Stretching and conformal bonding of organic solar cells to hemispherical surfaces
Timothy F.
O'Connor
,
Aliaksandr V.
Zaretski
,
Bijan A.
Shiravi
,
Suchol
Savagatrup
,
Adam D.
Printz
,
Mare Ivana
Diaz
and
Darren J.
Lipomi
*
Department of NanoEngineering, University of California, San Diego, 9500 Gilman Drive, Mail Code 0448, La Jolla, CA 92093-0448, USA. E-mail: dlipomi@ucsd.edu
First published on 15th November 2013
Abstract
This paper describes the stretching and conformal bonding (i.e., decal-transfer printing) of organic solar cells in both the “conventional” and “inverted” configurations to hemispherical glass surfaces with radii of 8 mm. This action produces equivalent biaxial tensile strains of 24%, which many materials used in organic electronic devices cannot accommodate without fracture. Consideration of the mechanical properties of conjugated polymers reveals a surprising effect of a single structural parameter—the length of the alkyl side chain—on the elasticity and ductility of regioregular polythiophene. This analysis enables selection of materials that can accommodate sufficient tensile strain for non-planar applications. For polymer–fullerene solar cells, devices based on the elastic and ductile poly(3-octylthiophene) (P3OT) exhibit typical photovoltaic properties when bonded to hemispherical glass substrates, while those based on the relatively brittle poly(3-hexylthiophene) (P3HT) exhibit extensive cracking, which degrades the photovoltaic effect significantly. The results suggest that mechanical properties should be taken into account when designing and selecting organic semiconductors for applications that demand significant deformation.
Broader contextOne of the prominent selling points of organic electronic materials and devices is the potential that they could be elastically and plastically deformed without fracture. In the context of organic solar cells, this mechanical compliance would facilitate roll-to-roll fabrication, robustness to the stresses encountered in the outdoor environment and in portable applications, integration with the moving parts of machines or the body, and bonding to non-planar surfaces such as vehicles and buildings. Organic semiconductors, however, are almost never optimized on the basis of mechanical properties, and most crack at modest strains. This paper demonstrates (1) the stretching and conformal bonding of whole organic solar cells to hemispherical surfaces and (2) the surprising influence of the length of the alkyl solubilizing group on the plasticity of the semiconducting layer. This result suggests that seemingly minor differences in the structure of an organic semiconductor can have drastic consequences on the suitability of a particular material for applications demanding tensile deformation. |
Organic electronic devices—e.g., solar cells, transistors, and displays based on π-conjugated polymers and small molecules—are treated as potentially low-cost and mechanically compliant alternatives to their inorganic counterparts.1,2 The high flexibility of organic electronic devices, however, is largely a function of the thinness of the active materials and substrates; it is not necessarily a function of the intrinsic mechanical compliance of organic semiconductors. In fact, the peak strains imposed on the active materials while bent are rarely greater than 2%—a modest level of deformation that nonetheless fractures some of the most well known organic semiconductors3 and polymer–small molecule composites.4,5 This paper demonstrates two major effects: (1) the ability to stretch and bond an especially ductile organic electronic device to hemispherical surfaces and (2) the unexpectedly large effect of a single structural parameter—the length of the alkyl side chain—on the compliance and ductility of thin films comprising poly(3-alkylthiophenes) (P3ATs) and P3AT
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :fullerene composites. We demonstrate that an analysis of the mechanical properties of organic semiconductors is necessary to select materials for applications that will require appreciable (>2%) strains. Such applications include those that demand resistance to mechanical failure (for portability)6,7 and integration with moving parts,8 textiles,9 and curved surfaces other than cylinders.10 We use the results of our investigation to show that the most popular active layer for organic photovoltaics, poly(3-hexylthiophene) (P3HT) mixed with [6,6]-phenyl C61 butyric acid methyl ester (PCBM), cannot be stretched to conform to a hemispherical surface without cracking, but that a substitution of materials informed by our analysis enables sufficient deformation and transfer (Fig. 1). We believe that the mechanical properties of an organic semiconductor should be given a high priority when selecting materials for applications that require one-time or repeated deformation. Our observations may suggest the design of new materials that combine state-of-the-art electronic properties with favorable mechanical ones.
:fullerene composites. We demonstrate that an analysis of the mechanical properties of organic semiconductors is necessary to select materials for applications that will require appreciable (>2%) strains. Such applications include those that demand resistance to mechanical failure (for portability)6,7 and integration with moving parts,8 textiles,9 and curved surfaces other than cylinders.10 We use the results of our investigation to show that the most popular active layer for organic photovoltaics, poly(3-hexylthiophene) (P3HT) mixed with [6,6]-phenyl C61 butyric acid methyl ester (PCBM), cannot be stretched to conform to a hemispherical surface without cracking, but that a substitution of materials informed by our analysis enables sufficient deformation and transfer (Fig. 1). We believe that the mechanical properties of an organic semiconductor should be given a high priority when selecting materials for applications that require one-time or repeated deformation. Our observations may suggest the design of new materials that combine state-of-the-art electronic properties with favorable mechanical ones.
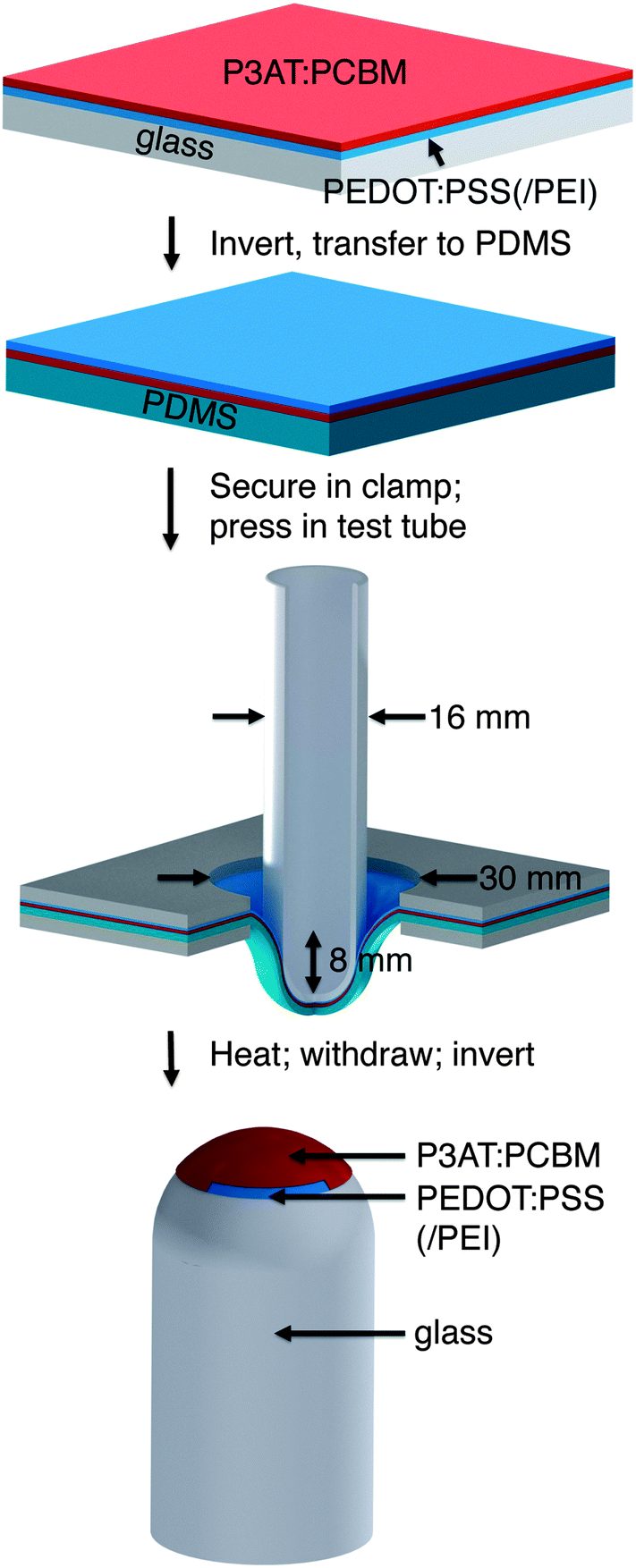 | ||
| Fig. 1 Summary of the process used to transfer multilayer films of conjugated polymers to a hemispherical glass substrate. Passivated glass is spin-coated with poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) and P3AT:PCBM. The multilayer film is transferred to a poly(dimethylsiloxane) (PDMS) slab and secured in a clamp bearing a circular aperture. A test tube is pressed into the suspended PDMS/P3AT:PCBM/PEDOT:PSS substrate, heated, and withdrawn. Slow withdrawal separates the conjugated polymer films and leaves it attached to the hemispherical surface of the test tube. | ||
