 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGastric-floating microcapsules provide controlled and sustained release of multiple cardiovascular drugs†
Wei Li
Lee‡
,
Peixin
Wee‡
,
Chandra
Nugraha
and
Say Chye Joachim
Loo
*
School of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, 639798 Singapore. E-mail: joachimloo@ntu.edu.sg; Fax: +65 6790-9081; Tel: +65 6790-4603
First published on 8th January 2013
Abstract
Floating polymeric microcapsules that simultaneously entrap multiple drugs were prepared using a solid/water/oil/water emulsion solvent evaporation method, based on harnessing interfacial phenomena and manipulation of the solvent removal process. The fabricated microcapsules exhibited excellent buoyancy in simulated gastric fluid and provided controlled and sustained release of multiple drugs for up to 24 h, thus revealing their potential as a rate-controlled oral drug delivery system.
Despite tremendous advancement in drug delivery, oral administration still remains the most convenient and preferable means of drug delivery due to its low cost, ease of administration and flexibility in formulation. However, conventional oral dosage forms (e.g. tablets, capsules) have several inherent limitations, including a limited control over drug release rates,1 lack of complete absorption of oral drugs due to variable and short gastrointestinal (GI) transit time,2 and high fluctuation in plasma drug levels due to multiple dosing frequency. Dosage forms with a prolonged gastric residence time (GRT) have been used to circumvent some of these problems.2,3 Gastro-retentive delivery systems have the potential to improve bioavailability and reduce drug wastage that show preferential solubility, stability or absorption in the stomach or the proximal part of the GI tract.2
Several approaches to prolong the GRT have been proposed based on various mechanisms, such as buoyancy (floating drug dosage systems, FDDS),4 swelling/plug type,5 high density6 and mucoadhesion.7 The constant renewal and high turnover of mucus may reduce the effectiveness of mucoadhesive systems. Unlike the retention of swelling systems and high density systems in the pylorus and the pyloric antrum, respectively, floating systems do not adversely affect the motility of the stomach. As such, FDDS have been extensively studied to retain drug forms in the stomach. With this, various floating systems have thus been developed using different materials (e.g.lipids,8hydroxypropyl methylcellulose (HPMC)-based hydrodynamically balanced systems9–11 and alginate beads containing gas-generating agents12) for controlled delivery of drugs.
While conventional oral drug delivery devices involve monolithic systems whereby only one drug is loaded, basic economics and patient compliance would favor the realization of a single microcapsule that simultaneously entraps and releases multiple drugs in a controlled manner. A single carrier or “tablet” that encapsulates multiple drugs would allow for a reduction in the number of oral tablets to be taken, as well as in the dosing frequency (i.e. pill burden), which in turn may improve patient compliance. Drug–drug interactions can also be avoided if individual drug substances are further protected within particles. Such a technology could potentially be used to treat diseases that require multiple drug combinations, such as HIV, cancer, tuberculosis and lupus. Another therapeutic area whereby floating microcapsules could be explored is for the treatment of chronic gastritis and peptic ulcers.13
Though floating single drug dosage forms have already been well-established,4,12,14 there is a dearth of studies demonstrating the encapsulation of particles loaded with different drugs, while maintaining good floatability. It is thus hypothesized that the encapsulation of drug-loaded particles within a larger hollow capsule would provide good buoyancy, along with controlled release of multiple drugs. The addition of oil into the capsule shell can provide this extra buoyancy.15,16 The manipulation of capsule/particle parameters, such as shell thickness, particle sizes, and polymer types can be tuned to control drug release rates. Herein, we first devised a modified emulsion solvent evaporation technique to encapsulate smaller drug-loaded particles (nano/micron-sized) within a larger floating microcapsule. In this present technique, high compression forces and elevated temperatures are not required, as compared with other common methods of producing floating tablets.17 In addition, vegetable oil and particles loaded with a variety of drugs can be concurrently encapsulated into a single microcapsule through a single step, thus improving process yield and quality control. In this study, three model drugs (i.e. metoprolol tartrate salt, metformin HCl, fenofibrate) were encapsulated within larger microcapsules. These drug choices would allow this delivery system to be used for the treatment of chronic cardiovascular diseases, i.e. high blood pressure, blood glucose and cholesterol.18
Multidrug-loaded gastric-floating microcapsules were prepared using the solid/water/oil/water (S/W1/O/W2) emulsion solvent evaporation method, as illustrated in Scheme 1. Pre-synthesized drug-loaded particles (either sub-micron or nano-sized) were first suspended in an aqueous phase. Subsequently, the particles/water suspension (S/W1) was introduced into a polymer solution (O) to form the S/W1/O phase before the addition of a vegetable oil (i.e. olive oil). The polymer solution (O) was prepared by dissolving the polymer in a volatile organic solvent (i.e.dichloromethane (DCM)). The resultant S/W1/O phase was poured into the water bath (W2) containing both poly(vinyl alcohol) (PVA) (as a surfactant) and DCM, and emulsified to form a S/W1/O/W2 emulsion using an overhead stirrer. After which, the hardening of the shell of the capsule was achieved under reduced pressure using a rotary evaporator with an addition of extra surfactant solution. Finally, the microcapsules produced were centrifuged, rinsed with de-ionized water, lyophilized and stored in a desiccator.
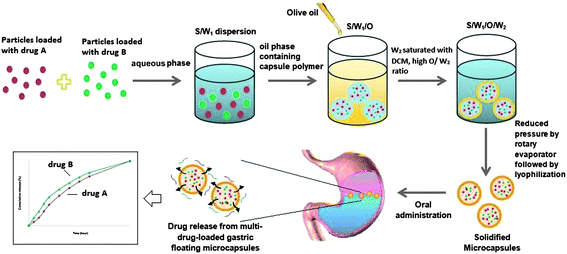 | ||
| Scheme 1 Schematic illustration of the encapsulation procedure for the preparation of floating microcapsules and the release of multiple drugs within the gastric region. | ||
The addition of olive oil to the shell of the microcapsule while achieving the ability to load drug-loaded particles together into the hollow cavity of the microcapsule is certainly not trivial. The inclusion of olive oil would unsettle the original formulation devised by Khung et al.,19 leading to a change in capsule structure, morphology, encapsulation capability of the microcapsule, and particle localization within the microcapsule. To verify this point, a reference set of microcapsules (capsule R) with the inclusion of olive oil (a non-solvent for capsule polymer) was fabricated through a conventional method.19 In this conventional method, a higher amount of PVA solution (without DCM) was used during emulsification and no rotary evaporator was employed to harden the capsules.19 The cross-sectional view (Fig. S1†) of the microcapsules reveals that the encapsulated particles were agglomerated and embedded within the capsule shell. This is believed to be caused by a change in the solvent quality of the oil phase (i.e. poly(D,L-lactide-co-glycolide, 53/47) (PLGA 53/47) capsule polymer, DCM and olive oil), and the subsequent formation of an adhesive gel layer at the inner water–oil interface. As PLGA is insoluble in olive oil, the solvent quality of the oil phase is reduced and PLGA is not completely solubilized during DCM extraction and evaporation. On the other hand, the surface activity of PLGA has been demonstrated by the fact that PLGA reduces the interfacial energy between the water and oil phases.20 As such, the W/O/W emulsion is in a kinetically stable state due to the absorption of PLGA molecules to the inner and outer water–oil interfaces and the presence of PVA as a surfactant at the water–oil interface. As the solvent quality decreased, PLGA molecular chains at the interface would form an adhesive gel layer, which has been similarly reported in copolymer-stabilized double emulsions.21,22 Encapsulated particles within the inner water phase subsequently adhered to this gel layer, resulting in the localization of agglomerated particles within the shell of the microcapsule. The embedment of drug-loaded particles within the shell would cause the formation of pores in the shell matrix upon drug release, which would then decrease the buoyancy as a result of higher water ingress.1 In addition, hydrophilic drug-loaded particles embedded within the shell of the capsule would promote water influx and decrease the buoyancy.15
Tweaking some process parameters therefore allows for the successful fabrication of olive oil-containing microcapsules, as evidenced from the scanning electron microscopy (SEM) (Fig. 1a and b). The size of the microcapsule was 621 ± 91 μm. Close-up examination of cross-sectioned microcapsules showed many smaller particles (6.8 ± 2.2 μm) on the interior walls of the microcapsules. The formation efficiency of this capsule structure was estimated to be at ∼100% (Fig. S2†). In order to encapsulate the drug-loaded particles within the hollow cavities of the floating capsules, diffusional mass transport processes of solvent molecules have to be manipulated. DCM diffuses into the continuous aqueous phase before evaporating at the water–air interface during the solvent removal process. The solubility of DCM in water is about 1–2% v/v,23 therefore, increasing the oil-to-water ratio and saturating the continuous aqueous phase with DCM during the emulsification step would result in a decrease in the solvent extraction rate. This would reduce the probability of the formation of an adhesive gel layer as the solvent quality remains unchanged. Subsequently, during the hardening process, the solvent removal rate can be accelerated through the use of a rotary evaporator under reduced pressure, and with the addition of extra water. As a result, an increased removal rate of DCM from the emulsion droplets to the external aqueous phase reduces the time allowed for the adhesion process between the encapsulated particles and inner water–oil interface to take place. This would give rise to a true encapsulation of particles within the microcapsule, instead of the embedment of these particles within the microcapsule shell.
 | ||
| Fig. 1 SEM images of (a) cross-sectional view and (b) close-up view of a microcapsule where smaller particles were encapsulated within the hollow cavity. | ||
To mimic the in vivo gastric residence behavior, the floating ability of the microcapsules was investigated in simulated gastric fluid (SGF) at 37 °C under magnetic stirring at 250 rpm for 24 h.15 The buoyancy profiles are shown in Fig. 2. Olive oil-containing microcapsules (596 ± 84 μm) were able to float continuously for over 24 h, while almost all the non-oil-loaded microcapsules (618 ± 93 μm) (refer to Fig. S3† for morphological analysis) sank. In contrast to the conventional gas-generating floating devices,24 these oil-loaded microcapsules floated immediately upon contact with the SGF, without any lag time in buoyancy. This excellent floating property could be due to the combined effects of the capsule's low density provided by both the hollow cavity (Fig. S4†) and the hydrophobic nature (i.e. reduced medium uptake) of the capsule shell with olive oil.1,16 It was found that larger microcapsules (596 ± 84 μm) allowed for better floatability as the buoyancy decreased with decreasing capsule size due to reduced cavity volume (Fig. S5†).25 Also, it was observed that cracks on the surface of smaller capsules (224 ± 53 μm) would further decrease their buoyancy (Fig. S5†). In addition, it was also noted that drug incorporation and subsequent release of drugs did not impair the buoyancy of the microcapsules.
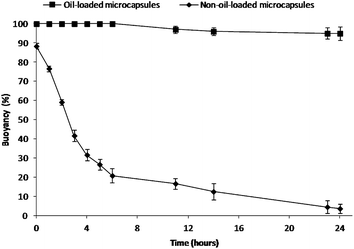 | ||
| Fig. 2 Percent buoyancy vs. time profiles of the present floating formulation compared with the non-oil-loaded microcapsules. | ||
Metoprolol tartrate-loaded PLGA 75/25 particles (4.9 ± 1.8 μm) and fenofibrate-loaded PCL particles (9.7 ± 3.8 μm) together with free metformin HCl were encapsulated into the microcapsules (596 ± 84 μm) for the drug release study conducted in SGF medium. Comparable sizes were also observed for the other microcapsule groups. Hydrophilic metformin HCl can be easily dissolved in water, and thus allowed for direct encapsulation into the microcapsule.12 The SEM image (Fig. S6†) shows similar results whereby the drug-loaded particles and free metformin HCl were found to be encapsulated within the hollow cavities of the microcapsules. For the microcapsules, the actual loading of metoprolol tartrate, fenofibrate and metformin HCl were 0.82 ± 0.02 wt%, 1.89 ± 0.03 wt%, 10.92 ± 0.16 wt%, respectively. The amount of each loaded drug in the floating microcapsules (800 mg) was determined at a dose of approximately 10% equivalent to marketed formulations, i.e. 5 mg for metoprolol tartrate, 20 mg for fenofibrate and 50 mg for metformin HCl. It is noted that delivery devices with prolonged GRT would increase the bioavailability of drugs, thus requiring a lower dosage.9,25 Furthermore, when used for preventive measures, dosages are relatively lower compared to therapeutic applications.
The polymer type of the drug-loaded particles was selected such that 100% release from free particles was obtained within 1 h in SGF at 37 °C. When these particles are encapsulated within the hollow cavities of the microcapsules, it is hypothesized that the shell of the microcapsule would serve as an additional diffusion barrier, thus limiting initial burst and controlling release rates. It was observed that PLGA 75/25 particles alone exhibited nearly 100% metoprolol tartrate release within 1 h (Fig. S7a†). The fast release could be due to the high drug loading (∼25% w/w) and the high aqueous solubility of the drug,26 which caused a huge burst release from the PLGA particles upon SGF ingress. On the other hand, there was 100% release of fenofibrate from poly(caprolactone) (PCL) particles within 1 h, whereas fenofibrate release from PLGA 75/25 particles proceeded relatively slower (cumulative release of 25% for 24 h) (Fig. S7b†). The more rapid release of hydrophobic fenofibrate from PCL particles could be due to the highly flexible rubbery state of PCL with a glass transition temperature of −60 °C. At the drug release condition of 37 °C and surrounded by SGF, PCL chains are in a highly mobile state with increased free volume, which allows for better dissolution of the hydrophobic drug and promotes further release.27,28
The drug-loaded particles, in their respective polymer types, were now encapsulated within the microcapsules. The release profiles of fenofibrate (log P = 5.2, highly hydrophobic),29 metoprolol tartrate salt (log P = 1.6, hydrophilic)26 and metformin HCl (log P = −2.64, highly hydrophilic)30 from non-floating marketed tablets and floating microcapsules of various shell polymer types are shown in Fig. 3. From the release studies, each marketed tablet exhibited relatively rapid drug release, while the microcapsules provided a more controlled release. The suppression of release from microcapsules was highly evident for fenofibrate (Fig. 3a). It is noted that the dissolution process of fenofibrate occurs with great difficulty due to its neutral and lipophilic nature.29 This fact highlights the importance of a substantial amount of SGF uptake required for fenofibrate release. The marketed fenofibrate tablets (capsule) (Lipanthyl®) allowed for direct access of drug powder to SGF after the rapid dissolution of the gelatin shell, thus leading to a huge burst release upon quick SGF influx. The shell of the microcapsule, on the other hand, would limit the rate of SGF influx, and serve as a rate-limiting layer that impedes rapid drug loss and controls drug diffusion rates. However, for the release of metoprolol tartrate and metformin HCl from PLGA 53/47 microcapsules (Fig. 3b and c), the retardation of release by the PLGA shell was observed to be insignificant. This result could be due to the fact that the relatively hydrophilic PLGA 53/47, arising from its fully amorphous structure, is permeable to water-soluble molecules.31 As such, metoprolol tartrate and metformin HCl drug molecules diffused rapidly through PLGA. Changing the polymer type of the capsule's shell had an effect on drug release kinetics (Fig. 3a–c). It is clear that the semi-crystalline morphology and relatively hydrophobic nature of the PLLA shell retard appreciable medium influx and limits drug diffusion,32 thus giving rise to the slowest drug release rates. In contrast to the pure PLLA shell, the incorporation of PCL nanoparticulates (25 wt%) into the PLLA shell (Fig. S8†) was found to accelerate the drug release and to result in a more consistent release (Fig. 3), which would be advantageous in achieving sustained release. The PLLA–PCL shell was less dense due to the presence of rubbery PCL, which resulted in a relatively faster release.28
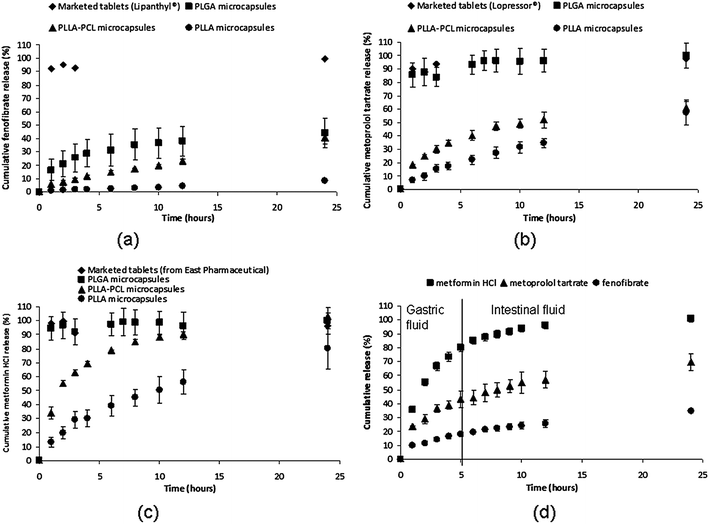 | ||
| Fig. 3 Release profiles of (a) fenofibrate, (b) metoprolol tartrate salt and (c) metformin HCl from various floating microcapsule groups and marketed tablets in SGF. (d) Release profiles of three different drugs from a single PLLA–PCL microcapsule in SGF for 5 h followed by release into SIF at 37 °C. It should be noted that the release profile of each drug from microcapsules had been derived from the microcapsules that co-encapsulated three different drugs. | ||
The drug release profiles of three different drugs from a single PLLA–PCL microcapsule for 5 h in SGF (pH 1) followed by release in simulated intestinal fluid (SIF, pH 6.8) at 37 °C are shown in Fig. 3d. Multiple drugs, each with a different hydrophilicity, were observed to be released from the microcapsules in a sustained manner for 24 h. It has been reported that floating microspheres can exhibit prolonged GRT of 5 h in humans.14,33 The release data demonstrate that certain amounts of drugs would be released in the stomach depending on the gastric emptying time and the remaining incorporated drugs would be released in the intestinal region, as similarly reported by other groups.4,25 The release of metformin HCl was observed to be the fastest. This could be explained by the fact that the highly hydrophilic drug was encapsulated into the capsule in the form of free drug, while the encapsulation of fenofibrate and metoprolol tatrate within the particles was deemed to further control the release kinetics. For the release from drug-loaded particles within microcapsules, metoprolol tartrate was released more rapidly as a result of its highly water-soluble nature, which facilitates drug dissolution, in comparison with the fenofibrate release. Surface and cross-sectional views of microcapsules (Fig. S9†) show that the capsule shell and encapsulated particles still remained intact after 24 h in vitro, suggesting diffusional drug release.32 In addition, the absence of metformin HCl free drug was observed within the cavities of the microcapsules after 24 h, which concurs with its complete release. The main absorption site of metformin HCl was reported to be the proximal small intestine.12 Thus, its substantial release from a gastroretentive system in the upper GI tract (5–7 h) would clearly be advantageous. On the other hand, although a relatively slow release was observed for fenofibrate and metoprolol tartrate within the first 5 h (in gastric fluid), the remaining drugs after gastric emptying process would still be absorbed throughout the intestinal tract.34,35 For a drug with low release rate (usually for hydrophobic drugs) or high dosage demand, microcapsules given along with a small loading dose (free drug) would provide a higher plasma drug concentration over a prolonged period.4 It is also worth mentioning that the integrity of the shell of microcapsules exiting the stomach may be impaired, which would then accelerate the drug release in the intestinal fluid. Therefore, part of the future plan is to analyze the GI transit behavior of the dosage form in vivo to examine the relationship between GI transit and drug absorption.
In conclusion, the microencapsulation of drug-loaded particles within the hollow cavity of a floating polymer-based microcapsule through a modified emulsion solvent evaporation method was demonstrated. Simply by designing and tailoring the delivery system, multiple drugs can be loaded, and their release profiles can be fine-tuned accordingly. As a result, this system may be a promising platform to deliver multiple drugs orally, while providing controlled and sustained release, which would be further evaluated in vivo as part of the future studies.
Acknowledgements
The authors would like to acknowledge the financial support from the Agency for Science, Technology and Research (A*STAR) (Project no: 102 129 0098), and the National Medical Research Council (NMRC/EDG/0062/2009 and IRG10may017).References
- Y. Sato, Y. Kawashima, H. Takeuchi and H. Yamamoto, Eur. J. Pharm. Biopharm., 2004, 57, 235 CrossRef CAS.
- B. N. Singh and K. H. Kim, J. Controlled Release, 2000, 63, 235 CrossRef CAS.
- A. Streubel, J. Siepmann and R. Bodmeier, Expert Opin. Drug Delivery, 2006, 3, 217 CrossRef CAS.
- N. J. Joseph, S. Lakshmi and A. Jayakrishnan, J. Controlled Release, 2002, 79, 71 CrossRef CAS.
- J. Chen, W. E. Blevins, H. Park and K. Park, J. Controlled Release, 2000, 64, 39 CrossRef CAS.
- S. J. Hwang, H. Park and K. Park, Crit. Rev. Ther. Drug Carrier Syst., 1998, 15, 243 Search PubMed.
- Y. Liu, J. Zhang, Y. Gao and J. Zhu, Int. J. Pharm., 2011, 413, 103 CrossRef CAS.
- S. K. Jain and A. Gupta, AAPS PharmSciTech, 2009, 10, 1128 CrossRef CAS.
- J. Ali, S. Arora, A. Ahuja, A. K. Babbar, R. K. Sharma, R. K. Khar and S. Baboota, Eur. J. Pharm. Biopharm., 2007, 67, 196 CrossRef CAS.
- L. Danki, A. Sayeed and S. Salger, Int. J. Pharma Bio Sci., 2010, 1, 1 Search PubMed.
- C. Narendra, M. Srinath and G. Babu, AAPS PharmSciTech, 2006, 7, 23 CrossRef.
- A. Nayak, S. K. Jain and R. S. Pandey, Mol. Pharmaceutics, 2011, 8, 2273 CrossRef CAS.
- L. Yang, J. Eshraghi and R. Fassihi, J. Controlled Release, 1999, 57, 215 CrossRef CAS.
- Y. Sato, Y. Kawashima, H. Takeuchi, H. Yamamoto and Y. Fujibayashi, J. Controlled Release, 2004, 98, 75 CrossRef CAS.
- Y. D. Tang, S. S. Venkatraman, F. Y. C. Boey and L. W. Wang, Int. J. Pharm., 2007, 336, 159 CrossRef CAS.
- J. H. Lee, T. Park, Y. B. Lee, S. C. Shin and H. K. Choi, J. Microencapsulation, 2001, 18, 65 CrossRef CAS.
- A. Singh, S. Singh, S. Puthli and R. Tandale, WO Pat., WO/2008/062440, 2008.
- N. J. Wald and M. R. Law, Br. Med. J., 2003, 326, 1419 CrossRef CAS.
- Y. L. Khung, W. L. Lee, K. L. Chui, Y. Liu, M. P. Lim, C. L. Huang and S. C. J. Loo, Adv. Healthcare Mater., 2012, 1, 159 CrossRef CAS.
- M. H. Lee, K. C. Hribar, T. Brugarolas, N. P. Kamat, J. A. Burdick and D. Lee, Adv. Funct. Mater., 2012, 22, 131 CrossRef CAS.
- H. C. Shum, Y. Zhao, S. H. Kim and D. A. Weitz, Angew. Chem., Int. Ed., 2011, 123, 1686 CrossRef.
- H. C. Shum, E. Santanach-Carreras, J. W. Kim, A. Ehrlicher, J. Bibette and D. A. Weitz, J. Am. Chem. Soc., 2011, 133, 4420 CrossRef CAS.
- S. R. Mao, Y. Shi, L. Li, J. Xu, A. Schaper and T. Kissel, Eur. J. Pharm. Biopharm., 2008, 68, 214 CrossRef CAS.
- L. Meka, B. Kesavan, K. M. Chinnala, V. Vobalaboina and M. R. Yamsani, AAPS PharmSciTech, 2008, 9, 612 CrossRef CAS.
- Y. Sato, Y. Kawashima, H. Takeuchi and H. Yamamoto, Int. J. Pharm., 2004, 275, 97 CrossRef CAS.
- L. Narasimham and V. D. Barhate, Eur. J. Chem., 2011, 2, 36 CrossRef CAS.
- L. L. Lao, S. S. Venkatraman and N. A. Peppas, Eur. J. Pharm. Biopharm., 2008, 70, 796 CrossRef CAS.
- W. L. Lee, C. Loei, E. Widjaja and S. C. J. Loo, J. Controlled Release, 2011, 151, 229 CrossRef CAS.
- M. Van Speybroeck, R. Mellaerts, R. Mols, T. D. Thi, J. A. Martens, J. Van Humbeeck, P. Annaert, G. Van den Mooter and P. Augustijns, Eur. J. Pharm. Sci., 2010, 41, 623 CrossRef CAS.
- A. Liu and S. P. Coleman, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2009, 877, 3695 CrossRef CAS.
- X. Wang, S. S. Venkatraman, F. Y. C. Boey, J. S. C. Loo and L. P. Tan, Biomaterials, 2006, 27, 5588 CrossRef CAS.
- A. Frank, S. K. Rath and S. S. Venkatraman, J. Controlled Release, 2005, 102, 333 CrossRef CAS.
- N. Ma, L. Xu, Q. Wang, X. Zhang, W. Zhang, Y. Li, L. Jin and S. Li, Int. J. Pharm., 2008, 358, 82 CrossRef CAS.
- T. Zhu, J. C. Ansquer, M. T. Kelly, D. J. Sleep and R. S. Pradhan, J. Clin. Pharmacol., 2010, 50, 914 CrossRef CAS.
- G. Jobin, A. Cortot, J. Godbillon, M. Duval, J. P. Schoeller, J. Hirtz and J. J. Bernier, Br. J. Clin. Pharmacol., 2012, 19, 97S CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, experimental methods, and sample characterization details. See DOI: 10.1039/c2tb00495j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2013 |