 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent-induced conformational changes in cyclic peptides: a vibrational circular dichroism study†
Christian
Merten
*ab,
Fee
Li
b,
Kenny
Bravo-Rodriguez
c,
Elsa
Sanchez-Garcia
c,
Yunjie
Xu
*a and
Wolfram
Sander
b
aDepartment of Chemistry, University of Alberta, Edmonton, Alberta, T6G2G2, Canada. E-mail: yunjie.xu@ualberta.ca
bRuhr-Universität Bochum, Fakultät für Chemie und Biochemie, 44801 Bochum, Germany. E-mail: christian.merten@ruhr-uni-bochum.de
cMax-Planck-Institut für Kohlenforschung, 45470 Mülheim an der Ruhr, Germany
First published on 11th February 2014
Abstract
The three-dimensional structure of a peptide is strongly influenced by its solvent environment. In the present study, we study three cyclic tetrapeptides which serve as model peptides for β-turns. They are of the general structure cyclo(Boc-Cys-Pro-X-Cys-OMe) with the amino acid X being either glycine (1), or L- or D-leucine (L- or D-2). Using vibrational circular dichroism (VCD) spectroscopy, we confirm previous NMR results which showed that D-2 adopts predominantly a βII turn structure in apolar and polar solvents. Our results for L-2 indicate a preference for a βI structure over βII. With increasing solvent polarity, the preference for 1 is shifted from βII towards βI. This conformational change goes along with the breaking of an intramolecular hydrogen bond which stabilizes the βII conformation. Instead, a hydrogen bond with a solvent molecule can stabilize the βI turn conformation.
Introduction
Besides periodic secondary structures like α-helices and β-sheets, turns are the most common structural motifs in peptides and as such they establish the directional reversal in peptide strands. Most attention in studies on turns has been paid to the tight β-turns which link the strands of antiparallel β-sheet structures. They are also exposed to the environment of peptides, i.e. the solvent, ions, and substrates. β-Turns consist of four amino acid residues linked by three amide bonds and show a high structural flexibility. Initially, β-turns were assumed to form a hydrogen bond between the carbonyl group of the first (position i) and the amide N–H of the fourth amino acid residue (position i + 3).1 However, it was later shown that β-turns without this hydrogen bond exist as well.2 Nowadays, eight different tight β-turns are distinguished and characterized mainly by the dihedral angles adopted by the amino acid residues i + 1 and i + 2.3 The amino acid sequences and its peptidic and non-peptidic environments determine which β-turn conformation is adopted under certain conditions.The most common experimental techniques used to study peptide conformations are infrared and Raman spectroscopy,3 electronic circular dichroism (ECD) and two-dimensional NMR spectroscopy. The chiroptical versions of infrared and Raman spectroscopy, vibrational circular dichroism (VCD) spectroscopy and Raman optical activity (ROA), which measure vibrational optical activity (VOA),4 have also become well-known and powerful techniques to study peptides, proteins5–8 and carbohydrates9 as well as other chiral molecules in solution.10–17
Small peptides can serve as structural model systems for examining the influence of the peptide environment on the conformations of turns. They can easily be synthesized and studied in much greater detail using spectroscopic and theoretical methods. In many cases, small peptides have also been used to establish structure–spectra relationships. For instance, the CD spectra of pure βI and βII turns have been determined by Perczel et al. in their studies on cyclic hexapeptides18 confirming the theoretical predictions by Woody.19,20 Diem et al. used cyclic tri-, tetra-, and pentapeptides to establish the VCD spectral pattern of the amide I vibrations of β-turns.21–23
Recently, we introduced the model peptide cyclo(Boc-Cys-Pro-Gly-Cys-OMe) 1 (Fig. 1),24 a cyclic disulfide-bridged tetrapeptide of which the two derivatives cyclo(Cys-Pro-Gly-Cys)23 (1a) and cyclo(Boc-Cys-Pro-Gly-Cys-NHMe)25 (1b) have been studied before. For the unprotected form 1a of the tetrapeptide, Diem et al. assumed a mixture of βI and βII turn structures in DMSO. This assignment was based on a comparison of the amide I VCD pattern with the ones measured for cyclo(Cys-Pro-Phe-Cys) and cyclo(Cys-Pro-D-Phe-Cys) in DMSO and DMSO/CDBr3, which contain mainly either βI or βII turn structures, respectively. The cyclo(Boc-Cys-Pro-Gly-Cys-NHMe) derivative 1b, on the other hand, was assigned to a βI structure based on the lack of experimental evidence for nuclear Overhauser effects (NOE) between the Pro-CαH and Gly-NH.