The field of stretchable electronics encompasses and extends the field of flexible electronics.11–14 The use of buckled,15 wavy,16 or serpentine microstructures,17 stretchable interconnects,18 controlled fracture,19 or dispersion of conductive particles in a deformable insulator20,21 are all methods used to fabricate devices that are, on the whole, stretchable. The outcome of most of these strategies is that the active material is not subjected to significant strain.22 Designing materials—semiconductors in particular—that retain their electronic properties when deformed represents a significant scientific challenge.23,24 There are, however, very few examples in the literature where whole organic electronic devices have accommodated significant tensile deformation with little loss in function. Pei and coworkers, for example, demonstrated a plastically deformable light-emitting device comprising a blue emitter based on a polyfluorene backbone. When this emissive layer was sandwiched between films of carbon nanotubes as electrodes, the whole device could be heated and deformed plastically by ≥45% with its ability to emit light intact.24 Another successful example is that of Müller et al., who demonstrated that a diblock copolymer having P3HT and polyethylene segments with weight fractions of the insulating block up to 90% maintains significant charge mobility when stretched by 600%,23 but there are many applications (e.g., solar cells), for which it will not be desirable to have a large insulating component in the active layer. Most pure films of organic semiconductors and devices composed of multiple layers are significantly less compliant than the two examples cited above.
Measurements of the mechanical properties of conjugated polymers have revealed that not all of these materials can be treated as equally deformable. For example, Tahk et al. measured the tensile modulus of polyaniline to be 30 MPa (though likely plasticized by residual solvent), and that of the molecular semiconductor pentacene to be ∼15 GPa.3 These authors and others5 have shown that a 1:1 mixture of poly(3-hexylthiophene) (P3HT) and [6,6]-phenyl C61 butyric acid methyl ester (PCBM) had a tensile modulus five times greater than that of the pure polymer.3 O'Connor et al. have observed an apparent trade-off between electronic performance and mechanical compliance.25 These investigators observed that increasing the crystallinity of pure films of PBTTT by thermal annealing4 and of P3HT:PCBM blends by the speed of solvent evaporation25 increased the charge-carrier mobility and photovoltaic efficiency while rendering the films substantially more brittle (the best-performing PBTTT and P3HT:PCBM films fractured at strains <2.5%).4,25 The pioneering work of Smith and Heeger studied the mechanical properties of early conjugated polymers such as polyacetylene,26 poly(2,5-thienylene vinylene),27 and regiorandom polythiophene.28 The demands of applications such as organic solar cells and field-effect transistors, however, have led to a dramatic increase in the number of soluble, regioregular, low-bandgap, donor–acceptor, and structurally complex polymers available,29 and these polymers are rarely optimized on the basis of mechanical properties. General guidelines that link the structural characteristics of these polymers to their mechanical properties would be useful for any device intended for portability,30 prolonged outdoor service,31 or biological integration.10,32
While unsubstituted conjugated polymers such as poly(acetylene) can exhibit high stiffness (tensile modulus 50 GPa) and strength (ultimate tensile strength 900 MPa) when oriented,26 unsubstituted materials are generally not processable from solution. Alkyl pendant groups impart solubility to the polymer, but also drastically change the mechanical properties of the material.28 These side chains reduce the number of covalently bonded units along the axis of the main chain per unit volume and also reduce the secondary interactions between the main chain.33 Both effects tend to reduce the modulus and strength (and conductivity) of the polymer.33 While these effects are deleterious for materials intended for use in structural applications,33 renewed interest in stretchable semiconductors holds compliance and ductility in high regard.4,23,34 In applications demanding reversible deformation or one-time bonding to non-planar substrates, the goal is not to maximize electronic performance, but to find materials exhibiting an acceptable compromise between mechanical and optoelectronic properties.
We looked to the regioregular poly(3-alkylthiophene)s (P3ATs) as a model system to investigate the influence of alkyl side chain on the intrinsic stretchability of conjugated polymers. While P3ATs are well known, their properties have not been compared in a context relevant to stretchable devices. In a series of P3ATs where A = butyl (B), hexyl (H), octyl (O), and dodecyl (DD), the compliance and ductility increased with increasing length of the alkyl chain from P3BT to P3OT, while the properties of P3OT and P3DDT were similar.35 These measurements were corroborated by theoretical calculations—which elaborated on earlier methods described by Seitz36 and Tahk3—that accounted for differences in the glass transition temperatures along with structural parameters.35 The largest differences in mechanical properties between adjacent polymers in the series occurred between P3HT and P3OT, whose tensile moduli differed by nearly an order of magnitude: 1.09 GPa for P3HT and 0.15 GPa for P3OT.
The incorporation of fullerenes in films of conjugated polymers has several important consequences for the mechanical properties and interfacial adhesion of bulk heterojunction films. Previous reports have shown that the tensile modulus of P3HT:PCBM (1:1) films (4.3 GPa) was nearly five times greater than that of the pure polymer (0.92 GPa).3,5 We have also measured the crack-onset strains of P3HT (9%) and P3HT:PCBM (3%) on PDMS substrates, and found that fullerenes increase the brittleness of the films relative to the pure polymers.35 Other deleterious effects of PCBM on the compliance of devices include lowered cohesive energy of the bulk heterojunction (from 2.5 J m−2 to 0.5 J m−2 for P3HT:PCBM blends containing from 25% to 100% PCBM)7 and lowered adhesion of bulk heterojunction films to the PEDOT:PSS electrode (1.6 J m−2 for the pure polymer and 0.1 J m−2 for the pure fullerene).6 The influence of PCBM on the moduli of conjugated polymers is predictable in some circumstances by composite theory, as described by Tahk et al.,3 but the exact dependence is a strong function of the identity of the polymer5 and processing conditions, especially the way in which the rate of solvent evaporation affects the crystallinity of the polymer.25
Blends of P3HT and P3OT with [6,6]-phenyl C61 butyric acid methyl ester (PCBM) also exhibited dramatically different propensities to fracture, which can be treated as a measure of ductility. P3HT:PCBM cracked at an average strain of 3%, while P3OT:PCBM did not crack until 47% strain.35 We reasoned that substitutions of materials based on an analysis of the structural parameters that influence the mechanical properties of conjugated polymers could enable applications in stretchable and conformable electronics not accessible by standard materials. Thus, we developed procedures to stretch and transfer organic solar cells based on P3HT and those based on P3OT to hemispherical substrates in order to compare the performance in this mechanically demanding geometry.
Results
Dependence of molecular weight on mechanical compliance of P3ATs
Most polymeric materials exhibit an increase in tensile modulus with molecular weight.37 This effect, however, tends to saturate above a sufficiently high molecular weight.37 Another trend predicts that increased polydispersity index (PDI) produces increased compliance.37 Given that different synthetic methods and levels of purification can yield different molecular weights and polydispersities, we measured the tensile modulus of a commercial sample of P3HT (Mw = 29000 g mol−1, PDI = 2.0, as determined by gel permeation chromatography using polystyrene standards). We then synthesized a sample of P3HT in our laboratory by the Grignard Metathesis polymerization38 and quenched the reaction after approximately 10 s to produce a sample with low molecular weight (Mw = 7500 g mol−1, PDI = 1.2). The tensile moduli of both samples were similar: 1.09 ± 0.15 GPa for the commercial sample and 1.05 ± 0.35 GPa for the low-Mw sample (synthesized in-house). The P3OT (obtained commercially) had a greater Mw, 108000 g mol−1 (PDI = 2.5), but a much smaller tensile modulus (0.15 ± 0.05 GPa) than the P3HT samples. These data suggest that the Mw and PDI do not significantly affect the mechanical properties of the materials in the range of Mw and PDI of the samples studied, and that the Mw of P3OT cannot explain its significantly greater compliance compared to that of P3HT. We believe that the length of the alkyl chain was the principal determinant of the difference in mechanical properties observed between the P3HT and P3OT.
Determination of onset of plastic deformation
We note that the descriptor “stretchability” can be subdivided into elasticity and plasticity. For form factors that require one-time bonding to curved substrates, tensile modulus can be used as a metric for overall compliance, but it does not predict the ductility and cracking behavior of films. We measured the approximate yield strain of P3HT and P3OT using the strain at which buckles first appeared (“buckling-onset” strain) as a proxy. In this experiment, we bonded the P3AT films to PDMS substrates and stretched the PDMS/P3AT samples using a computer-controlled linear actuator and applied cyclic strains in increments of 1%, that is 0% → 1% → 0% → 2% → 0% etc. Upon each return to 0% strain, we examined the films with a microscope and took the minimum strain at which buckles formed to be an approximation of the onset of plastic deformation. P3OT exhibited a greater range of elastic behavior before the onset of plastic deformation: the onset of buckling occurred after stretching to 11% strain for P3OT and after stretching to 4% strain for P3HT.Conformal bonding to hemispherical substrates
To bond a thin film (or stack of films) conformally to a hemispherical surface requires the film to accommodate compressive or tensile deformation so that wrinkles are not generated.39Fig. 1 summarizes the process—based on kinetically controlled transfer printing40—we used to fabricate the devices. Briefly, the devices were fabricated on passivated glass substrates, transferred to poly(dimethylsiloxane) (PDMS) membranes, mounted in a stage bearing a circular aperture, and transferred to the hemispherical terminus of a glass test tube by pushing the test tube into the PDMS membrane bearing the conjugated polymer layers. A computer-controlled linear actuator was used to impose precise displacements of the test tube. Heating and slow withdrawal of the test tube transferred the part of the film in contact with the hemispherical terminus of the tube to its surface. We used two configurations, which are summarized in Fig. 2. In the conventional geometry, PEDOT:PSS behaved as the bottom, high-work-function electrode, on top of which the P3AT:PCBM film was deposited. Eutectic gallium–indium (EGaIn), deposited after bonding of the PEDOT:PSS/P3AT:PCBM films to the hemispherical surface, served as the low-work-function top electrode. In the inverted geometry, PEDOT:PSS treated with polyethyleneimine (PEI) behaved as the bottom, low-work-function electrode.41 A film of untreated PEDOT:PSS, laminated to the device in a step subsequent to the procedure depicted in Fig. 1, served as the high-work-function electrode.42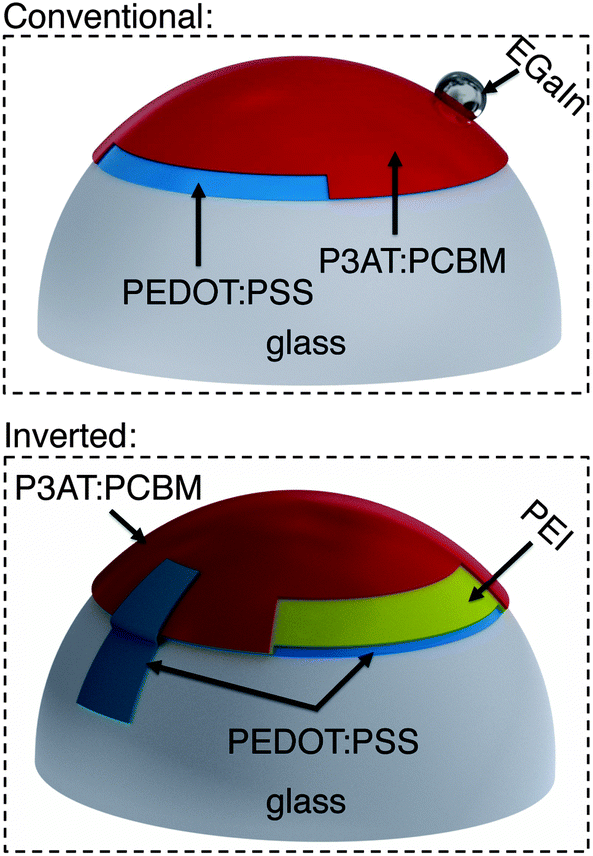 | ||
| Fig. 2 Conventional (top) vs. inverted (bottom) geometries of organic solar cells discussed in the text. In the conventional geometry, a droplet of eutectic gallium–indium (EGaIn) provides the low-work-function top contact (cathode). In the inverted geometry, a layer of PEDOT:PSS bearing a thinner layer of polyethyleneimine (PEI), applied to the glass substrate, behaves as the low-work-function electrode. | ||
Computational analysis of strain
We selected materials for this experiment based on a computational analysis whose goal was to predict the minimum strain required to stretch a planar sheet over a hemispherical surface without generating wrinkles. Fig. 3 shows the results of our simulation. It assumes no slippage of the film against the hemispherical surface during the process of transfer from the PDMS to the hemisphere. This assumption is justified by a visual inspection of the films during the transfer: once the film made contact with the hemispherical surface, it did not continue to crack with additional displacement of the test tube into the PDMS membrane. Moreover, when we drove the test tube into the substrate from the opposite direction (such that the conjugated polymer films were on the convex surface of the deflected PDMS membrane) we observed cracking patterns consistent with the simulation on the convex surface, as shown in Fig. 3. That is, the density of cracks was greatest at the apex. According to our simulation of the strain on the concave surface, the maximum strain of 24% occurred at an arc-length of 4.42 mm from the apex of the hemisphere (r = 8 mm). Our analysis of the mechanical properties of P3ATs suggested that a film of P3HT:PCBM would be severely damaged as a result of the transfer, while P3OT:PCBM would survive the transfer. Our placement of the PEDOT:PSS top contact in the inverted geometry (as shown schematically in Fig. 2) was chosen to overlap with the region of the greatest predicted deformation.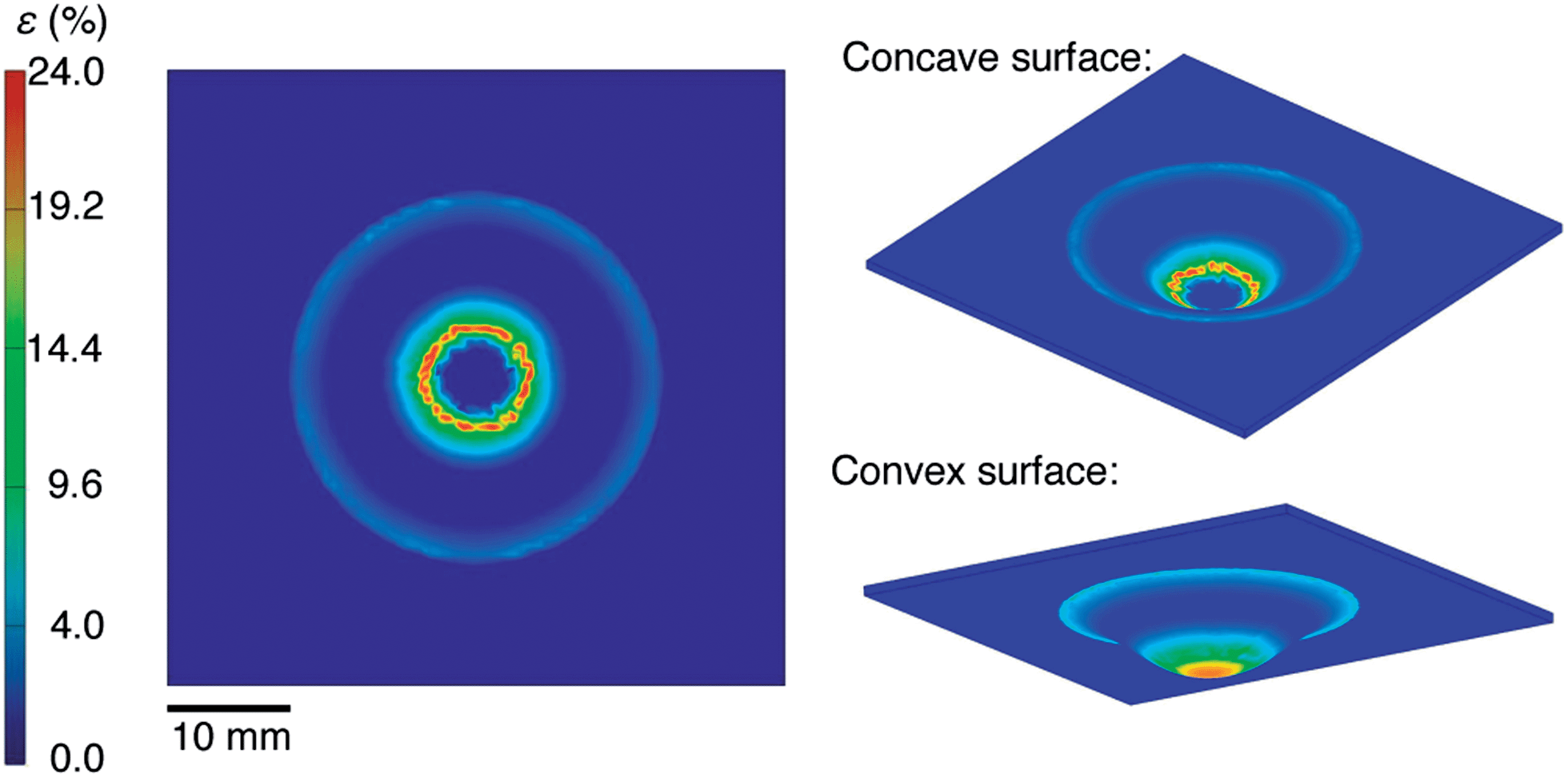 | ||
| Fig. 3 Computational analysis of the strain produced in the thin films when they are transferred to the hemispherical surfaces. The 2D map (left) represents the strain generated at the surface of the PDMS membrane bearing the multilayered conjugated polymer film. This is the same view shown in the 3D rendering of the concave surface (top right). The distribution of strain observable in the convex surface (bottom right), which is dissimilar from that of the concave surface, is a consequence of the finite thickness of the PDMS membrane, and that the convex surface is not pinned by contact with the glass hemisphere during the process of deformation, as is the concave surface. | ||
Cracking behavior
Photographs of films comprising a single layer of the two P3ATs blended with PCBM are shown in Fig. 4a and b. The P3HT:PCBM film exhibited extensive rupturing; the P3OT:PCBM film, in contrast, was undamaged. We also observed striking differences in the cracking behavior of P3AT:PCBM films on PDMS substrates when stretched with and without an intervening layer of PEDOT:PSS. We attribute the apparent increase in ductility of the P3HT:PCBM film when PEDOT:PSS was used to the behavior of PEDOT:PSS as a layer that promotes adhesion between PDMS and conjugated polymer films.5 Improved adhesion between a thin film and a stretchable substrate increase the effective ductility of the film by distributing strain uniformly. In a poorly adhered film, global tensile strains localize to delaminated regions and thus form cracks at much smaller strains than for systems with better adhesion.43 This effect has been observed in stretched films of copper on polyimide substrates with and without chromium adhesion layers.43 The presence of PEDOT:PSS significantly reduced the extent of cracking—though still enough to cause catastrophic failure of the device—in PEDOT:PSS/P3HT:PCBM films (Fig. 4c). The bilayer film comprising PEDOT:PSS/P3OT:PCBM was undamaged (Fig. 4d). Interestingly, while PEDOT:PSS improved the effective ductility of P3HT:PCBM in these experiments, it had a deleterious effect on the effective ductility of P3OT:PCBM. PEDOT:PSS on PDMS substrates has a crack-onset strain of ∼12% (ref. 44) which is intermediate between that of P3HT:PCBM (3%)35 and P3OT:PCBM (47%).35 The cracking of PEDOT:PSS underneath P3OT:PCBM thus drives the formation of cracks in the top layer.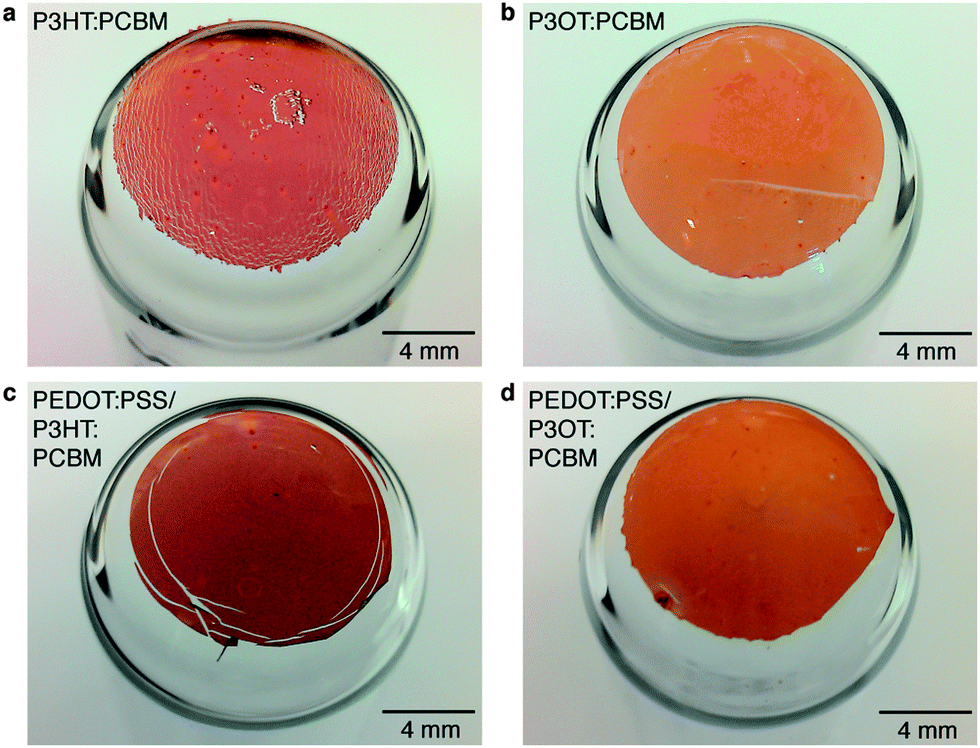 | ||
| Fig. 4 Photographs of conjugated polymer films stretched and bonded to hemispherical surfaces. (a) P3HT:PCBM film exhibiting extensive fracturing. (b) Intact P3OT:PCBM film. (c) Bilayer film comprising PEDOT:PSS/P3HT:PCBM exhibiting concentric cracking pattern which is not observed in the PEDOT:PSS/P3OT:PCBM film (d). | ||
Given that the presence of residual solvent can reduce the tensile modulus and increase the ductility of conjugated polymer samples (as Tahk et al. observed for polyaniline3), we measured the crack-onset strain for an as-cast sample of P3HT and found it to be indistinguishable to that of a sample that was dried in vacuum (<200 mtorr) for 60 min. This experiment suggests that the length of the alkyl chain, rather than the plasticizing effects of residual solvent, is the principal determinant of the mechanical properties of the two P3ATs examined in this work.
Measurement of photovoltaic properties
To test the effect of the deformation on the optoelectronic behavior of the devices, we measured the photovoltaic properties using a solar simulator (Fig. 5). Fig. 5a demonstrates that the damage caused by cracking limits the applicability of P3HT:PCBM in devices demanding mechanical compliance. In this device, a droplet of EGaIn was positioned in the region on the film that experienced the maximum strain. The current density vs. voltage (J–V) for these devices resembled resistors in parallel with solar cells. We attribute this behavior to areas in which the EGaIn penetrated through cracks in the active layer and made direct contact with the PEDOT:PSS. In contrast, the devices comprising undamaged P3OT:PCBM films in the conventional geometry exhibited behavior characteristic of devices on planar substrates (Fig. 5a). The J–V characteristics of an inverted device with the structure glass/PEDOT:PSS/PEI/P3OT:PCBM with a laminated PEDOT:PSS top contact are shown in Fig. 5b. The footprint of the EGaIn droplet and the laminated PEDOT:PSS top contact42 defined the active regions of the conventional and inverted devices, respectively. Optical microscopy did not reveal any cracks in the PEDOT:PSS top electrode after transfer in the inverted geometry.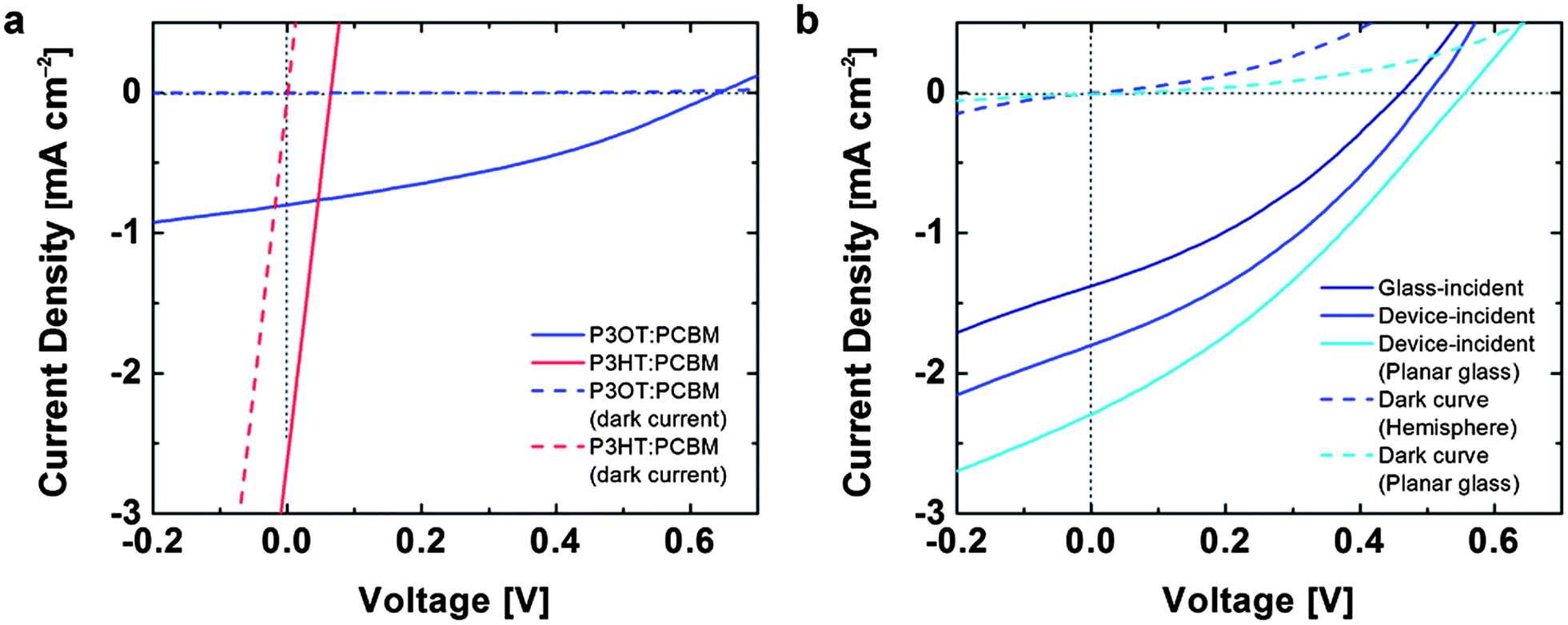 | ||
| Fig. 5 Representative photovoltaic characteristics of organic solar cells bonded to hemispherical substrates. (a) Current density vs. voltage (J–V) for devices in the conventional geometry: glass/PEDOT:PSS/P3AT:PCBM/EGaIn and P3OT:PCBM devices stretched and bonded to hemispherical surfaces. Extensive cracking produced a J–V plot resembling a short circuit. (b) Current density vs. voltage for an all-organic device in the inverted geometry: glass/PEDOT:PSS/PEI/P3OT:PCBM/PEDOT:PSS. The chart includes three plots: the device on the hemispherical surface with the light impinging on the concave surface (glass-incident) and the convex surface (device-incident) and a separate device prepared on planar glass (also device-incident). | ||
Compared to the device in the conventional geometry with the EGaIn top electrode, the all-organic device exhibited a reduction in open-circuit voltage (VOC) but an increase in short-circuit current density (JSC) by approximately 27%. While we selected P3OT on the basis of its ability to survive conformal transfer, we did not optimize devices for efficiency, which we estimate to be around 0.36% for the inverted devices. There are several possible sources of inefficiency, the most prominent of which is the inferior charge-transport properties of P3OT, whose hole mobility as measured in field-effect transistors is ten to fifty times lower than that of P3HT.45 Additionally, our process for transferring the films to hemispherical substrates proceeded in ambient air, and required a heating step. The geometry in which we measured these devices—with the cylindrical shaft of the test tube oriented toward the light source—probably reduced the diffuse contribution to the AM 1.5G spectrum impinging on the active area. The curved geometry also deflected the path of the light in ways we did not model. For the inverted device, which has a transparent “top” anode, it was possible to measure the photovoltaic properties in two configurations: with the light incident on the glass surface (glass-incident) and the light incident directly on the device layers (device-incident, Fig. 5b). We attribute the greater current density (27%) in the device-incident configuration to reduced reflective losses due to the glass surface and increased contributions from diffuse photons impinging on the active area.
We attempted to fabricate inverted devices using P3HT:PCBM as the active material, but were unsuccessful, because the process of transferring the PEDOT:PSS top contact inadvertently delaminated the PEDOT:PSS/PEI/P3HT:PCBM film already present on the hemispherical substrate. This inadvertent delamination was initiated at edges (as shown schematically in Fig. 6 in the case of planar glass). The extensive cracking of the brittle P3HT:PCBM film caused extensive delamination of the active material in every attempt at transferring the top contact. In the uncracked P3OT:PCBM devices, the transfer was facilitated by the physical contiguity of the active layer.
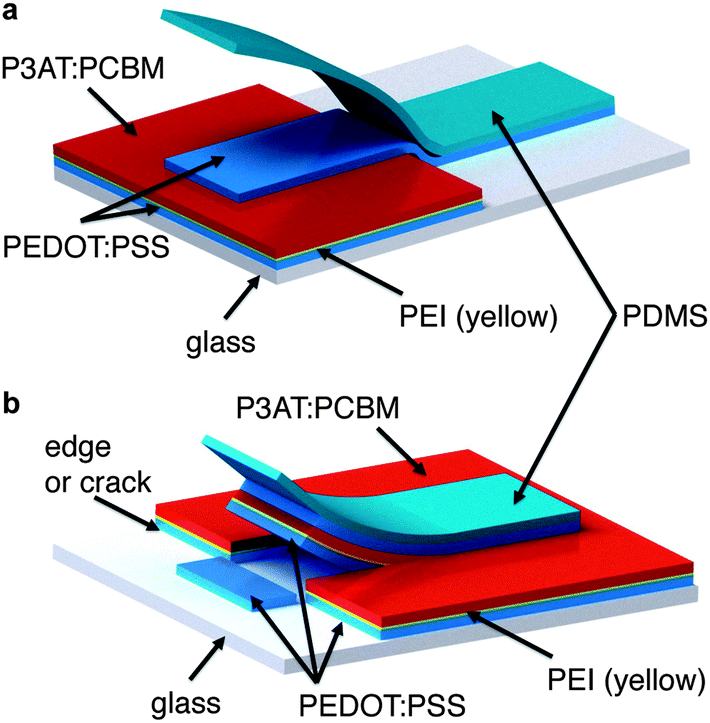 | ||
| Fig. 6 Schematic drawings of the successful (a) and unsuccessful (b) processes intended to transfer the PEDOT:PSS top contact to the glass/PEDOT:PSS/PEI/P3AT:PCBM device bonded to a glass substrate. The process is shown on planar glass, but the same effects were observed on hemispherical glass. In image (a) the transfer proceeded from a physically continuous region of the active layer to a region of bare glass, and the PEDOT:PSS film is transferred intact. In image (b), the transfer is initiated from—or passes over—an edge (which could be produced by a crack). Instead of transferring the PEDOT:PSS top contact, the layers already present on the glass substrate are inadvertently delaminated. We attribute successful transfer in mode (a) to the physical contiguity of the active layer (as in P3OT:PCBM devices) and unsuccessful transfer in mode (b) to extensive cracking (as in P3HT:PCBM devices). | ||
Yield
We estimate a yield in transferring films comprising active layer material, P3AT:PCBM, and partial devices, PEDOT:PSS/(PEI)/P3AT:PCBM from FOTS-treated glass to PDMS of >90% (N > 50), and the yield of transferring these films from PDMS to the hemispherical glass surface was >95% (N > 45). In inverted architectures, which require the transfer of a PEDOT:PSS top contact, the yield of transfer of the top contacts was ≥50% (N > 10). Using the most successful version of the process we developed, we measured a photovoltaic effect in 100% (7/7) of conventional cells employing EGaIn as a top contact, and ∼28% (2/7) of inverted cells employing PEDOT:PSS as both the top and bottom electrodes. We believe that sub-optimal yields and efficiencies are in part a consequence of the manual processes used, most of which proceeded in ambient air. Automated processing in an inert environment would, we believe, improve the yields and efficiencies substantially.Conclusion
This paper described a process to stretch and transfer conjugated polymer films and complete, all-organic solar cells to hemispherical surfaces. The process was enabled by a consideration of the mechanical properties of conjugated polymers, in particular, that the elasticity of regioregular polythiophenes increases by a factor of seven with an increase in the length of the alkyl pendant groups, from hexyl to octyl, with a corresponding increase in ductility. A computational simulation of the strain required to bond a thin film to a hemispherical substrate enabled our prediction that the increased ductility of P3OT:PCBM over P3HT:PCBM would permit bonding of the more ductile polymer–fullerene composite without wrinkling or cracking. Our experiments and analysis suggest that mechanical properties must be considered when selecting materials destined to experience mechanical deformation, and that seemingly minor structural variations—i.e., the addition of two methylene units in the pendant group—can either enable or prevent a material from performing in an application. Our results may suggest the design of new materials that maximize both electronic performance and mechanical compliance.There are two classes of unconventional form factors to which stretchable electronic systems are applicable: those that require reversible response to strain for integration with moving parts of machines or the body and those that require one-time bonding to a non-planar surface. The former category requires elasticity, while for the latter ductility is sufficient. The work described falls into the latter category, as the films accommodated the strain principally by plastic deformation. Applications of one-time bonding of plastically deformable electronic devices to non-planar surfaces could include integration with windshields or eyeglasses for semitransparent heads-up displays, integration with the curved surfaces of biomedical implants (as in electronic eye cameras39 or artificial retinas10), or bonding to nonplanar surfaces for applications requiring biofeedback (e.g., fatigue-sensing steering wheels and human-interactive robotics). Deformable organic solar cells in particular may find use in similar applications and also when bonded to architectural elements or the exteriors of vehicles in ways that neither compromise aesthetics nor aerodynamics. The selection of materials on the basis not only of electronic but also of mechanical properties is required. We believe our observations may provide some insights into the realization of truly “plastic” electronics.
Experimental methods
Materials
For the hemispherical substrates, we used borosilicate test tubes (d = 16 mm, l = 125 mm) from Fischer Scientific. Planar glass substrates were microscope slides obtained from Premiere. PEDOT:PSS (Clevios PH1000) was purchased from Heraeus. The solid content of the PH 1000 solution was 1–1.3% and had a ratio of PEDOT to PSS of 1:2.5 by weight. (Tridecafluoro-1,1,2,2-tetrahydrooctyl)-trichlorosilane (FOTS) was purchased from Gelest. Zonyl FS-300 (Zonyl), DMSO, ortho-dichlorobenzene (ODCB), poly(3-hexylthiophene) (P3HT), poly(3-octylthiophene) (P3OT), [6,6]-phenyl C61 butyric acid methyl ester (PCBM, >99%), and eutectic gallium–indium (EGaIn, ≥99.99%) were purchased from Sigma-Aldrich and used as received. The PDMS was prepared by puddle-casting a mixed and degassed PDMS prepolymer with a thickness of 1 mm (Dow Corning Sylgard 184, with a ratio of base to crosslinker of 10:1 by mass) in a polystyrene Petri dish. It was then cured at ambient temperature for 48 h and cut into 4 cm × 4 cm squares. The surface cured at the air interface was used for all experiments.
Fabrication of devices and bonding to hemispherical surfaces
We began by cutting 2.5 cm × 2.5 cm glass slides and sonicating them for 10 min each in deionized water with Alconox, pure deionized water, acetone, and isopropanol. In between each sonication step the slides were blown dry with compressed air. After the final sonication and drying step, the slides were treated in a plasma cleaner (30 W, 200 mtorr ambient air, 3 min) and subsequently placed in a vacuum desiccator containing a vial of ∼100 μL FOTS. Dynamic vacuum was applied for a minimum of 3 h. The PEDOT:PSS was filtered through a syringe filter (1 μm glass microfiber filter) to remove large particles. The composition of PEDOT:PSS was 94% by weight PEDOT:PSS, 5% by weight DMSO, and 1% by weight Zonyl. We spin-coated the solution onto the FOTS-treated glass at 700 rpm for 60 s then 2k rpm for 60 s. The slides were then dried on a hot plate in ambient air for 30 min at 150 °C. For devices to be measured in the inverted architecture, we spin-coated PEI at a concentration of 1% in methoxyethanol at a speed of 3 krpm for 60 s atop the dried PEDOT:PSS layer. This layer was annealed on a hot plate in air at 110 °C for 10 min.The active layer solution was prepared with a 1:1 solution of P3HT:PCBM or P3OT:PCBM in ODCB and stirred overnight at room temperature (40 mg mL−1 total). We then filtered these solutions through a 0.2 μm PTFE syringe filter and spin-coated them onto the FOTS-treated slides bearing a film of PEDOT:PSS(/PEI) at 500 rpm for 180 s then 2k rpm for 20 s. Dust was removed from the 4 cm × 4 cm PDMS squares using Scotch tape. We placed the clean surface of the PDMS against the PEDOT:PSS/P3AT:PCBM bilayer and established a conformal seal with the aid of gentle pressure from tweezers, such that the film was centered in the PDMS stamp. One edge of the PDMS was held and lifted at a rate of ∼10 cm s−1so as to quickly peel the PDMS off; this fast action transferred the film from the glass to the PDMS.
The PDMS/P3AT:PCBM/(PEI/)PEDOT:PSS substrates were mounted in a clamp bearing a circular aperture (d = 30 mm). The hemispherical bottoms of borosilicate test tubes were wiped with isopropanol and plasma treated (30 W, 500 mtorr air, 3 min). The tubes were subsequently mounted onto the computer-controlled linear actuator. The test tube was advanced toward the immobilized PDMS membrane until it made contact, and then driven 8 mm into the PDMS membrane at a rate of 0.25 mm s−1. This action deformed the PDMS membrane, which conformed to the hemispherical bottom of the test tube. Once at its maximum displacement, heat was applied to the convex side of the PDMS substrate for 30 s, which reached a temperature of 75 °C, as measured with a thermocouple. The test tube was then withdrawn from the suspended PDMS membrane at a rate of 0.25 mm s−1. Slow withdrawal transferred the PEDOT:PSS(/PEI)/P3AT:PCBM film to the hemispherical surface in the area of contact with the test tube.
Fabrication of devices with the conventional architecture
Starting with a test tube whose hemispherical bottom surface bore a PEDOT:PSS/P3AT:PCBM bilayer film, a region of PEDOT:PSS was exposed by wiping away the P3AT:PCBM layer using a swab soaked with chloroform. Silver paint was then used to facilitate contact between the PEDOT:PSS and the copper wire. A droplet of EGaIn was placed manually onto a portion of the active layer ∼4.5 mm from the apex of the hemisphere. A second copper wire was then inserted into the EGaIn droplet and secured to the glass tube with tape.Fabrication of devices with the inverted architecture
For devices with the inverted architecture, we laminated a top contact of PEDOT:PSS on top of the three-layer structure PEDOT:PSS/PEI/P3OT:PCBM. We used a modification of the procedure described by Janssen and coworkers to transfer the top PEDOT:PSS electrode to the P3OT:PCBM surface.42 Briefly, partially cured PDMS substrates (10:1 base to crosslinker) were prepared by puddle casting and cured at 70 °C for 25 min, then immediately placed in a refrigerator (8 °C) to reduce the rate of crosslinking. These PDMS substrates were treated in a plasma cleaner (30 W, 500 mtorr ambient air, 10 s) and PEDOT:PSS (containing 5% DMSO) was subsequently spin-coated. The PDMS/PEDOT:PSS substrates were allowed to dry in air for 5 min, after which they were cut into strips. The substrates were then immediately placed onto the partially completed hemispherical devices with the PEDOT:PSS layer in contact with the P3OT:PCBM layer, heated to ∼80 °C for 30 s with a heat gun, then allowed to cool to room temperature. After cooling, the PDMS was mechanically peeled from the hemisphere. This action left the PEDOT:PSS top contacts in place. We made electrical contact with the PEDOT:PSS top contacts by attaching a copper wire using silver paint as a conductive adhesive. Wiping away a few square mm of the P3AT:PCBM film exposed a region of PEDOT:PSS/PEI film, to which we also made contact by attaching a copper wire with silver paint.
Computational modeling of strain
We modeled the distribution of strain needed for the conjugated polymer films to conform to the hemispherical substrates (Fig. 3) using finite element analysis static linear mechanical simulations. A representative 3D model of the experimental set-up (Fig. 1) was created in Autodesk Inventor 2014 software suite. We input the corresponding dimensions and mechanical properties of each component of the experimental system into the software, as follows. The end of the glass test tube was modeled as a solid glass hemisphere with the radius of 8 mm. A square PDMS membrane (50 mm × 50 mm × 1 mm) was sandwiched between two steel plates, each having a 30 mm circular aperture through the center. The contacts between the membrane and the plates were setup as bonded (no slipping). Translation of the glass hemisphere was constrained to the axis orthogonal to the plane of the membrane through the center of the circular apertures in the steel plates. The contact between the glass hemisphere and the PDMS membrane was setup as to have no slipping (in the experimental system, the conjugated polymer films were on the side of the PDMS membrane facing the glass hemisphere). These contact parameters generated simulated results that were most representative of our experimental data. Further, the orthogonal load of 0.75 N was applied to the flat surface of the hemisphere. The load resulted in the axial displacement of the hemisphere by 8 mm into (and deforming) the PDMS membrane. The simulation assumed that the PDMS substrate accommodated all of the deformation, and that the glass hemisphere remained rigid.Photovoltaic measurements
We performed all photovoltaic measurements in a nitrogen-filled glovebox using a solar simulator approximating the AM 1.5G spectrum with a flux of 100 mW cm−2 (ABET Technologies 11016-U up-facing unit calibrated with a reference cell with a KG5 filter).Acknowledgements
This work was supported by the Air Force Office of Scientific Research (AFOSR) Young Investigator Program, grant number FA9550-13-1-0156. Additional support was provided by laboratory startup funds from the University of California, San Diego. A. Z. acknowledges a fellowship from SoCal Clean Energy Technology Acceleration Program from the von Liebig Center at UCSD sponsored by the US Department of Energy. S. S. acknowledges a fellowship from the National Science Foundation Graduate Research Fellowship Program under Grant no. DGE-1144086. M. I. D. acknowledges support from the Calit2 High School Summer Scholars Program at UCSD. The authors thank Dr Ryan Chiechi and Thomas Voortman for measurements of molecular weight of the polymers.References
- M. Kaltenbrunner, M. S. White, E. D. Glowacki, T. Sekitani, T. Someya, N. S. Sariciftci and S. Bauer, Nat. Commun., 2012, 3, 770 CrossRef PubMed.
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS.
- D. Tahk, H. H. Lee and D. Y. Khang, Macromolecules, 2009, 42, 7079–7083 CrossRef CAS.
- B. O'Connor, E. P. Chan, C. Chan, B. R. Conrad, L. J. Richter, R. J. Kline, M. Heeney, I. McCulloch, C. L. Soles and D. M. DeLongchamp, ACS Nano, 2010, 4, 7538–7544 CrossRef PubMed.
- D. J. Lipomi, H. Chong, M. Vosgueritchian, J. G. Mei and Z. N. Bao, Sol. Energy Mater. Sol. Cells, 2012, 107, 355–365 CrossRef CAS.
- S. R. Dupont, M. Oliver, F. C. Krebs and R. H. Dauskardt, Sol. Energy Mater. Sol. Cells, 2012, 97, 171–175 CrossRef CAS.
- V. Brand, C. Bruner and R. H. Dauskardt, Sol. Energy Mater. Sol. Cells, 2012, 99, 182–189 CrossRef CAS.
- D. J. Lipomi, B. C.-K. Tee, M. Vosgueritchian and Z. N. Bao, Adv. Mater., 2011, 23, 1771–1775 CrossRef CAS PubMed.
- F. C. Krebs, M. Biancardo, B. Winther-Jensen, H. Spanggard and J. Alstrup, Sol. Energy Mater. Sol. Cells, 2006, 90, 1058–1067 CrossRef CAS.
- D. Ghezzi, M. R. Antognazza, R. Maccarone, S. Bellani, E. Lanzarini, N. Martino, M. Mete, G. Pertile, S. Bisti, G. Lanzani and F. Benfanati, Nat. Photonics, 2013, 7, 400–406 CrossRef CAS.
- S. Wagner and S. Bauer, MRS Bull., 2012, 37, 207–213 CrossRef.
- Z. Suo, MRS Bull., 2012, 37, 218–225 CrossRef CAS.
- T. Sekitani and T. Someya, Adv. Mater., 2010, 22, 2228–2246 CrossRef CAS PubMed.
- J. A. Rogers, T. Someya and Y. G. Huang, Science, 2010, 327, 1603–1607 CrossRef CAS PubMed.
- Y. G. Sun, W. M. Choi, H. Q. Jiang, Y. G. Y. Huang and J. A. Rogers, Nat. Nanotechnol., 2006, 1, 201–207 CrossRef CAS PubMed.
- J. Jones, S. P. Lacour, S. Wagner and Z. G. Suo, J. Vac. Sci. Technol., A, 2004, 22, 1723–1725 CAS.
- D. H. Kim, N. S. Lu, R. Ma, Y. S. Kim, R. H. Kim, S. D. Wang, J. Wu, S. M. Won, H. Tao, A. Islam, K. J. Yu, T. I. Kim, R. Chowdhury, M. Ying, L. H. Xu, M. Li, H. J. Chung, H. Keum, M. McCormick, P. Liu, Y. W. Zhang, F. G. Omenetto, Y. Huang, T. Coleman and J. A. Rogers, Science, 2011, 333, 838–843 CrossRef CAS PubMed.
- J. Lee, J. A. Wu, M. X. Shi, J. Yoon, S. I. Park, M. Li, Z. J. Liu, Y. G. Huang and J. A. Rogers, Adv. Mater., 2011, 23, 986–991 CrossRef CAS PubMed.
- I. M. Graz, D. P. J. Cotton and S. P. Lacour, Appl. Phys. Lett., 2009, 98, 071902 CrossRef.
- T. Sekitani and T. Someya, MRS Bull., 2012, 2012, 236–245 CrossRef.
- T. Sekitani, Y. Noguchi, K. Hata, T. Fukushima, T. Aida and T. Someya, Science, 2008, 321, 1468–1472 CrossRef CAS PubMed.
- D. J. Lipomi and Z. N. Bao, Energy Environ. Sci., 2011, 4, 3314–3328 CAS.
- C. Muller, S. Goffri, D. W. Breiby, J. W. Andreasen, H. D. Chanzy, R. A. J. Janssen, M. M. Nielsen, C. P. Radano, H. Sirringhaus, P. Smith and N. Stingelin-Stutzmann, Adv. Funct. Mater., 2007, 17, 2674–2679 CrossRef CAS.
- Z. B. Yu, X. F. Niu, Z. Liu and Q. B. Pei, Adv. Mater., 2011, 23, 3989–3994 CrossRef CAS PubMed.
- O. Awartani, B. Lemanski, H. W. Ro, L. J. Richter, D. M. DeLongchamp and B. T. O'Connor, Adv. Energy Mater., 2013, 3, 399–406 CrossRef CAS.
- Y. Cao, P. Smith and A. J. Heeger, Polymer, 1991, 32, 1210–1218 CrossRef CAS.
- S. Tokito, P. Smith and A. J. Heeger, Synth. Met., 1990, 36, 183–194 CrossRef CAS.
- J. Moulton and P. Smith, Polymer, 1992, 33, 2340–2347 CrossRef CAS.
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- F. C. Krebs, M. Jorgensen, K. Norrman, O. Hagemann, J. Alstrup, T. D. Nielsen, J. Fyenbo, K. Larsen and J. Kristensen, Sol. Energy Mater. Sol. Cells, 2009, 93, 422–441 CrossRef CAS.
- F. C. Krebs, T. D. Nielsen, J. Fyenbo, M. Wadstrom and M. S. Pedersen, Energy Environ. Sci., 2010, 3, 512–525 CAS.
- C. J. Bettinger and Z. N. Bao, Adv. Mater., 2010, 22, 651–655 CrossRef CAS PubMed.
- A. R. Postema, K. Liou, F. Wudl and P. Smith, Macromolecules, 1990, 23, 1842–1845 CrossRef CAS.
- D. J. Lipomi, M. Vosgueritchian, B. C.-K. Tee, C. H. Fox, J. A. Lee and Z. N. Bao, Nat. Nanotechnol., 2011, 6, 788–792 CrossRef CAS PubMed.
- S. Savagatrup, A. S. Makaram, D. J. Burke and D. J. Lipomi, Adv. Funct. Mater., 2013 DOI:10.1002/adfm.201302646.
- J. T. Seitz, J. Appl. Polym. Sci., 1993, 49, 1331–1351 CrossRef CAS.
- L. E. Nielsen and R. R. Landel, Mechanical Properties of Polymers and Composites, CRC Press, New York, 1994 Search PubMed.
- R. D. McCullough, Adv. Mater., 1998, 10, 93–116 CrossRef CAS.
- H. C. Ko, M. P. Stoykovich, J. Z. Song, V. Malyarchuk, W. M. Choi, C. J. Yu, J. B. Geddes, J. L. Xiao, S. D. Wang, Y. G. Huang and J. A. Rogers, Nature, 2008, 454, 748–753 CrossRef CAS PubMed.
- M. A. Meitl, Z. T. Zhu, V. Kumar, K. J. Lee, X. Feng, Y. Y. Huang, I. Adesida, R. G. Nuzzo and J. A. Rogers, Nat. Mater., 2006, 5, 33–38 CrossRef CAS.
- Y. H. Zhou, C. Fuentes-Hernandez, J. W. Shim, J. Meyer, A. J. Giordano, H. Li, P. Winget, T. Papadopoulos, H. Cheun, J. Kim, M. Fenoll, A. Dindar, W. Haske, E. Najafabadi, T. M. Khan, H. Sojoudi, S. Barlow, S. Graham, J. L. Bredas, S. R. Marder, A. Kahn and B. Kippelen, Science, 2012, 336, 327–332 CrossRef CAS PubMed.
- D. Gupta, M. M. Wienk and R. A. J. Janssen, Adv. Energy Mater., 2013, 3, 782 CrossRef CAS.
- N. S. Lu, X. Wang, Z. G. Suo and J. Vlassek, Appl. Phys. Lett., 2007, 91, 221909 CrossRef.
- D. J. Lipomi, J. A. Lee, M. Vosgueritchian, B. C.-K. Tee, J. A. Bolander and Z. N. Bao, Chem. Mater., 2012, 24, 373–382 CrossRef CAS.
- A. Babel and S. A. Jenekhe, Synth. Met., 2005, 148, 169–173 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |