Gold: a versatile tool for in vivo imaging
Vanessa W. K.
Ng
a,
Romain
Berti
b,
Frédéric
Lesage
*b and
Ashok
Kakkar
*a
aDepartment of Chemistry, McGill University, 801 Sherbrooke St. W., Montréal, Canada. E-mail: ashok.kakkar@mcgill.ca; Fax: +1 514-398-3797; Tel: +1 514 389-6912
bÉcole Polytechnique, 2900, boul Édouard-Montpetit, Montréal, Canada. E-mail: frederic.lesage@polymtl.ca; Fax: +1 514-340-4611; Tel: +1 514 340-4711
First published on 24th August 2012
Abstract
Imaging for diagnostics or for evaluating the efficacy of a particular drug constitutes a key challenge, and a topical area of research in nanomedicine. There has been a tremendous effort devoted to the evaluation of a variety of contrast agents, and gold nanomaterials due to their inherent and geometrically induced optical properties, have offered significant potential for in vivo imaging. The gold based nanostructures that are most commonly employed for biological imaging include nano-spheres, -rods, -shells, -cages and -stars. This feature article provides an overview of the current state of research in utilizing these gold nano-architectures in imaging, with particular emphasis on modalities such as two-photon luminescence, computed tomography, optical coherence tomography, near infrared and photoacoustic imaging.
 Vanessa W. K. Ng | Vanessa Ng received her BASc degree in Honours Nanotechnology Engineering at the University of Waterloo in 2010. She is currently pursuing a Master's degree in Chemistry at McGill University under the joint supervision of Dr Ashok Kakkar, Dr Frédéric Lesage, Dr Eric Rhéaume and Dr Jean-Claude Tardif. Her interests are in using gold nanoshells for applications in biology. |
 Romain Berti | Romain Berti is a post-doctoral fellow at the École Polytechnique de Montréal in Frédéric Lesage's Molecular and Optical Imaging Laboratory. He received his PhD in Paris 6 at the Parametric Imaging Laboratory with Wladimir Urbach and Nicolas Taulier. His present interests are in imaging using gold nanoparticles and diffuse optical imaging. |
 Frédéric Lesage | Dr Frédéric Lesage is an Associate Professor in the Department of Electrical Engineering at École Polytechnique Montréal. He did his PhD in theoretical physics at Université Paris-Sud and Saclay, and subsequently did a post-doctoral fellowship at USC. He then worked for Advanced Research Technologies in molecular imaging. His research interests include molecular imaging based on fluorescence and photoacoustic, and brain imaging using hemodynamic signals and diverse techniques; diffuse optical imaging, OCT, two-photon microscopy and intrinsic optical imaging. |
 Ashok Kakkar | Dr Ashok Kakkar is an Associate Professor in the Department of Chemistry at McGill University, Montreal, Canada. He obtained training in Chemistry under the directions of Professor Todd B. Marder (PhD, University of Waterloo), Professor The Lord Lewis (NSERC Post-doctoral Fellow, University of Cambridge) and Professor Tobin Marks (NSERC Post-doctoral Fellow, Northwestern University). His research interests include developing methodologies to complex architectures (hyperbranched macromolecules including dendrimers, miktoarm polymers, metal nanoparticles), and studying their efficacy in drug delivery and theranostics. |
Introduction
As a precious metal, gold has played a significant role in human civilization throughout history, both in its bulk form and as a nanomaterial. For example, gold nanomaterials have been incorporated into human inventions from as early as the creation of the 4th century Lycurgus cup.1 The beauty of this cup lies in the duality of transmitted red and reflected green light, an effect that was created using embedded gold nanoparticles. At the time, this was likely made without a true scientific explanation of how or why this phenomenon was occurring. It was not until approximately a hundred and fifty years ago that scientific inquiry of gold materials began, initiated by pioneers such as Faraday2 and Mie.3 Their work founded the fundamentals of colloidal gold chemistry used today. Since then, the scientific investigation of gold has evolved into the ability to precisely control the synthesis of gold nanomaterials in terms of size, shape, and uniformity. Research groups have now developed gold nanomaterials beyond simple improvements of the original colloidal gold nanospheres (AuNS), leading to the creation and control of increasingly complex structures, such as gold nanoshells (AuNSh),4,5 nanobowls,6 nanorods (AuNR),7,8 nanorice,9 bipyramids,10 tetrahedras,11 octahedras,12 cuboctahedras,13 nanocubes,14 nanocages (AuNC),15 nanostars (AuNSt)16 and nanocrescents,17 (Fig. 1).18 | ||
| Fig. 1 Gold nanostructures including (a) gold nanospheres (b) gold nanorods, (c) gold bipyramids, (d) gold nanorods with silver shells, (e) nanorice, (f) SiO2/Au nanoshells (inset is a hollow nanoshell), (g) nanobowls with bottom cores), (h) spikey SiO2/Au nanoshells, (inset is a gold nanostar), (i) gold tetrahedra, octahedra and cuboctahedra, (j) gold nanocubes, (k) silver nanocubes (insets are gold nanocages), (l) gold nanocrescents. Reproduced from ref. 18. | ||
The motivation behind manipulating dimensional and conformational properties lies in the intended applications of these materials. The nanoscale sizes of gold structures offer a substantial increase of available surface area for modifications, while remaining dimensionally comparable to biological units. Coupling this with the fact that elemental gold is known to be biologically inert19 and resistant to oxidation,20 it is justified that the primary focus of gold nanomaterials is for applications in biology. While a great deal of research has employed gold nanomaterials as a biocompatible platform for functionalization in biosensing or therapeutic applications, there is also a significant interest in taking advantage of their other unique characteristics, namely, their optical properties. High absorption and scattering cross-section features can be easily designed into gold nanomaterials, both of which can be attributed to a phenomenon known as surface plasmon resonance (SPR).21 SPR occurs due to the interaction between the surface conduction electrons and applied electromagnetic waves, producing a collectively amplified coherent resonance. Optical tunability of these resonances enables researchers to exploit the use of what is known as the “biological window” (650–900 nm), the wavelength range in which the least blood and tissue attenuation occurs. Thus, a high visible contrast can be obtained with particles absorbing in this region.22 This is especially important for imaging targeted areas located deep within biological tissues that cannot be imaged by simpler, superficial methods.23 In addition, the high absorption of these nanomaterials gives rise to high photothermal conversion efficiencies.24 As these characteristics can now be manipulated by defining the size and shape of the nanomaterial, it allows appropriate selectivity of a system for a particular application.
The ability to combine multiple functions related to therapy is amongst one of the potential benefits of modern day nanoscience. In particular, the ability to quantitatively and qualitatively visualize the effects of a delivered therapeutic agent in vivo has been as important as the development of the therapy itself. As such, this feature article aims to identify and consolidate current research that has incorporated the use of gold nanomaterials in imaging for in vivo studies. Gold nanomaterials have been functionalized with a range of surface modifiers that permit further imaging enhancements; however, this review will limit its scope to those imaging modalities that solely utilize the properties of gold nanomaterials themselves. The syntheses and optical properties of gold nano-spheres, -rods, -shells, -cages and -stars are first described. The role of these nanostructures is then discussed in what the authors deem as important advances in the field of in vivo imaging using gold nanostructures. They are presented according to the imaging modality used, including two-photon luminescence (TPL), computed tomography (CT), near infrared absorption (NIR), optical coherence tomography (OCT), and photoacoustic tomography (PAT).
Gold nanomaterial synthesis
Nanospheres (AuNS)
Gold nanospheres, which are commonly referred to as colloidal gold, have the longest history and thus, are the most well established of gold nanomaterials used today. The most frequently used synthetic route, now known as the “Turkevich method”, was introduced in 1951 (ref. 25) in which Turkevich modified a previously published method by Hauser and Lynn.26 Using a sodium citrate reduction of solvated aurochloric acid, monodisperse AuNS of approximately 15 nm diameter were synthesized. This methodology has also formed the basis of subsequent research regarding many different shapes of gold nanomaterials that can now be constructed. Though most synthetic schemes use the gold salt method, there are two important synthetic variants that have been explored in the modifications: the chosen reducing agent, and the stabilizing ligand. While Turkevich's method used citrate as both the reducing and stabilizing agent,25 the evolution of this method now incorporates the use of other reducing agents such as trisodium citrate or sodium borohydride (NaBH4), and a range of stabilizing ligands.27 Fine-tuning of these parameters is the primary means by which a wide range of nanospheres with variable sizes and uniformity can now be synthesized. Frens published an in depth study in 1973, refining the Turkevich method,28 while an update was published by Kimling et al. in 2006 that expanded on this work.29 Another regularly used synthetic method for nanospheres was introduced in 1994, using a high yielding two-phase water–toluene method with a thiol as the stabilizing ligand, and tetraoctylammonium bromide (TOAB) as the phase transfer agent.30 This “Brust–Schiffrin” method, as it is now referred to, was an important achievement as it provided a direct “one-pot” alternative for functionalization of a gold colloid. Previously, AuNS functionalization required a separate ligand exchange step after synthesizing the nanospheres by replacing the original colloidal stabilizing ligand with the desired ligand. As an alternative, this improved method uses the final decorating ligand as the stabilizing ligand during synthesis. This is achieved by using a thiol terminated ligand and exploiting strong thiol–gold interactions. This also means that any desired ligand, once thiolized, can be a viable candidate in the Brust–Schiffrin biphasic scheme.30Optically, it is well known that solid gold nanospheres produce a single resonance typically around 520 nm,31 which can be tuned to a maximum of approximately 600 nm over size variations of 9–99 nm diameters.32 This is the major disadvantage to the use of gold nanospheres in biological applications, as contrast is significantly decreased outside the biological window.15,22
Nanorods (AuNR)
The second most well established member of the gold nanostructure family is the gold nanorod. While energy conservation dictates that small particle synthesis will lean towards a spherical conformation, the incorporation of certain additives can induce an elongated growth direction, in this case, forming nanorods. Two of the most common methods for the synthesis of nanorods are by either electrochemical means or seed mediated growth. In the electrochemical method, which was published and refined by Wang et al.,33,34 the synthesis requires the use of a sacrificial gold metal anode plate and a platinum cathode, submerged in an electrolytic solution. The solution includes a stabilizing cationic surfactant, cetyltrimethylammonium bromide (CTAB), a co-surfactant, TOAB which is responsible for inducing rod formation, acetone to facilitate the TOAB incorporation, and cyclohexane to augment the formation of micellular CTAB rods. The electrolysis initiates the formation of AuBr4− ions from the gold anode which can then complex with the CTAB surfactant and eventually become reduced at the platinum cathode. Sonication is also required to separate nanorods from the cathode plate. To specifically control the size of the AuNR, a silver plate is incorporated during the reaction as a source of silver ions, where concentration and release rate are the determining factors.34Undoubtedly, the most popular means used today to produce gold nanorods is the seed-mediated growth method, which began as early as 1989.35 The development of this method has led to its current form which is simple and efficient, ensuring uniformity, high yields and control over sizes and aspect ratios (defined below). The synthesis of the initial seed is essentially that of small gold nanospheres (∼3 to 4 nm), where a gold salt is reduced by a strong reducing agent such as NaBH4, in the presence of a stabilizing or capping agent such as citrate, or CTAB. The seeds are then introduced into a growth medium typically containing more of the gold salt, a shape-controlling agent such as CTAB, a weak reducing agent such as ascorbic acid, and optionally, a metal additive to improve the yield (>99%) such as silver nitrate (AgNO3).36,37 The generally accepted hypothesis is that CTAB is able to direct growth by capping