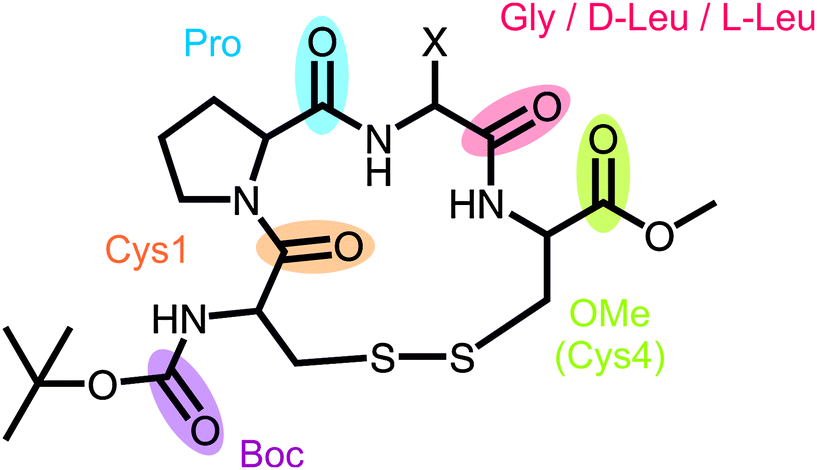 | ||
| Fig. 1 General structure of the investigated cyclic tetrapeptides with X = H for 1 and X = iso-butyl for D- and L-2. | ||
In contrast to these previously studied derivatives, 1 is highly soluble in non-polar chloroform as well as in polar solvents such as acetonitrile and DMSO. This allowed us to investigate the effect of solvents and temperature on the turn conformation with different techniques such as 2D-NMR, IR, and CD spectroscopy, as well as Car–Parrinello molecular dynamics.24 It has been shown that 1 adopts a βII structure in solution and in the solid state and that a hydrogen bond between Cys1-C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and Cys4-NH stabilizes the turn conformation of the cyclic peptide. While Cys1-CO forms another hydrogen bond with the NH-bond of Gly of the neighboring molecule in solid state, in solution, however, the carbonyl group competes with solvent molecules for the hydrogen bonding donor.
O and Cys4-NH stabilizes the turn conformation of the cyclic peptide. While Cys1-CO forms another hydrogen bond with the NH-bond of Gly of the neighboring molecule in solid state, in solution, however, the carbonyl group competes with solvent molecules for the hydrogen bonding donor.
The present work complements this initial investigation by applying VCD spectroscopy to investigate the preferred conformations of the cyclic peptide 1 in solvents of different polarities, i.e. chloroform, acetonitrile and DMSO. Additionally, two related peptides (L-2 and D-2) with L- or D-leucine instead of glycine are studied here. Unlike NMR and ECD, VCD spectroscopy can be used to compare the peptides in all three solvents. For instance, the peaks of some NH protons could not be resolved in the reported NMR spectra of 1 in chloroform. The UV and ECD bands of the peptides were also masked by bands from chloroform or DMSO.
In this paper, the VCD analysis of the three tetrapeptides is carried out first without making any assumptions based on the previous studies (except the crystal structure of 1). Afterwards, the obtained results are compared to the NMR experiments and molecular dynamics simulations. Our VCD analysis thus provides new evidence to establish more conclusively the β-turn structures of 1 and 2 in acetonitrile and the role of the solvent on the conformation of the peptides.
Experimental and computational details
The synthesis procedures of 1, L-2, and D-2 were reported elsewhere.24,26 Deuterated solvents were purchased from Sigma-Aldrich and Cambridge Isotopes.The IR and VCD spectra of 1, L-2, and D-2 were measured at a resolution of 4 cm−1 in the fingerprint region (1800–1100 cm−1) using a Bruker Vertex 70 FTIR spectrometer equipped with a PMA50 module for polarization modulated measurements. The photoelastic modulator was set to an optimum working frequency of 1500 cm−1. The spectra were recorded in the solvents CDCl3, CD3CN, and DMSO-d6 under otherwise identical conditions, i.e. a concentration of 45 mg ml−1 (∼0.1 mM), a path length of 100 μm, and an accumulation time of 4 hours (∼25k scans). Background-correction by subtracting solvent spectra has been carried out for the IR spectra while the VCD spectra are shown as obtained.
Geometry optimizations and spectra calculations were performed at the B3LYP/6-311++G(2d,p) level of theory using the Gaussian 09 (Rev. C.01) software package.27 Dispersion-correction has been taken into account at the DFT-D level using the iop(3/124 = 3) setting; however, the correction did not significantly affect the calculated VCD pattern in the amide I and amide II regions. Implicit solvation effects have been accounted for by using the polarizable continuum model (IEFPCM) for chloroform and acetonitrile.28–30 The normal mode frequencies obtained were scaled by a factor of 0.98 for a better visual comparison with the experimental spectra. Line broadening has been taken into account by assigning a Lorentzian band shape with a half-width at half-height of 8 cm−1 to the calculated dipole and rotational strengths.
