PCB153-induced oxidative stress and cell apoptosis on cultured rat Sertoli cells
Ming
Gao
,
Nanxiang
Wu
*,
Yang
Song
,
Lingzhi
Jin
,
Jianlin
Lou
and
He
Tao
Institute of Hygiene, Zhejiang Academy of Medical Sciences, Hangzhou 310013, China. E-mail: gm19851105@126.com; wunanxiang@hotmail.com; Fax: +86-571-88085911; Tel: +86-571-88215595
First published on 9th January 2013
Abstract
In this study, we explored the temporal characteristics of superoxide dismutase (SOD) activity, malondialdehyde (MDA) concentration, and the effect of oxidative stress on DNA damage, and cell apoptosis after exposure to 2,4,5,2′,4′,5′-hexachlorobiphenyl (PCB153) on cultured rat Sertoli cells (SC). SOD activity and MDA concentration were determined after cell exposure to 10, 20, 30 μmol L−1 PCB153 for 6, 12, 24, 36 or 48 h. Antioxidant N-acetyl-L-cysteine (NAC) was added to one group as a pre-treatment at the time point having the best dose–effect relationship. DNA damage, cell apoptosis, and morphological observation were determined by comet assay, flow cytometry, and fluorescence photomicrographs, respectively. After exposure to PCB153, SOD activity increased initially, but decreased subsequently. Each group reached a respective maximum value after 24 h when MDA concentration started increasing. No significant difference was observed in DNA damage after 24 h exposure, but cell apoptosis increased with the level of PCB153, although NAC-pretreatment provided protection. These were corroborated by the morphological observation. These findings suggest that 24 h was the best time point to determine oxidative stress indicators after exposure to PCB153 on cultured rat SC. Furthermore, PCB153-induced oxidative stress could increase cell apoptosis, while DNA damage was not evident.
1. Introduction
Polychlorinated biphenyls (PCBs) belong to a family of synthetic chlorinated organic compounds,1 which are included in the controlled list of the “Stockholm Convention on Persistent Organic Pollutants (POPs)”. They are considered as environmental contaminants in the modern world, causing health hazards in human beings.2–4 According to WHO (World Health Organization) statistics, the industrial production of PCBs began in the late 1920s. Since 1929, about 2 × 109 kg industrial PCBs have been produced. Meanwhile, 2 × 108 kg of PCBs were estimated to remain in the environment in 2003.5PCBs have well-known biological consequences, in particular endocrine disrupting effects.6,7 The testes are among the major target organs, which malfunction upon PCB exposure.8,9 In recent years, oxidative stress has become an important factor underlining the mechanism of PCB-induced reproductive toxicity.10,11 However, the detailed mechanism of oxidative stress induced by PCBs is still not clear. Microsomal monooxygenase has been indicated to be one major player.12,13 Furthermore, numerous studies have confirmed that PCBs were able to induce testicular oxidative stress both in vivo11 and in vitro.14–16 Indicators include superoxide dismutase (SOD), catalase, glutathione peroxidase, glutathione reductase, lipid peroxidation, glutathione, hydrogen peroxide. However, most studies just determined the level of oxidative stress induced on the entire testis10 or germ cells,17 which are rarely associated with Sertoli cells (SC). SC, germ cells, and Leydig cells in the testis have a clear division of labor, and carry out their respective functions.18 SC play an important role in spermatogenesis, the blood–testis barrier, phagocytosis, and immune exemption.19,20 They are sensitive to several environmental toxicants. Once these cells are injured, the entire spermatogenic process will be affected.21–23 To clarify how the environmental pollutants adversely affect SC, we need to have a more comprehensive and accurate understanding of the mechanisms involved in inducing male reproductive toxicity. Meanwhile, recent studies of PCB-induced reproductive toxicity pay less attention to the mechanisms following afte oxidative stress.10,15
In the present study, we focused on the temporal characteristics of oxidative stress after exposing the cultured rat SC to PCB153. SOD activity and malondialdehyde (MDA) concentration were considered as the indicators for observation. It was also undertaken to delineate the effects of oxidative stress on DNA damage and cell apoptosis of SC and to explore the intervention function of the antioxidant N-acetyl-L-cysteine (NAC).
