 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermodynamic stability of various phases of zinc tin oxides from ab initio calculations†
Joohwi Leeab,
Seung-Cheol Leea,
Cheol Seong Hwangb and
Jung-Hae Choi*a
aElectronic Materials Research Center, Korea Institute of Science and Technology, Seoul 136-791, Korea. E-mail: choijh@kist.re.kr; Fax: +82 2 958 6658; Tel: +82 2 958 5488
bWCU Hybrid Materials Program, Department of Materials Science and Engineering, Inter-university Semiconductor Research Center, Seoul National University, Seoul 151-744, Korea
First published on 5th August 2013
Abstract
Thermodynamic stabilities of various phases in ZnO–SnO2 systems were investigated based on the Gibbs energy obtained from density functional theory (DFT) calculations. The pressure–temperature (p–T) phase diagram was determined; the coexistence of ZnO and SnO2 was the most stable phase in the low temperature region at zero external pressure, while Zn2SnO4 with the inverse spinel structure and ZnSnO3 with the lithium niobate structure were stable at the high temperature and high pressure region. Various octahedral configurations of the inverse spinel structures of Zn2SnO4 were considered. The calculated results showed feasible agreement with experimental data on the phase stability and explained well the experimental observation of the mixed state of Zn2SnO4, ZnO and SnO2 at mid-range temperatures and pressures. Considering the atomic structures, bulk moduli and thermodynamic stabilities, the local density approximation calculations were found to describe experimental observations more accurately than the generalized gradient approximation calculations. The phase transitions in the ZnO–SnO2 system were found to be dominated by the changes in both the Zn–O bond length and the coordination number of Zn, rather than changes in the bond length of Sn–O and the coordination number of Sn.
1 Introduction
Zinc tin oxide is one of the most promising transparent amorphous oxide semiconductors (AOSs) which are used in electronic devices such as displays, solar cells, photocatalysts and gas sensors due to their high transparency, low resistivity and high chemical selectivity.1–9 Additionally, it has the industrial and eco-friendly advantages of not including expensive indium or toxic cadmium, as opposed to InGaZnO4 or Cd2SnO4.10,11 Recently, the adoption of AOS materials as the channel layer of thin film transistors (TFTs) for semiconductor memory devices has attracted a great deal of interest.9 Most of these promising applications rely upon high carrier (electron) mobility (>∼10 cm2 V−1 s−1), even with an amorphous structure, which is not the case for the most common amorphous Si semiconductor (<∼1 cm2 V−1 s−1). This has been attributed to the fact that the lower edge of the conduction band (or energy states near the mobility edge) in these AOSs is mostly the result of the In 5s orbitals whose electronic structure is only marginally influenced by the bond distortions related to the amorphous structure compared with the crystalline counterpart. In zinc tin oxide systems, Sn 5s orbitals are expected to play a similar role, and thus zinc tin oxide is expected to show promising electrical properties as the channel material in TFTs, which was indeed proved experimentally.8Making AOS with high mobility basically depends on several principles; firstly, it is necessary to mix component oxides whose crystal structures are quite distinctive. Secondly, the use of as many component oxides as possible, such as in InGaZnO4, is desirable. Thirdly, s orbitals originating from metal ions must be the main components of the lower edge (or mobility edge) of the conduction band, considering their non-directionality and spatial delocalization. Therefore, zinc tin oxide is a feasible candidate for high performance TFTs. However, there could be several complications in the thermodynamic stability of the various stable (metastable) phases in the ZnO–SnO2 system.
An oxide containing Zn and/or Sn can exist as a binary oxide and as a ternary oxide, such as Zn2SnO4 (ZTO214) and ZnSnO3 (ZTO113). The inverse spinel phase of ZTO214 (ref. 12–15) was reported to exist as a single phase at 1275 °C.16,17 Polycrystalline ZTO214 thin films were also reported, which were deposited by sputtering onto glass substrates.12,13 Additionally, two rhombohedral phases of ZTO113 were observed: a lithium niobate phase (LN) and an ilmenite phase (IL). LN-ZTO113 was synthesized from a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of ZnO and SnO2 by quenching from a pressure greater than 7 GPa and from a high temperature of 1000 °C.18 Meanwhile, IL-ZTO113 was synthesized by the ion-exchange reaction of Li2SnO3, which has the same structure as IL-ZTO113.19 Zinc tin oxide has various phases and shows a tendency to form an amorphous phase.3,4,7,8 Therefore, understanding the crystalline phases and their thermodynamic stability is a prerequisite to figure out the amorphous structure and nanostructure.
:1 mixture of ZnO and SnO2 by quenching from a pressure greater than 7 GPa and from a high temperature of 1000 °C.18 Meanwhile, IL-ZTO113 was synthesized by the ion-exchange reaction of Li2SnO3, which has the same structure as IL-ZTO113.19 Zinc tin oxide has various phases and shows a tendency to form an amorphous phase.3,4,7,8 Therefore, understanding the crystalline phases and their thermodynamic stability is a prerequisite to figure out the amorphous structure and nanostructure.
Previous ab initio calculations reported that pressurization changes the thermodynamic stability from the coexistence of ZnO and SnO2 to the coexistence of ZTO214 and SnO2.20–22 These calculations only investigated the pressure effect at 0 K based on the enthalpy, and the reported transition pressure varies from 0 to 12 GPa. However, temperature is in fact a much more important thermodynamic variable for determining the stability of materials and for understanding their properties under practical synthesis and working conditions. Furthermore, the space group of the inverse spinel structure varies according to the occupancy of the octahedral sites by Zn and Sn at the unit cell scale when an inverse spinel is assumed to be ordered.15 Seko et al. reported that the most stable structure of ZTO214 belongs to the space group P4122,15 while Gracia et al. calculated the stability of the ZTO214 phase that belongs to the space group Imma.22 Nakayama et al. reported the most stable structure of ZTO214 from their calculations but did not specify the space group.20 Additionally, the different exchange-correlation functionals (xc-functionals) used in the previous calculations led to different optimized structures and energetics, which could affect the thermodynamic stability.
In this paper, ab initio calculations were performed for various zinc tin oxide phases using two xc-functionals. The ab initio phonon calculations were carried out to obtain the enthalpy and Gibbs energy, which were used to investigate the effects of both pressure and temperature on the thermodynamic stabilities. The pressure–temperature (p–T) phase diagram was obtained for the composition with an atomic ratio of Zn to Sn to O of 1:1:3 and compared with previous synthesis or existence conditions (stability range) of various zinc tin oxide phases. The composition of Zn to Sn to O of 2:1:4 was also considered. For these two compositions, several space groups of the ordered and disordered inverse spinel structure were examined. Finally, the changes in the bond length of cation–oxygen and coordination numbers (CN) of the cations were discussed.
2 Background theory
Thermodynamic stability can be described using several thermodynamic variables. The Gibbs energy describes the thermodynamic stability of a state at a certain pressure and temperature. Fig. 1 shows the schematic illustration for obtaining the Gibbs energy taking into account both pressure and temperature effects.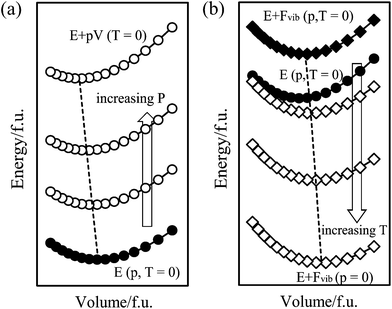 | ||
| Fig. 1 Schematic illustrations of the Gibbs energy as a function of the cell volume. The energy can be extracted using (a) E + pV to consider the pressure effect (T = 0) and (b) E + Fvib to consider the temperature effect (p = 0). | ||
The thermodynamic stability under external pressure can be determined by the enthalpy, H(p), obtained from the energy–volume curve (E–V curve) calculated from ab initio calculations at 0 K. Fig. 1a illustrates how to add the pV term to the total energy of the electronic structure for a given volume at 0 K, E(V),23 as shown in eqn (1), to consider the external hydrostatic pressure.
| H(p) = E(V) + pV | (1) |
The solid circles indicate the E–V curve at p = 0 at 0 K, which are the total energies of the electronic structure for a given volume. The open circles indicate the curves under external p. Note that a positive pressure denotes hydrostatic compression. The minimum thermodynamic values for the given pressures were found and linked by a dashed line, which indicates that the higher the external compressive pressure, the smaller the equilibrium volume.
The temperature effects on the thermodynamic stability can be included by including phonon calculations in the ab initio study. For any point on the E–V curve, the phonon frequency can be obtained using the linear response method based on density functional perturbation theory (DFPT).24 Then, the Helmholtz phonon vibrational energy, Fvib, can be calculated for a given volume and temperature using the quasi-harmonic approximation, as shown in eqn (2),
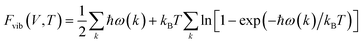 | (2) |
Fig. 1b shows how to calculate the temperature effect shown in eqn (2). The curve composed of solid circles is identical to that in Fig. 1a, whereas the curve composed of solid rhombi was obtained by adding the first term of eqn (2) to account for the temperature-independent vibrational property. The other E–V curves, represented by open rhombi, were obtained for various temperatures, and the minimum energy values for the given temperatures were found and linked by a dashed line. It is noted that the magnitude of the Fvib(T) term increases as the temperature increases, and that this increasing tendency is more significant at larger volumes. These behaviors result in an increase in the equilibrium volume, which describes the thermal expansion.
In addition, for the disordered inverse spinel, the configurational energy is given by eqn (3),15
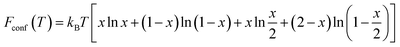 | (3) |
2. In an ordered inverse spinel, by contrast, Fconf(T) is not considered because the configurations of cations at the octahedral sites are fixed.
By combining eqn (1)–(3), the Gibbs energy is defined as the minimum value with respect to the volume for a given p and T, as in eqn (4).25,26
| G(p, T) = minV[E(V) + pV + Fvib(V, T) + Fconf(T)] | (4) |
The points on the E–V curve were fitted to the Birch–Murnaghan equation of state (BM-EOS)29 before determining the minimum value. The Gibbs energy can be used to determine the phase stability and phase transition. In this paper, the term “transition” is used to describe the onset of changing thermodynamic stability, which is the prerequisite for the real occurrence of a phase transformation.
3 Computational methods
Calculations were performed using the Vienna ab initio Simulation Package (VASP).30,31 The projector augmented wave (PAW) method,32 within both the local density approximation (LDA) parameterized by Ceperley and Alder33 and the generalized gradient approximation (GGA) parameterized by Perdew et al. (PBE),34 was used, with a cutoff energy of 500 eV. The 3d and 4s orbitals for Zn, the 4d, 5s and 5p orbitals for Sn, and the 2s and 2p orbitals for O were treated as the valence electrons in the non-spin-polarized calculations. The cutoff energy and k-grid were set to guarantee the convergence of the total energy below 1 meV f.u.−1.To obtain the E–V curves, the energies at 15 different volumes under external pressure were calculated for all phases considered. The calculations were performed in two steps, as shown schematically in Fig. 2a. In the first step, full relaxation of the cell parameters and internal atomic coordinates was performed with an applied external p, ranging from −14 GPa to 14 GPa with an interval of 2 GPa. In this step, the reduced or expanded volumes of the unit cells were obtained, which are indicated as the open circles with x-marks in Fig. 2a. Electronic iterations were performed until the energy difference was less than 10−6 eV, and atomic relaxations were performed until the maximum force between atoms was less than 10−3 eV Å−1. All the phases considered in this study show an anisotropic change in their cell parameters under hydrostatic pressure due to their non-cubic symmetry. In the second step, static calculations were performed without changing the volume and shape of the unit cell and the atomic coordinates. The external pressure was eliminated in this step. Then, the total energy of the electronic structure for a given V, E(V), was obtained, which is shown by the solid circles in Fig. 2a. Finally, the E–V curve with 15 points was fitted to the BM-EOS to calculate the optimized unit cell volume and the bulk modulus of each phase at 0 K.
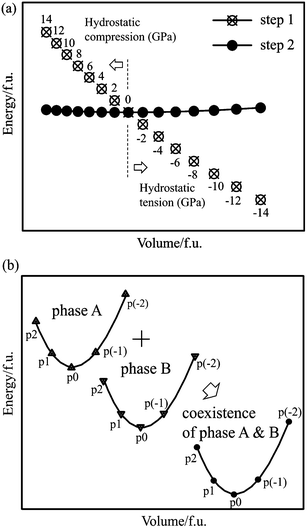 | ||
| Fig. 2 (a) Schematic illustration of the E–V curve. The volume at step 1 is maintained at step 2. (b) The summation of energy and volume were performed at the same hydrostatic pressure. The positive and negative values correspond to the hydrostatic compressive and tensile stresses, respectively. | ||
The coexistence of two phases under the same external pressure was obtained by the summation of the E–V curves of the two phases, as shown schematically in Fig. 2b. The x- and y-axes represent the sum of the unit cell volumes and the sum of the electronic structure total energies of the two phases, respectively. The summation of the unit cell volume and the total energy was performed at each external hydrostatic pressure. For example, the energy of the coexistence of phases A and B at pressure p2 is the sum of the energies of phases A and B at pressure p2.
The linear response method based on DFPT was used to calculate the force constants of the supercells.24 Iterations of the electronic steps were performed until the energy differences were less than 10−8 eV. The Helmholtz vibrational energies were used to confirm the convergence of supercells and k-points, which are less than 1 meV f.u.−1. The phonon vibrational frequency and phonon density of states were calculated from the force constants using Phonopy.26,27 Table 1 summarizes the space groups, number of atoms in the unit cells, supercell sizes for the phonon calculations and k-points mesh for the unit cells and supercells.
| Composition | Space group (abbreviation) | Unit cell | Number of atoms in unit cell (Zn/Sn/O) | k-Points for unit cell (for supercell) | Supercell size for phonon calculation |
|---|---|---|---|---|---|
a Γ-centered.b Monkhorst–Pack.28c This cell is slightly deviated from the primitive cell of the normal spinel, which belongs to the space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. m. |
|||||
| ZnO | P63mc | Hexagonal (primitive) | 2/0/2 | 7 × 7 × 5 − Γa (3 × 3 × 3 − Γ) | 3 × 3 × 2 |
| SnO2 | P42/mnm | Tetragonal (primitive) | 0/2/4 | 4 × 4 × 6 − MPb (2 × 2 × 2 − MP) | 2 × 2 × 3 |
| ZnSnO3 | R3c (LN) | Hexagonal | 6/6/18 | 5 × 5 × 2 − Γ (3 × 3 × 2 − Γ) | 2 × 2 × 1 |
| R (IL) |
Hexagonal | 6/6/18 | 5 × 5 × 2 − Γ (3 × 3 × 2 − Γ) | 2 × 2 × 1 | |
| Zn2SnO4 | P4122 (tetra) | Tetragonal (primitive) | 8/4/16 | 4 × 2 × 4 − MP (2 × 2 × 2 − MP) | 2 × 1 × 2 |
| Imma (ortho) | FCC-likec (primitive) | 4/2/8 | 5 × 5 × 5 − Γ (3 × 3 × 3 − Γ) | 2 × 2 × 2 | |
| P1 (mono) | Monoclinic | 16/8/32 | 2 × 2 × 2 − MP (2 × 2 × 2 − MP) | 1 × 1 × 1 | |
The Gibbs energy was determined in the range of 0–14 GPa with an interval of 0.1 GPa and in the range of 0–1600 K with an interval of 5 K. Then, the p–T phase diagram was constructed.
4 Results and discussion
4.1 Structural properties
Most of the calculation results are for the composition having the atomic ratio of Zn:Sn:O = 1:1:3. This is referred to as the ZTO113 composition, which is different from ZTO113, denoting the ZnSnO3 phase. Some results will also be described for the composition having the atomic ratio of Zn:Sn:O = 2:1:4, which will be referred to as the ZTO214 composition.
For ZTO113, two phases were examined: the lithium niobate phase (R3c, space group 161; LN-ZTO113) and the ilmenite phase (R, space group 148; IL-ZTO113). They have rhombohedral primitive cells;38 however, hexagonal unit cells were used in the ab initio calculations for convenience. In the hexagonal cell, Zn, Sn and a vacancy occupy the octahedral sites in distinctive ways. In IL-ZTO113, a plane normal to the 〈0001〉 direction is occupied by one cation species or a vacancy, whereas it is occupied by the mixture of Zn, Sn and vacancies in LN-ZTO113.
The stable phase of ZTO214 was reported to be an inverse spinel.12–15 If all of the octahedral sites are treated as symmetrically identical due to cation disordering, it is called a disordered inverse spinel ZTO214 (DIS-ZTO214), which belongs to the space group Fdm as the normal spinel.15 However, on the atomic scale, the occupancy of octahedral sites by two different metal elements might lower the symmetry of the structure, which is called an ordered inverse spinel ZTO214 (OIS-ZTO214).
For OIS-ZTO214, three configurations were investigated: a tetragonal phase (P4122, space group 91; tetra-ZTO214),15 an orthorhombic phase (Imma, space group 74; ortho-ZTO214)22 and a monoclinic phase (P1, space group 1; mono-ZTO214). Other unit cells, belonging to the space groups P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) m2 and Cm, were found to change into ortho-ZTO214 via relaxation of the shape and volume of the cells. Meanwhile, the volume, energy, and the vibrational properties of DIS-ZTO214 were obtained by averaging over eight different OIS-ZTO214 cells. Six of them were constructed by generating random numbers to have different fillings of the octahedral sites by Zn and Sn, where each individual structure was a mono-ZTO214. Tetra-ZTO214 and ortho-ZTO214 were also included as samples to obtain the average properties of the DIS-ZTO214. Note that tetra-ZTO214 was calculated to have the lowest energy,15 and this was also confirmed by this calculation. The primitive cell of each phase was defined in comparison with the conventional unit cell of the cubic spinel, as shown in Fig. 3.
m2 and Cm, were found to change into ortho-ZTO214 via relaxation of the shape and volume of the cells. Meanwhile, the volume, energy, and the vibrational properties of DIS-ZTO214 were obtained by averaging over eight different OIS-ZTO214 cells. Six of them were constructed by generating random numbers to have different fillings of the octahedral sites by Zn and Sn, where each individual structure was a mono-ZTO214. Tetra-ZTO214 and ortho-ZTO214 were also included as samples to obtain the average properties of the DIS-ZTO214. Note that tetra-ZTO214 was calculated to have the lowest energy,15 and this was also confirmed by this calculation. The primitive cell of each phase was defined in comparison with the conventional unit cell of the cubic spinel, as shown in Fig. 3.
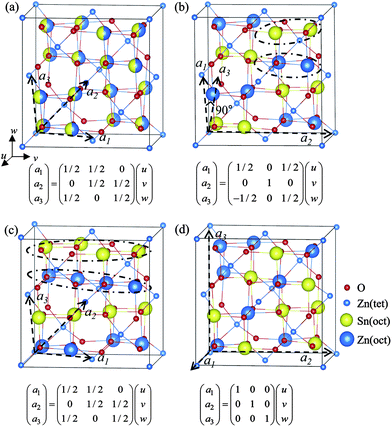 | ||
| Fig. 3 Unit cells of various inverse spinel structures of ZTO214. u, v and w are the lattice vectors of the conventional unit cell of the normal spinel structure. Dashed vectors of a1, a2, and a3 indicate the unit vectors of the primitive cell for (a) DIS-ZTO214 and several OIS-ZTO214 structures, which are (b) tetra-ZTO214 (P4122), (c) ortho-ZTO214 (Imma) and (d) mono-ZTO214 (P1), respectively. | ||
Tables 2 and 3 show the volume of the unit cell and the bulk modulus for each phase from the LDA and GGA calculations, respectively. They also show the comparison with experimental values and some previous calculation results. ZnO in the wurtzite structure and SnO2 in the rutile structure were also calculated as references. The volumes and bulk moduli were obtained from the BM-EOSs.29 The difference between the energy obtained using the relaxed volume and that obtained using the volume fitted to the EOS was negligible, below 1 meV f.u.−1. The calculated volumes of the optimized cells agree well with the experimental data, to within an error of 5%. The volumes obtained from the LDA calculations are slightly smaller and those obtained from the GGA calculations are slightly larger than the experimental values, as observed for many other materials.25 The volumes from the LDA calculations were closer to the experimental data than those from the GGA calculations.
| Composition | Space group (abbreviation) | Volume (Å3 f.u.−1) | |||||
|---|---|---|---|---|---|---|---|
| This work | References | ||||||
| LDAa | GGAb | Experimental | LDA | GGA | B3LYPd | ||
| a LDA parameterized by Ceperley and Alder.33b GGA parameterized by Perdew et al.34c LDA parameterized by Vosko et al.36d B3LYP parameterized by Lee et al.37e Disordered inverse spinel.f Ordered inverse spinel. | |||||||
| ZnO | P63mc | 22.86 | 24.88 | 23.80 [ref. 45] | 22.87a [ref. 23] | 24.83 [ref. 23] | 24.38 [ref. 22] |
| SnO2 | P42/mnm | 35.78 | 37.80 | 35.75 [ref. 40] | 34.94c [ref. 39] | 35.50 [ref. 44] | |
| ZnSnO3 | R3c (LN) | 54.99 | 58.72 | 55.95 [ref. 18] | 58.60a [ref. 42] | 60.08 [ref. 41] | |
| 54.15c [ref. 43] | |||||||
| R (IL) |
55.50 | 59.68 | 56.78 [ref. 19] | 56.29c [ref. 43] | 60.82 [ref. 41] | ||
| Zn2SnO4 | Fdm (DIS)e |
79.15 | 85.34 | 81.11 [ref. 14 and 46] | |||
| P4122 (tetra)f | 78.89 | 84.96 | |||||
| Imma (ortho)f | 78.91 | 85.02 | 83.74 [ref. 22] | ||||
| P1 (mono)f | 79.12 | 85.39 | |||||
| Composition | Space group (abbreviation) | Bulk modulus (GPa) | |||||
|---|---|---|---|---|---|---|---|
| This work | References | ||||||
| LDAa | GGAb | Experimental | LDA | GGA | B3LYPd | ||
| a LDA parameterized by Ceperley and Alder.33b GGA parameterized by Perdew et al.34c LDA parameterized by Vosko et al.36d B3LYP parameterized by Lee et al.37e Disordered inverse spinel.f Ordered inverse spinel.g No method was reported. | |||||||
| ZnO | P63mc | 161 | 128 | 183 [ref. 45] | 162a [ref. 23] | 134 [ref. 23] | 114 [ref. 22] |
| SnO2 | P42/mnm | 202 | 173 | 205 [ref. 40] | 245c [ref. 39] | 221 [ref. 44] | |
| ZnSnO3 | R3c (LN) | 195 | 167 | 161 [ref. 41] | |||
| R (IL) |
173 | 137 | 141 [ref. 41] | ||||
| Zn2SnO4 | Fdm (DIS)e |
169 | 135 | 169g [ref. 51] | |||
| P4122 (tetra)f | 173 | 142 | |||||
| Imma (ortho)f | 170 | 139 | 186 [ref. 22] | ||||
| P1 (mono)f | 167 | 133 | |||||
To the best of the authors' knowledge, the bulk moduli of the ternary oxides of ZTO have scarcely been reported experimentally, making the verification of the calculation results by comparing them with experimental results improbable. In order to verify the validity of the calculation of the bulk moduli, therefore, the calculated bulk moduli of the binary oxides, ZnO and SnO2, were compared with experimental data. The bulk moduli from the LDA calculations were close to the experimental values, however, those from the GGA calculations were quite a bit lower than the experimental values. The bulk moduli of the three OIS-ZTO214 phases and DIS-ZTO214 are similar regardless of the space group, i.e., regardless of the configuration of the octahedral sites within the same exchange-correlation functionals. In contrast, the LN-ZTO113 phase shows a slightly higher bulk modulus than the IL-ZTO113 phase.
4.2 Thermodynamic stability from the enthalpy
Fig. 4a and b show the calculated total energies of the electronic structures with respect to the volumes of various zinc tin oxides using the LDA and GGA functionals, respectively, for the ZTO113 composition. As explained in Fig. 2b, the simultaneous presence of two phases was described by the summation of the volume and total energy under the given external pressure. The thermodynamic variables of IL-ZTO113 are not reliable at a volume higher than the arrow in Fig. 4b and so are not shown. The transition pressure can be acquired from the slope of the negative common tangent of the E–V curve fitted to the BM-EOS because the compression is defined as a positive value. The enthalpies of the two coexisting phases are the same at the transition pressure and are represented by the intercept of the extrapolated tangent line with the energy axis.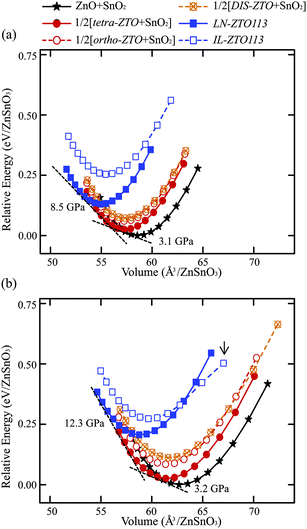 | ||
| Fig. 4 E–V curves obtained from the (a) LDA and (b) GGA calculations. The negative common tangent corresponds to the transition pressure. The arrow in (b) indicates that the curve becomes meaningless for IL-ZTO113 under an external pressure over −10 GPa. | ||
The general tendencies of the E–V curve and the thermodynamic stability are the same for both exchange-correlation functionals: a transition from the coexistence of ZnO and SnO2 at zero external pressure to the coexistence of tetra-ZTO214 and SnO2, and then to LN-ZTO113 as the external hydrostatic pressure increases. The transition reactions for the ZTO113 composition were defined as follows:
| Transition A: ZnO + SnO2 → ½(ZTO214 + SnO2) | (5) |
| Transition B: ½(ZTO214 + SnO2) → LN-ZTO113 | (6) |
| Transition C: ZnO + SnO2 → LN-ZTO113 | (7) |
The transition reactions for the ZTO214 composition were also defined:
| Transition A′: 2ZnO + SnO2 → ZTO214 | (8) |
| Transition D: ZTO214 → LN-ZTO113 + ZnO | (9) |
| Transition C′: 2ZnO + SnO2 → LN-ZTO113 + ZnO | (10) |
4.3 Phonon density of states
Fig. 5 shows the phonon density of states (PhDOSs) obtained from the LDA calculations under zero external, −14 GPa of tensile hydrostatic and 14 GPa of compressive hydrostatic pressures. The maximum frequency of each phase under compression is higher than those at zero and tensile pressure, and the maximum frequency of the oxides examined in this study is of the same sequence as the bulk modulus in Table 3. The PhDOSs of tetra-ZTO214 in Fig. 5c and of the ortho-ZTO214 in Fig. 5d are very similar, implying that the phonon properties of inverse spinel phases having different octahedral occupancies are comparable. It should be noted that peaks in the negative frequency region indicate the instability of a material.25 Among the several oxides examined, only IL-ZTO113 under high tensile hydrostatic pressure shows non-negligible PhDOS at negative frequencies in Fig. 5f, indicating that this phase is unstable in the tensile region and cannot exist even as a metastable phase.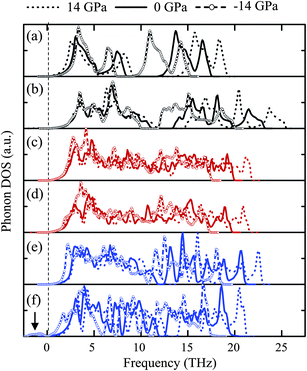 | ||
| Fig. 5 PhDOSs of (a) ZnO, (b) SnO2, (c) tetra-ZTO214, (d) ortho-ZTO214, (e) LN-ZTO113 and (f) IL-ZTO113 at −14, 0 and 14 GPa. The arrow in (f) indicates the negative phonon frequency in IL-ZTO113 at −14 GPa. | ||
4.4 Thermodynamic stability from the Gibbs energy
The Gibbs energy was calculated for all phases of the zinc tin oxides except for IL-ZTO113. IL-ZTO113 was excluded because it had much higher energy state than the phases at 0 K and it exhibited an imaginary phonon frequency under tensile pressure, as shown in Fig. 5f. Fig. 6 shows the Gibbs energies as a function of temperature for the various phases of the ZTO113 composition from LDA calculations. A temperature range from 0 K to 1600 K was selected according to the report that 1600 K is the maximum temperature at which ZTO214 can exist.16,17 The state of simultaneous presence of ZnO and SnO2 was taken as the Gibbs energy reference. At zero external pressure in Fig. 6a, it is clear that the coexistence of ZnO and SnO2 is the most stable in the low temperature region, and the Gibbs energy of DIS-ZTO214 at 0 K is much higher than those of tetra-ZTO214 and ortho-ZTO214, because DIS-ZTO214 includes several mono-ZTO214 structures having relatively higher energies. However, the addition of the configurational energy terms in DIS-ZTO214 accelerates the decrease of its Gibbs energy as the temperature increases. As a result, the temperature of transition A is 1040 K for DIS-ZTO214 (upwards arrow in Fig. 6a) while it is 1365 K for tetra-ZTO214 (downwards arrow in Fig. 6a), and the stable temperature region for ortho-ZTO214 was not found. Overall, ZTO214 is more stable than the simultaneous presence of ZnO and SnO2 at high temperature.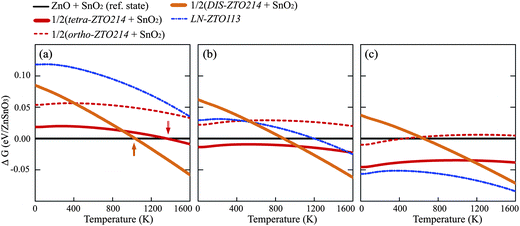 | ||
| Fig. 6 Gibbs energies of various phases as a function of temperature for the ZTO113 composition using the LDA calculations when the external pressure is (a) p = 0, (b) p = 4 GPa and (c) p = 8 GPa. | ||
Fig. 6b and c show the pressure effects on the variation of Gibbs energy as a function of temperature. Fig. 6b at 4 GPa shows that the coexistence of tetra-ZTO214 and SnO2 is the most stable below 1035 K, while DIS-ZTO214 replaces tetra-ZTO214 above that temperature. Note that when the external pressure is 4 GPa, the Gibbs energy of LN-ZTO113 drastically decreases, and becomes comparable to the energies of the other phases. In contrast, at 8 GPa in Fig. 6c, LN-ZTO113 is the most stable phase for the whole temperature range, which agrees well with the experimental observation that LN-ZTO113 is the high-pressure phase.18
4.5 p–T phase diagram
Fig. 7 shows the p–T phase diagrams obtained from the LDA calculations. The results for OIS-ZTO214s and DIS-ZTO214 are presented separately in order to show the effects of cation disordering in the spinel structure on the phase stability. Fig. 7a shows the p–T phase diagram for the ZTO113 composition, assuming that ZTO214 is tetra-ZTO214, which is the most stable OIS-ZTO214. The thermodynamically stable regions of the coexistence of ZnO and SnO2, the coexistence of tetra-ZTO214 and SnO2, and the LN-ZTO113 phases are represented. At 0 K, transition A occurs at 2.3 GPa, which is approximately 0.8 GPa lower than that calculated from the enthalpy, shown in Fig. 4. This difference comes from the temperature-free term, 1/2kϖ, expressed as the first term in eqn (2). The pressure for transition A decreases as the temperature increases. At 0 K, transition B was calculated to occur at 7.3 GPa, which is also lower than that obtained from the enthalpy. This transition pressure also decreases as the temperature increases.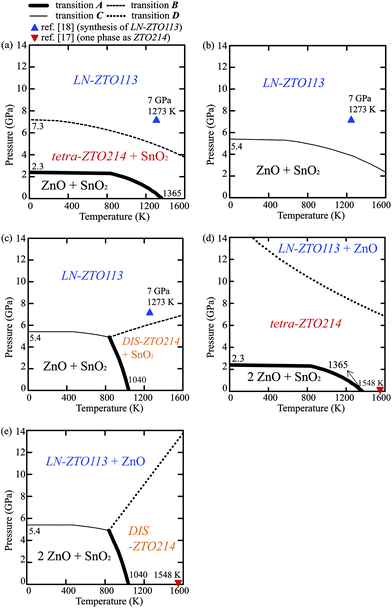 | ||
| Fig. 7 p–T phase diagrams for (a) the ZTO113 composition treating ZTO214 as tetra-ZTO214, (b) the ZTO113 composition treating ZTO214 as ortho-ZTO214, (c) the ZTO113 composition considering DIS-ZTO214 instead of OIS-ZTO214s, (d) the ZTO214 composition treating ZTO214 as tetra-ZTO214, and (e) the ZTO214 composition considering DIS-ZTO214 instead of OIS-ZTO214s. | ||
Fig. 7b shows the p–T phase diagram of the ZTO113 composition when ZTO214 was assumed to be ortho-ZTO214, which is another OIS-ZTO214. As Fig. 6 shows, the Gibbs energy of ortho-ZTO214 was higher than that of tetra-ZTO214 by approximately 0.04 eV f.u.−1. As a result, the coexistence region of ZTO214 and SnO2 does not appear and a phase boundary between the simultaneous presence of ZnO and SnO2 phases and the LN-ZTO113 phase appears (transition C).
Fig. 7c shows the p–T phase diagram of the ZTO113 composition assuming that ZTO214 is DIS-ZTO214. The coexistence region of ZTO214 and SnO2 does not appear at low temperatures due to the high energy state of DIS-ZTO214. However, the additional configurational energy enhances transition A as the temperature increases, therefore, the temperature of transition A at zero external pressure is lower than that of tetra-ZTO214 by more than 300 K. This agrees with the previous simulation result which reported that DIS-ZTO214 is more stable than OIS-ZTO214 at high temperatures over ∼1000 K.15 The phase boundary between the coexistence of ZnO and SnO2 phase and the LN-ZTO113 phase appears as the pressure increases to 5.4 GPa at 0 K.
Fig. 7d shows the p–T phase diagram for the ZTO214 composition when ZTO214 was assumed to be tetra-ZTO214. The thermodynamically stable regions for the coexistence of ZnO and SnO2, tetra-ZTO214, and the coexistence of LN-ZTO113 and ZnO phases are presented. The general tendency of Fig. 7d is similar to that of Fig. 7a, except for the much higher transition pressure required for LN-ZTO113 phase to appear, suggesting that the synthesis pressure should be increased at this ZTO214 composition (transition D compared to transition B). The single phase region of ZTO214 reported in previous phase diagrams16,17 is shown as an inverted triangle, which agrees with this p–T phase diagram.
Fig. 7e shows the p–T phase diagram for the ZTO214 composition when ZTO214 was assumed to be DIS-ZTO214. In the low temperature region, the minimum pressure for LN-ZTO113 to be stable decreased obviously compared to Fig. 7d (from transition D to transition C′). However, due to the stability of DIS-ZTO214 at high temperature, the transition pressure for LN-ZTO113 drastically increases as the temperature increases, which is a remarkable contrast to transition B in Fig. 7c. The transition pressures determined from our calculation results are summarized in comparison with the previous reports in Table 4.
| Thermodynamic variable | Ref. | Space group of ZTO214 | xc-functional | Minimum pressure for ZTO214a (GPa) | Minimum pressure for LN-ZTO113b (GPa) |
|---|---|---|---|---|---|
| a Transition A.b Transtition B or C.c LDA parameterized by Ceperley and Alder.33d GGA parameterized by Perdew et al.34e B3LYP parameterized by Lee et al.37f NA: not available.g The authors of this reference reported that they calculated the most stable structure of ZTO214 but did not specify the space group. | |||||
| G | This work | P4122 | LDAc | 2.3 (0 K), 0.6 (1273 K) | 7.3 (0 K), 5.1 (1273 K) |
| GGAd | 2.4 (0 K), 0 (1273 K) | 11.7 (0 K), 9.9 (1273 K) | |||
| Fdm |
LDAc | No transition (0 K), 0 (1273K) | 5.4 (0 K), 6.2 (1273K) | ||
| H | This work | P4122 | LDAc | 3.1 (0 K) | 8.5 (0 K) |
| GGAd | 3.2 (0 K) | 12.3 (0 K) | |||
| H | 20 | NAf,g | GGA | ∼0 (0 K) | 0.8 (0 K) |
| H | 21 | NAf | GGA | 5.9 (0 K) | 7.1 (0 K) |
| H | 22 | Imma | B3LYPe | 12c (0 K) | |
Our calculations describe well the experimental observations; at zero-pressure and low temperature, the simultaneous presence of ZnO and SnO2 is the most stable irrespective of the overall composition and atomic configuration in ZTO214. Experimentally, the mixture films of ZnO and SnO2 were found in an amorphous state below the crystallization temperature of ZTO214, which was reported to be approximately 900 K.7,12,19,47 In other words, they do not form any ternary crystalline compounds in the usual film growth temperature range (<900 K) according to thermodynamic considerations. When the amorphous mixture film was heated up to ∼900 K, they seemed to be separated into the ZnO and SnO2 binary oxides by atomic force microscopy and energy dispersive X-ray spectroscopy.7 In contrast, in the high temperature region, ZTO214 is stable regardless of the disordering of ZTO214. For the ZTO113 composition, Inaguma et al. reported that LN-ZTO113 decomposed into mixed phases comprised of ZTO214 and SnO2 above 1073 K.18 The decomposition of IL-ZTO113 into the coexistence of ZTO214 and SnO2 was also observed for a film above 873 K (ref. 47) and in the bulk above 1273 K.18 Additionally, for the ZTO214 composition, Wang et al. reported that the simultaneously present ZnO and SnO2 phases gradually changed into ZTO214 at approximately 1000 K and finally disappeared at 1200 K.49,50 At the high temperature and high pressure region, the synthesis condition of LN-ZTO113 (ref. 18) was represented as a triangle in Fig. 7a–c. The results also agree with the calculation results, regardless of the local atomic configuration of ZTO214.
There are certain discrepancies between the theoretical and experimental results for the phase transition temperatures and pressures, which could be attributed to several factors, although the general trends are well matched. One of the reasons is that the thermodynamic calculations using the ab initio method cannot account for the occurrence of a transformation route due to the kinetic activation barrier. In addition, our assumption to describe the cation disordering for DIS-ZTO214 includes the averaging effect with a limited number of structures, which may bring about some deviation of transition temperature and pressure. As shown in Fig. 7, the thermodynamically stable region can change with the atomic distribution such that ZTO214 can be the stable phase for mid-range pressures depending on the distribution of Zn and Sn. In fact, it seems plausible that actually ZTO214 has a mixture of a disordered inverse spinel structure and several ordered inverse spinel structures.
Another factor is that the experimental data were almost always obtained from thin films, whose structures were amorphous in most cases. During crystallization, they are under biaxial compressive or tensile stress depending on the mismatch of both the lattice parameters and the thermal expansion with the substrates. The compressive stress on the order of GPa (about 104 atm) is practically quite high. However, such a level of stress is often observed in thin film systems. For example, 6 GPa corresponds to approximately 2% biaxial strain in LN-ZTO113 thin films, which is not an uncommon level of strain in several thin film systems. The diverse experimental observations and the coexistence of multi-phases can be explained by the various configurations of ZTO214 and kinetics. In fact, Hoel reported the simultaneous presence of ZTO214, ZnO and SnO2 phases in a polycrystalline film when the film was formed at a high temperature of over 927 K.47,48
Table 4 shows the transition pressures from our calculation results in comparison with the previous reports. For the ZTO113 composition, the sequence of transition A and B along the pressure increase was the same as the previous reports which only considered the enthalpy.20,21 Nakayama et al. calculated that the transition pressures of transitions A and B are 0 and 0.8 GPa, respectively.20 However, they mentioned three plausible reasons for their underestimation of the transition pressures: the softness of the GGA functional, the exclusion of the temperature effect, and the method of selecting the occupancy of Zn and Sn in the unit cell. It should be pointed out that this study took these three drawbacks into account by testing the LDA functional as well as the GGA functional, by using the Gibbs energy to estimate the thermodynamic stability, and by investigating several atomic configurations in both ZTO113 and ZTO214.
The GGA results in Table 4 overestimated the transition pressure and temperature and disagreed with this experimental observation. The use of the exchange-correlation functional of the DFT+U method35 was also attempted. However, it was not helpful for the thermodynamic calculation of zinc tin oxide systems; when the LDA+U scheme was used, all the volumes of the zinc tin oxide phases decreased slightly. This means that the differences between the calculated and experimental values of the bond lengths and the cell volumes increased. Because the bond lengths are related to the phonon vibrations, the LDA+U scheme was not appropriate to use. Conversely, when the GGA+U scheme was adopted, the calculated volume became closer to the experimental value. However, the failure to describe the tensile region of IL-ZTO113 and the stable region of LN-ZTO113 also remained in the GGA+U calculation.
4.6 Bond length and CN
Fig. 8a and b show the bond lengths of Zn–O and Sn–O as a function of the external pressure at 0 K. Black, blue and red symbols correspond to the bonds in the binary phases, tetra-ZTO214 and LN-ZTO113, respectively. The squares and stars in Fig. 8a represent the Zn–O bonds where the CNs of Zn are 4 and 6, respectively, whereas the hexagons in Fig. 8b represent the Sn–O bonds where the CN of Sn is 6. The multiple bond lengths for a given CN in one phase were induced by the atomic relaxation of cations from the ideal tetrahedral or octahedral sites. The closed symbols and solid lines denote the bonds in the stable phases, while the open symbols and dotted lines denote the bonds in the unstable phases in Fig. 7a. The detailed bond lengths and the CNs of the cations under zero external pressure at 0 K are summarized in Table 5.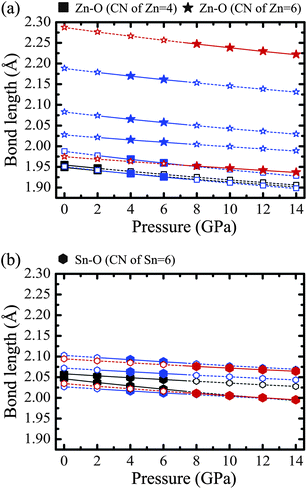 | ||
| Fig. 8 Bond lengths of (a) Zn–O and (b) Sn–O as a function of the external pressure at 0 K. Black, blue and red symbols indicate the bonds in the binary phases, tetra-ZTO214 and LN-ZTO113 phases, respectively. The squares and stars in (a) represent the Zn–O bonds where the CNs of Zn are 4 and 6, respectively. The hexagons in (b) represent the Sn–O bonds where the CN of Sn is 6. | ||
| Composition | Space group (abbreviation) | xc-functional | Bond length of Zn–O (Å) | CN of Zn | Bond length of Sn–O (Å) | CN of Sn |
|---|---|---|---|---|---|---|
| a LDA parameterized by Ceperley and Alder.33b GGA parameterized by Perdew et al.34 | ||||||
| ZnO | P63mc | LDAa | 4 × 1.951 (±0.003) | 4 | — | — |
| GGAb | 4 × 2.008 (±0.004) | — | ||||
| SnO2 | P42/mnm | LDA | — | — | 6 × 2.052 (±0.006) | 6 |
| GGA | — | 6 × 2.093 (±0.001) | ||||
| ZnSnO3 | R3c (LN) | LDA | 3 × 1.97, 3 × 2.29 | 6 | 3 × 2.03, 3 × 2.09 | 6 |
| GGA | 3 × 2.03, 3 × 2.35 | 3 × 2.07, 3 × 2.13 | ||||
| Zn2SnO4 | P4122 (tetra) | LDA | 2 × 1.95, 2 × 1.99 | 4 | 2 × 2.03, 2 × 2.07, 2 × 2.10 | 6 |
| 2 × 2.03, 2 × 2.08, 2 × 2.19 | 6 | |||||
| GGA | 2 × 2.01, 2 × 2.05 | 4 | 2 × 2.06 2 × 2.11, 2 × 2.14 | 6 | ||
| 2 × 2.08, 2 × 2.15, 2 × 2.25 | 6 | |||||
In Fig. 8a, all Zn–O bond lengths decrease almost linearly as the pressure increases, and the longer bonds decrease slightly more in length than the shorter bonds. In the low pressure region, all Zn atoms in the stable phase, ZnO, are located at tetrahedral sites, while in the high pressure region, all Zn atoms in the stable LN-ZTO113 are located at octahedral sites. At mid-range pressure, in contrast, half of the Zn atoms occupy the tetrahedral sites and the other half occupy the octahedral sites in tetra-ZTO214. This increasing tendency of the CN of Zn with increasing pressure is the same as for ZnO. In fact, the CN of Zn in ZnO increases from four to six with the change in the stable phase from the wurtzite to the rock salt structure at 9 GPa.23
The Sn–O bond lengths in Fig. 8b also decrease as the external pressure increases, however, they show a very different tendency compared to the Zn–O bonds. All Sn atoms are located at the octahedral sites in all the phases considered, hence their CN is kept at six. In addition, the discrepancy in the bond length of Sn–O in each phase is remarkably small compared to the case of Zn. It is also noted that the rutile structure SnO2 with the CN of 6 is stable up to 21 GPa,40 which is much higher than the transition pressure of ZnO. This means that Zn–O bonds are more sensitive to the pressure change compared to Sn–O bonds, and this can be inferred from the lower bulk modulus of ZnO than of SnO2 in Table 3. A similar tendency was also observed in ZTO as shown in Fig. 8a and b. Therefore, the phase stabilities and phase transitions in the ZTO system are dominated by changes in both the bond length of Zn–O and the CN of Zn, rather than changes in the bond length of Sn–O and the CN of Sn.
5 Summary
We examined the thermodynamic stability of various phases of zinc tin oxides based on the Gibbs energy using ab initio calculations. The pressure–temperature phase diagram was obtained, and the transition pressures and temperatures were determined and compared to previous synthesis and/or existence conditions for the ZTO113 and ZTO214 compositions. In the low pressure region, the simultaneous presence of ZnO and SnO2 phases was the most stable configuration at low temperature, but ZTO214 became more stable as the temperature increased. The atomic configurations of ZTO214 in the unit cell affected the thermodynamic stability of LN-ZTO113 as well as that of ZTO214. As the external pressure increased, the thermodynamic stability changed from the coexistence of ZnO and SnO2 to the coexistence of ZTO214 and SnO2 and then to LN-ZTO113 if tetra-ZTO214 was assumed. Meanwhile, a direct transition from the coexistence of ZnO and SnO2 to LN-ZTO113 was predicted if DIS-ZTO214 was assumed. The LDA functional was found to describe the experimental observations better than the GGA functional when considering the atomic structures, bulk moduli and transition pressures and temperatures. The phase transitions were found to occur mainly due to the changes of Zn–O bond lengths and the CN of Zn. The discrepancies between the experimental observations of the various phases of zinc tin oxide and the coexistence of multi-phases can be attributed to the possible kinetic barriers which were not considered in these calculations, and the local occupancy of the octahedral sites by Zn and Sn in ZTO214.Acknowledgements
This work was supported by the IT R&D program of MOTIE/KEIT [10035320], the Converging Research Center Program through the Ministry of Science, ICT and Future Planning (2013K000159) and the Institutional Research Program of KIST (2E24001). The authors would also like to acknowledge the support from the KISTI Supercomputing Center through the Strategic Support Program for Supercomputing Application Research (KSC-2010-C2-0005) and Prof. Atsushi Togo at Kyoto University, Japan for helpful discussions.References
- P. Görrn, M. Sander, J. Meyer, M. Kröger, E. Becker, H.-H. Johannes, W. Kowalsky and T. Riedl, Adv. Mater., 2006, 18, 738 CrossRef.
- P. Görrn, F. Ghaffari, T. Riedl and W. Kowalsky, Solid-State Electron., 2009, 53, 329 CrossRef PubMed.
- H. Q. Chiang, J. F. Wager, R. L. Hoffman, J. Jeong and D. A. Keszler, Appl. Phys. Lett., 2005, 86, 013503 CrossRef.
- W. B. Jackson, R. L. Hoffman and G. S. Herman, Appl. Phys. Lett., 2005, 87, 193503 CrossRef.
- P. Song, Q. Wang and Z. Yang, Sens. Actuators, B, 2011, 156, 983 CrossRef CAS PubMed.
- B. Tan, E. Toman, Y. Li and Y. Wu, J. Am. Chem. Soc., 2007, 129, 4162 CrossRef CAS PubMed.
- J. Heo, S. B. Kim and R. G. Gordon, Appl. Phys. Lett., 2012, 101, 113507 CrossRef.
- B. S. Yang, S. Park, S. Oh, Y. J. Kim, J. K. Jeong, C. S. Hwang and H. J. Kim, J. Mater. Chem., 2012, 22, 10994 RSC.
- S. H. Rha, J. Jung, Y. S. Jung, Y. J. Chung, U. K. Kim, E. S. Hwang, B. K. Park, T. J. Park, J.-H. Choi and C. S. Hwang, Appl. Phys. Lett., 2012, 100, 203510 CrossRef.
- T. Kamiya, K. Nomura and H. Hosono, Sci. Technol. Adv. Mater., 2010, 11, 044305 CrossRef.
- S. B. Zhang and S.-H. Wei, Appl. Phys. Lett., 2002, 80, 1376 CrossRef CAS.
- D. L. Young, H. Moutinho, Y. Yan and T. J. Coutts, J. Appl. Phys., 2002, 92, 310 CrossRef CAS.
- D. L. Young, D. L. Williamson and T. J. Coutts, J. Appl. Phys., 2002, 91, 1464 CrossRef CAS.
- S.-H. Wei and S. B. Zhang, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 045112 CrossRef.
- A. Seko, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 054114 CrossRef.
- G. B. Palmer and K. R. Poeppelmeier, Solid State Sci., 2002, 4, 317 CrossRef CAS.
- S. Harvey, K. R. Poeppelmeier and T. O. Mason, J. Am. Ceram. Soc., 2008, 91, 3683 CrossRef CAS.
- Y. Inaguma, M. Yoshida and T. Katsumata, J. Am. Chem. Soc., 2008, 130, 6704 CrossRef CAS PubMed.
- D. Kovacheva and K. Petrov, Solid State Ionics, 1998, 109, 327 CrossRef CAS.
- M. Nakayama, M. Nogami, M. Yoshida, T. Katsumata and Y. Inaguma, Adv. Mater., 2010, 22, 2579 CrossRef CAS PubMed.
- H. Gou, J. Zhang, Z. Li, G. Wang, F. Gao, R. C. Ewing and J. Lian, Appl. Phys. Lett., 2011, 98, 091914 CrossRef.
- L. Gracia, A. Beltrán and J. Andrés, J. Phys. Chem. C, 2011, 115, 7740 CAS.
- J. E. Jaffe, J. A. Snyder, Z. Lin and A. C. Hess, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 1660 CrossRef CAS.
- S. Baroni, P. Giannozzi and A. Testa, Phys. Rev. Lett., 1987, 58, 1861 CrossRef CAS.
- R. P. Stoffel, C. Wessel, M.-W. Lumey and R. Dronskowski, Angew. Chem., Int. Ed., 2010, 49, 5242 CrossRef CAS PubMed.
- A. Togo, L. Chaput, I. Tanaka and G. Hug, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 174301 CrossRef.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188 CrossRef.
- F. Birch, Phys. Rev., 1947, 71, 809 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- D. M. Ceperley and B. J. Alder, Phys. Rev. Lett., 1980, 45, 566 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200 CrossRef CAS PubMed.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- J. B. Goodenough and J. J. Stickler, Phys. Rev., 1967, 164, 768 CrossRef CAS.
- B. Zhu, C.-M. Liu, M.-B. Lv, X.-R. Chen, J. Zhu and G.-F. Ji, Phys. B, 2011, 406, 3508 CrossRef CAS PubMed.
- J. Haines and J. M. Léger, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 11144 CrossRef CAS.
- H. Gou, F. Gao and J. Zhang, Comput. Mater. Sci., 2010, 49, 552 CrossRef CAS PubMed.
- H. Wang, H. Huang and B. Wang, Solid State Commun., 2009, 149, 1849 CrossRef CAS PubMed.
- N.-N. Ge, C.-M. Liu, Y. Cheng, X.-R. Chen and G.-F. Ji, Phys. B, 2011, 406, 742 CrossRef CAS PubMed.
- L. Gracia, A. Beltrán and J. Andrés, J. Phys. Chem. B, 2007, 111, 6479 CrossRef CAS PubMed.
- H. Karzel, W. Potzel, W. Schiessl, M. Steiner, U. Hiller, G. M. Kalvius, D. W. Mitchell, T. P. Das, P. Blaha, K. Schwarz and M. P. Pasternak, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 11425 CrossRef CAS.
- R. J. Hill, J. R. Craig and G. V. Gibbs, Phys. Chem. Miner., 1979, 4, 317 CrossRef CAS.
- Y. Hayashi, K. Kondo, K. Murai, T. Moriga, I. Nakabayashi, H. Fukumoto and K. Tominaga, Vacuum, 2004, 74, 607 CrossRef CAS PubMed.
- C. A. Hoel, T. O. Mason, J.-F. Gaillard and K. R. Poeppelmeier, Chem. Mater., 2010, 22, 3569 CrossRef CAS.
- C. Wang, X. Wang, J. Zhao, B. Mai, G. Sheng, P. Peng and J. Fu, J. Mater. Sci., 2002, 37, 2989 CrossRef CAS.
- C. Wang, X. Wang, B.-Q. Xu, J. Zhao, B. Mai, P. Peng, G. Sheng and J. Fu, J. Photochem. Photobiol., A, 2004, 168, 47 CrossRef CAS PubMed.
- F. Birch, J. Geophys. Res., 1952, 57, 227 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3tc30960f |
| This journal is © The Royal Society of Chemistry 2013 |