The dynamical behaviour of 1 in acetonitrile was investigated by Replica Exchange Molecular Dynamics (REMD) simulations carried out for 60 ns using the Gromacs4.6 code31–33 and the OPLSA force field.34 The temperature range was 290–400 K and the temperature distribution was established according to the Patriksson and van der Spoel approach.35 Although originally intended for water, this approach allowed for a good exchange ratio between replicas in acetonitrile too (see the ESI† for replica exchange statistics). The reported crystal structure of 124 was used as the starting point for the simulations. For the treatment of electrostatic and van der Waals interactions the PME method36 and a twin range cut-off37 were used, respectively, with cut-off distances of 1.2 nm in both cases. The exchange of replicas was attempted every 1 ps. The time step for all simulations was 2 fs. Bonds containing hydrogen atoms were kept fixed using the LINCS algorithm.38 The cluster analysis of the results was performed using the Gromos method.39
Results and discussion
Experimental IR and VCD spectra
The fingerprint region of the infrared spectrum of a peptide can be divided into the carbonyl/amide I region (1800–1600 cm−1), the amide II region (1600–1480 cm−1) which is characterized by N–H bending and C–N stretching vibrations, and the region below 1480 cm−1 which contains the amide III region (1350–1250 cm−1) and arises from complex peptide backbone as well as side group vibrations. For the present study, we focus on the amide I and amide II regions since they are very characteristic for the hydrogen bonding network of the peptide, and comment only briefly on the region below 1480 cm−1.Fig. 2 compares the experimental IR and VCD spectra of 1, L-2, and D-2 measured in the three different solvents CDCl3, CD3CN, and DMSO-d6. Direct comparisons for the IR spectra of 1 and D-2 are provided in the supporting information (Fig. S1, ESI†). For the spectra of 1, it has been ensured that the spectral pattern did not show any concentration dependence. For all three peptides, distinct changes in the amide I and II regions of the IR spectra can be observed when the solvent is changed from non-polar chloroform to either of the polar solvents. The band assignments discussed below are summarized in Table 1.
 | ||
| Fig. 2 Experimental VA and VCD spectra of 1 (top), D-2 (middle), and L-2 (bottom) in CDCl3, CD3CN, and DMSO-d6. | ||
| 1 (X = Gly) | D-2 (X = D-Leu) | L-2 (X = L-Leu) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| A | B | C | A | B | C | A | B | C | |
| Ester CO |
1751 | 1753 | 1747 | 1749 | 1753 | 1749 | 1745 | 1753 | 1751 |
| Boc CO |
1703 | 1711 | 1703 | 1706 | 1711 | 1703 | 1703 | 1713 | 1701 |
| Amide I X | 1691 | 1689 | 1682 | 1686 | |||||
| Amide I Pro | 1682 | 1680 | 1672 | 1678 | 1670 | 1666 | 1672 | 1676 | 1668 |
| Amide I Cys1 | 1630 | 1639 | 1637 | 1628 | 1637 | 1637 | 1649 | 1641 | 1637 |
| Amide II | 1548 | 1537 | 1539 | 1529 | 1535 | 1544 | 1524 | 1534 | 1534 |
| 1512 | 1515 | 1520 | 1505 | 1516 | 1520 | 1514 | 1519 | ||
| 1496 | 1491 | 1498 | 1494 | 1496 | |||||
First, we examine the amide I regions of the IR spectra of the peptides and the solvent-induced changes. The carbonyl vibration of the methyl ester group of Cys(4) appears to be unaffected by the different solvents and is found to be at 1750 cm−1 in all spectra. The carbonyl vibration of Cys(1) observed at 1627 cm−1 in chloroform-d1 is blue-shifted by about 10 cm−1 in the polar solvents and is slightly narrower. The most prominent changes occur in the region 1750–1650 cm−1. In the spectrum of 1 in chloroform-d1, this region is dominated by a broad band with an intensity maximum at 1680 cm−1 and shoulders at the higher wavenumber side. In acetonitrile-d3, a sharp feature at 1712 cm−1 separates from the broad band which is still found to be centred at 1680 cm−1 (1703 and 1672 cm−1 in DMSO-d6). The amide I region of L-2 behaves quite similarly. For peptide D-2, an intense band with two maxima at 1678 and 1691 cm−1 and a shoulder at the higher wavenumber side is observed in chloroform which splits into three bands found at 1670, 1689, and 1711 cm−1 in the spectra measured in acetonitrile-d3 (1666, 1682, and 1703 cm−1 in DMSO-d6). From higher to lower wavenumbers, these three bands are assigned to the carbonyl vibration of the Boc-group, Pro, and the amino acid Gly respectively D/L-Leu.
Second, we consider the VCD spectra of the amide I region of the three peptides. The experimental spectra taken in chloroform feature a strong positive band for the carbonyl vibration of the methyl ester group of Cys(4) which becomes weaker in polar solvents. For the Cys(1) carbonyl vibration, a strong positive VCD is observed for 1 and D-2 which shows as two closely spaced positive bands in chloroform. For L-2, the intensity of the VCD of Cys(1) in chloroform is much weaker and becomes stronger in polar solvents. The VCD pattern in the 1750–1650 cm−1 region is rather complex. The VCD band assigned to the carbonyl vibration of the variable amino acid residue, Gly or Leu, is in all cases negative. However, for 1 the negative band at 1678 cm−1 features a small negative shoulder at 1669 cm−1. For the carbonyl vibration of the Boc-group, a weak negative VCD is observed, while the VCD band of the carbonyl vibration of Pro can only be clearly identified as the positive band in the spectra of L-2.
For the amide I region, it can be concluded that the IR and VCD spectra do not show clear evidence for significant conformational changes due to solute–solvent interactions. Furthermore, a direct comparison with the amide I VCD spectra reported by Diem et al. for the unprotected version 1a is difficult because of the unknown influence of the Boc-group on the amide I vibrations.
Therefore, we now turn to the amide II region which features three vibrational modes assigned to the Cys(4), the variable amino acid residue Gly respectively D/L-Leu, and Cys(1). In all sets of IR spectra, the amide II vibrations are found to blend into one very broad band. However, a clear blue-shift of the bands can be observed when going from CDCl3 over CD3CN towards DMSO-d6. The corresponding VCD spectra of the amide II vibrations show very significant changes upon substitution of the solvent. The region features a dominant −/+/+ pattern in all spectra of peptide D-2, and a +/− pattern for L-2. The spectral pattern of peptide 1, however, clearly changes from −/+/+ in chloroform-d1 to +/−/+ in the two polar solvents. This observation is a strong indicator for a solvent-induced conformational change of the tetrapeptide's cyclic backbone which is only possible for 1 but not for D- or L-2.
Below 1480 cm−1, no significant changes can be observed in the IR spectra. In light of the above mentioned spectral changes of 1, it is noteworthy that the amide III region of 1 is similar to that of L-2. The VCD pattern found that the 1385–1330 cm−1 region of the chloroform measurements of 1 and 2 compared to the lack of VCD bands in polar solvents could carry further interesting structural information which, however, we will not address any further in this study.
Our experimental spectra thus suggest that the conformational equilibria in D- and L-2 are much less sensitive to solvent-induced conformational changes than in 1. By contrast, we can clearly establish that the conformational preferences of 1 very much depend on the solvent environment.
Conformational analysis
In order to further interpret the experimental VCD spectra, density functional theory (DFT) based spectra calculations were carried out.40–42 Since VCD spectroscopy is very sensitive to conformational changes, a thorough conformational analysis of 1 and subsequent spectra calculations were performed.The starting point for the analysis was the crystal structure of 1,24 which has been optimized at the B3LYP-D/6-311++G(2d,p) level of theory in the gas phase. The obtained optimized geometry, denoted r1–c1, is shown in Fig. 3. As the comparison of the ϕ and ψ angles in Table 2 shows, a βII turn structure is adopted in the solid state and in the optimized geometry. The hydrogen bond between Cys(1)-CO and Cys(4)-NH which has been found in the crystal structure is also found in the optimized geometry. Additionally, the gas phase optimized structure features a hydrogen bond between Cys(1)-CO and Gly-NH.
 | ||
| Fig. 3 Optimized geometry of 1 in the gas phase (r1–c1). The arrows indicate conformational changes in the side group conformers c2 to c8. | ||
| Structure | ϕ (i + 1) | ψ (i + 1) | ϕ (i + 2) | ψ (i + 2) |
|---|---|---|---|---|
| βI ideal | −60 | −30 | −90 | 0 |
| βII ideal | −60 | 120 | 80 | 0 |
| 1 crystal | −62.1 | 135.6 | 60.4 | 29.4 |
| βII r1–c1 (g(−)g(−)t) | −77.7 | 103.0 | 78.5 | 9.6 |
| βII r7–c1 (g(−)g(+)g(+)) | −75.6 | 105.4 | 113.1 | −16.8 |
| βII r6–c1 (tg(+)g(+)) | −83.3 | 81.3 | 125.9 | −6.7 |
| βI r2–c1 (g(−)g(−)t) | −85.7 | −6.7 | −116.5 | 26.6 |
| βI r4–c1 (tg(+)g(+)) | −83.1 | −3.8 | −107.2 | 10.0 |
For the first step of the conformational analysis, the ring conformation was kept essentially unchanged, while the relative conformations of the terminating Boc and OMe ester groups as well as the proline ring were modified. This procedure yielded a total of eight different side group conformations (denoted c1 to c8, shown in Fig. S3 of the ESI†). In all but conformers c1 and c2, the hydrogen bond between Cys(1)-CO and Gly-NH was found to be cleaved.
Raman measurements of 1 and D-2 indicate that the S–S bridge adopts different conformations (cf. ESI†). The conformation of disulfide bridges are typically described by the three torsional angles of the Cα–C–S–S–C–Cα unit as gauche–gauche–gauche (ggg), trans–gauche–gauche (tgg or ggt), and trans–gauche–trans (tgt). In its crystal structure and in the optimized gas phase structure (Fig. 3), the tetrapeptide 1 adopts a g(−)g(−)t conformation. For 1 with a βII turn structure and the side groups adopting conformation c1, additional ring conformations with g(+)g(−)t, g(+)g(−)g(+), g(−)g(+)g(+), tg(+)g(+), tg(−)g(+) geometries of the disulfide bridge were generated (Fig. S4, ESI†).
For two of the SS bridge conformations, namely the g(−)g(−)t, and the tg(+)g(+) conformations, the βII has been changed to a βI turn structure (Table 2 and Fig. S5, ESI†). For both βI-tgg conformations, the effects of conformational changes of the side groups have been evaluated as well by generating all eight side group conformations.
After this comprehensive conformational analysis, implicit solvation effects of chloroform and acetonitrile have been taken into account by repeating the geometry optimizations for the βII ring geometries r1 and r6 as well as for the corresponding βI structures r2 and r4, applying the integral equation formalism of the polarizable continuum model (IEFPCM).28–30 Unlike for the gas phase structures, the hydrogen bond between Cys(1)-CO and Gly-NH was found to be cleaved for the r1–c1 conformer (Fig. S6, ESI†). For all the obtained optimized structures, IR and VCD spectra calculations were carried out.
A conformational analysis of the highly flexible molecule 1 based on static structures certainly cannot cover all possible conformations. Furthermore, explicit solute–solvent interactions, especially with the polar solvents, will also influence the relative population of the conformers and completely change energetic preferences. Therefore, the calculated energy differences between the conformers are of low significance, although some trends can be derived. For instance, the side-group conformations c5–c8 are typically far less favoured than the first four. All energy differences are given in the ESI† (Table S1). Therefore, instead of the analysis of the VCD spectra of 1 based on energetic differences, we focus on similarities between the experimental and calculated spectral patterns.
Fig. 4 exemplifies the clear trend in the calculated VCD spectral pattern for the βII structures r1–c1 and r6–c1 and their corresponding βI structures r2–c1 and r4–c1. The complete set of calculated VCD spectra is shown in Fig. S7–S9 of the ESI.† In general, the amide I region (1800–1600 cm−1) is found to be much more sensitive to conformational changes of the side group than the amide II region (1600–1480 cm−1) which contains mostly contributions of the terminal Boc and methyl ester groups.
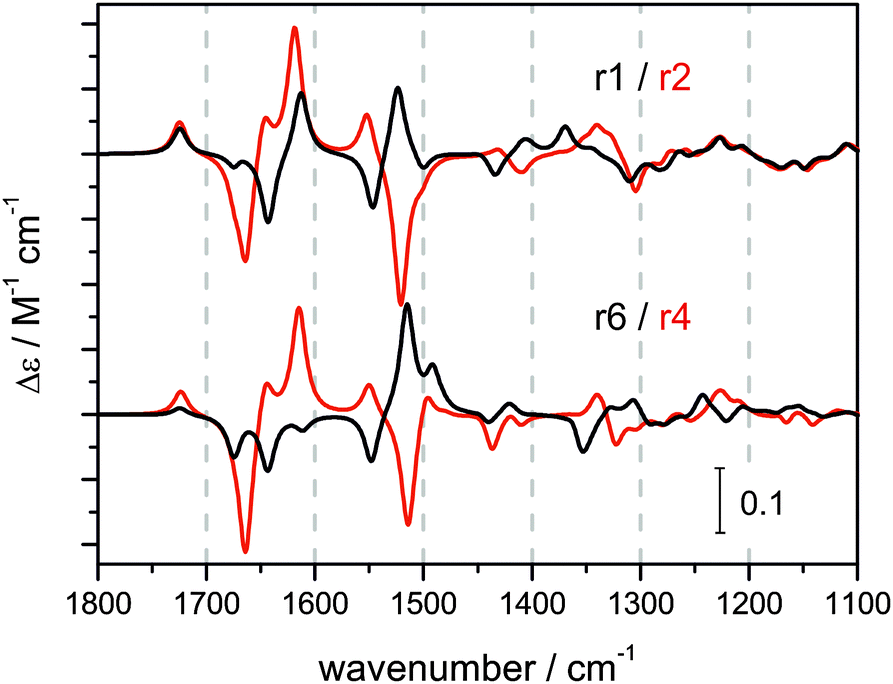 | ||
| Fig. 4 Calculated VCD spectra of the βII structures r1–c1 and r6–c1 and the corresponding βI structures r2–c1 and r4–c1 (IEFPCM acetonitrile). | ||
Overall, most of the calculated VCD spectra resemble the strong experimental −/+ VCD couplet found in the amide I region reasonably well. However, there are no obvious characteristics in the spectra which could be assigned to either βII or βI turn structures. On the other hand, a significant and more characteristic change is observed for the amide II region when comparing βII and βI structures. The amide II of the βII structures features a −/+/(+) pattern which resembles the experimentally observed amide II region of 1 in chloroform and D-2 in all three solvents. In contrast, the amide II pattern is inverted for βI structures showing a +/−/− or +/−/+ pattern. This pattern is very similar to the experimental amide II of L-2 and of 1 in polar solvents. This comparison of spectral patterns suggests that D-2 adopts predominantly a βII turn structure, while L-2 preferably resides in a βI turn. For peptide 1, significant contributions of both βI and βII type structures to the overall conformational equilibrium in polar solvents has to be assumed.
Explicit solute–solvent interactions can affect not only the energetic preferences of certain conformers, but, in general, also influence the calculated IR and VCD intensities. Therefore, we briefly considered explicit solvation for r1–c1 and r2–c1. One acetonitrile molecule was placed in a hydrogen bond with the Gly-NH in r1–c1 and r2–c1. This, however, only affected the negative VCD band of the amide II region of the βI structure, so that the spectra obtained look very similar to those without solvent molecules. The corresponding calculated spectra are shown in the ESI† (Fig. S11).
Replica exchange MD of 1
In our initial study on 1, we only carried out short MD calculations (500 ps) without replica exchange.24 The study on D-2 and L-2, however, showed that REMD can help to ensure that the calculations do not get trapped in the local energy minimum of the βII turn structure.26 Therefore, we carried out additional REMD simulations on 1 for the present study. Starting from a crystal structure like βII conformation, the simulations show that the βII motif is basically conserved for 1 in acetonitrile, not only at 300 K but also at higher temperatures (Table 3). A small contribution of βI structures was found representing only 7% of the total number of structures in the 300 K simulation and about 14% in the 400 K runs. These conformers rapidly converted back to the preferred βII motif. This was corroborated by additional simulations with a βI structure as starting point (Fig. S14, ESI†). This observation proves that the energy barrier for the conversion from βII to βI is much higher than in the opposite direction. Furthermore, the new REMD simulations show that 1 does indeed adopt the βI turn structure, although only to a very small extent. On the other hand, this small amount of βI structures in 1 may increase with longer simulation times.| ϕ (i + 1) | ψ (i + 1) | ϕ (i + 2) | ψ (i + 2) | CαCys1–CαCys2 | |
|---|---|---|---|---|---|
| 300 K | −79 | 102 | 110 | −16 | 5.77 |
| 400 K | −75 | 92 | 116 | −9 | 5.63 |
| REMD | −76 ± 8 | 103 ± 16 | 106 ± 10 | −11 ± 15 | 5.7 ± 0.2 |
| Ideal βII | −60 | 120 | 80 | 0 | <7.0 |
Comparison with NMR results and MD data
For the tetrapeptide D-2, the VCD analysis points to a predominant βII turn structure. This is consistent with the previous experimental findings from 2D-NMR studies and theoretical REMD simulations.26 For the peptide L-2, the interpretation of the VCD spectra suggests a dominant βI turn conformation, while in 2D-NMR the presence of both βII and βI turns was observed,26 although it was not possible to use the NMR data to quantify the relative preferences of the turn structures.For the glycine-containing peptide 1, the previous 2D-NMR of 1 in CD3CN showed some clear indications for a βII turn, while the cross-peaks, characteristic for βI turns, were unfortunately overlapping with other signals.24 Therefore, the presence or absence of βI could not be proven experimentally with NMR spectroscopy.
It is noted that the VCD band of 1 in the amide II region is relatively weak in comparison to those of D- and L-2. Furthermore, the VCD spectrum resulting from a simple average of the experimental VCD spectra of D- and L-2 in either polar solvent is remarkably similar to the spectrum of 1 in the same solvent (cf. Fig. S2, ESI†). These two observations are important since they allow us to conclude that 1 indeed adopts a βI structure to a certain extent in polar solvents although this preference for βI is estimated to be only half as much as for L-2. In chloroform, the conformational equilibrium of 1 is slightly shifted towards βII.
The REMD simulations also qualitatively support these conclusions. The relative trend – L-2 prefers the βI turn twice as much as 1 – is also established.
Conclusions
In the present work, we apply VCD spectroscopy to study the β-turn conformations of three cyclic tetrapeptides:24,261, D-, and L-2, in the three different solvents, namely chloroform-d1, acetonitrile-d3, and DMSO-d6. The comparison of experimental and theoretical VCD spectra supports the previous finding24,26 that D-2 preferably features a βII turn structure. For L-2, the VCD analysis clearly identifies the βI turn as the major conformation. This advances the previous studies where evidence for both βII and βI turn structures was reported although no relative preferences could be quantified. The most important and surprising finding in the present study, however, is that peptide 1 preferably adopts a βI structure in polar solvents, whereas previous NMR evidence for βI was obscured. The current study further demonstrates that this equilibrium between βII and βI turns for 1 depends on the polarity of the solvent. These results therefore reveal a high flexibility of the peptide 1.In summary, this study shows that not only can the chirality of a single amino acid change conformational preferences from βII to βI, but also solvent polarity can have significant effects. Furthermore, VCD spectroscopy can provide characteristic spectral patterns of βII and βI turns to complement NMR studies of secondary structural preferences, especially when unresolved or overlapping NMR peaks prevent one from reaching a conclusion.
In future work, we plan to investigate other cyclic tetrapeptides in order to establish a generalized correlation between the VCD spectral pattern and the adopted secondary structure. We will further study the solvent-dependence of the conformational preferences and also the structural changes caused by ring opening reactions.
Acknowledgements
C.M. acknowledges financial support from the Alexander von Humboldt foundation for a Feodor Lynen Postdoctoral fellowship during his stay at the University of Alberta. E.S.-G. and C.M. thank the Fonds der chemischen Industrie for a Liebig fellowship. This research was supported by the University of Alberta, the Natural Sciences and Engineering Research Council of Canada, and the Canada Research Chairs Program, as well as the Deutsche Forschungsgemeinschaft (DFG) through the Cluster of Excellence RESOLV (EXC 1069). We also gratefully acknowledge access to the computing facilities provided by the Western Canada Research Grid (Westgrid).Notes and references
- C. M. Venkatachalam, Biopolymers, 1968, 6, 1425–1436 CrossRef CAS PubMed.
- P. N. Lewis, F. A. Momany and H. A. Scheraga, Biochim. Biophys. Acta, 1973, 303, 211–229 CrossRef CAS.
- E. Vass, M. Hollósi, F. Besson and R. Buchet, Chem. Rev., 2003, 103, 1917–1954 CrossRef CAS PubMed.
- L. A. Nafie, Vibrational Optical Actvity, John Wiley & Sons Ltd, UK, 2011 Search PubMed.
- T. A. Keiderling, in Circular Dichroism. Principles and Applications, ed. K. Nakanishi, N. Berova and R. W. Woody, VCH Publishers Inc., Weinheim and New York and Cambridge, 1994, pp. 497–522 Search PubMed.
- T. A. Keiderling, Curr. Opin. Chem. Biol., 2002, 6, 682–688 CrossRef CAS.
- E. W. Blanch, L. Hecht and L. D. Barron, Methods, 2003, 29, 196–209 CrossRef CAS.
- J. Hudecová, J. Horníček, M. Buděšínský, J. Šebestík, M. Šafařík, G. Zhang, T. A. Keiderling and P. Bouř, ChemPhysChem, 2012, 13, 2748–2760 CrossRef PubMed.
- C. Johannessen, R. Pendrill, G. Widmalm, L. Hecht and L. D. Barron, Angew. Chem., Int. Ed., 2011, 50, 1–4 CrossRef PubMed.
- C. Merten, M. Amkreutz and A. Hartwig, Phys. Chem. Chem. Phys., 2010, 12, 11635–11641 RSC.
- C. Merten, L. D. Barron, L. Hecht and C. Johannessen, Angew. Chem., Int. Ed., 2011, 50, 9973–9976 CrossRef CAS PubMed.
- C. Merten, K. Hiller and Y. Xu, Phys. Chem. Chem. Phys., 2012, 14, 12884–12891 RSC.
- J. João Marcos Batista, A. N. L. Batista, D. Rinaldo, W. Vilegas, Q. B. Cass, V. S. Bolzani, M. J. Kato, S. N. López, M. Furlan and L. A. Nafie, Tetrahedron: Asymmetry, 2010, 21, 2402–2407 CrossRef PubMed.
- A.-C. Chamayou, S. Lüdeke, V. Brecht, T. B. Freedman, L. A. Nafie and C. Janiak, Inorg. Chem., 2011, 50, 11363–11374 CrossRef CAS PubMed.
- H. Sato and A. Yamagishi, Int. J. Mol. Sci., 2013, 14, 964–978 CrossRef CAS PubMed.
- V. P. Nicu, E. Debie, W. Herrebout, B. V. der Veken, P. Bultinck and E. J. Baerends, Chirality, 2010, 21, E287–E297 CrossRef PubMed.
- E. D. Gussem, P. Bultinck, M. Feledziak, J. Marchand-Brynaert, C. V. Stevens and W. Herrebout, Phys. Chem. Chem. Phys., 2012, 14, 8562–8571 RSC.
- A. Perczel, M. Hollosi, B. M. Foxman and G. D. Fasman, J. Am. Chem. Soc., 1991, 113, 9772–9784 CrossRef CAS.
- R. W. Woody, Peptides, Polypeptides & Proteins, Wiley, New York, 1974 Search PubMed.
- R. W. Woody, Biopolymers, 1978, 17, 1451–1467 CrossRef CAS.
- H. R. Wyssbrod and M. Diem, Biopolymers, 1992, 32, 1237–1242 CrossRef CAS PubMed.
- P. Xie, Q. Zhou and M. Diem, J. Am. Chem. Soc., 1995, 117, 9502–9508 CrossRef CAS.
- P. Xie, Q. Zhou and M. Diem, Faraday Discuss., 1994, 99, 233–243 RSC.
- F. Li, K. Bravo-Rodriguez, C. Phillips, R. Seidel, F. Wieberneit, R. Stoll, N. L. Doltsinis, E. Sanchez-Garcia and W. Sander, J. Phys. Chem. B, 2013, 117, 3560–3570 CrossRef CAS PubMed.
- B. N. N. Rao, A. Kumar, H. Balaram, A. Ravi and P. Balaram, J. Am. Chem. Soc., 1983, 105, 7423–7428 CrossRef CAS.
- F. Li, K. Bravo-Rodriguez, M. Fernandez-Oliva, J. M. Ramirez-Anguita, K. Merz, M. Winter, C. W. Lehmann, W. Sander and E. Sanchez-Garcia, J. Phys. Chem. B, 2013, 117, 10785–10791 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- B. Mennucci, C. Cappelli, R. Cammi and J. Tomasi, Chirality, 2011, 23, 717–729 CrossRef CAS.
- B. Mennucci, J. Tomasi, R. Cammi, J. R. Cheeseman, M. J. Frisch, F. J. Devlin, S. Gabriel and P. J. Stephens, J. Phys. Chem. A, 2002, 106, 6102–6113 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS.
- T. Okabe, M. Kawata, Y. Okamoto and M. Mikami, Chem. Phys. Lett., 2001, 335, 435–439 CrossRef CAS.
- E. Guàrdia, R. Pinzón, J. Casulleras, M. Orozco and F. J. Luque, Mol. Simul., 2001, 26, 287–306 CrossRef.
- G. A. Kaminski, R. A. Friesner, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 2001, 105, 6474–6487 CrossRef CAS.
- A. Patriksson and D. van der Spoel, Phys. Chem. Chem. Phys., 2008, 10, 2073–2077 RSC.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS PubMed.
- M. Tuckerman, B. J. Berne and G. J. Martyna, J. Chem. Phys., 1992, 97, 1990–2001 CrossRef CAS PubMed.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- X. Daura, K. Gademann, B. Jaun, D. Seebach, W. F. van Gunsteren and A. E. Mark, Angew. Chem., Int. Ed., 1999, 38, 236–240 CrossRef CAS.
- J. R. Cheeseman, M. J. Frisch, F. J. Devlin and P. J. Stephens, Chem. Phys. Lett., 1996, 252, 211–220 CrossRef CAS.
- P. J. Stephens, J. Phys. Chem., 1985, 89, 748–752 CrossRef CAS.
- F. J. Devlin, P. J. Stephens, J. R. Cheeseman and M. J. Frisch, J. Phys. Chem. A, 1997, 101, 6322–6333 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional comparative plots for experimental spectra, Raman spectra of 1 and D-2, calculated VCD spectra. See DOI: 10.1039/c3cp55018d |
| This journal is © the Owner Societies 2014 |