2. Materials and methods
2.1 Animals
18- to 20-day-old male Sprague–Dawley rats were purchased from the Experimental Animal Center of Zhejiang Province (Zhejiang, China). Rats were utilized in accordance with the Guide for the Care and Use of Laboratory Animals published by the Ministry of Health of China. This study was approved by the Zhejiang Academy of Medical Sciences Animal Care and Use Committee.2.2 Cell culture and treatment
SC were isolated by enzymatic digestion according to the method of Shi with some modifications.24 In brief, after removing the testes aseptically, they were decapsulated, and washed in Hanks’ balanced salt solution (HBSS). Finally, they were cut into small fragments. The fragments were then transferred into a 50 mL conical flask and sequentially digested in 0.25% trypsin (Amresco, OH, USA) in a shaking water bath (37 °C, 150 rpm, 15 min). The isolated testicular fragments were centrifuged at 1000 rpm for 5 min and washed twice in HBSS. Then, they were subjected to further digestion in 0.1% collagenase I (Invitrogen, CA, USA) (37 °C, 100 rpm, 30 min). Digested fragments were filtered through a 200 mesh stainless steel filter. The cell suspension was then centrifuged at 1000 rpm for 5 min and washed twice in no-phenol red-Dulbecco's modified Eagle's medium nutrient mixture F-12 HAM (DMEM/F-12) (Gibco, Invitrogen, CA, USA). Finally, the isolated cells were seeded into culture dishes at a density of 1.5 × 106 cells per mL in DMEM/F-12 medium with 5% fetal bovine serum (Hyclone, UT, USA) and maintained in a humidified atmosphere of 95% air/5% CO2 (v/v) at 35 °C.After 24 hours, these cells were extensively washed twice with HBSS solution to remove unattached cells. Then, they were treated with 20 mmol L−1 of Tris-HCl (pH 7.4) for 5 minutes. Thereafter, these cells were cultured in a serum-free medium for 24 hours.
PCB153 (AccuStandard, CT, USA) was dissolved in dimethyl sulfoxide (DMSO) to form the stock solution. Then, this stock solution was diluted with culture medium to different concentrations before being added to the cells in culture. The final DMSO concentration in the medium did not exceed 0.5% (v/v). Thus, DMSO did not affect the viability of SC at such a low concentration. Cells were cultured with 0.5% DMSO in the control group for determining cell viability. For performing other experiments, cells were cultured in 0.3% DMSO.
2.3 Cell viability study
In this experiment, a cell counting kit-8 (CCK-8) (Dojindo, Tokyo, Japan) was employed to quantitatively evaluate SC viability. In 96-well plates, purified SC were treated with concentrations of PCB153 (0, 10, 20, 30, 40 or 50 μmol L−1) and cultured for 24 h or 48 h. Approximately 100 μL DMEM/F-12 medium and 100 μL CCK-8 solution was added to each sample. These samples were incubated at 35 °C for 3–4 h. The optical density was determined at 450 nm using a microplate reader (Sunrise, Tecan, Switzerland). Five parallel experiments were performed with each sample to assess the cell viability.2.4 Determination of SOD and MDA
Purified SC in the 35 mm cell culture dishes were treated with concentrations of PCB153 (0, 10, 20 or 30 μmol L−1), and collected after 6 h, 12 h, 24 h, 36 h, or 48 h exposure. Cells were lysed in ice-cold lysis buffer (Beyotime, Jiangshu, China) with 1% phenylmethylsulfonyl fluoride,25 followed by a 15 min incubation on ice. Samples were centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g for 5 min at 4 °C. The protein concentrations of the supernatant were determined by the bicinchoninic acid (BCA) protein assay kit (Beyotime, Jiangshu, China).
000 g for 5 min at 4 °C. The protein concentrations of the supernatant were determined by the bicinchoninic acid (BCA) protein assay kit (Beyotime, Jiangshu, China).
According to the method of Mockett et al.,26 the assay of SOD activity was based on its ability to inhibit the oxidation of WST by superoxide anion, which was produced from the xanthine–xanthine oxidase system. Its activity was expressed as U mg−1 protein. One unit of SOD activity was defined as the amount that caused a 50% reduction in the absorbance at 450 nm. Lipid peroxidation was evaluated by measuring MDA concentrations according to the method of Mihara and Uchiyama.27 The method was based on the spectrophotometric measurement of the color produced during the reaction of thiobarbituric acid with MDA. MDA concentrations were calculated by the absorbance of the reactive substances of thiobarbituric acid at 532 nm. MDA concentrations were expressed in terms of nmol mg−1 protein.
2.5 Assay for SOD activity after NAC pretreatment
According to the results of MDA and SOD at different time points, cells were treated with concentrations of PCB153 (0, 10, 20 or 30 μmol L−1) and antioxidant NAC (Sigma, MO, USA). The NAC group was pretreated with 300 μmol L−1 NAC for 1 h. Then, it was exposed to 30 μmol L−1 PCB153. After the 24 h exposure, total SOD activities of all groups were evaluated according to the method described earlier.2.6 Comet assay
Purified SC in the 12-well plates were treated with NAC and different concentrations of PCB153 (0, 10, 20 or 30 μmol L−1). Then, they were harvested and washed with HBSS after 24 h exposure. Alkaline comet assay was under taken according to the protocols of Singh et al.28 with minor modifications.29 Frosted microscope slides were coated previously with normal melting agarose (NMA, 1%) (Sigma, MO, USA) in PBS. For each slide, 75 μL low melting agarose (LMA, 0.9%) (Sigma, MO, USA) was mixed with 7–10 μL cell suspension (106 cells) at 37 °C. All the slides were covered with coverslips and refrigerated to gel the agarose. After approximately 20 min, the coverslips were removed. The slides were then placed in a closed box containing freshly prepared lysis solution (2.5 mol L−1 NaCl, 0.1 mol L−1 EDTA, 0.01 mol L−1 Tris, 1% sodium N-lauroyl sarcosinate, pH set to 10 with NaOH, 1% TritonX-100 and 10% DMSO) at 4 °C, and the cells were lysed for more than 1 h. Alkaline unwinding was carried out for 20 min in a horizontal electrophoresis chamber, which was filled with freshly prepared cold alkaline buffer (0.3 mol L−1 NaOH, 1 mmol L−1 EDTA, pH > 13), and electrophoresis was performed in the same buffer for 20 min at 300 mA, 22 V. The slides were then neutralized two times with a neutralization buffer (0.4 mmol L−1 Tris, pH 7.5) for 5 min. Thereafter, the slides were drained and stained with gel-red. Then, the slides were covered with coverslips and analyzed immediately. All these steps were carried out in darkness. The presence of comets was examined at 200× using a BX51 fluorescence microscope (Olympus, Tokyo, Japan), which was equipped with an excitation filter at 530–560 nm. Images of the comets were captured by an Olympus DP50 digital camera. An image analysis system CASP was employed to measure various comet parameters.30 DNA damage was determined in each slide (three replicates for each treatment) by evaluating relative tail DNA (%TailDNA), tail length, tail moment and Olive tail moment in 100 randomly chosen comet cells.2.7 Cell apoptosis and morphological observation
The extent of apoptosis was measured through FITC annexin-V apoptosis detection kit (BD, NJ, USA) according to the manufacture's instructions. Purified SC were treated and collected in a manner described previously in the comet assay. Then, they were gently suspended again in annexin-V binding buffer and incubated with annexin V-FITC/PI in darkness for 15 min. The analysis was performed by flow cytometry using cell quest software (BD FACSCalibur, NJ, USA). Cells undergoing early stage apoptosis are stained with only annexin V-FITC, whereas cells at late stage apoptosis or necrotic cells are stained with both annexin V-FITC and PI. Moreover, the morphological alterations of apoptosis of SC were observed by LSM710 confocal laser scanning microscope (Zeiss, Jena, Germany) using a Hoechst33342/PI apoptotic cell detection kit (KeyGen, Jiangshu, China).2.8 Statistical analysis
Statistical analysis was performed with SPSS 17.0 software. Data were expressed as mean ± standard deviation (SD) of at least three independent experiments. Comparison of multiple groups was conducted using homogeneity test of variance and one-way ANOVA. The Student–Newman–Keuls test was used in case of homoscedasticity. In other cases, the Games–Howell test was used. Differences were considered to be significant when p < 0.05.3. Results
3.1 CCK-8 assay for cell viability
Cell viability by CCK-8 assay showed that PCB153 directly affected SC viability in vitro in a concentration- and time-dependent manner (Fig. 1). After 24 h, the survival rates of groups which were treated with 10, 20, 30 μmol L−1 PCB153 were more than 90%. No significant difference was found between these treated groups and the control group. On the other hand, the survival rates of groups treated with 40 μmol L−1 and 50 μmol L−1 of PCB153 were significantly reduced to under 80% (P < 0.05). After 48 h, cell viability dropped significantly at concentrations of >20 μmol L−1 of PCB153 (P < 0.05): the minimum survival rate was under 50%.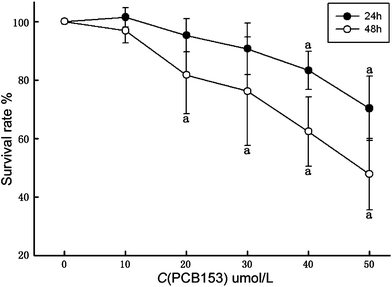 | ||
| Fig. 1 Determination of cell viability in cultured rat Sertoli cells treated with PCB153 for 24 h and 48 h by CCK-8 assay. Data were expressed as mean ± SD for four individual experiments. ap < 0.05 vs. control. | ||
3.2 Levels of SOD and MDA at different time
Purified SC were exposed to PCB153 (0, 10, 20 or 30 μmol L−1) for 6, 12, 24 36 or 48 h. Results obtained for the SOD activity and MDA concentration are presented in Tables 1 and 2. After 24 h exposure to SC, the PCB153 (20, 30 μmol L−1) group showed significantly decreased SOD activity and increased MDA concentration comparing to the solvent control (P < 0.05), with a dose–response relationship. As a comparison to the previous time point with the same dose, SOD activity significantly increased after a 12 h exposure to 30 μmol L−1 of PCB153 and 24 h exposure to 20 μmol L−1 of PCB153 (P < 0.05), but significantly decreased after 36 h exposure to 10, 20 μmol L−1 of PCB153 (P < 0.05). Meanwhile, MDA concentration significantly increased after 24 h exposure to 30 μmol L−1 of PCB153 and 36 h exposure to 20 μmol L−1 of PCB153 (P < 0.05), but significantly decreased after 48 h exposure to 20 μmol L−1 PCB153 (P < 0.05). Both of them illustrated a time–response relationship at time points of 12, 24, 36 h.| C (PCB153) μmol L−1 | 6 h | 12 h | 24 h | 36 h | 48 h |
|---|---|---|---|---|---|
| a p < 0.05 vs. control at the same time point. b p < 0.05 vs. previous time point at the same concentration. | |||||
| 0 | 7.07 ± 1.15 | 52.75 ± 42.25 | 74.36 ± 6.75 | 42.96 ± 23.56 | 26.43 ± 5.83 |
| 10 | 6.45 ± 1.37 | 37.64 ± 30.31 | 68.77 ± 8.58 | 23.45 ± 8.39b | 30.67 ± 3.67 |
| 20 | 6.75 ± 2.88 | 31.39 ± 29.62 | 59.18 ± 14.27a,b | 20.94 ± 9.67b | 36.84 ± 15.32 |
| 30 | 6.67 ± 2.11 | 32.02 ± 22.32b | 48.64 ± 11.14a | 29.57 ± 13.95 | 42.03 ± 13.94 |
| C (PCB153) μmol L−1 | 6 h | 12 h | 24 h | 36 h | 48 h |
|---|---|---|---|---|---|
| a p < 0.05 vs. control at the same time point. b p < 0.05 vs. previous time point at the same concentration. | |||||
| 0 | 6.67 ± 0.36 | 7.28 ± 3.18 | 5.79 ± 1.35 | 19.69 ± 9.02 | 18.23 ± 4.88 |
| 10 | 6.90 ± 1.37 | 7.10 ± 1.41 | 7.86 ± 1.70 | 22.19 ± 4.44 | 23.96 ± 14.08 |
| 20 | 8.02 ± 1.24 | 9.29 ± 1.66 | 11.21 ± 1.68a | 31.47 ± 7.40b | 19.50 ± 8.37b |
| 30 | 7.81 ± 0.27 | 8.98 ± 2.30 | 17.51 ± 1.92a,b | 40.65 ± 20.18 | 35.05 ± 7.63 |
3.3 SOD activity after 1 h pretreatment with 300 μmol L−1 of NAC
The 24 h exposure to 20 or 30 μmol L−1 of PCB153 leads to a significant (P < 0.05) decrease in SOD activity (69.04 ± 2.19 and 62.90 ± 1.73 U mg−1 protein, respectively) comparing to the control group (84.56 ± 1.59 U mg−1 protein) (Fig. 2). The SOD activity of NAC pre-treated group (71.15 ± 3.58 U mg−1 protein) also decreased significantly (P < 0.05) comparing to the control group. On the other hand, SOD activity of NAC pre-treated group significantly (P < 0.05) increased comparing to the 30 μmol L−1 PCB153 treated group (Fig. 2).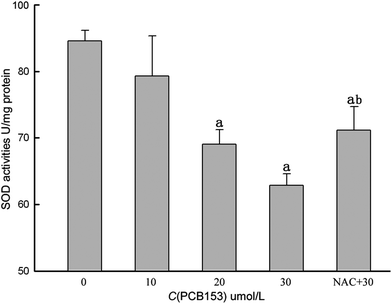 | ||
| Fig. 2 SOD activity after exposing cultured rat Sertoli cells with PCB153 for 24 h. NAC + 30: 300 μmol L−1 NAC pre-treatment for 1 h followed by incubation with 30 μmol L−1 PCB153 for 24 h. ap < 0.05 vs. control, bp < 0.05 vs. 30 μmol L−1 PCB153. | ||
3.4 PCB153-induced DNA damage measured by comet assay
The result of the comet assay of SC which were treated with NAC and PCB153 for 24 h showed that there was no significant difference between control and experimental groups on the indexes of DNA damage, such as TailDNA%, TailLength, TailMoment and OliveTailMoment (Table 3).| C (PCB153) μmol L−1 | %TailDNA | TailLength | TailMoment | OliveTailMoment |
|---|---|---|---|---|
| a NAC + 30: 300 μmol L−1 NAC pre-treated 1 h and followed by incubation with 30 μmol L−1 PCB153 for 24 h. | ||||
| 0 | 5.13 ± 1.85 | 8.60 ± 3.22 | 0.63 ± 0.40 | 0.72 ± 0.31 |
| 10 | 5.00 ± 1.90 | 8.21 ± 3.14 | 0.65 ± 0.38 | 0.71 ± 0.30 |
| 20 | 4.15 ± 1.56 | 7.17 ± 2.23 | 0.44 ± 0.24 | 0.56 ± 0.21 |
| 30 | 4.71 ± 2.26 | 8.81 ± 3.25 | 0.66 ± 0.48 | 0.72 ± 0.38 |
| NAC + 30a | 3.71 ± 0.73 | 6.23 ± 1.70 | 0.33 ± 0.12 | 0.49 ± 0.10 |
3.5 PCB153-induced apoptosis examined by flow cytometry
The apoptotic rate of SC stained with annexin V-FITC/PI was defined as the sum of early apoptosis and late apoptosis (Fig. 3F). The percentages of control, PCB153 (0, 10, 20 or 30 μmol L−1) and NAC pre-treated groups were 5.50 ± 0.98, 8.17 ± 1.84, 10.21 ± 0.85, 13.87 ± 2.07 and 11.74 ± 0.74, respectively (Fig. 3A–E). Each experimental group showed a significant (P < 0.05) increase in apoptotic rate by comparing to the control group. On the other hand, the NAC pre-treated group was significantly (P < 0.05) decreased when compared with the 30 μmol L−1 PCB153 treated group.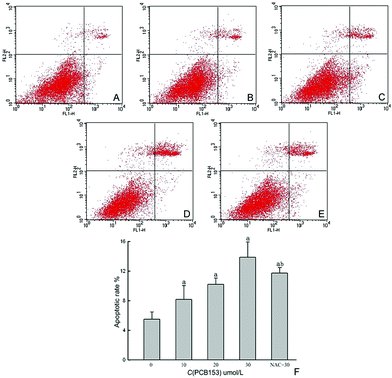 | ||
| Fig. 3 Representative plots of annexin V-FITC/PI staining of cultured rat Sertoli cells treated with PCB153 for 24 h. (A) 0.3% DMSO as control, (B) 10 μmol L−1 PCB153, (C) 20 μmol L−1 PCB153, (D) 30 μmol L−1 PCB153, (E) 300 μmol L−1 NAC pre-treated for 1 h and followed by incubation with 30 μmol L−1 PCB153 for 24 h, (F) the apoptotic rate of SC after exposure to PCB153 for 24 h. Data were expressed as mean ± SD of three independent experiments, ap < 0.05 vs. control, bp < 0.05 vs. 30 μmol L−1 PCB153. | ||
3.6 PCB153-induced apoptosis measured by Hoechst/PI staining fluorescence imaging
The fluorescence photomicrographs of SC stained with Hoechst33342 and PI showed that the apoptotic cells, characterized by condensed nuclei, were observed after being exposed to PCB153 for 24 h (Fig. 4). The number of apoptotic cells increased with higher concentration of PCB153. Various cavities appeared at low concentration, while cell shrinkage and detachment were detected at high concentration. NAC pretreatment could partially reverse these morphological changes induced by PCB153.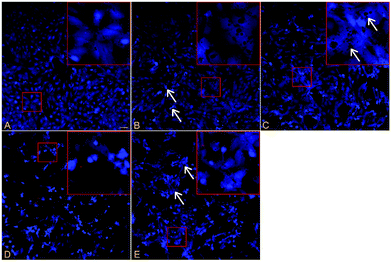 | ||
| Fig. 4 The fluorescence photomicrographs of cell apoptosis stained with Hoechst33342/PI after exposing cultured rat Sertoli cells with PCB153 for 24 h. (A) 0.3% DMSO as control, (B) 10 μmol L−1 PCB153, arrows indicate apoptotic cells, (C) 20 μmol L−1 PCB153, arrows indicate a variety of holes, (D) 30 μmol L−1 PCB153, (E) pre-treated for 1 h with 300 μmol L−1 NAC and followed by incubation with 30 μmol L−1 PCB153 for 24 h, arrows indicate cell shrinkage. | ||
4. Discussion
PCBs are a class of POPs which show estrogen-like effects and which exist widely in the environment.31 Thus, they have a tendency to accumulate in steroid producing organs, such as the testes,8 ovaries,32 and adrenal glands.33 The reproductive toxicity of PCBs mediated by oxidative stress has been gaining quite a lot of attention recently.11,15 In this study, PCB153 was selected as an indicator of the PCB family, because its detection rate and level are in the forefront on a global scale.34–36 Meanwhile, primary cultured rat SC were selected as the experimental carrier. Although 18- to 20-day-old male rats were immature, the quantity and function of SC are similar to that of adult rats. The separation process was such that they could not be easily polluted with other cells. The method of their separation, purification, and cultivation was successfully formulated in 1975,37 and it has been modified and improvised after further exploration.38–40 Based on the results of cell viability of cultured rat SC, 10, 20, 30 μmol L−1 of PCB153 were chosen as the appropriate dosages to measure SOD activity and MDA concentration.SOD is a scavenger of superoxide.41 It provides protection against oxygen free radicals as it catalyzes the removal of superoxide radical (O2−), which damages the membrane and biological structures.42 Oxidized lipids are able to produce MDA in the form of a decomposition product. It is postulated that the mechanism involves the formation of prostaglandin-like endoperoxides from polyunsaturated fatty acids having two or more double bonds.43 SOD activity and MDA concentration are usually measured together. SOD activity indirectly reflects the ability to eliminate oxygen free radicals, whereas MDA concentration indirectly reflects the level of cellular damage caused by attacking free radicals. In our study, a 24 h exposure upon 20 or 30 μmol L−1 of PCB153 caused a significant decrease in SOD activity and a significant increase in MDA concentration, with a dose-dependent manner. SOD activity and MDA concentration had a time–response relationship at time points of 12, 24, 36 h. In all groups, SOD activities increased initially and then decreased. All of the groups showed their respective maximum SOD activities at 24 h time point. MDA concentrations of all groups were relatively lower at 6, 12 h and relatively higher at 36, 48 h. An increase with a significant gradient was observed at 24 h. All of these observations prove that 24 h is the best time point to determine oxidative stress indicators after exposing cultured rat SC with PCB153.
At this moment, little attention is focused on the mechanisms of PCB-induced reproductive toxicity following oxidative stress. On the basis of the above results, we selected the 24 h time point and included an NAC pretreatment group to perform preliminary investigation of the follow-up mechanisms. NAC is a precursor to the amino acid cysteine.44 Through its metabolic contribution to glutathione production, cysteine participates in the general antioxidant activities of the body.45 The present results showed that exposure to 20 or 30 μmol L−1 of PCB153 for 24 h resulted in a significant decrease of SOD activity, whereas NAC pretreatment could partially reverse this effect. The damage caused by oxidative stress is attributed to the free radicals which occur during the process of oxidative stress.46 The free radicals produce lipid peroxides which are toxic to cells. In addition, they also diffuse into the nucleus easily, and directly attack DNA. This causes DNA base modification and the DNA strand breaks, ultimately leading to apoptosis.47 Comet assay, also known as single cell gel electrophoresis, was first proposed as a technology that examined the DNA strand breaks to determine the genetic toxicity.48 It can effectively detect and quantify the injury caused to DNA single or double strand breaks. Our comet assay results revealed that exposure to PCB153 for 24 h did not cause DNA damage in SC, because no significant difference was reported between control and experimental groups on the comet assay indexes. It corroborated the findings that exposure to PCB153 alone would not produce DNA damage.49 However, apoptotic rate of SC was increased with an increasing dosage of PCB153. NAC pretreatment had a protective effect as it significantly reduced the cell apoptosis. These findings were corroborated by the morphological observation of apoptosis by staining with Hoechst33342 and PI. Accordingly, it suggested that free radicals originating from oxidative stress might not enter the nucleus to attack DNA. In other words, there was another pathway to mediate apoptosis of SC. p,p′-DDE was able to induce oxidative stress on cultured rat SC. Moreover, it could also decrease the mitochondrial membrane potential, increase cytochrome C release, and activate the activity of caspase-3 and caspase-9, thereby leading to mitochondria-mediated apoptosis.50 Further studies are needed to confirm whether PCB153 has a similar way of inducing apoptosis of SC.
In conclusion, the present in vitro study showed that 24 h was the best time point to determine oxidative stress indicators after exposing cultured rat SC to PCB153. In the concentration range of 10–30 μmol L−1, PCB153 was able to induce oxidative stress and increase the cell apoptotic rate of cultured rat SC. However, it did not cause any apparent damage to DNA. NAC pretreatment showed a protective effect. These data provide reference for further studies on the mechanism of PCB-induced toxicity in the testes.
Acknowledgements
This work was supported by a grant from the Department of Science and Technology of Zhejiang Province [2007F30015].References
- J. W. Lloyd, R. M. Moore, B. S. Woolf and H. P. Stein, J. Occup. Environ. Med., 1976, 18, 109–113 CAS.
- W. J. Crinnion, Altern. Med. Rev., 2011, 16, 5–13 Search PubMed.
- A. Goncharov, M. Pavuk, H. R. Foushee and D. O. Carpenter, Environ. Health Perspect., 2011, 119, 319–325 CrossRef CAS.
- L. W. Robertson and G. Ludewig, Gefahrst. Reinhalt. Luft, 2011, 71, 25–32 CAS.
- CICAD 55, Polychlorinated Biphenyls: Human Health Aspects, WHO publication, 2003, 11 Search PubMed.
- S. M. Dickerson, S. L. Cunningham and A. C. Gore, Toxicol. Appl. Pharmacol., 2011, 252, 36–46 CrossRef CAS.
- K. Selvakumar, B. L. Sheerin, G. Krishnamoorthy, P. Venkataraman, P. Elumalai and J. Arunakaran, Exp. Toxicol. Pathol., 2011, 63, 105–112 CrossRef CAS.
- P. Elumalai, G. Krishnamoorthy, K. Selvakumar, R. Arunkumar, P. Venkataraman and J. Arunakaran, Reprod. Toxicol., 2009, 27, 41–45 CrossRef CAS.
- D. Y. Han, S. R. Kang, O. S. Park, J. H. Cho, C. K. Won, H. S. Park, K. I. Park, E. H. Kim and G. S. Kim, Toxicol. Ind. Health, 2010, 26, 287–296 CrossRef.
- H. A. Aly, O. Domenech and N. A. B. Abdel, Food Chem. Toxicol., 2009, 47, 1733–1738 CrossRef CAS.
- A. Atessahin, G. Turk, S. Yilmaz, M. Sonmez, F. Sakin and A. O. Ceribasi, Basic Clin. Pharmacol. Toxicol., 2010, 106, 479–489 CrossRef CAS.
- E. J. Lim, Z. Majkova, S. Xu, L. Bachas, X. Arzuaga, E. Smart, M. T. Tseng, M. Toborek and B. Hennig, Chem. Biol. Interact., 2008, 176, 71–78 CrossRef CAS.
- S. H. Park, J. H. Jang, C. Y. Chen, H. K. Na and Y. J. Surh, Toxicology, 2010, 278, 131–139 CrossRef CAS.
- Y. L. Mi, C. Q. Zhang, W. D. Zeng, J. X. Liu and H. Y. Liu, Poult. Sci., 2007, 86, 2008–2012 CAS.
- P. Murugesan, M. Balaganesh, K. Balasubramanian and J. Arunakaran, J. Endocrinol., 2007, 192, 325–338 CrossRef CAS.
- P. Murugesan, T. Muthusamy, K. Balasubramanian and J. Arunakaran, Reprod. Toxicol., 2008, 25, 447–454 CrossRef CAS.
- G. Krishnamoorthy, P. Venkataraman, A. Arunkumar, R. C. Vignesh, M. M. Aruldhas and J. Arunakaran, Reprod. Toxicol., 2007, 23, 239–245 CrossRef CAS.
- P. J. O'Shaughnessy and P. A. Fowler, Reproduction, 2011, 141, 37–46 CrossRef CAS.
- T. L. Nel, C. W. Jang, M. D. Stewart, H. Akiyama, R. S. Viger and R. R. Behringer, Biol. Reprod., 2011, 84, 342–350 CrossRef.
- L. Su, D. D. Mruk and C. Y. Cheng, J. Endocrinol., 2011, 208, 207–223 CAS.
- H. A. Aly and R. M. Khafagy, Toxicol. Appl. Pharmacol., 2011, 252, 273–280 CrossRef CAS.
- J. S. Taylor, B. M. Thomson, C. N. Lang, F. Y. Sin and E. Podivinsky, J. Toxicol. Environ. Health, Part A, 2010, 73, 1075–1089 CrossRef CAS.
- M. Zhang, Z. He, H. Yuan, L. Zhu, C. Guo, L. Yin, J. Wu, S. Deng, L. Yuan and L. Wen, J. Vet. Med. Sci., 2011, 73, 199–203 CrossRef CAS.
- Y. Shi, Y. Song, Y. Wang, X. Liang, Y. Hu, X. Guan, J. Cheng and K. Yang, J. Biomed. Biotechnol., 2009, 2009, 181282 CrossRef.
- V. Sekar and J. H. Hageman, Biochem. Biophys. Res. Commun., 1979, 89, 474–478 CrossRef CAS.
- R. J. Mockett, A. C. Bayne, B. H. Sohal and R. S. Sohal, Methods Enzymol., 2002, 349, 287–292 CAS.
- M. Mihara and M. Uchiyama, Anal. Biochem., 1978, 86, 271–278 CrossRef CAS.
- N. P. Singh, M. T. McCoy, R. R. Tice and E. L. Schneider, Exp. Cell Res., 1988, 175, 184–191 CrossRef CAS.
- R. R. Tice, E. Agurell, D. Anderson, B. Burlinson, A. Hartmann, H. Kobayashi, Y. Miyamae, E. Rojas, J. C. Ryu and Y. F. Sasaki, Environ. Mol. Mutagen., 2000, 35, 206–221 CrossRef CAS.
- K. Konca, A. Lankoff, A. Banasik, H. Lisowska, T. Kuszewski, S. Gozdz, Z. Koza and A. Wojcik, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2003, 534, 15–20 CrossRef CAS.
- M. Calo, D. Alberghina, A. Bitto, E. R. Lauriano and C. P. Lo, Food Chem. Toxicol., 2010, 48, 2458–2463 CrossRef CAS.
- K. Yoshizawa, A. E. Brix, D. M. Sells, M. P. Jokinen, M. Wyde, D. P. Orzech, G. E. Kissling, N. J. Walker and A. Nyska, Toxicol. Pathol., 2009, 37, 921–937 CrossRef CAS.
- L. A. Li, Toxicol. in Vitro, 2007, 21, 1087–1094 CrossRef CAS.
- R. J. Szlinder, I. Barska, J. Mazerski and Z. Usydus, Mar. Pollut. Bull., 2009, 58, 85–92 CrossRef.
- T. Todaka, T. Hori, H. Hirakawa, J. Kajiwara, D. Yasutake, D. Onozuka, T. Iida and M. Furue, Chemosphere, 2009, 74, 902–909 CrossRef CAS.
- H. Wang, Q. An, Y. H. Dong, D. C. Li and B. Velde, J. Hazard. Mater., 2010, 176, 1027–1031 CrossRef CAS.
- M. J. Welsh and J. P. Wiebe, Endocrinology, 1975, 96, 618–624 CrossRef CAS.
- A. G. Davidson, R. J. Bell, G. E. Lees and K. E. Murphy, In Vitro Cell. Dev. Biol.: Anim., 2007, 43, 324–327 CrossRef CAS.
- J. P. Mather, K. M. Attie, T. K. Woodruff, G. C. Rice and D. M. Phillips, Endocrinology, 1990, 127, 3206–3214 CrossRef CAS.
- S. S. Raychoudhury, A. F. Flowers, C. F. Millette and M. F. Finlay, J. Androl., 2000, 21, 964–973 CAS.
- S. I. Liochev and I. Fridovich, Free Radical Biol. Med., 2010, 48, 1565–1569 CrossRef CAS.
- I. A. Abreu and D. E. Cabelli, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 263–274 CrossRef CAS.
- J. Lykkesfeldt, Clin. Chim. Acta, 2007, 380, 50–58 CrossRef CAS.
- R. A. Sansone and L. A. Sansone, Innov. Clin. Neurosci., 2011, 8, 10–14 Search PubMed.
- A. Maheshwari, M. M. Misro, A. Aggarwal, R. K. Sharma and D. Nandan, Mol. Reprod. Dev., 2011, 78, 69–79 CrossRef CAS.
- A. R. Collins, Mutat. Res., Rev. Mutat. Res., 2009, 681, 24–32 CrossRef CAS.
- N. Chiaverini and L. M. De, Free Radical Res., 2010, 44, 605–613 CrossRef CAS.
- O. Ostling and K. J. Johanson, Biochem. Biophys. Res. Commun., 1984, 123, 291–298 CrossRef CAS.
- Y. C. Jeong, N. J. Walker, D. E. Burgin, G. Kissling, M. Gupta, L. Kupper, L. S. Birnbauma and J. A. Swenberg, Free Radical Biol. Med., 2008, 45, 585–591 CrossRef CAS.
- Y. Song, X. Liang, Y. Hu, Y. Wang, H. Yu and K. Yang, Toxicology, 2008, 253, 53–61 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |