Jones, Nanotoxicology, 2011, 5, 79–90 CrossRef CAS PubMed.
- B. Gulson, M. McCall, M. Korsch, L. Gomez, P. Casey, Y. Oytam, A. Taylor, M. McCulloch, J. Trotter, L. Kinsley and G. Greenoak, Toxicol. Sci., 2010, 118, 140–149 CrossRef CAS PubMed.
- J. Gigault and V. A. Hackley, Anal. Chim. Acta, 2013, 763, 57–66 CrossRef CAS PubMed.
- S. A. Cumberland and J. R. Lead, J. Chromatogr. A, 2009, 1216, 9099–9105 CrossRef CAS PubMed.
- I. Römer, T. A. White, M. Baalousha, K. Chipman, M. R. Viant and J. R. Lead, J. Chromatogr. A, 2011, 1218, 4226–4233 CrossRef PubMed.
- F. Larner and M. Rehkämper, Environ. Sci. Technol., 2012, 46, 4149–4158 CrossRef CAS PubMed.
- M. Baalousha and J. R. Lead, Environ. Sci. Technol., 2012, 46, 6134–6142 CrossRef CAS PubMed.
- A. Litzen, Anal. Chem., 1993, 65, 461–470 CrossRef CAS.
- M. Baalousha and J. R. Lead, Nat. Nanotechnol., 2013, 8, 308–309 CrossRef CAS PubMed.
- M. Baalousha and J. R. Lead, Environ. Sci. Technol., 2007, 41, 1111–1117 CrossRef CAS.
- M. Baalousha, A. Manciulea, S. Cumberland, K. Kendall and J. R. Lead, Environ. Toxicol. Chem., 2008, 27, 1875–1882 CrossRef CAS.
- M. Tejamaya, I. Römer, R. C. Merrifield and J. R. Lead, Environ. Sci. Technol., 2012, 46, 7011–7017 CrossRef CAS PubMed.
- J. H. Chen and G. J. Wasserburg, Geochim. Cosmochim. Acta, 1990, 54, 1729–1743 CrossRef CAS.
- K. G. Heumann, S. M. Gallus, G. Radlinger and J. Vogl, J. Anal. At. Spectrom., 1998, 13, 1001–1008 RSC.
- M. Schönbächler, R. W. Carlson, M. E. Horan, T. D. Mock and E. H. Hauri, Int. J. Mass Spectrom., 2007, 261, 183–191 CrossRef PubMed.
- S. J. Woodland, M. Rehkämper, A. N. Halliday, D. C. Lee, B. Hattendorf and D. Gunther, Geochim. Cosmochim. Acta, 2005, 69, 2153–2163 CrossRef CAS PubMed.
- A. V. Chugaev and I. V. Chernyshev, Geochem. Int., 2012, 50, 899–910 CrossRef CAS.
- E. Hauri, R. Carlson and J. Bauer, presented in part at the 31st Lunar and Planetary Science Conference, Houston, March, 2000.
- A. M. Desaulty, P. Telouk, E. Albalat and F. Albarede, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9002–9007 CrossRef CAS PubMed.
- Y. Luo, E. Dabek-Zlotorzynska, V. Celo, D. C. G. Muir and L. Yang, Anal. Chem., 2010, 82, 3922–3928 CrossRef CAS PubMed.
- T. Arnold, M. Schönbächler, M. Rehkämper, S. Dong, F. J. Zhao, G. J. Kirk, B. J. Coles and D. J. Weiss, Anal. Bioanal. Chem., 2010, 398, 3115–3125 CrossRef CAS PubMed.
- S. Ripperger, M. Rehkämper, D. Porcelli and A. N. Halliday, Earth Planet. Sci. Lett., 2007, 261, 670–684 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3en00100h |
| This journal is © The Royal Society of Chemistry 2014 |