 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanostructure of an ionic liquid–glycerol mixture†
Thomas
Murphy
a,
Robert
Hayes
a,
Silvia
Imberti
b,
Gregory G.
Warr
c and
Rob
Atkin
*a
aDiscipline of Chemistry, The University of Newcastle, Callaghan, NSW 2308, Australia. E-mail: rob.atkin@newcastle.edu.au
bSTFC, Rutherford Appleton Laboratory, Didcot, UK
cSchool of Chemistry, The University of Sydney, NSW 2006, Australia
First published on 20th May 2014
Abstract
The nanostructure of a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 vol% mixture of glycerol and ethylammonium formate (EAF), a protic ionic liquid (IL), has been investigated using neutron diffraction and empirical potential structure refinement (EPSR) fits. EPSR fits reveal that the mixture is nanostructured. Electrostatic interactions between IL charge groups leads to the formation of ionic regions. These solvophobically repel cation alkyl groups which cluster together to form apolar domains. The polar glycerol molecules are preferentially incorporated into the charged domains, and form hydrogen bonds with EAF groups rather than with other glycerol molecules. However, radial distribution functions reveal that glycerol molecules pack around each other in a fashion similar to that found in pure glycerol. This suggests that a glycerol channel runs through the ionic domain of EAF. The absence of significant glycerol–glycerol hydrogen bonding indicates that glycerol molecules are able to span the polar domain, bridging EAF charge groups. Glycerol can adopt six distinct conformations. The distribution of conformers in the EAF mixture is very different to that found in the pure liquid because hydrogen bonds form with EAF rather than with other glycerol molecules, which imparts different packing constraints.
:50 vol% mixture of glycerol and ethylammonium formate (EAF), a protic ionic liquid (IL), has been investigated using neutron diffraction and empirical potential structure refinement (EPSR) fits. EPSR fits reveal that the mixture is nanostructured. Electrostatic interactions between IL charge groups leads to the formation of ionic regions. These solvophobically repel cation alkyl groups which cluster together to form apolar domains. The polar glycerol molecules are preferentially incorporated into the charged domains, and form hydrogen bonds with EAF groups rather than with other glycerol molecules. However, radial distribution functions reveal that glycerol molecules pack around each other in a fashion similar to that found in pure glycerol. This suggests that a glycerol channel runs through the ionic domain of EAF. The absence of significant glycerol–glycerol hydrogen bonding indicates that glycerol molecules are able to span the polar domain, bridging EAF charge groups. Glycerol can adopt six distinct conformations. The distribution of conformers in the EAF mixture is very different to that found in the pure liquid because hydrogen bonds form with EAF rather than with other glycerol molecules, which imparts different packing constraints.
Introduction
ILs are solvents composed entirely of ions, with melting points below 100 °C.1 ILs often boast an array of desirable physiochemical properties, including high electrochemical stability,2 high thermal stability,3 low vapour pressure2,4,5 and the ability to dissolve both organic and inorganic substances.6 ILs are ‘designer solvents’ because their properties can be tuned via alteration of their chemical structures.4,7,8Many ILs are nanostructured, meaning that the bulk liquid is structurally inhomogeneous.9–11 IL nanostructure usually arises due to the solvophobic segregation of cation alkyl chains.9,10,12–14 IL nanostructure was first predicted by molecular dynamics (MD) simulations15–17 and later confirmed by neutron10 and X-ray diffraction.9 Over the past several years the nanostructure of a suite of ILs has been elucidated,10,12–14 enabling the molecular origins of nanostructure to be discerned, and structure–property relationships to be explained.
IL nanostructure has attracted considerable research attention. It influences solvent strength,18–20 rheological properties,21–23 interfacial structure,8,24–27 IL–solid interface tribological properties,28–32 the stability of nanoparticle dispersions in ILs33,34 and the ability of the IL to support amphiphilic self-assembly.18,19 IL nanostructure can be used to template the synthesis of structured materials.35,36 As many IL applications require the addition of solutes and co-solvents, understanding the interactions and structure of solvent–IL mixtures is thus critical; solvents can induce changes in nanostructure.19,37
The structure and interactions in IL–solvent mixtures has been probed using molecular dynamic simulations,20,38–40 dielectric spectroscopy,41–43 FT-IR,38,44,45 optical Kerr effect spectroscopy,40,46 and X-ray19,47,48 and neutron diffraction.37,49 Solutes added to ILs are solvated by the domain for which they have greatest affinity; non-polar solutes are solvated by the uncharged alkyl domains17,19 while polar solutes are solvated by the ionic regions.17,20,37,39 At low concentrations solutes only effect the IL nanostructure weakly, but at higher concentrations solutes can have a marked effect on the IL native bulk structure,17,20 leading to one of two outcomes. The solute and the IL can mix homogeneously,50,51 resulting in a smooth transition between the properties of the IL and solute, or structural heterogeneity can be retained up to high concentration,37,52 with a step change in properties when the nanostructure is broken resulting in a homogeneous mixture.
Studies of IL–solvent mixtures have focused largely on aprotic (especially imidazolium) ILs.20,38,40,42,47,53 Comparatively few studies have examined protic ionic liquid mixtures19,37,41 despite applications including as enzyme stabilisation agents54,55 or for electrolytes in hydrogen fuel cells.56,57 Previously, the structure of a 1:6 mole fraction ethylammonium nitrate (EAN)–water mixture was examined using neutron diffraction coupled with EPSR simulations.37 Pure EAN has a sponge-like nanostructure12 with alkyl and ionic groups segregated into bicontinuous domains throughout the bulk liquid. When water is added, it is incorporated into EAN's polar domains and interacts with the charge groups to transform the structure into a locally cylindrical mesh morphology.37 Spatial density functions reveal that the packing of water molecules around each other in the mixture is similar to pure water, and that packing of EAN ions in the mixture is similar to pure EAN. This revealed that the water and IL formed distinct domains in the liquid, with a well-defined interface between them consisting of the nonpolar ethyl groups on one side, and ammonium cation, nitrate anion, and water on the other side.37
Glycerol has long been used as a cryoprotective agent.58 When added to water, glycerol prevents the formation of percolating hydrogen bond networks, hindering the formation of large crystallites which would otherwise damage cryopreserved tissues.59 The liquid structure of pure glycerol, and its mixtures with water, has been elucidated using neutron diffraction and EPSR simulations by Towey et al.59–61 Neat glycerol adopts one of six distinct molecular conformations, 83% of which are in the “αβ” conformation. Addition of water to glycerol at lower concentrations (0.25 mol fraction) has little effect on the interactions between glycerol molecules.63 However, when glycerol is the minor component in water (0.05 mol fraction glycerol), both the preferred glycerol conformation, and the conformer distribution, change. This is attributed to incorporation of glycerol into water's hydrogen bonding network in the most energy efficient fashion.61
In this work we examine how glycerol affects protic IL nanostructure. Like water, glycerol has extensive hydrogen bonding capacity, but glycerol has a much larger molecular volume and significant conformational freedom. As the combination of EAN and glycerol has the potential to form explosive mixtures, ethylammonium formate (EAF) is used in place of EAN.
Experimental
A series of 5 chemically identical, but isotopically different samples at the same EAF:Gly concentration (50:50 vol:vol%) were prepared: H-EAF/H-glycerol (CH3CH2NH3HCO2/HOCH2CH(HO)CH2OH), d3-EAF/d3-glycerol (CH3CH2ND3 HCO2/DOCH2CH(DO)CH2OD), H-EAF/d5-glycerol (CH3CH2NH3HCO2/HOCD2CD(HO)CD2OH), d3-EAF/d8-glycerol (CH3CH2ND3HCO2/DOCD2CD(DO)CD2OD) and d8-EAF/d8-glycerol (CD3CD2ND3HCO2/DOCD2CD(DO)CD2OD). Selective deuteration enables the contribution of different correlations to the structure factor function, S(q), to be highlighted via contrast variation.
H-EAF was prepared via the drop-wise addition of formic acid (HCOOH) (Sigma-Aldrich, 95 w/w%) to a chilled solution (<5 °C) of ethylamine (Sigma-Aldrich 70 wt%) and distilled water. Excess water was removed firstly by rotor evaporation for several hours at 25 °C and then under high vacuum conditions. The final water content of the IL was determined to be <0.5% by Karl-Fischer titration.
d 3-EAF was synthesised by performing the acid–base reaction in deuterium oxide (D2O (99% Sigma Aldrich)). 1H-NMR experiments reveal that, on average, 2.5 out of 3 amino hydrogen atoms are replaced with deuterium. d5-EAF and d8-EAF were synthesised using 1,1,1,2,2-d5-ethylamine (CD3CD2NH2) (CDN isotopes) via the above procedures for H-EAF and d3-EAF respectively. Protiated and deuterated samples of anhydrous glycerol (Sigma-Aldrich) were used without additional purification.
Neutron diffraction measurements were performed on the SANDALS instrument at the ISIS research facility, Rutherford Appleton Laboratories, UK. The neutron wavelength range is 0.05–4.95 Å, and data was collected over the Q range 0.1 to 50 Å−1.
Samples were contained in chemically inert, null scattering Ti0.68Zr0.32 flat plate cans sealed with PTFE O-rings during the neutron diffraction experiment. The can dimensions are 35 × 35 mm2 with a 1 mm path length and 1 mm wall thicknesses of known atom density, 0.0541 atoms Å−3. Prior to loading, diffraction measurements were made on the empty cans, empty instrument and a vanadium standard sample for data correction and normalization.
Diffraction experiments were conducted at 298 K under vacuum. The sample chamber was left to equilibrate for 10 min prior to measurements and the temperature was maintained at 25 ± 0.1 °C by a Julabo FP50 temperature controller. The combined weight of the can and sample for each contrast was measured before and after the diffraction measurement to ensure evaporation had not occurred in the vacuum chamber. The net run time for each system was at least 8 h.
Data analysis was carried out using GUDRUN, as described in the ATLAS manual.64 This performed corrections including normalisation to the incident flux, absorption and multiple scattering corrections, Ti–Zr can subtraction and normalisation to absolute units by dividing the measured differential cross section by the scattering of a vanadium standard of known thickness. Corrections for single atom scattering and hydrogen inelasticity were also applied. Fitting to the normalised diffraction data was conducted using empirical potential structure refinement (EPSR). The optimized three-dimensional geometries of both EAF and glycerol have been determined previously. An EPSR model was developed consisting of 500 anions, 500 cations and 613 glycerol molecules in a simulation box, consistent with the measured diffraction data, and specified physical constraints including molecular structure, sample composition, atomic overlaps, and liquid density. Atomic and molecular translations and rotations were governed by the standard rules for a Monte Carlo simulation with respect to a reference potential of Lennard-Jones plus Coulombic interactions. The potential was refined via an iterative algorithm until convergence between the simulated and experimental structure factors was reached. All five EAF/glycerol contrasts were modelled simultaneously, normalising for isotope populations.
Results and discussion
Neutron diffraction spectra were obtained for five isotopomeric 50:50 vol% mixtures of EAF and glycerol, and EPSR simulations performed to fit the diffraction data. The structures of EAF and glycerol, and the labels used to identify specific atoms, are shown in Fig. 1. The Lennard-Jones parameters for EAF and glycerol60 used in the fitting routine have been published previously.
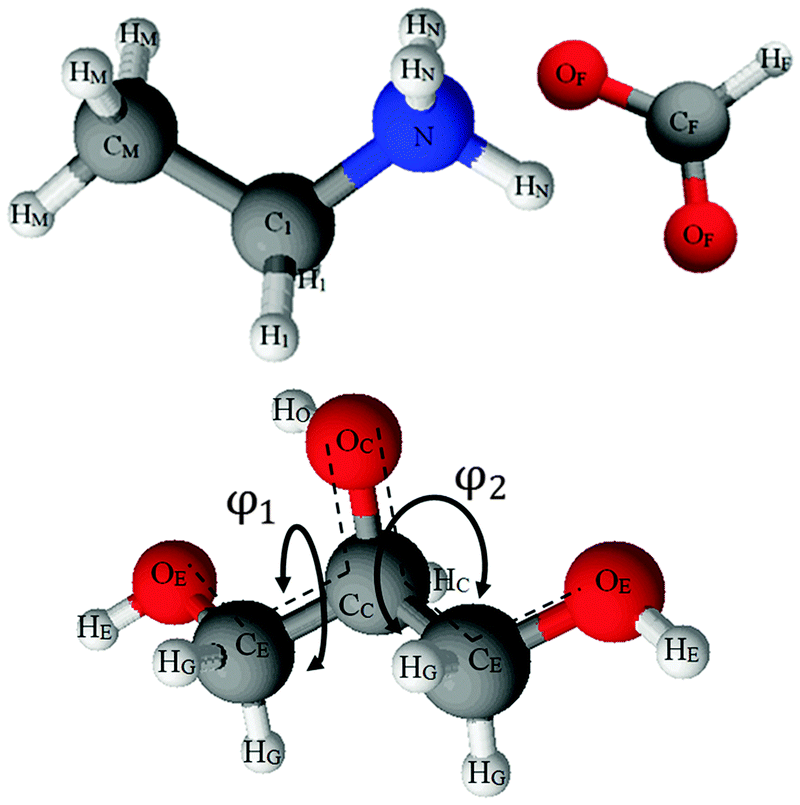 | ||
| Fig. 1 Structure of ethylammonium formate (top) and glycerol (bottom). The atoms; carbon (black) oxygen (red) nitrogen (blue) and hydrogen (white) are labelled with the notations used in the EPSR simulation and throughout this paper. The conformation of glycerol is defined by a set of two dihedral angles, φ1 and φ2. φ1 is defined by the angle between the planes containing the bonds OE–CE–CC and CE–CC–CE while φ2 is defined by the angle between the planes containing the bonds CE–CC–CE and CC–CE–OE. | ||
The experimental diffraction data and EPSR fits for the five 50:50 vol% EAF–glycerol contrasts examined are shown in Fig. 2. Unequivocally assigning diffraction peaks in ILs is often problematic because multiple atom–atom correlations contribute to the structure factor function, S(q).65 However, for neutron based experiments contrast variation enables extraction of detailed structural information.66 For the systems studied here, the isotopically pure contrasts, H-EAF + H-glycerol and d8-EAF + d8-glycerol, produce dissimilar spectra because scattering lengths of H and D are different. Systematic substitution of hydrogen for deuterium in ethylammonium and glycerol emphasise the contribution of different correlation lengths to S(q). The d3-EAF + d3-glycerol contrast emphasises correlations between exchangeable hydrogens in the mixture, the H-EAF + d5-glycerol contrast highlights correlations between glycerol alkyl chains, and the d3-EAF + d8-glycerol contrast probes interactions between glycerol molecules and the ionic regions of EAF. In EPSR, all of these spectra are fit simultaneously using the same model of the liquid structure. This provides confidence that the model is correct.
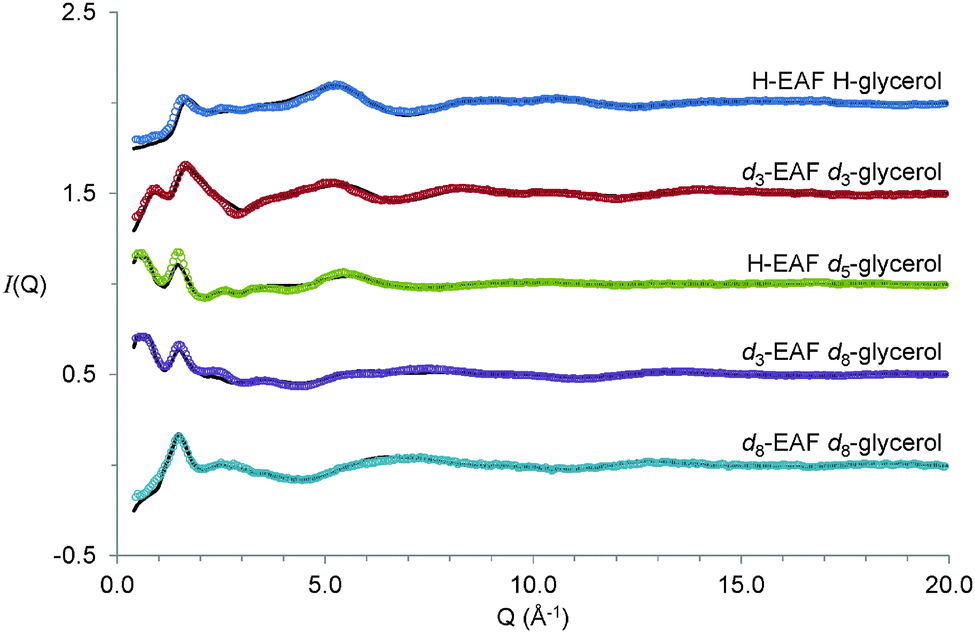 | ||
| Fig. 2 Experimental (open circles) and EPSR fitted (solid lines) scattering intensity as a function of Q (Å−1) for 50:50 v.v.% EAF–glycerol isotopomer mixtures at 298 K. Data are offset for clarity. | ||
A striking feature of the diffraction spectra in Fig. 2 is the presence of low-Q peaks. These occur at 0.6 Å−1 in both H-EAF + d5-glycerol and d3-EAF + d8-glycerol and at ∼0.85 Å−1 for d3-EAF + d3-glycerol. These peak positions correspond to repeat spacings of 10.5 Å and ∼7.4 Å respectively. Additionally, all five spectra feature a peak at ∼1.5 ± 0.1 Å−1, consistent with a repeat spacing of ∼4 Å. The molecular dimensions of EAF and glycerol (from the pure liquid densities67) are 5.0 Å and 5.3 Å, respectively. While the 4 Å could be due to short range ion–ion, or ion–glycerol correlations, the larger 10.5 Å and 7.4 Å distances clearly indicate the presence of larger scale nanostructure in this mixture.
In pure ILs, low Q peaks in both neutron diffraction and X-ray scattering spectra11–13,68–71 indicate intermediate-range order resulting from the bulk IL self-assembled nanostructure.9–13,70 Partial structure factor analysis72 and molecular dynamic simulations73–75 have elucidated the origin of the first peak in aprotic and protic ILs. In pure EAF the first peak is at 0.625 Å−1, corresponding to a repeat spacing of 10.1 Å,76 slightly less than the 10.5 Å spacing found in the EAF–glycerol mixture. As such, the larger repeat spacing in the mixture is broadly consistent with glycerol swelling the pre-existing EAF sponge structure, rather than the EAF nanostructure being broken to form a homogenous mixture, which would result in the low Q peak disappearing. However, from the scattering data alone it is not possible to tell how glycerol and EAF pack together to produce a self-assembled structure. To resolve these issues, EPSR fits to the diffraction data have been performed, and are shown as solid lines in Fig. 2. Excellent agreement between the data and fits is obtained across the entire angular (Q) range. The very slight deviations between the data and fit at low Q is attributed to residual inelastic scattering.
Snapshots of the front face of the equilibrated EPSR simulation box corresponding to the data fits are shown in Fig. 3. When all atoms are shown, discerning the self-assembled nanostructure is difficult because of the system complexity (Fig. 3A). To reveal the nanostructure more clearly, in Fig. 3B–D only the atoms of the glycerol molecules, ethylammonium cations and formate anions (respectively) are shown, with the other atoms rendered invisible. Fig. 3C shows that the alkyl groups of the ethylammonium cations are aggregated together; the grey carbon atoms orient towards each other with the blue ammonium groups facing away. The arrangement of formate anions in Fig. 3D corresponds to the cation charge group positions, (Fig. 3C) consistent with the formation of polar domains. In Fig. 3B, voids between glycerol molecules correspond to the space occupied by EAF.
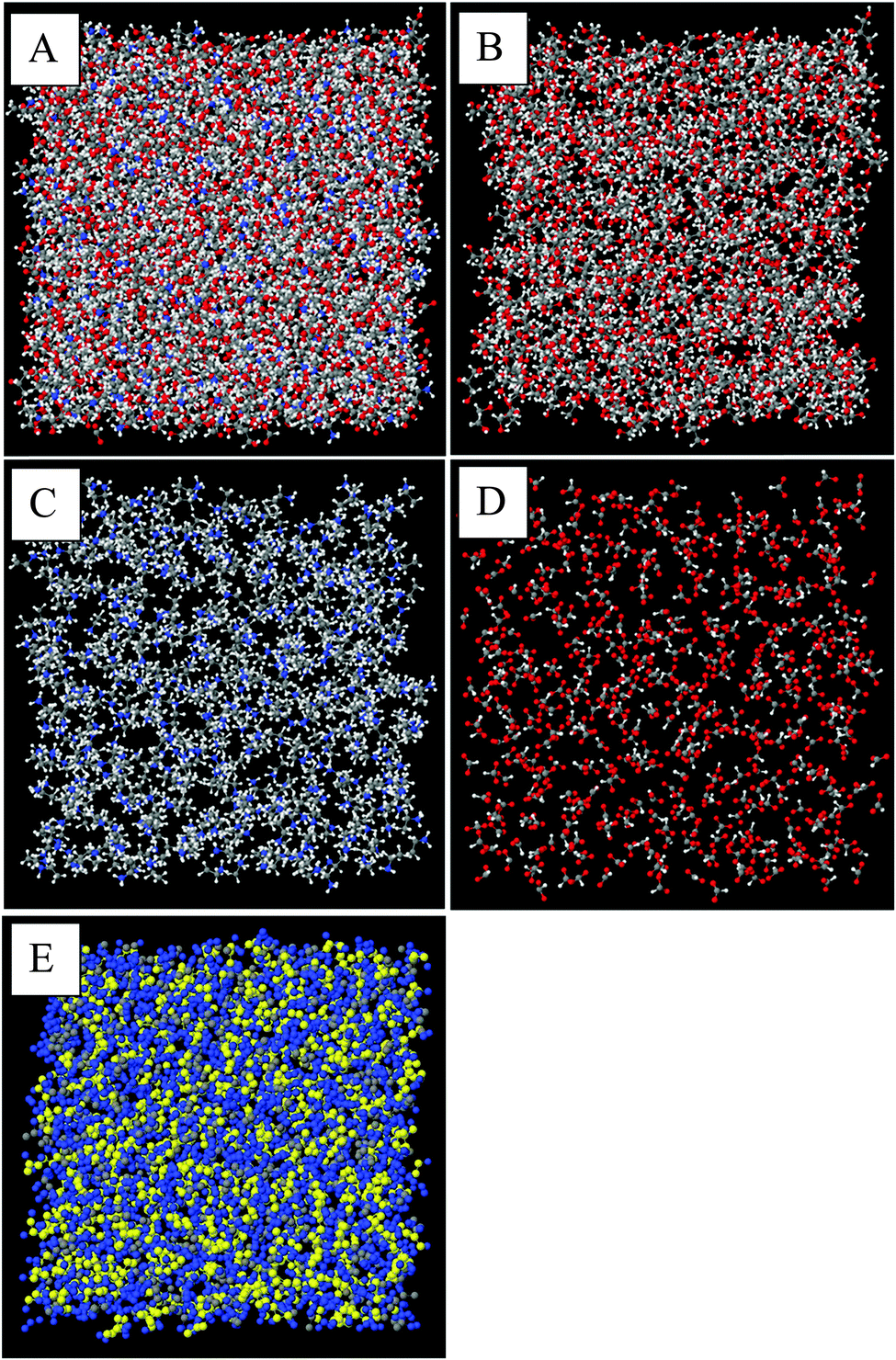 | ||
| Fig. 3 Snapshots of the fitted bulk structure of a 50:50 vol% EAF–glycerol mixture equilibrated at 300 K. In A–E, carbon atoms are shown in grey, hydrogen in white, oxygen in red and nitrogen in blue. (A) Shows all atoms, (B) glycerol molecules only (C) ethylammonium cations only (D) formate anions only. In figure (E) the position of the glycerol carbon atoms are highlighted. Glycerol carbons are coloured yellow, cation alkyl chain chains are grey, polar groups (cation nitrogens, anions, and glycerol alcohols) are blue, and hydrogen atoms are invisible. | ||
The alkyl chain clustering noted in Fig. 3C suggests that solvophobic self-assembly occurs in the EAF–glycerol mixture, as per pure EAF, to create an apolar domain. The remainder of the liquid volume consists of EAF charged groups and the glycerol molecules, forming a polar domain, cf.Fig. 3E. The periodic order in these well-defined regions produces the low Q (large correlation length) peaks in the diffraction data. In the mixture, the volume fraction ratio of the polar domain to the apolar domain, based on liquid densities,67 is 0.73:0.27 whereas in pure EAF, it is 0.44:0.56. Simply swelling the polar domains of a bicontinuous nanostructure with a sponge-like morphology would shift the low Q peak down by the swelling ratio to around 0.4 Å−1, or a repeat spacing of 16–17 Å. Accommodating this marked increase in the polar domain volume and the small change in primary peak position observed requires a change in liquid nanostructure so that the “interface” between the two domains becomes more curved.37 The manner in which the ions arrange around each other to facilitate this change is elucidated using radial distribution functions, gij(r), and spatial density function (SDF) plots determined from the EPSR simulation box.
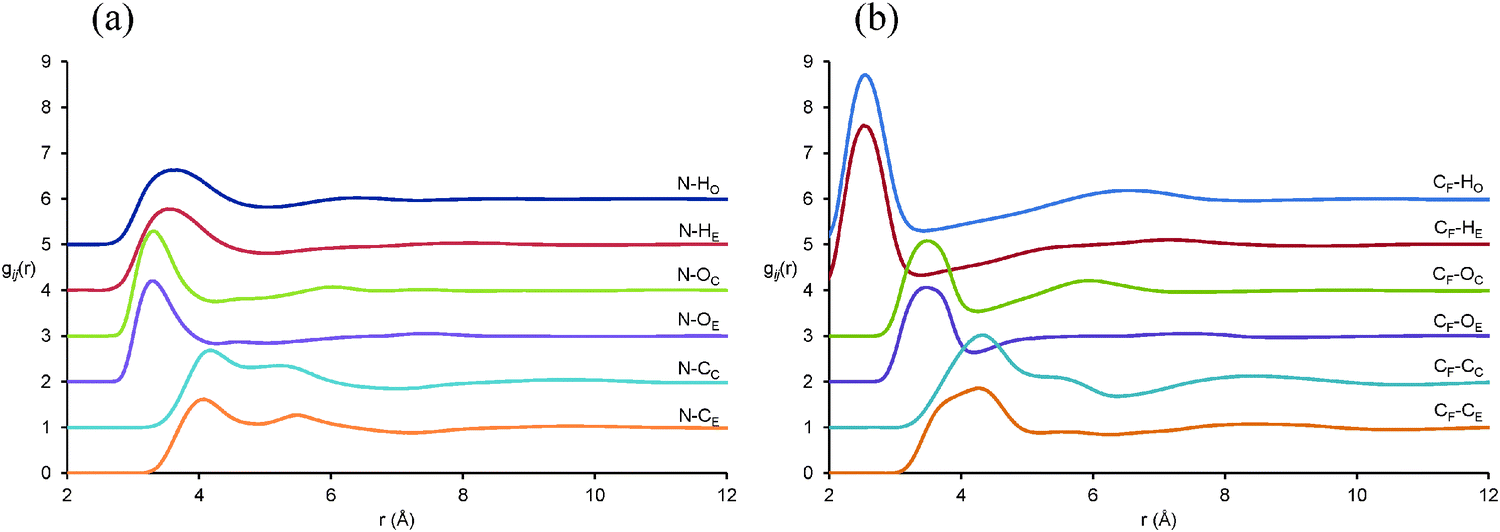
Pair correlation functions, gij(r), reveal the positions of atom–atom pairs as a function of their radial separation. The first peaks in the gij(r) functions correspond to the first coordination shell of nearest neighbours. gij(r) functions for key atom–atom correlations between EAF ions in the EAF–glycerol mixture (solid lines) are presented in Fig. 4. The corresponding data for pure EAF (dotted lines) obtained previously76 is shown for comparison. In the mixture, the first peaks for the cation–anion (N⋯CF), alkyl chain–alkyl chain (CM⋯CM) and anion–anion (CF⋯CF) correlations are remarkably similar to those found in pure EAF.76 This suggests similar arrangements of these ions in the first coordination shell in the pure IL and the mixture; The N⋯CFgij(r) data is similar because strong electrostatic interactions between the cation and anion are retained when glycerol is present and the CM⋯CM is nearly identical because solvophobic segregation of alkyl groups occurs in both systems.76
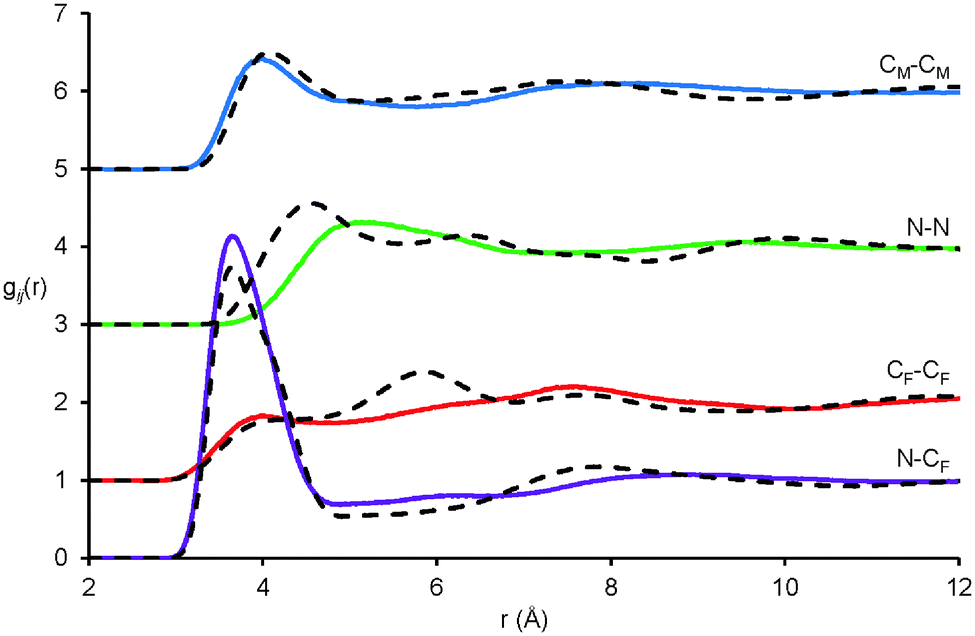 | ||
| Fig. 4 Key EPSR-derived atom–atom pair correlations, gij(r), for EAF ions in 50:50 v.v.% EAF–glycerol. Solid lines represent data for the EAF–glycerol mixture and dotted lines data previously reported for pure EAF.76 | ||
The N⋯N gij(r) is markedly different in the mixture compared to the pure IL. In pure EAF, the N⋯N probability begins to increase at 3.5 Å and reaches a peak at 4.6 Å followed by additional oscillations at larger separations. In the mixture, the probability does not begin to increase until 3.8 Å, the (much broader) peak is not reached until 5.2 Å, and subsequent oscillations are weak or absent. This data shows that in the mixture the cation charge centres are further apart, and occupy less well-defined positions. As the pair correlation functions for other IL groups are largely unchanged in the first coordination shell, this must mean that (parts of) the glycerol molecules occupy the space between the cation charge centres and effectively push them apart. This will increase the curvature of the self-assembled nanostructure in the mixture compared to pure EAF, allowing the polar volume to increase without forcing the nonpolar domains further apart and shifting the long-range correlation peak. The decrease in the intensity of the 2nd peak (2nd coordination shell) for the CF⋯CF data is also consistent with increased curvature, as anions interacting electrostatically with the “next” cation are further away than in the pure liquid. A similar change in the N⋯N gij(r) was noted when water was added to EAN.37
Spatial density function (SDF) plots provide a 3D reconstruction of the gij(r) functions, thereby revealing the most probable spatial arrangements of atoms. Selected SDF plots for EAF in the EAF–glycerol mixture are given in Fig. 5, together with the corresponding plots for pure EAF. There is little change in the CM@CM spatial distribution when EAF is mixed with glycerol (Fig. 5A and D), consistent with the gij(r) data, confirming solvophobic segregation of EA+ alkyl chains. The other SDF plots have been selected because they indicate significant differences in the local arrangements of EAF ions upon mixing with glycerol.
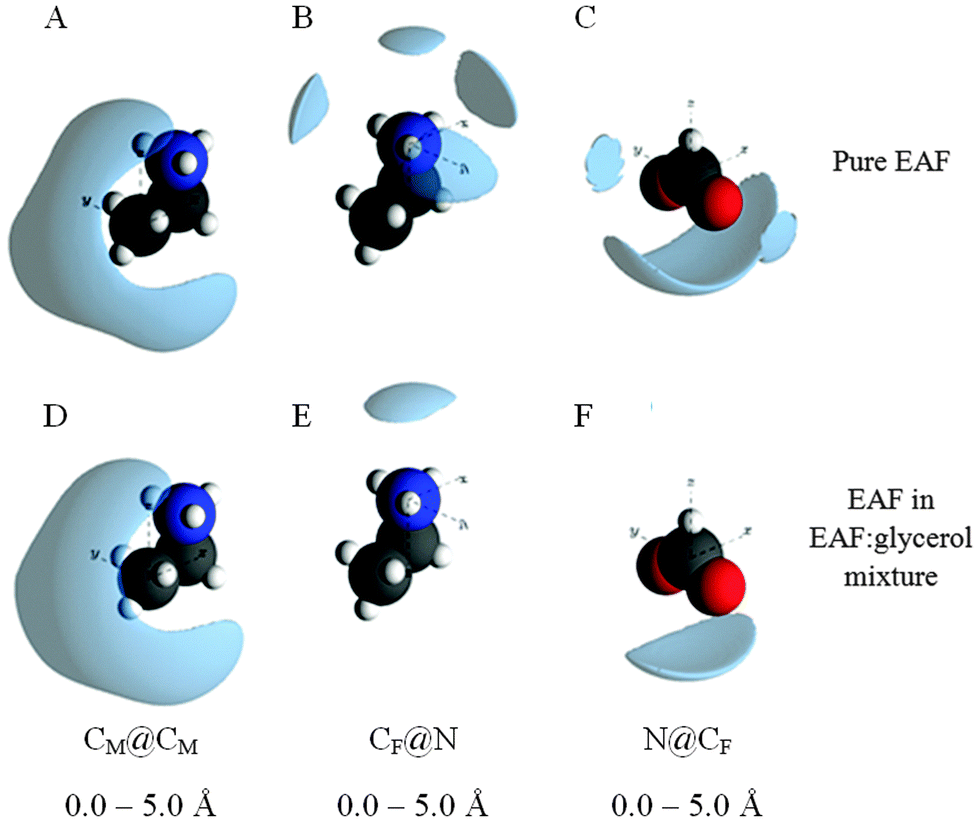 | ||
| Fig. 5 EPSR sdf reconstructions of the gij(r) data for key atom–atom correlations between EAF ions. The lobes represent the 20% probability isosurfaces for the orientation of a selected atom around a central atom. Sdf plots for pure EAF were sourced from ref. 76. | ||
The most likely positions of the formate carbon in the first co-ordination shell around the cation charge centre is shown by the CF@N sdf plot. In pure EAF the formate carbon occupies one of 4 positions around and above the cation charge; formate anions around (but not above) the cation charge group are shared with the next cation along in the locally planar structure. In the mixture over the same distance only one lobe is present, above the cation charge. The lateral (surrounding) probability lobes are lost because neighbouring cations are further away. For the same reason, the lateral probability lobes for the cation charge groups around the formate carbon in the N@CF SDF plot are not present in the mixture.
Interactions between glycerol and EAF are elucidated by the gij(r) functions between glycerol atoms and the (a) cation and (b) anion charge centres shown in Fig. 6. For all of these gij(r) functions little difference is noted in the data for central (OC, HO, CC) and distal (OE, HE, CE) glycerol atoms of the same type (refer Fig. 1), i.e. the N⋯HO data is very similar to the N–HE data, etc. Note that the position of the first peak in the N⋯CC and N⋯CE data are both at shorter distances than the peak for cation charge centre to cation charge centre (N–N). This is consistent with glycerol molecules being interdigitated between cation charge groups but not penetrating into the apolar domain.
 | ||
| Fig. 6 Key EPSR-derived atom–atom pair correlations, gij(r) data, for (a) cation charge centre (N)–glycerol molecules and (b) anion charge centre (CF)–glycerol molecules. | ||
For glycerol–cation interactions, peaks in the N⋯O data occur at the shortest distances, followed by the N⋯H then N⋯C peaks. For glycerol–formate interactions, C⋯H peaks are at the smallest separations, followed by C⋯O then C⋯C. Together, these data sets suggest that glycerol alcohol groups are strongly associated with the charge groups of EAF. The glycerol alcohol oxygen has the capacity to accept hydrogen bonds from cation ammonium hydrogens, and glycerol alcohol hydrogens can hydrogen bond with the oxygen of the formate anion. Packing constraints then dictate that the glycerol carbon is farthest from the cation and anion charge centres.
This correlation data suggests that hydrogen bonding between the glycerol and EAF is critical for understanding the structure of the mixture. Table 1 presents the average hydrogen bond lengths and angles for the various hydrogen bond types in the mixture, along with the associated co-ordination numbers; the coordination number is the number of atoms of a given type within the first coordination sphere, which is defined by a distance that captures the first peak in the gij(r) data. Fig. A in the ESI† shows the complete triplet-bond angle distributions for EAF–glycerol mixtures compared to pure EAF.
| Hydrogen bond | Length (Å) | Angle (°) | Coord. number (β/Å) | Classification | |
|---|---|---|---|---|---|
| EAF–EAF | HN–OF | 2.43 (2.43) | 109 (109) | 1.53 ± 1.39 (3.25) | Long/bent |
| EAF–Gly | HN–OE | 2.61 | 109 | 1.34 ± 0.96 (3.60) | Long/bent |
| HN–OC | 2.55 | 109 | 0.63 ± 0.69 (3.60) | Long/bent | |
| HE–OF | 1.65 | 175 | 0.87 ± 0.81 (2.30) | Short/straight | |
| HO–OF | 1.65 | 175 | 0.43 ± 0.60 (2.30) | Short/straight | |
| Gly–Gly | HE–OE | 1.83 (1.80) | 175 (175) | 0.16 ± 0.37 (2.30) | Short/straight |
| HE–OC | 1.83 (1.80) | 175 (175) | 0.08 ± 0.26 (2.30) | Short/straight | |
| HO–OE | 1.83 (1.80) | 175 (175) | 0.12 ± 0.33 (2.30) | Short/straight | |
| HO–OC | 1.83 (1.80) | 175 (175) | 0.08 ± 0.27 (2.30) | Short/straight | |
Form the coordination numbers in Table 1, EAF–glycerol hydrogen bonds occur most frequently in the mixture. When multiple hydrogen bonds of the same types are accounted for (e.g. there are two OE atoms for each glycerol), there are 11 glycerol–EAF hydrogen bonds and 6.8 EAF–EAF hydrogen bonds for each glycerol–glycerol hydrogen bond.
In pure EAF,14 hydrogen bonds are long (>2 Å) and predominately bent (<165°).77 The situation is the same for EAF–EAF hydrogen bonds in the EAF glycerol mixture, but the coordination number is reduced substantially from 2.6714 to 1.53. In pure glycerol, hydrogen bonds are short, straight and simple,62 meaning that the coordination numbers of alcohol hydrogens and oxygens with each other are near 1. In the glycerol–EAF mixture, hydrogen bonds remain short and straight, but the co-ordination numbers are drastically reduced to ∼0.1, meaning that each glycerol alcohol hydrogen has on average only 0.1 coordinated glycerol alcohol oxygens, and vice versa. That is, in the mixture, glycerol–glycerol hydrogen bonds are almost absent, and EAF–EAF hydrogen bonding is ∼50% reduced. Both have been replaced by glycerol–EAF hydrogen bonds, consistent with the gij(r) functions, cf.Table 1.
There are two main types of hydrogen bonding between the EAF and the glycerol. Glycerol can act as a hydrogen bond acceptor (to the alcohol oxygen) and form a hydrogen bond with the cation ammonium hydrogen (HN⋯OE and HN⋯OC). Alternatively, glycerol can donate a hydrogen bond from its alcohol hydrogens to the anion oxygen (HE⋯OF and HO⋯OF). When glycerol acts an acceptor, the resulting hydrogen bond is long and bent, similar to those formed in pure EAF, but when glycerol acts as a donor, the hydrogen bond is strong and straight, like in pure glycerol. This is likely because solvophobic segregation means that the position of the cation in the nanostructure is more constrained than the anion. As such, the anion is free to move to a position where a strong straight hydrogen bond can form with glycerol, but the (relative) immobility of the cation results in a bent, weak hydrogen bond. The coordination number associated with the weak, bent hydrogen bonds is 1.97 (1.34 + 0.63) and with the strong straight bonds is 1.30 (0.87 + 0.43).
Finally, we turn to the spatial arrangement and conformation of glycerol in the mixture. Fig. 7 shows key glycerol–glycerol gij(r) distributions in the mixture, along with those reported previously for the pure liquid.60 The glycerol C⋯C correlations in the glycerol EAF mixture are extremely similar to those for pure glycerol. This means that, relative to each other, the glycerol carbon atoms occupy spatially similar positions in the mixture and the pure liquid, i.e. the carbon atoms are arranged in a similar fashion. However, the first O⋯O correlation peaks have significantly reduced intensities in the mixture compared to the pure liquid. This is because most of the glycerol–glycerol hydrogen bonds have been replaced by glycerol–EAF hydrogen bonds, cf.Table 1.
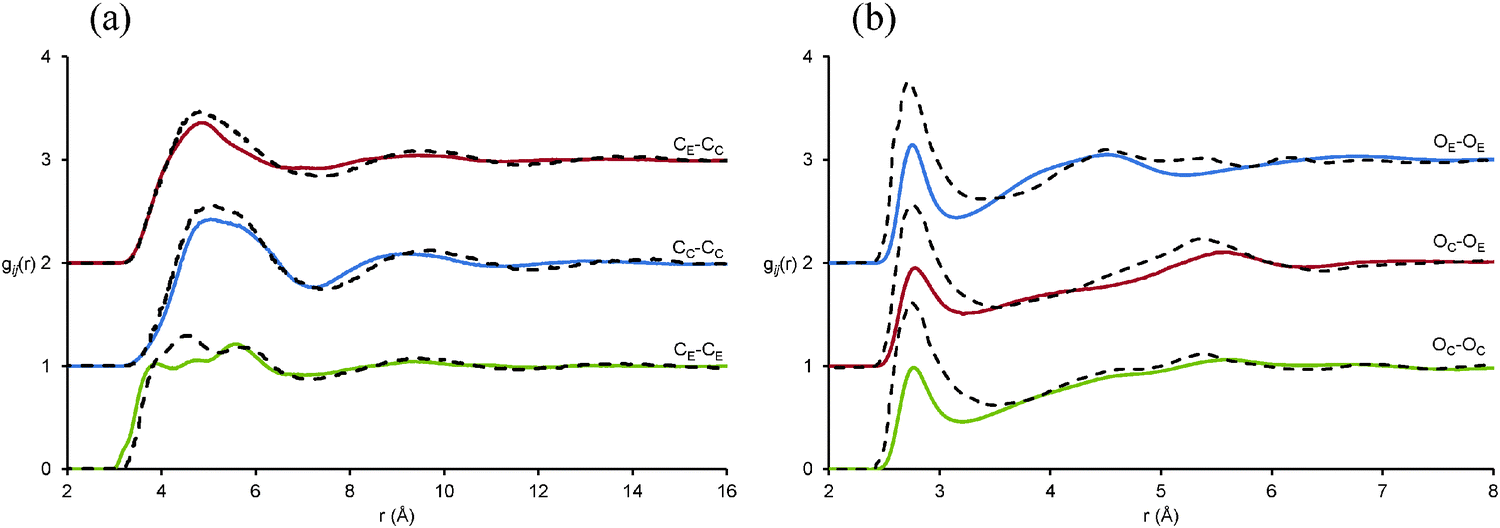 | ||
| Fig. 7 Key EPSR-derived atom–atom pair correlations, gij(r) data, for (a) glycerol C–C correlations and (b) O–O correlations in a 50:50.vol% EAF/glycerol mixture. Solid lines represent data for the EAF–glycerol mixture and dotted lines data previously reported for pure glycerol.60 | ||
The picture emerging is that glycerol carbon atoms are packed together in a fashion similar to that in pure glycerol, but that glycerol alcohol groups preferentially interact with EAF charged groups resulting in conformations different to the pure liquid (expanded upon below). Conceptually, this can be visualised as a glycerol channel, structurally reminiscent of pure glycerol in terms of the carbon packing within the polar domain, as shown in Fig. 3E. The absence of glycerol–glycerol hydrogen bonds means that the glycerol molecule bridges from one side of the polar domain to the other. This is markedly different to results obtained previously for pure water at the same volume fraction in EAN, where water molecules hydrogen bonded extensively.37
Glycerol molecules have significant conformational freedom. The conformers available to glycerol are conventionally described by two dihedral angles, φ1 and φ2 (see Fig. 1).78 These are commonly divided into three angular regions, α, β and γ, where α = 120 < φ < 140, β = 240 < φ < 360 and γ = 0 < φ < 120.79 Assignment of α, β or γ to the two dihedrals yields six possible conformers (heterogeneous pairs are equivalent i.e. αβ = βα). The conformer populations in pure glycerol determined by neutron diffraction and EPSR simulations60 and those calculated for the EAF–glycerol mixture are compared in Table 2.
In pure glycerol, the αβ conformation is by far the most populous, at 83.5%. In the EAF–glycerol mixture it accounts for only 24% of the observed conformations, while the population of every other conformer is increased. The largest increase is for the αα conformer, which makes up 30% of the conformers in the mixture but is not observed to occur in the pure liquid according to the EPSR fit to the neutron diffraction data (other methods have suggested it is the αα population which dominates in the pure glycerol at 70%80). The broader conformer distribution in the glycerol–EAF mixture compared to pure glycerol is a consequence of its preferential hydrogen bonding with EAF charged groups rather than with other glycerol molecules. The position of these hydrogen bonds in the mixture is more random than in pure glycerol, and a significant proportion of these hydrogen bonds are weak and bent, whereas in pure glycerol all of the hydrogen bonds are short and straight. This allows the oxygen atoms to occupy a wider range of positions in space in the mixture, making the conformer distribution more diverse.
Conclusions
Neutron diffraction measurements combined with EPSR fits have enabled the nanostructure of a glycerol–EAF mixture to be elucidated in detail. In the mixture, just as in pure EAF, cation alkyl chains are solvophobically segregated into apolar domains. EAF charged groups and glycerol molecules associate together to form polar domains; glycerol molecules are not appreciably incorporated into the apolar domain. The glycerol alcohol groups preferentially hydrogen bond with the EAF charged groups. This leaves few glycerol–glycerol hydrogen bonds in the mixture, and the number of EAF–EAF hydrogen bonds is also markedly reduced. Hydrogen bonding leads to the alcohol groups intruding between EAF cations, increasing the curvature of the self-assembled nanostructure. In pure glycerol, glycerol–glycerol hydrogen bonds are strong (short and straight) and one of the six available conformers is dominant. In the mixture, hydrogen bonds between the cation ammonium hydrogens to the glycerol oxygens are weak (long and bent) while those between the glycerol alcohol hydrogens and the formate oxygens are strong. This is attributed to the ability of the anion to move within the cations coordination shell (such that electrostatic interactions are retained) to a position where a strong hydrogen bond can form. By contrast, the position of the cation is relatively fixed by the requirement for alkyl chains to solvophobically associate, and weaker, bent hydrogen bonds result. A much broader range of glycerol conformers is found in the mixture compared to the pure liquid. This is because the position of the hydrogen bonding sites in the mixture is more diverse, and the fact that a large proportion of the hydrogen bonds are weak and thus relatively flexible. However, the packing of glycerol carbon atoms around each other is similar in the mixture and pure glycerol. This suggests that a glycerol channel, structurally reminiscent of pure glycerol in terms of the carbon packing, is present within the polar domain, and the absence of glycerol–glycerol hydrogen bonds means that the glycerol molecule bridges from one side of the polar domain to the other.Acknowledgements
TM thanks the University of Newcastle for a PhD stipend. RA thanks the ARC for a Future Fellowship. RH thanks AINSE for a PGRA. This research was supported by an ARC Discovery Project and an ISIS beamtime grant.References
- F. Endres and S. Z. El Abedin, Phys. Chem. Chem. Phys., 2006, 8, 2101 RSC.
- W. Lu and S. A. Tassou, Appl. Energy, 2012, 91, 366 CrossRef CAS PubMed.
- D. R. MacFarlane and K. R. Seddon, Aust. J. Chem., 2007, 60, 3 CrossRef CAS.
- M. J. Earle and K. R. Seddon, Pure Appl. Chem., 2000, 72, 1391 CrossRef CAS.
- C. A. Angell, N. Byrne and J.-P. Belieres, Acc. Chem. Res., 2007, 40, 1228 CrossRef CAS PubMed.
- M. Freemantle, Chem. Eng. News, 1998, 76, 32 Search PubMed.
- H. Weingaertner, Angew. Chem., Int. Ed., 2008, 47, 654 CrossRef CAS PubMed.
- R. Hayes, G. G. Warr and R. Atkin, Phys. Chem. Chem. Phys., 2010, 12, 1709 RSC.
- A. Triolo, O. Russina, H. J. Bleif and E. Di Cola, J. Phys. Chem. B, 2007, 111, 4641 CrossRef CAS PubMed.
- R. Atkin and G. G. Warr, J. Phys. Chem. B, 2008, 112, 4164 CrossRef CAS PubMed.
- T. L. Greaves, D. F. Kennedy, S. T. Mudie and C. J. Drummond, J. Phys. Chem. B, 2010, 114, 10022 CrossRef CAS PubMed.
- R. Hayes, S. Imberti, G. G. Warr and R. Atkin, Phys. Chem. Chem. Phys., 2011, 13, 3237 RSC.
- R. Hayes, S. Imberti, G. G. Warr and R. Atkin, Phys. Chem. Chem. Phys., 2011, 13, 13544 RSC.
- R. Hayes, S. Imberti, G. G. Warr and R. Atkin, Angew. Chem., Int. Ed., 2013, 52, 4623 CrossRef CAS PubMed.
- S. M. Urahata and M. C. C. Ribeiro, J. Chem. Phys., 2004, 120, 1855 CrossRef CAS PubMed.
- Y. T. Wang and G. A. Voth, J. Am. Chem. Soc., 2005, 127, 12192 CrossRef CAS PubMed.
- J. Lopes and A. A. H. Padua, J. Phys. Chem. B, 2006, 110, 3330 CrossRef PubMed.
- T. L. Greaves and C. J. Drummond, Chem. Soc. Rev., 2013, 42, 1096 RSC.
- T. L. Greaves, D. F. Kennedy, N. Kirby and C. J. Drummond, Phys. Chem. Chem. Phys., 2011, 13, 13501 RSC.
- A. A. H. Padua, M. F. Gomes and J. Lopes, Acc. Chem. Res., 2007, 40, 1087 CrossRef CAS PubMed.
- J. A. Smith, G. B. Webber, G. G. Warr and R. Atkin, J. Phys. Chem. B, 2013, 117, 13930 CrossRef CAS PubMed.
- G. L. Burrell, N. F. Dunlop and F. Separovic, Soft Matter, 2010, 6, 2080 RSC.
- A. Takada, K. Imaichi, T. Kagawa and Y. Takahashi, J. Phys. Chem. B, 2008, 112, 9660 CrossRef CAS PubMed.
- J. J. Segura, A. Elbourne, E. J. Wanless, G. G. Warr, K. Voitchovsky and R. Atkin, Phys. Chem. Chem. Phys., 2013, 15, 3320 RSC.
- D. Wakeham, A. Nelson, G. G. Warr and R. Atkin, Phys. Chem. Chem. Phys., 2011, 13, 20828 RSC.
- P. Niga, D. Wakeham, A. Nelson, G. G. Warr, M. Rutland and R. Atkin, Langmuir, 2010, 26, 8282 CrossRef CAS PubMed.
- H. Li, F. Endres and R. Atkin, Phys. Chem. Chem. Phys., 2013, 15, 14624 RSC.
- H. Li, M. W. Rutland and R. Atkin, Phys. Chem. Chem. Phys., 2013, 15, 14616 RSC.
- H. Li, R. J. Wood, M. W. Rutland and R. Atkin, Chem. Commun., 2014, 50, 4368 RSC.
- R. A. Asencio, E. D. Cranston, R. Atkin and M. W. Rutland, Langmuir, 2012, 28, 9967 CrossRef CAS PubMed.
- O. Werzer, E. D. Cranston, G. G. Warr, R. Atkin and M. W. Rutland, Phys. Chem. Chem. Phys., 2012, 14, 5147 RSC.
- J. Sweeney, F. Hausen, R. Hayes, G. B. Webber, F. Endres, M. W. Rutland, R. Bennewitz and R. Atkin, Phys. Rev. Lett., 2012, 109, 155502 CrossRef.
- K. Ueno and M. Watanabe, Langmuir, 2011, 27, 9105 CrossRef CAS PubMed.
- J. A. Smith, O. Werzer, G. B. Webber, G. G. Warr and R. Atkin, J. Phys. Chem. Lett., 2010, 1, 64 CrossRef CAS.
- Z. F. Chen, T. L. Greaves, R. A. Caruso and C. J. Drummond, J. Mater. Chem., 2012, 22, 10069 RSC.
- M. Antonietti, D. B. Kuang, B. Smarsly and Z. Yong, Angew. Chem., Int. Ed., 2004, 43, 4988 CrossRef CAS PubMed.
- R. Hayes, S. Imberti, G. G. Warr and R. Atkin, Angew. Chem., Int. Ed., 2012, 51, 7468 CrossRef CAS PubMed.
- C. Roth, A. Appelhagen, N. Jobst and R. Ludwig, ChemPhysChem, 2012, 13, 1708 CrossRef CAS PubMed.
- W. Jiang, Y. T. Wang and G. A. Voth, J. Phys. Chem. B, 2007, 111, 4812 CrossRef CAS PubMed.
- F. Bardak, D. Xiao, L. G. Hines, P. Son, R. A. Bartsch, E. L. Quitevis, P. Yang and G. A. Voth, ChemPhysChem, 2012, 13, 1687 CrossRef CAS PubMed.
- M. A. Neouze, J. Mater. Chem., 2010, 20, 9593 RSC.
- J. Dupont and J. D. Scholten, Chem. Soc. Rev., 2010, 39, 1780 RSC.
- M. Bester-Rogac, A. Stoppa, J. Hunger, G. Hefter and R. Buchner, Phys. Chem. Chem. Phys., 2011, 13, 17588 RSC.
- L. Cammarata, S. G. Kazarian, P. A. Salter and T. Welton, Phys. Chem. Chem. Phys., 2001, 3, 5192 RSC.
- A. Pal, B. Kumar and T. S. Kang, Fluid Phase Equilib., 2013, 358, 241 CrossRef CAS PubMed.
- A. L. Sturlaugson, K. S. Fruchey and M. D. Fayer, J. Phys. Chem. B, 2012, 116, 1777 CrossRef CAS PubMed.
- Y. Imai, H. Abe and Y. Yoshimura, J. Phys. Chem. B, 2009, 113, 2013 CrossRef CAS PubMed.
- T. L. Greaves, D. F. Kennedy, A. Weerawardena, N. M. K. Tse, N. Kirby and C. J. Drummond, J. Phys. Chem. B, 2011, 115, 2055 CrossRef CAS PubMed.
- T. Shimomura, K. Fujii and T. Takamuku, Phys. Chem. Chem. Phys., 2010, 12, 12316 RSC.
- M. Y. Lui, L. Crowhurst, J. P. Hallett, P. A. Hunt, H. Niedermeyer and T. Welton, Chem. Sci., 2011, 2, 1491 RSC.
- J. M. Andanson, M. J. Beier and A. Baiker, J. Phys. Chem. Lett., 2011, 2, 2959 CrossRef CAS.
- R. Katoh, M. Hara and S. Tsuzuki, J. Phys. Chem. B, 2008, 112, 15426 CrossRef CAS PubMed.
- H. Niedermeyer, J. P. Hallett, I. J. Villar-Garcia, P. A. Hunt and T. Welton, Chem. Soc. Rev., 2012, 41, 7780 RSC.
- N. Byrne and C. A. Angell, J. Mol. Biol., 2008, 378, 707 CrossRef CAS PubMed.
- J. P. Mann, A. McCluskey and R. Atkin, Green Chem., 2009, 11, 785 RSC.
- H. Nakamoto and M. Watanabe, Chem. Commun., 2007, 2539 RSC.
- S. Y. Lee, A. Ogawa, M. Kanno, H. Nakamoto, T. Yasuda and M. Watanabe, J. Am. Chem. Soc., 2010, 132, 9764 CrossRef CAS PubMed.
- J. Nordstrom, L. Aguilera and A. Matic, Langmuir, 2012, 28, 4080 CrossRef PubMed.
- D. B. Williams, M. E. Stoll, B. L. Scott, D. A. Costa and W. J. Oldham, Chem. Commun., 2005, 1438 CAS.
- Y. Z. Su, J. W. Yan, M. G. Li, Z. X. Xie, B. W. Mao and Z. Q. Tian, Z. Phys. Chem., 2012, 226, 979 CrossRef CAS.
- J. J. Towey and L. Dougan, J. Phys. Chem. B, 2012, 116, 1633 CrossRef CAS PubMed.
- J. J. Towey, A. K. Soper and L. Dougan, Phys. Chem. Chem. Phys., 2011, 13, 9397 RSC.
- J. J. Towey, A. K. Soper and L. Dougan, J. Phys. Chem. B, 2011, 115, 7799 CrossRef CAS PubMed.
- A. K. Soper, W. S. Howells and A. C. Hannon, ATLAS-Analysis of Time-of-Flight Diffraction Data from Liquid and Amorphous Samples RAL-89-046, 1989 Search PubMed.
- H. V. R. Annapureddy, H. K. Kashyap, P. M. De Biase and C. J. Margulis, J. Phys. Chem. B, 2010, 114, 16838 CrossRef CAS PubMed.
- B. Hammouda, S. Krueger and C. J. Glinka, J. Res. Natl. Inst. Stand. Technol., 1993, 98, 31 CrossRef CAS.
- R. Hayes, N. Borisenko, B. Corr, G. B. Webber, F. Endres and R. Atkin, Chem. Commun., 2012, 48, 10246 RSC.
- O. Russina, A. Triolo, L. Gontrani and R. Caminiti, J. Phys. Chem. Lett., 2012, 3, 27 CrossRef CAS.
- O. Russina and A. Triolo, Faraday Discuss., 2012, 154, 97 RSC.
- C. Hardacre, J. D. Holbrey, C. L. Mullan, T. G. A. Youngs and D. T. Bowron, J. Chem. Phys., 2010, 133, 7 CrossRef PubMed.
- M. Macchiagodena, L. Gontrani, F. Ramondo, A. Triolo and R. Caminiti, J. Chem. Phys., 2011, 134, 15 CrossRef PubMed.
- H. K. Kashyap, J. J. Hettige, H. V. R. Annapureddy and C. J. Margulis, Chem. Commun., 2012, 48, 5103 RSC.
- H. K. Kashyap, C. S. Santos, N. S. Murthy, J. J. Hettige, K. Kerr, S. Ramati, J. Gwon, M. Gohdo, S. I. Lall-Ramnarine, J. F. Wishart, C. J. Margulis and E. W. Castner, J. Phys. Chem. B, 2013, 117, 15328 CrossRef CAS PubMed.
- K. Shimizu, C. E. S. Bernardes and J. N. C. Lopes, J. Phys. Chem. B, 2014, 118, 567 CrossRef CAS PubMed.
- A. A. Freitas, K. Shimizu and J. N. Canongia Lopes, J. Chem. Eng. Data, 2014 DOI:10.1021/je500197x.
- R. Hayes, S. Imberti, G. G. Warr and R. Atkin, J. Phys. Chem. C, 2014 Search PubMed , submitted.
- J. Kroon and J. A. Kanters, Nature, 1974, 248, 667 CrossRef CAS.
- O. Bastiansen, Acta Chem. Scand., 1949, 3, 415 CrossRef CAS PubMed.
- O. Bastiansen, H. Borgiel and E. Saluste, Acta Chem. Scand., 1949, 3, 415 CrossRef CAS PubMed.
- R. Chelli, F. L. Gervasio, C. Gellini, P. Procacci, G. Cardini and V. Schettino, J. Phys. Chem. A, 2000, 104, 5351 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp01570c |
| This journal is © the Owner Societies 2014 |