 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preventing viral infections with polymeric virus catchers: a novel nanotechnological approach to anti-viral therapy†
Niranjani
Sankarakumar‡
a and
Yen Wah
Tong‡
*ab
aDepartment of Chemical & Biomolecular Engineering, National University of Singapore, Singapore. E-mail: chetyw@nus.edu.sg; Tel: +65-65168467
bDepartment of Bioengineering, National University of Singapore, 21 Lower Kent Ridge Road, Singapore 119077
First published on 20th February 2013
Abstract
Infectious diseases caused by viruses are among the most vexing medical issues that we are yet to solve, even with billions of dollars being spent on drugs and vaccines to combat and prevent such infections. Finding a cure or vaccine for some of the most common or the most deadly viruses such as the influenza or the Ebola virus has been a daunting task due to their constant mutation and adaptation to our attempts. Existing therapeutics that lose their effectiveness with time due to mutations have brought about the need to consider alternative treatment approaches. To cater to the ever-increasing demand for new antiviral strategies, a novel approach of using high affinity polymeric receptors prepared by a molecular imprinting technique to “catch” viruses is presented. After three hours of mixing with the template virus, a very significant reduction in infectivity titers was observed with the virus imprinted particles. The virucidal action of the imprinted particles was rapid, dose dependent on virus and polymer concentration and occurred due to specific adsorption. The fabricated nanoparticles displayed remarkable positive anti-viral results that significantly hindered viral infections as compared to the controls. This work thus successfully demonstrates the application of miniemulsion polymerization to surface imprinting of viruses and the novel application of such imprinted materials in infectious disease treatment. The findings represent a significant breakthrough in the field of molecular imprinting and antiviral therapy and we anticipate this work to be a starting point for path-breaking research in the near future.
Viruses are pathogens that have the capability to infect all types of organisms. In particular for humans, viral infections and diseases such as AIDS, hepatitis, dengue and influenza pose significant social and economic problems in all nations. At present, the most common routes of controlling viral diseases are through vaccinations and anti-virals. Current antiviral drugs include channel and transcription blocking compounds, immunoglobulins, nucleoside analogs, polymerase, fusion, neuraminidase, proteinase and virion assembly inhibitors each targeting different stages of viral infection.1 On the other hand, viral vaccines have been very successful in preventing infections, for example, against smallpox, polio and measles, producing an enormous positive impact on world health over the last 50 years. Due to the increasing threat of pandemics and mortality rates, the need for an effective antiviral therapy is of great urgency. In this regard, we propose polymeric virus catchers prepared by molecular imprinting as a cheaper, faster, more stable, and safer mode of therapy.
Molecular imprinting is a technique that can create tailor-made artificial materials that possess comparable molecular recognition and binding capabilities as with natural biomolecules like antibodies. It involves the creation of three dimensional highly specific binding sites in a synthetic matrix (‘the lock’) that are complementary to the size, shape and functional group orientation of the ‘substrate’ molecule (‘the key’). In contrast to natural receptors, these synthetic materials, commonly made of polymers, are mechanically and chemically more robust, reusable and easily reproducible on a large scale. Thus, molecularly imprinted polymers (MIPs) have been extensively used as substitutes for antibodies or enzymes in a wide range of technologies such as separation and purification,2 sensing,3 catalysis4 and drug delivery.5 In the past few decades, the imprinting technique has been applied from small molecular templates6 to larger biomolecules like proteins7–10 and even whole cells.11 For large particles like viruses, many groups have focused on imprinting on two-dimensional (2D) surfaces mainly for virus detection.12 In addition to using the entire viral protein as a template, an epitope (a short amino acid sequence) approach has also been employed.13 For example, a molecularly imprinted film was prepared in the presence of an epitope of NS1 (nonstructural) protein, excreted from infected cells into the blood stream when there is a dengue virus infection. The binding of the epitope or whole protein to the film was studied based on quartz-crystal microbalance (QCM) frequency shifts.
To date, there have been very few studies on molecular imprinting of whole viruses as templates and most of these were on 2D virus imprints used in sensing technologies.14 Biosensors were developed that can detect viruses by integrating molecular imprinting and quartz-crystal microbalances (QCMs).15 Imprinted polymeric films were produced using the soft gel stamping technique and were found to bind the tobacco mosaic virus (TMV) target on the electrode of the QCM. Different polymers such as polyurethanes and polyacrylates were used as the polymer matrix, and the stamping technique was further applied to more viral strains such as human rhinovirus, foot and mouth disease virus and the parapox ovis virus, a harmless surrogate for smallpox virus. In addition to these 2D imprinting methods, others have focused on using polymeric gels as bulk imprinting matrices using whole viral templates.16 The imprinted gels displayed enhanced TMV uptake compared to a differently shaped non-target tobacco necrosis virus. Though this method was successful in creating polymers having cavities that mimicked the shape of TMV, the study was not extended to the prevention of viral infections.
Miniemulsion polymerization is a technique that involves the dispersion of monomers in a continuous phase with application of high shear forces. A co-surfactant is added to suppress diffusion processes occurring in the continuous phase in order to create a stable emulsion with homogeneous sizes. This technique produces a nearly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 transfer of monomer nanodroplets into regularly shaped polymerized beads as small as 50 nm.17 With excellent heat dispersal and consistent product quality this polymerization technique is highly suited for large-scale production. For molecular imprinting, miniemulsion polymerization was studied for targets such as drugs,18 amino acids19 and proteins,9 but there are no known reports on its application to viruses.
:1 transfer of monomer nanodroplets into regularly shaped polymerized beads as small as 50 nm.17 With excellent heat dispersal and consistent product quality this polymerization technique is highly suited for large-scale production. For molecular imprinting, miniemulsion polymerization was studied for targets such as drugs,18 amino acids19 and proteins,9 but there are no known reports on its application to viruses.
At first, we have successfully fabricated surface imprinted nanoparticles using a one-stage miniemulsion polymerization,9 while defining an optimized condition for its effective application to protein imprinting.10 This surface imprinting approach served to solve the limitations posed by the traditional bulk imprinting methodology in addition to making it suitable for large-scale commercial production. Following this, a two-stage core–shell miniemulsion polymerization surface imprinting approach was developed using immobilized templates7 and its general applicability was illustrated for a whole class of protein molecules.20 All of the imprinted polymeric nanoparticles displayed excellent template recognition, superior imprinting efficiency, favorable adsorption kinetics and selectivity in an aqueous medium for proteins. As viruses have an outer capsid with abundant surface proteins, this molecular imprinting technique was used to create complementary polymeric mimics of these protein-based viral shells. In order to accomplish this, a one-stage and a two-stage redox-initiated miniemulsion polymerization processes were employed and the imprinting process was carried out by the addition of whole viral particles into the polymerization system. Subsequently, the anti-viral activity of imprinted polymers was investigated both in the absence and presence of host cells. To the best of our knowledge, this is the first study on the application of imprinted materials as anti-viral agents targeted at infectious disease treatment. The novelty of this work lies in the unique combination of surface imprinting of whole viral templates using miniemulsion polymerization producing imprinted polymers that are applied as anti-virals in a model host–viral system.
Results and discussion
A series of inhibition assays of viral infection were performed in a model host–virus system in order to replicate real-time infection conditions. For this proof of concept study, a simple bacteriophage was used as the model virus as they are increasingly being used as surrogates for human viruses. Phages are viruses that infect bacteria and an fr bacteriophage (ATCC® 15767-B1™) was chosen as the model virus in this study. Fr is a small enteric phage that belongs to the family Leviviridae and is specific to its bacterial host Escherichia coli (ATCC® 19853™).21 The infective fr virion is icosahedral in shape composed of a coat protein, a maturation (or A) protein and a single-stranded RNA. The coat protein and other maturation proteins assemble to form the phage of about 23 nm in size. It has an isoelectric point ranging from 8.9 to 922 and thus the virus has an overall positive charge in the naturally occurring pH range of 6–8. Bacteriophages such as the fr phage have been recently used as “indicator viruses” owing to their similarity to many medically important human viruses such as enteroviruses in terms of size, composition, structure and morphology. In addition, the absence of replication in the environment similar to mammalian viruses that do not replicate outside the human body makes them an ideal human surrogate virus in this work. The fr phages used in our studies were prepared in-house (see Fig. S1 in the ESI†). For details on the host and phage propagation, purification and phage enumeration assay used, please refer Materials and methods section in the ESI.†
Virus surface imprinted nanoparticles (vMIPs)
A one-stage redox-initiated miniemulsion polymerization was employed in the preparation of surface imprinted nanoparticles. The selectivity of any imprinted polymer is determined by the functional monomers used with methacrylate and acrylic acid monomers being used previously for imprinting of biomolecules through non-covalent assembly.23 In the first set of experiments, nanoparticles were prepared using a hydrophobic cross-linker ethylene glycol dimethacrylate (EGDMA) mixed with different functional monomers with a fixed molar ratio of 1:4. Virus imprinted nanoparticles (vMIPs) were obtained by the addition of the whole viral template virus (fr phages) into the monomer phase prior to the polymerization step. Non-imprinted particles (NIPs) were also prepared without the addition of the template virus and were used as controls in all the experiments. X-ray photoelectron spectroscopy (XPS) was used to confirm the inclusion of the template during miniemulsion polymerization and template extraction by SDS/acetic acid (see Results section in the ESI†). The one-stage mini-emulsion polymerization system for virus surface imprinting is illustrated in Fig. 1.
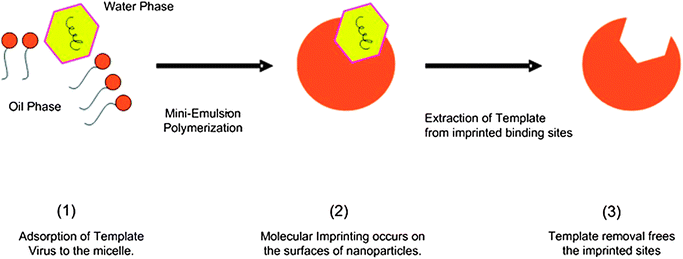 | ||
| Fig. 1 An illustration of virus surface imprinting via one-stage miniemulsion polymerization. | ||
Surface imprinted nanoparticles using immobilized virus template (viMIP)
An alternative viral imprinting method, a two-stage miniemulsion polymerization, was also used to improve viral recognition. In this strategy, the whole viral templates were covalently immobilized onto support particles before the surface imprinting was performed. The advantage of template immobilization can be realized in cases where the template molecules are not soluble in the imprinting polymerization mixture and has previously been proven to be effective for imprinting protein molecules.7 Virus immobilized molecularly imprinted polymeric nanoparticles (viMIPs) and the corresponding non-imprinted particles (iNIPs) were prepared and the results of the characterization studies have been discussed in the Results section in the ESI.†Anti-phage activity of imprinted particles
The effect of the imprinted particles on phage viability was studied. Equal volumes of phage solutions of 6300 pfu mL−1 titer were aseptically mixed with imprinted and non-imprinted particles prepared using both of the polymerization methods above. After 24 h of mixing at 4 °C, the phage concentration in the supernatant was established by standard plaque assays as described in the Experimental methods section. Controls containing only phages were also subjected to the test. The virucidal effect was expressed in terms of Log Reduction Value (LRV):| LRV = log(CINITIAL) − log(CSUPERNATANT) | (1) |
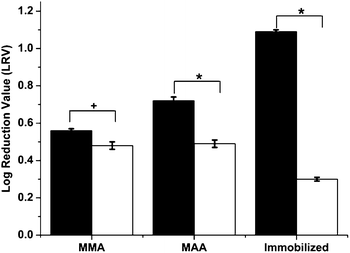 | ||
Fig. 2 Virucidal effect of nanoparticles on phage viability. Bars represent standard error. Statistical significance was denoted by +/*. Student's t-test: *p < 0.01; +p < 0.05 ( – virus imprinted particles; – virus imprinted particles;  – non-imprinted particles). – non-imprinted particles). | ||
The reason for this virucidal action was due to the adsorption of viruses during the mixing procedure and the difference in anti-viral activity can be attributed to the imprinting effect as explained below. After imprinting and template removal, the vMIPs contain template-specific imprinted cavities that exhibit greater affinity towards the template virus (fr phages). Hence, when these polymeric nanoparticles come in close proximity to the template viruses, preferential binding occurs on the particle surface resulting in an overall reduction in virus viability. As mentioned above, hydrophobic and electrostatic interactions dominate the phage binding processes of vMIPMMA and vMIPMAA respectively. On the other hand, the non-imprinted particles have been prepared in the absence of the template and hence any virus uptake by these particles would be via non-specific adsorption. Additionally, the vMIPMAA displayed 28% higher log reduction in phage titers than the vMIPMMA. This difference could be attributed to the fact that a greater amount of template molecules were imprinted during the synthesis of MAA imprinted polymers (see ESI Table 1†).
The influence of virus concentration and polymer dose on the virucidal effect of the imprinted particles using one-stage miniemulsion polymerization was also investigated (Fig. 3). Increase in polymer dose had an obvious positive influence on the virucidal activity due to higher viral adsorption by the particles. On the other hand, at an equal polymer dose, a one log increase in phage concentration resulted in 12% decrease in LRV and hence viral loading on the vMIPs. It can thus be seen that poly(MMA-co-EGDMA) and poly(MAA-co-EGDMA) virus surface imprinted nanoparticles were successfully fabricated using one-stage redox-initiated miniemulsion polymerization. In addition, the vMIPMAA particles displayed a greater virucidal effect as compared to vMIPMMA particles due to favorable hydrogen bond formation and electrostatic interactions between the template phages and particles resulting in increased recognition and specific binding in the aqueous environment.
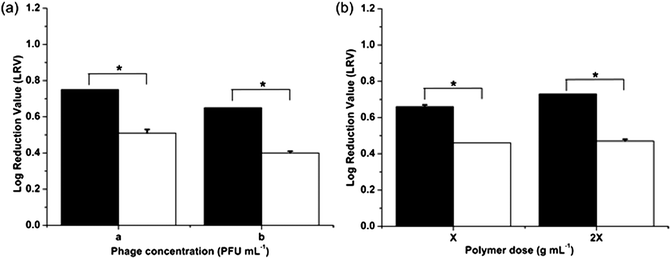 | ||
Fig. 3 Virucidal effect of nanoparticles on phage viability at different (a) phage concentrations; a: 6300 pfu mL−1; b: 63000 pfu mL−1 and (b) polymer doses; X: 0.005 g mL−1. Statistical significance was denoted by *. Student's t-test: p < 0.01 ( – vMIPMAA; – vMIPMAA;  – NIPMAA). – NIPMAA). | ||
A more interesting and effective result was observed with the alternative viral imprinting method. At equal polymer doses of 0.01 g mL−1, a maximum reduction in phage infectivity titers was obtained for the viMIPs as compared to all the polymeric particles (Fig. 2), with an LRV of 3.25 times higher for the viMIPs than for the control particles. This signifies that the two-stage imprinting strategy not only improved the virucidal behavior due to higher specific virus uptake of the viMIPs but also effectively diminished the non-specific viral binding by the iNIPs, proving the successful creation of imprinted sites on the viMIPs. Although there is no prior work on using imprinted polymers as anti-viral therapies, these results are comparable to anti-viral drug screening studies performed using phages as model viruses.24,25
RNA phages such as MS2 or fr were found to be least susceptible to natural anti-viral extracts compared to other human surrogate viruses.26 Additionally, non-enveloped phages like fr or MS2 belonging to the Leviviridae family have been illustrated to be relatively more resistant to disinfection and other treatment processes exhibiting higher survival rates than human viruses such as enteroviruses.27 This implies that the virucidal action of the imprinted particles on phages may not represent the general viral population behavior and in fact the anti-viral effect of particles will be magnified when applied to human viruses. In addition to hydrophobic interactions occurring between the functional monomer (MMA) and template virus, shape complementarity of the binding cavities was also responsible for the superior recognition, adsorption ability and virucidal activity of the viMIP. Even so, there may be concerns over the suitability of using MMA as the functional monomer instead of MAA during the second stage of polymerization, because hydrophobic interactions are generally considered less specific than other non-covalent interactions like ionic and hydrogen bonds. However, its ease of applicability in aqueous medium and its effective application in imprinting of biomolecules has been previously demonstrated.28,29 Although MAA proved to be a better functional monomer for the one-stage imprinting polymerization of free viral templates, the superior anti-viral effects of viMIPs indefinitely prove that imprinting of viruses is also possible exclusively based on hydrophobic interactions.
The kinetics of virus adsorption by the MIPs needs to be known if they are to be used as anti-viral agents, and therefore the virucidal effect of a single dose of imprinted and non-imprinted particles on phage viability was determined over time. Equal volumes of phage suspensions at a concentration of 6300 pfu mL−1 were mixed with the particles for 3, 6, 22.5, and 24 h at 4 °C. At each time point the phage concentration in the supernatant was evaluated by standard plaque assays. Controls without MIPs, containing only phages were similarly tested (Fig. 4). After just 3 h of mixing with phages, higher LRV values of 0.67 and 0.56 were observed for samples containing vMIPMAA and viMIP respectively compared to their non-imprinted counterparts. A maximum of 1.09 log reduction in phage concentration was observed after 6 h of mixing with viMIPs. There were no significant differences in the initial and supernatant concentrations of the controls without MIPs indicating that the reduction in phage titers was caused due to specific phage adsorption by the imprinted particles. The virus imprinted particles displayed fast and favorable phage titer reduction kinetics reaching equilibrium within 3 h. The phage adsorption by non-imprinted particles is non-specific resulting in a significantly lower LRV after 24 h. Additionally, it was also found that the viruses adsorbed by the imprinted particles lose their ability to infect the host cells (see Adsorbed virus infectivity study in the Results section in the ESI†).
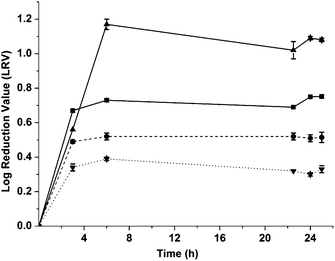 | ||
Fig. 4 Kinetic study of polymer dose response. Differences between the imprinted and control samples were statistically significant. Student's t-test: p < 0.01 ( : vMIPMAA; : vMIPMAA;  : NIPMAA; : NIPMAA;  : viMIP; : viMIP; 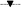 : iNIP). : iNIP). | ||
Preventing viral infections using imprinted particles
Even though the virucidal effects were successfully demonstrated, these conditions do not replicate the actual viral infection conditions. Therefore, a study on the antiviral activity of imprinted polymeric nanoparticles in the presence of host cells was conducted with a bacterial growth assay. Bacterial cells were infected with phage suspensions of a multiplicity of infection (MOI or ratio of viruses to cell numbers) of 1 and treated with a single dose of vMIP or viMIP together with the controls. The bacterial growth of these samples was monitored and this concentration (expressed in log numbers) was enumerated after 4 and 8 h of infection (Fig. 5). Phage infected bacteria incubated with imprinted particles displayed the highest bacterial concentration and faster growth rates at all time, as compared to those with and without non-imprinted particles. At a single polymer dose of 0.01 g mL−1 after 4 h of infection, a 0.59 and 1.08 log higher bacterial concentrations compared to phage infected bacteria were observed for samples containing vMIPMAA and viMIP respectively. After 8 h of infection, a 15% reduction of this higher concentration was observed for the imprinted particles. In contrast, bacterial growth with the non-imprinted particles was sluggish and displayed lower concentrations that were similar to the phage infected bacterial control samples. It can also be observed that for a single MIP dose, the virus infection was not completely blocked by the imprinted particles when compared to the non-infected controls. | ||
Fig. 5 Antiviral effect of particles in a host–virus system after a single polymer dose. Differences between the imprinted and control samples were statistically significant. Single-factor ANOVA: p < 0.01 ( : vMIPMAA; : vMIPMAA;  : NIPMAA; : NIPMAA;  : viMIP; : viMIP;  : iNIP; : iNIP;  : phage infected bacteria without particles; : phage infected bacteria without particles;  – non-infected bacteria without particles). – non-infected bacteria without particles). | ||
As a subsequent measure, a booster dose of 20% of the initial polymer concentration was applied to all test samples (see Fig. S2 in the ESI†). However, no statistically significant improvement in bacterial growth was observed even with the booster dose in all cases. Additionally, it was also found that the imprinted particles do not have any positive or negative effect on the host cell viability (see Influence of particles on bacterial growth in theResults section in the ESI†).
The fr phages infect the host E. coli in a lytic cycle and at the beginning of this infection, free phages, phages adsorbed on host cells, phage infected bacteria and uninfected cells all coexist together. Therefore, in a treatment cycle, the imprinted particles can target the fraction of extra-cellular phages. Anti-virals are currently classified based on the different stages of viral infection and the discovery of drugs that target the initial stages prior to viral attachment and entry is an effective therapeutic approach. There are very few reports on entry inhibitors with only one approved for use, which targets the surface envelope protein of HIV and prevents viral fusion.30 Instead of attacking a single protein residue or receptor, our imprinting process targets a larger portion of the viral protein shell thereby increasing the specificity and probability of whole viral surface recognition and adsorption by the imprinted particles. Assuming that the infected hosts do not divide together with the absence of multiple phage adsorption to a single infected host, the significant improvement in bacterial concentration in the presence of imprinted particles would be due to a proportional decrease in phage infectivity titers. As observed from the kinetics study, maximum phage adsorption occurred after 3 h of contact time between the imprinted particles and phages. Hence the virus imprinted particles target and adsorb the phages thereby preventing the phages from infecting the host E. coli cells.
However, phage growth is a function of two important parameters: latent period and burst size.31 Latent period is the time from the beginning of the infection cycle until cell lysis, and the number of phages produced per cell in a generation is termed as the burst size. The RNA-phages such as the fr have been reported to have a burst size ranging between 10 and 100 phages per cell with a latent period between 10 and 60 min.32 Therefore, any fraction of the phages not adsorbed by the particles can infect the cells to produce, on average, 55 phages per infected cell every 35 min thus rapidly increasing phage titers in a short time period, as would happen in a standard phage infection. As the percentage of non-infected colonies also doubles each generation, the phage replication cycle will reach an equilibrium once the bacteria approach the stationary phase. Therefore, the significantly higher percentage of non-infected colonies and 28.5% improvement in growth rate after 4 h of infection in the presence of the imprinted particles was due to the prevention of viral attachment and entry to the host cells. Although a complete cure was not observed since the rate of phage replication was much higher than the phage adsorption rate by the imprinted particles, the results strongly demonstrated the antiviral activity of the imprinted particles as well as a significant reduction of viral infection. This treatment effect is similar to common antiviral drugs and viral vaccines as any viral treatment can only aid in reducing the severity of illness but may not totally block or prevent viral infection.
This study represents a novel effort on the use of molecularly imprinted materials as anti-virals for disease treatment. It is also the first report on the application of miniemulsion polymerization to prepare nanoparticles having a specific surface imprinted memory of viruses. Virus imprinted polymeric nanoparticles were prepared using different functional monomers and surface imprinting strategies with fr phages employed as a model virus. The imprinted polymeric particles prepared were of sizes ranging from 40 nm to 570 nm for the one- to two-stage imprinting method. In comparison to standard antiviral drugs that target the viral protein residues, virus-infected cells or viral infection routes, which can be rendered non-effective by mutations, we put forward that the imprinted particles that target the whole virus shell would be an ideal antiviral agent for effective virus-capture and infection inhibition. In order to prove this non-conventional anti-viral therapy, a series of phage infectivity and infection inhibition assays were performed in the absence and presence of host cells respectively. The imprinted particles exerted greater virucidal action than the non-imprinted particles when incubated with only the phages due to interactions between the template phages and particles resulting in increased recognition, specific adsorption and virus capture. The virus imprinted particles also displayed fast phage titer reduction kinetics, reaching an equilibrium after only 3 h of contact time. Finally, a proof-of-concept study on the anti-viral action of the imprinted particles in the presence of the host cells showed that samples treated with imprinted particles had significantly higher bacterial growth rates and concentrations when dosed with phages, proving that the imprinted particles are able to significantly reduce phage infection in E. coli. Further characterization of captured virus viability together with optimization of viral concentrations, the timing of polymer doses, and host–virus ratio to maximize the antiviral activity of the particles are needed to better elucidate the mechanism of infection inhibition and ultimately testing with human viruses. The simple design, instantaneous fabrication and ease of use of the polymeric particles enable easy scale-up and large volume production suitable for pharmaceutical applications. The viral imprints may eventually be applied as antiviral agents in topical, extracorporeal or combination treatment therapies providing an economical solution to traditional antiviral strategies or even as disinfecting agents in water treatment facilities.
Experimental methods
Preparation of the virus surface imprinted nanoparticles (vMIPs)
Miniemulsion polymerization was used for the virus imprinting process as in our previous studies.9,10 Briefly, the aqueous phase was mixed with the oil phase and homogenized at 24000 rpm with a homogenizer (T25B, IKA Labortechnik, Germany) to create a miniemulsion. The model virus fr phages were added into the miniemulsion and mixed with a magnetic stirrer to allow interaction between template and functional monomers. The mixture was then diluted in a second aqueous phase and polymerized for 24 h at 40 °C (see Materials and methods section in the ESI†).
Preparation of surface imprinted nanoparticles using an immobilized virus template (viMIP)
Core–shell surface imprinted particles were prepared following the described method as in our previous work7 (for detailed procedures, see Materials and methods section in the ESI†). Briefly, poly(MMA-co-EGDMA) support beads were synthesized in a first-stage core–shell miniemulsion polymerization. Subsequently, aminolysis, aldehyde functionalization and virus immobilization were performed on the support particles. A second stage of miniemulsion polymerization was initiated to form a polymeric shell over the virus-immobilized core particles. The template virus was extracted through base hydrolysis, creating an outer imprinted shell having complementary binding cavities specific to the template virus producing virus immobilized molecularly imprinted polymers (viMIPs). The corresponding non-imprinted polymers (NIP and iNIP) to the above vMIP and viMIP respectively were prepared and washed in a similar manner, except without the addition of the template virus immobilization step. These particles served as control samples in the characterization and adsorption studies.Anti-phage activity of imprinted particles
Equal volumes of phage solutions and polymeric nanoparticle suspensions were mixed under aseptic conditions. The samples containing the phage–nanoparticle suspension were affixed onto a rotamix (RKVS, ATR Inc., Japan) and mixing was carried out at an ambient temperature of 4 °C. Positive control consisted of phage solutions without particles. At different time points from the start, the samples were centrifuged (Universal 32R, Hettich Zentrifugen, Germany) at 9000 rpm and the supernatant was titered by plaque assay to determine the supernatant phage concentration. Similar experiments were performed at different polymer doses and phage concentrations in order to study their influence on phage infectivity. The test was also carried out for the NIP and iNIP nanoparticles as control experiments.Preventing viral infections using imprinted particles
The anti-viral activity of the virus imprinted nanoparticles was studied in a model host–virus system. A fixed concentration of aerobically grown bacterial cells was inoculated with phage suspensions in the presence of vMIP or viMIP. The solutions were mixed thoroughly and incubated at 37 °C and 300 rpm. Controls included phage-infected bacteria with and without non-imprinted particles (NIP and iNIP) and non-infected bacteria without particles. At different time points, samples were drawn to determine the bacterial concentration established by colony counting on agar plates. The above experiment was further repeated to study the toxicity of the particles towards the host bacterium. The growth of non-infected bacteria with and without particles was monitored for 12 h at 37 °C.Statistical analysis
Triplicate samples were used for each sample set. OriginPro 8.1 (OriginLab, Northampton, MA) was the graphing software used. Standard deviation calculations, Student's t-tests and one-way analysis of variance (one way-ANOVA) were carried out using Microsoft Excel (Seattle, WA) for statistical comparisons between pairs of samples and multiple sample analysis.Acknowledgements
This work was made possible through grants from the Bill and Melinda Gates Foundation Grand Challenges for Global Health Explorations Grant and the National University of Singapore (R279000293272). The authors thank Dr Justin J. H. Chu (YLL School of Medicine, NUS), Dr Tan Chau Jin, Mr Anjaneyulu Kodali and Mr Lee Tian Jonathan for their helpful suggestions and thoughtful comments.References
- F. Zoulim, Antiviral Res., 2006, 71, 206–215 Search PubMed.
- H. Q. Shi, W. B. Tsai, M. D. Garrison, S. Ferrari and B. D. Ratner, Nature, 1999, 398, 593–597 CrossRef CAS.
- D. Cai, L. Ren, H. Zhao, C. Xu, L. Zhang, Y. Yu, H. Wang, Y. Lan, M. F. Roberts, J. H. Chuang, M. J. Naughton, Z. Ren and T. C. Chiles, Nat. Nanotechnol., 2010, 5, 597–601 CrossRef CAS.
- B. Sellergren, Nat. Chem., 2010, 2, 7–8 CrossRef CAS.
- F. Puoci, F. Iemma and N. Picci, Curr. Drug Delivery, 2008, 5, 85–96 Search PubMed.
- Y. Hoshino, H. Koide, T. Urakami, H. Kanazawa, T. Kodama, N. Oku and K. J. Shea, J. Am. Chem. Soc., 2010, 132, 6644–6645 CrossRef CAS.
- C. J. Tan, M. G. Chua, K. H. Ker and Y. W. Tong, Anal. Chem., 2008, 80, 683–692 CrossRef CAS.
- C. J. Tan and Y. W. Tong, Anal. Chem., 2007, 79, 299–306 CrossRef CAS.
- C. J. Tan and Y. W. Tong, Langmuir, 2007, 23, 2722–2730 CrossRef CAS.
- C. J. Tan, S. Wangrangsimakul, R. Bai and Y. W. Tong, Chem. Mater., 2008, 20, 118–127 CrossRef CAS.
- A. Seifner, P. Lieberzeit, C. Jungbauer and F. L. Dickert, Anal. Chim. Acta, 2009, 651, 215–219 CrossRef CAS.
- F. L. Dickert, P. A. Lieberzeit, O. Hayden, S. Gazda-Miarecka, K. Halikias, K. J. Mann and C. Palfinger, Sensors, 2003, 3, 381–392 CrossRef CAS.
- D. F. Tai, C. Y. Lin, T. Z. Wu and L. K. Chen, Anal. Chem., 2005, 77, 5140–5143 CrossRef CAS.
- Y. Wang, Z. Zhang, V. Jain, J. Yi, S. Mueller, J. Sokolov, Z. Liu, K. Levon, B. Rigas and M. H. Rafailovich, Sens. Actuators, B, 2010, 146, 381–387 CrossRef.
- O. Hayden, P. A. Lieberzeit, D. Blaas and F. L. Dickert, Adv. Funct. Mater., 2006, 16, 1269–1278 CrossRef CAS.
- L. D. Bolisay, J. N. Culver and P. Kofinas, Biomaterials, 2006, 27, 4165–4168 CrossRef CAS.
- C. D. Anderson, E. D. Sudol and M. S. El-Aasser, Macromolecules, 2002, 35, 574–576 CrossRef CAS.
- F. Priego-Capote, L. Ye, S. Shakil, S. A. Shamsi and S. Nilsson, Anal. Chem., 2008, 80, 2881–2887 CrossRef CAS.
- D. Vaihinger, K. Landfester, I. Krauter, H. Brunner and G. E. M. Tovar, Macromol. Chem. Phys., 2002, 203, 1965–1973 CrossRef CAS.
- N. Sankarakumar and Y. W. Tong, RSC Adv., 2013, 3, 1519–1527 RSC.
- P. Pushko, T. Kozlovskaya, I. Sominskaya, A. Brede, E. Stankevica, V. Ose, P. Pumpens and E. Grens, Protein Eng., Des. Sel., 1993, 6, 883–891 Search PubMed.
- M. Abbaszadegan, B. K. Mayer, H. Ryu and N. Nwachuku, Environ. Sci. Technol., 2007, 41, 971–977 Search PubMed.
- J. Wiklander, B. C. Karlsson, T. Aastrup and I. A. Nicholls, Anal. Bioanal. Chem., 2011, 400, 1397–1404 Search PubMed.
- I. Cock and F. R. Kalt, Pharmacogn. Res., 2010, 2, 221–228 Search PubMed.
- X. Su, A. B. Howell and D. H. D'Souza, Food Microbiol., 2010, 27, 985–991 CrossRef CAS.
- X. Su and D. H. D'Souza, Appl. Environ. Microbiol., 2011, 77, 3982–3987 Search PubMed.
- L. Mocé-Llivina, M. Muniesa, H. Pimenta-Vale, F. Lucena and J. Jofre, Appl. Environ. Microbiol., 2003, 69, 1452–1456 Search PubMed.
- A. Rachkov and N. Minoura, J. Chromatogr., A, 2000, 889, 111–118 CrossRef CAS.
- A. Mollnelli, J. O'Mahony, K. Nolan, M. R. Smyth, M. Jakusch and B. Mizaikoff, Anal. Chem., 2005, 77, 5196–5204 CrossRef CAS.
- T. Matthews, M. Salgo, M. Greenberg, J. Chung, R. DeMasi and D. Bolognesi, Nat. Rev. Drug Discovery, 2004, 3, 215–225 CrossRef CAS.
- E. L. Ellis and M. Delbruck, J. Gen. Physiol., 1939, 22, 365–384 Search PubMed.
- S. T. Abedon, T. D. Herschler and D. Stopar, Appl. Environ. Microbiol., 2001, 67, 4233–4241 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed procedures on host and virus propagation and purification and enumeration assay, synthesis of vMIPs and viMIPs have been provided. Results and discussion on the characterization of the virus imprinted particles (vMIP and viMIP), adsorbed virus infectivity study and influence of particles on bacterial growth study. Electron micrographs of the phages used and the imprinted particles and the results of the booster dose effects have been included. See DOI: 10.1039/c3tb00009e |
| ‡ Y.W.T. conceived the idea of viral imprinting. N.S. and Y.W.T. designed the experimental methods. N.S. carried out the majority of experiments and data analysis. N.S. is the main author of the paper with significant input and comments from Y.W.T. The authors declare no competing financial interests. |
| This journal is © The Royal Society of Chemistry 2013 |