the high energy crystalline side faces of the gold seeds, thus allowing growth only at the rod ends.38,39 Another hypothesis by Mulvaney et al. suggests that the CTAB–Au ion complexes are formed and can subsequently collide with the gold seeds by means of a double layer interaction, preferentially occurring at the tips and thus inducing one directional growth.40 When AgNO3 is included, a proposed mechanism similar to that of the former CTAB hypothesis suggests that the silver ions deposit preferentially on certain faces, again encouraging growth at the seed ends. However, eventual deposition of the silver onto the rod ends leads to premature termination of growth, thus limiting the overall rod lengths to ∼100 nm.41 Therefore, by increasing the concentrations of gold ion or AgNO3 in the growth medium, tuning of the AuNR sizes can be achieved. This is valid only until a specific concentration threshold is reached after which nanorod sizes begin to decrease. In addition to chemical concentration, many other factors can also play a role in the synthetic process, including initial seed size, temperature, pH, solvent, and aging times.37,39,42 Finally, growth can be stopped at any point using sodium sulfide.43
Many other methods have been used to synthesize AuNR, but none with the same success in terms of simplicity, yield, reproducibility and geometric control. Some examples include an aluminium template method,44 photochemical reduction,45 lithography,46 and a seedless method47 that attempts to circumvent the known toxicity issues of the CTAB surfactant.19 The most recent advancement uses a one-pot synthetic method that employs lysine, a biocompatible amino acid that functions as a capping agent and can tune rod sizes by simple adjustment to the Lys![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Au ratio.48 Though a solid foundation of understanding has been built to control various parameters of gold nanoparticles synthesis, studies by Jiang et al. suggest otherwise. They argue that complete control is still out of reach and requires further research, as reproducibility between batches of gold nanorods remains an issue.49 Despite this, AuNR have found application in a myriad of fields, such as surface enhanced Raman scattering (SERS),50,51 photothermal therapy,52,53 biosensing,54,55 as drug carriers,56,57 and in molecular imaging,58–60 due to their distinctive optical properties. It is well known that these properties are defined by factors such as dielectric constant and the surrounding environment, as well as their overall geometry and size.32 The cylindrical conformation of AuNR generates a unique optical effect due to the presence of two critical dimensions, the long axis and the short axis. The ratio of these dimensions is referred to as the aspect ratio (length/width) and is commonly used in the literature to identify nanorod size.61 The oscillation of the long axes known as the longitudinal band, has been well studied and accepted to be characteristic of specific absorbance wavelengths while the transverse absorption band is relatively unaffected by rod size and is sustained at approximately 520 nm.32,62 With an increase of the aspect ratio, the absorbance of the longitudinal band shifts deeper into the near infrared (NIR) region as shown in Fig. 2. Though absorption peaks as high as 1300 nm have been attained,39 aspect ratios between 3 and 4 were found to be optimal for imaging, as they are known to absorb within the near infrared biological window.37,57 As with the AuNS, optical absorbance and scattering cross-sections are a function of size for nanorods. Therefore, larger nanorods produce increased scattering, and smaller nanorods produce increased absorbance. Likewise, small nanorods with high aspect ratios can be expected to absorb, while larger nanorods with lower aspect ratios will scatter light more prominently.31 For a more detailed study, in depth reviews have been written by Mulvaney et al.,7 Mannelli and Marco8 and El-Sayed et al.61
:Au ratio.48 Though a solid foundation of understanding has been built to control various parameters of gold nanoparticles synthesis, studies by Jiang et al. suggest otherwise. They argue that complete control is still out of reach and requires further research, as reproducibility between batches of gold nanorods remains an issue.49 Despite this, AuNR have found application in a myriad of fields, such as surface enhanced Raman scattering (SERS),50,51 photothermal therapy,52,53 biosensing,54,55 as drug carriers,56,57 and in molecular imaging,58–60 due to their distinctive optical properties. It is well known that these properties are defined by factors such as dielectric constant and the surrounding environment, as well as their overall geometry and size.32 The cylindrical conformation of AuNR generates a unique optical effect due to the presence of two critical dimensions, the long axis and the short axis. The ratio of these dimensions is referred to as the aspect ratio (length/width) and is commonly used in the literature to identify nanorod size.61 The oscillation of the long axes known as the longitudinal band, has been well studied and accepted to be characteristic of specific absorbance wavelengths while the transverse absorption band is relatively unaffected by rod size and is sustained at approximately 520 nm.32,62 With an increase of the aspect ratio, the absorbance of the longitudinal band shifts deeper into the near infrared (NIR) region as shown in Fig. 2. Though absorption peaks as high as 1300 nm have been attained,39 aspect ratios between 3 and 4 were found to be optimal for imaging, as they are known to absorb within the near infrared biological window.37,57 As with the AuNS, optical absorbance and scattering cross-sections are a function of size for nanorods. Therefore, larger nanorods produce increased scattering, and smaller nanorods produce increased absorbance. Likewise, small nanorods with high aspect ratios can be expected to absorb, while larger nanorods with lower aspect ratios will scatter light more prominently.31 For a more detailed study, in depth reviews have been written by Mulvaney et al.,7 Mannelli and Marco8 and El-Sayed et al.61
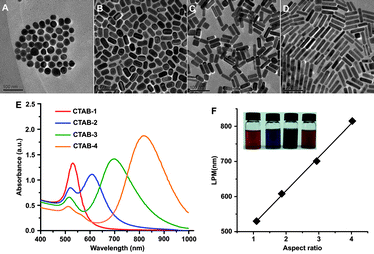 | ||
| Fig. 2 TEM images of CTAB capped AuNR of different aspect ratios (A) 1.1, (B) 1.9, (C) 2.9, (D) 4.0; (E) UV-Vis spectra of four samples; (F) linear correlation between longitudinal plasmonic resonance and aspect ratio (insets are representative samples of AuNR samples). Reproduced with permission from ref. 63. | ||
Nanoshells (AuNSh)
To address the limited tunability of optical absorbance found in gold nanospheres, the addition of gold nanoshells to the nanogold family provided another solution to this problem. The initial forays into AuNSh used a core–shell system, consisting of an inner Au2S core64 which quickly gave way to an improved silica core covered with either gold or silver.4,65,66 This was achieved by using silica nanoparticle core substrates67 functionalized with terminal amine groups. The latter are made available for immobilizing small pre-synthesized gold seed particles which then nucleate gold shell formation.65 Further studies were carried out to determine the optical tunability of these systems, deducing that thinner shells over larger cores increased the resonance shifts as observed by UV-Vis to between 800 and 2200 nm.68Fig. 3a displays the variation of UV-Vis peaks over the range of the afore-mentioned biological window.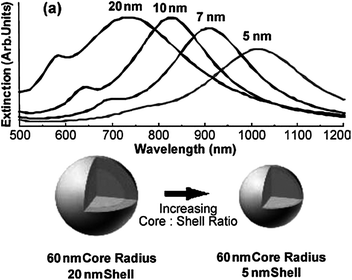 | ||
| Fig. 3 UV-Vis spectra of SiO2 cored AuNSh demonstrating the increase in the SPR peak with increasing core:shell ratio. Reproduced with permission from ref. 69. | ||
Since then, the Halas group has actively pursued the use of metal nanoshells in a ubiquitous manner, as demonstrated by applications in photo-oxidation reduction of semi-conducting polymers,70 immunoassays,71 improved surface enhanced Raman scattering (SERS) applications,72 NIR mediated drug delivery,73 thermoresponsive hydrogel–nanoshell composites,74 and thermoablation for cancer therapy.75,76 While many of these applications depend on the high absorption cross-section of the Si–Au nanoshell system, these particles can also be tuned to scatter light within the biological window so that visible contrast is enhanced. These properties have given rise to the use of silica cored gold nanoshells as imaging contrast agents for anatomical identification, or detection and diagnosis of cancer.77 The dual utility of larger silica cored nanoshells, as studied by Cole et al.,78 that possess both enhanced absorptive and scattering properties, translates into particles capable of both diagnosis via imaging, as well as therapy by thermoablation.79
Parallel to the study of the silica cored nanoshells, another class of nanoshells, which are hollow inside, have also been investigated. In 2002, the Xia group introduced a new template replacement method to synthesize hollow metal nanostructures. It significantly improved the ability to form shell walls with uniformity and face centred cubic (fcc) crystallinity,80 as evidenced by TEM. The foundation of this method used silver nanoparticles as a template upon which the gold salt is reduced, as per the following reaction:
| 3Ag(s) + AuCl4−(aq) → Au(s) + 3Ag+(aq) + 4Cl−(aq) |
As oxidation occurs, a porous layer is formed of partially oxidized silver and partially reduced gold, allowing for the resulting silver salt to diffuse out of the system throughout the exchange. As the system is held under reflux, the silver becomes completely consumed and the gold layer is able to reconstruct itself into smooth, crystalline face centred cubic facets. This period of reconstruction facilitates the complete sealing of the shell, while retaining the original templated shape.80 In 2004, Liang et al. took the Xia method and modified the procedure for applications in catalysis, using cobalt81 as the sacrificial template to synthesize platinum nanoshells stabilized by citrate.82 In the following year, Liang et al. extended this procedure to the synthesis of hollow AuNSh while investigating the tunability of their internal cavities.83 In 2006, Schwartzberg et al. augmented these findings by studying the synthetic techniques and chemical components that control the resulting nanoshell diameters and shell wall thicknesses. With the improved ability to control shell sizes and thicknesses, Schwartzberg also described the effect these two parameters had on nanoshell optical properties, and demonstrated the ease with which these nanostructures could be tuned into the NIR region (Fig. 4).5 In addition to the sensitivity to synthetic methods and surface modifications with regard to optical tuning,84 AuNSh are also adept in photothermal conversion, quickly and efficiently transforming light energy into heat.24,73 These properties have led to the employment of hollow AuNSh in the biological field for cancer treatment through photothermal ablation,73,85–87 photoinduced drug delivery vehicles,88 and photoacoustic imaging applications.89
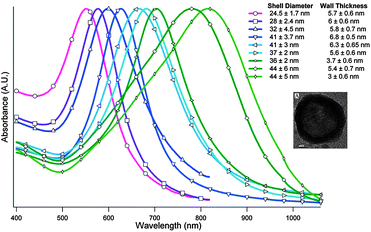 | ||
| Fig. 4 UV-Vis spectra of hollow gold nanoshells with various sizes and shell widths. The inset is a TEM image of a hollow gold nanoshell. Reproduced with permission from ref. 5. | ||
Nanocages (AuNC)
While investigating an improvement to their method of synthesizing hollow metal nanomaterials, Xia's group focussed on developing another category of nanostructure called gold nanocages.15 The construction of this unique gold nanostructure started with the synthesis of silver nanocubes that could be galvanically replaced with a gold salt solution, as is the case when synthesizing AuNSh. In this case however, an additional reaction also occurs where alloying takes place between the gold and silver to form the shell. With sufficient HAuCl4, the initiation of dealloying of the silver subtly reconstructs the alloyed shells into seamless boxes with evidence of truncation on the corners.90 Mechanistic studies were performed to determine the effect further dealloying had on the cages. It was reported that the early development of truncated corners and pinholes would eventually expand into larger pores, until complete degradation of the nanoboxes into pure gold fragments takes place.91 Utilizing this knowledge, an aqueous etchant such as Fe(NO3)3, NH4OH, or H2O2 can be used at an earlier stage to selectively remove the alloyed silver and thus control the creation of pores in the box walls, forming cages able to encapsulate and deliver chemical entities.92,93 A schematic of this process is shown in Fig. 5.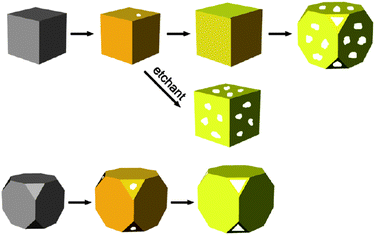 | ||
| Fig. 5 Schematic for structural changes during formation of gold nanocages with the possibility of using an etchant to selectively remove alloyed silver. Both sharp cornered nanocubes and nanocubes with truncated corners are shown. Reproduced with permission from ref. 15. | ||
More recently, research has been done on synthesizing double shelled nanocages using different combinations of gold, platinum, and palladium layers.94 The size of the silver nanocube template, in addition to the adjustment of the amount of HAuCl4 used, can fine-tune the SPR properties of the gold nanocages, as demonstrated in Fig. 6a. More explicitly, SPR is affected by the variation in cage sizes and wall thicknesses, in addition to the number of truncated corners.95
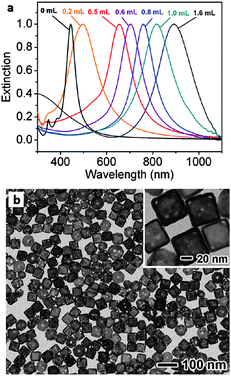 | ||
| Fig. 6 (a) UV-Vis spectra of gold nanocages with increasing concentrations of titrated aqueous HAuCl4; (b) TEM images of gold nanocages (∼50 nm edge length). Modified and reproduced with permission from ref. 15. | ||
The optical and structural advantages of these AuNC make them another viable candidate for biological applications, especially in theranostics, where the ability to encapsulate a payload is a distinguishing advantage. As outlined in a review by Xia et al. in 2011,15 AuNC have found success in therapeutics, as platforms for targeting,96 drug delivery97,98 and photothermal ablation,99 as well as cancer diagnostics using two- and three-photon luminescence (TPL and 3PL),100,101 and photoacoustic imaging.102–104 The advantages of these nanocage systems are their reproducibility, compact nature, where NIR absorbances are achieved at small sizes (35–40 nm nanocages for 800 nm peak absorbance), significantly enhanced absorbance cross-sections over conventional dyes (five times greater than indocyanine green),95 and encapsulation abilities.98
Nanostars (AuNSt)
The most recent gold nanostructures used in in vivo imaging thus far are spiked gold nanospheres, also known as nanostars,105,106 nanocrystals,107 nanourchins,108 and nanothorns.109,110 Typically two general approaches have been taken to synthesize branched gold nanospheres or gold nanostars.16 The first is a two-step seed mediated technique, requiring fabrication of a surfactant stabilized gold seed which is then introduced into a growth medium. The latter contains an aqueous gold salt and a reducing agent that induces anisotropic growth of gold into arms or branches, thus forming the star shape. The second is a “one-pot” method which simultaneously synthesizes the gold seeds while inducing branched growth.16In 2004, Sau and Murphy reported synthesizing star-like gold nanoparticles with a 70% yield, using the same seeded process as the one used for synthesizing gold nanorods.111 In 2006, Nehl et al. published a seed-based synthesis of AuNSt as a discovery of chance, when their group used a pre-made commercial colloid as their surfactant stabilized gold seed (Ted Pella), while following the routine procedure established by the Murphy group.105 TEM characterization showed a yield of 9% of the desired shape with a diversity of spiked nanoshells, from zero to seven spikes or branches. Most interestingly, they identified that nanostars produce multispectral signals due to the presence of multiple tips, which also serve to enhance plasmon resonances. Therefore, tuning the size and shape of the nanostar tips provides another means by which optical tunability could be achieved. Many groups have also supplemented this synthesis with the addition of AgNO3 to control branch growth, also a common additive similarly used in AuNR synthesis to control conformational aspect ratios.10,112,113 The effect of AgNO3 concentration on the synthesis of AuNSt was described in 2009, supporting the hypothesis that the Ag+ ions will preferentially form a complex with CTAB on particular crystalline faces of the gold nanoparticles, preventing further growth in those directions. A specific concentration of AgNO3 must be used, since an excess or insufficient AgNO3 results in a reduction of nanostar arm lengths. Organic additives can also play a role to either lengthen or shorten the branches of the nanostars.113 In 2008, Kumar et al. published a synthetic methodology which provided yields at nearly 100%. This was achieved using pre-formed poly(vinylpyrrolidone) (PVP) stabilized gold seeds in ethanol, while exploiting the reducing ability of a DMF growth solution containing high PVP surfactant concentrations on aqueous gold salts.12,114 Another group extensively studied this synthesis, elucidating the kinetic growth pathway and drawing conclusions that nanostar sizes could be controlled by adjusting the concentration of seed particles (Fig. 7).115 The search for the optimal synthetic strategy is still ongoing, as AuNSt constitute a relatively young field. Therefore, while both of the earlier methods involved the use of known toxic surfactants, CTAB or PVP, an alternative scheme has recently been published by Yuan et al. for a high yielding surfactant free synthesis.106
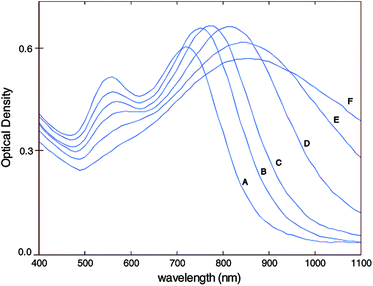 | ||
| Fig. 7 UV-Vis spectra of gold-nanostars demonstrating characteristic double peaking. A–F represent decreasing initial gold seed concentrations and thus increasing star sizes (45 to 116 nm). Reproduced with permission from ref. 115. | ||
This method used an in-house synthesized gold seed solution, made by boiling citrate with HAuCl4 with a growth solution of aqueous HAuCl4, and a rapid addition of a specified ratio of AgNO3 and ascorbic acid. The lack of CTAB or PVP overcomes the problem of toxicity as well as the difficulty of removing these surfactants for functionalization, improving the overall usability and versatility of this nanomaterial for biological applications. “One-pot” schemes have also been developed to facilitate the synthesis of AuNSt typically through the use of a weak reducing agent in the presence of a gold acid. Jose-Yacaman et al. used excess ascorbic acid to reduce an aqueous gold solution with 60% yield.116 Another group has proposed a “green” seedless, surfactantless method that uses a HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) buffer as the reducing and shape-directing agent to give high yields (92%) of branched nanocrystals.117 Liao et al. were able to produce pentagonal gold nanostars by using ascorbic acid as a reducing agent, by reacting the HAuCl4 in a deep eutectic solvent. Their synthetic method could, by simply adjusting the water content of the reaction solution, also produce nano snowflakes and nanothorns as shown in Fig. 8.109 Another synthetic scheme for branched nanocrystals was reported by Hao et al. in 2004 using a citrate solution containing hydrogen peroxide and a bis-(p-sulfonatophenyl) phenylphosphine dihydrate dipotassium (BSPP) stabilizer to which solvated HAuCl4 was added.107 This method was systematically studied and improved in 2011 by van de Broek et al., who also suggested an electroless plating method that could easily tune the branching length.118
 | ||
| Fig. 8 (a) Gold nanostars, (b) gold nanoflakes and (c) gold nanothorns. Modified and reproduced with permission from ref. 109. | ||
The unusual optical properties of AuNSt make them ideal candidates for many biomedical infrared absorption applications. The presence of branching and sharp tips translates into a dominating second resonance presence in addition to the resonance produced by the core of the particle.114 Increasing the number of branches, length of the branches and the sharpness of the tips increases the peak resonance, while the overall nanostar size is relatively irrelevant.107,119 This second plasmon can be tuned into the 1000–1800 nm range which is significantly higher than any of the previously reported nanostructures.120 While offering a larger reactive surface area, biocompatibility and far-ranging optical tunability, the potential of nanostars has encouraged a number of groups to take advantage of these properties in the fields of SERS,115,121 PAT,122 and TPL or 3PL;106 the mere beginnings of a growing field.
Biocompatibility for nanoparticles in vivo
Metal nanoparticles are a recognized foreign substance in the body, and will primarily be cleared by the liver and spleen if injected unmodified.123,124 The size of the particles plays an important role where it has been found that particles greater than 10 nm are more likely cleared in this manner.124 Thus, the tendency for bare nanoparticles to aggregate augments this problem by creating larger agglomerates that are more easily cleared by the body. Despite their inherent non-toxicity, a common synthetic step when using gold nanoparticles in vivo is the process of imparting further biocompatibility onto the nanostructures before injection into the body. This is a crucial step in maximizing the longevity, efficacy and efficiency of gold nanoparticles in the body. The most popular strategy to increase the circulation time is through conjugation to polyethylene glycol (PEG).123,125,126 Ligation with PEG has been studied by a number of groups to imbue “stealth” characteristics such as improved circulation time, reduced degradation and water solubility onto a number of structures including DNA,127,128 proteins,129,130 polymers,131 dendrimers,132,133 and inorganic materials.123,134,135 On gold nanostructures, PEGylation has been achieved by functionalization with single PEG units,123 branched PEG polymers,136 or used as a spacer or linker for another molecule.77 The increased circulation time that PEGylation offers translates into improved ability to arrive at specific in vivo destinations and extended time of exposure to the targeted areas. These two characteristics, in addition to the reduction of degradation which PEGylation offers, also work to improve the efficacy of the nanoparticle vehicle. Additionally, PEGylation can improve the solubility of a given system in water, which reduces its tendency to aggregate. While it is clear that PEGylation is the most commonly employed strategy, it is by no means the only method by which biocompatibility can be achieved.Often, the synthetic procedure itself can be modified to increase biocompatibility of the gold nanoparticles, by incorporating a dual functionality unit that can be both a biocompatible reducing and capping agent. Polymers possessing these abilities have been studied by Bhumkar et al. using a synthetic polymer, chitosan,137 or Yuan et al. who used a diblock of poly(2-(methacryloyloxy)ethyl phosphorylcholine) (PMPC) and poly(2-(dimethylamino)ethyl methacrylate) (PDMA).138 Lately, increased interest in using “green” biological materials has also been pursued. Studies have also included the use of phytochemicals present in cumin139 and soybeans,140 as well as ethanolic leaf extracts of Centella asiatica,141 and Rosa rugosa.142 More recently, the ethanolic extract of Fagopyrum esculentum has been explored, and its non-toxic nature implies that it could be used for future biomedical applications.143
Other methods, aside from PEGylation, to improve biocompatibility in gold nanostructures have also been investigated. For example, gold nanorods have been synthesized that possess a biocompatible multilayer polyelectrolyte.144 As mentioned earlier, the amino acid lysine has also been used as a capping agent during the synthesis of the gold nanorods.48 Gold nanocages can also be covered with polyvinylpyrrolidone, or PVP to impart biocompatibility.145
While biocompatibility within the body during circulation is important, surface agents can also affect the ability of a nanostructure to be taken up by a target cell. Cellular uptake of gold nanoparticles has also been a point of focus for the in vivo behaviour of gold nanostructures. Common strategies to enhance cellular uptake involve using folic acid,146,147 proteins such as transferrin,148 or cell penetrating peptides.149,150 More recently, Liang et al. have successfully demonstrated the use of an anionic group, meso-2,3-dimercaptosuccinic acid (DMSA), to enhance uptake by RAW 265.7 mouse macrophages, A543, human lung carcinoma and BEL-7402, human liver carcinoma.151
It is clear that the scientific community has recognized the importance of the role gold nanostructures play for in vivo applications. As such, this limited selection of articles demonstrates only a portion of the larger field of biocompatibility studies. It is a crucial step for in vivo applications and therefore must be considered when seeking in vivo imaging applications of gold.
Nanoparticles in imaging
To our knowledge there have only been a limited number of gold nanoparticle systems that have succeeded to the point of in vivo imaging. The examples cited here demonstrate the state of current imaging technology including NIR, TPL, optical and X-ray CT or PAT imaging using gold nanoparticles as contrast agents. An account of the known drawbacks and limitations will also be discussed.Surface plasmon resonance
Surface plasmon resonance is a phenomenon originating from the collective oscillation of conduction electrons in response to optical excitation. It may exist at the interface of two media with dielectric constants of opposite signs, for instance, a metal and a dielectric.152,153 Since gold is an excellent conductor and the media (biological for example) a dielectric material, a suitable interface is created for propagating surface electromagnetic waves. Resonance takes place when the frequency of light photons matches the natural frequency of oscillating surface electrons. Tunability of this resonance is dependent on changing the size, shape, composition, and surrounding medium of the nanostructure.154 The manipulation of these factors then offers a facile method to tune the nanomaterial SPR to the desired wavelength.84 In the case of nanoshells, SPR appears between the inner and the outer surface of gold. Theoretically, a thin shell geometry relative to the core dimension is associated with a strong resonant electric field at the outer surface nanoshell.119 As a consequence, it can be expected that shapes with sharp ends make them suited for enhanced plasmon resonance. In addition, plasmon resonance bands can be tuned from the visible to near-infrared region by variation of their geometric parameters (particle size or aspect ratio for nanorods).119The strong plasmon which enhances light scattering and absorption of plasmonic metal nanoparticles has been utilized recently for biological imaging and labeling.155 Complementarily, the strong plasmon absorption has also been employed in photothermal therapy of cancer and other theranostic applications.52 Fluorescence can also be modified by resonant coupling to the external electromagnetic environment, thus making dye functionalization onto a nanostructure more powerful with the SPR effect.
Two photon luminescence (TPL)
Two-photon luminescence is produced by the same mechanism as single photon imaging. However, the relatively weak signal of TPL can be amplified by several orders of magnitude when produced from metal substrates. This amplification is due to a resonant coupling with localized surface plasmons. Lately, plasmon-enhanced TPL from non-spherical gold nanoparticles (nano-rods, -shells, -cages, -stars) have been used as contrast agents in several reports.156,157 Among these, gold nanostars, and more often nanorods, are used for in vivo imaging.Wang et al.59 were the first to report in vivo imaging using gold nanorods in a mouse ear blood vessel in 2005. Many other groups have since achieved imaging with AuNR. For example, Motamedi et al.158 reported in vivo imaging of untargeted commercial AuNR in hamsters. Fig. 9 shows the microvasculature of the normal hamster buccal pouch prior to and after injection of AuNR and up to a depth of 230 microns. Oral carcinogenesis was induced in the buccal pouch of hamsters. It is a model that mimics the stages of precancers and oral squamous cell carcinoma in humans. A reconstructed 3D TPL image shows normal vasculature with enhanced contrast due to AuNR at low power compared to the same TPL image without contrast agents. Fig. 9a shows the confocal imaging as compared to the TPL images (b and c). Fig. 10 is a 3D reconstruction from cancer cell injected hamster vasculature obtained with 1 mW laser power. It shows a dense and disorganized network of blood vessels typically found in tumours for early stages of angiogenesis with an increased number of junctions compared to healthy vessels. This validates that non-targeted AuNR as contrast agents for TPL can potentially provide in vivo imaging to map microvasculature at any area with a shallow depth range.
 | ||
| Fig. 9 Representative two-photon microscopy images of intravenously delivered AuNR in a hamster model with accompanying confocal reflectance image. (a) Confocal reflectance image of tissue showing location of blood vessels which appear dark against the surrounding tissue. (b) High power (20 mW) two photon image of the same vascular region prior to intravenous injection of AuNR. (c) Two photon image using low incident power (1 mW) following intravenous injection of AuNR showing blood vessels in the tissue. Reproduced with permission from ref. 158. | ||
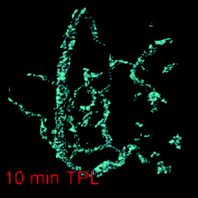 | ||
| Fig. 10 Two-photon 3D reconstructed image of a precancerous (dysplastic) lesion with 10 min post-injection. Modified and reproduced with permission from ref. 158. | ||
Recent work from Yuan et al.106 has demonstrated that gold nanostars can also serve as contrast agents with TPL. They injected a solution composed of S30 (10 branches) PEGylated gold nanostars concentrated to 50 nM in PBS, 50 μL of which are injected retro-orbitally in a nude mouse. Fig. 11a shows the in vivo images of vessels through a dorsal window chamber on nude mice within 5 minutes after injection. Because of the non-specificity of the nanostars, they travel along the blood vessels where the vasculature becomes clearly visible at low excitation power (1–5% transmission) with minimal autofluorescence background. Fig. 11b on the other hand is taken without a contrast agent which necessitated the use of a higher excitation power (20% transmission) to see the vasculature. The blue and green colours are a result of autofluorescence of collagen and blood that were induced by the higher power.
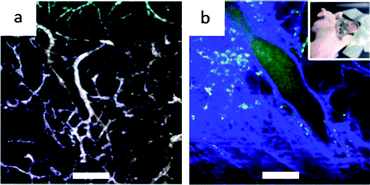 | ||
| Fig. 11 TPL imaging through a dorsal window chamber (inset) on a nude mouse. (a) With nanostars, (b) without nanostars. The blue and green colors were from the collagen and blood. Scale bar 100 μm. Reproduced with permission from ref. 106. | ||
AuNSt with TPL act as strong contrast agents and therefore show promising possibilities for biomedical imaging and diagnostic applications in the case of targeted particles. Limitations such as the melting and eventual destruction of nanorods under high laser power159 used in TPL still have to be addressed. Nevertheless, Motamedi et al.160 and others59,161 reported that low incident laser power (compared to fluorescence) is sufficient to induce a two-photon luminescence signal from nanorods and reduce autofluorescence from the tissue. Furthermore, when using standard dyes attached to gold, the distance between them must be precisely controlled to avoid quenching. This occurs when the entities are too close, causing the dye to transfer energy to the particle and thus become nonfluorescent.162 Therefore, imaging using gold nanoparticles as a contrast agent themselves circumvents these issues associated with dyes. Moreover, Park et al. demonstrated that the laser power used for standard dyes such as fluorescein, which is commonly used with TPL, would lead to the melting of nanoshells with silica cores, whereas the lower power needed for nanoshells or nanorods would keep them intact.163 TPL has also been reported to identify the resonance peak of a single nanorod (0.3 × 1 μm), detectable in real time through a mouse ear blood vessel.59 This exhibits the fine sensitivity TPL can provide for monitoring biological processes.
Computed tomography (CT)
Vascular angiogenesis is a trademark of tumour development, but its detection remains a challenge. X-ray can provide information on the vascular network when combined with contrast agents to discriminate between tissues and blood vessels. High density elements are good candidates and, as such, gold nanoparticles provide nanosized units that can be dispersed, while also possessing useful imaging contrast properties. Two particular types of gold nanoparticles are typically found in the literature for CT imaging: nanospheres and nanorods.Many authors have already reported the benefits of using plain gold nanospheres as contrast agents for CT tomography.164,165 Among them, Hainfeld et al. presented166 a whole body CT scan of a mouse where contrast agents can be seen enhancing the image in Fig. 12. They used untargeted gold nanospheres with intravenous injection and followed the diffusion of the nanospheres over time. Clearance occurred 60 min after injection when the particles gathered in the bladder, post filtration by the kidneys. In the same study, the authors also imaged a tumour in the left leg of a mouse (data not shown). Results showed nanosphere accumulation in a dense and anarchic vascular network due to the high perfusion of porous vessels which is characteristic of tumours. The resolution is such that 100 μm wide vessels can be distinguished.
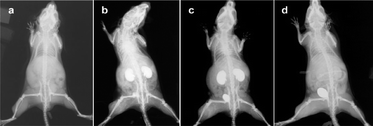 | ||
| Fig. 12 Pharmacokinetics of gold nanospheres (a–d) in mice. (a) Before injection; (b) 2 min after injection; (c) 10 min after injection; (d) 60 min after injection. Reproduced with permission from ref. 166. | ||
CT micrography uses unmonochromatized synchrotron X-rays. Removing the monochromator avoids losses in flux, and as a result, increases it on the object by several orders of magnitude.167 This leads not only to higher spatial resolution (<10 μm) but also to higher time resolution (∼100 ms). To compare, the full CT scan in Fig. 12 required 0.4 s to reach 100 μm resolution. Nevertheless, the need for contrast agents is still crucial.168 In Fig. 13, sequential images of bare-AuNS injected in the femoral artery can be seen. Images reported by Chien et al.168 were taken after injection of 200 μL of 15.5 ± 5.1 nm bare-AuNS in the mouse leg. The images (f)–(j) were processed with a background-flattening filter. The interval between images is ∼60 s. Yellow arrows indicate that the nanoparticles adhere to the vessel wall while forming clusters, eventually leading to complete blockage of flux within the vessels.
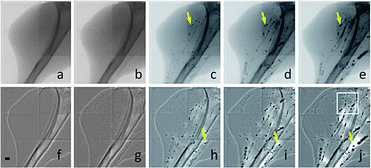 | ||
| Fig. 13 X-ray micrographies of vasculature of the leg of a mouse taken after injection of gold nanospheres. The interval between images is around 60 s. Scale bar: 500 μm. Reproduced with permission from ref. 168. | ||
Huang et al.169 used gold nanorods containing silica cores as CT contrast agents terminated with folic acid to target tumours. In Fig. 14, the coloured CT scan shows the accumulation of nanorods in the tumour. These two different studies confirm the feasibility of AuNS and AuNR as contrast agents for in vivo imaging with CT scans. Current work now aims to improve methods of coating gold nanoparticles with antibodies170 or High Density Lipoprotein (HDL)171 to address the target of interest.
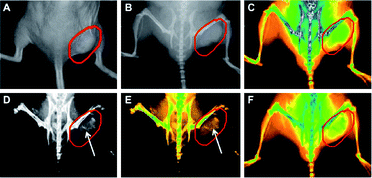 | ||
| Fig. 14 Real-time in vivo X-ray images after intravenous injection of AuNR–SiO2–FA in nude mice at different time points. (A) The photograph of the tumor tissue; (B) the X-ray image at 0 h; (C) the X-ray image at 0 h (in color); (D) the X-ray image at 12 h, (E) the X-ray image at 12 h (in color); (F) the X-ray image at 24 h (in color). Reproduced with permission from ref. 169. | ||
NIR absorption
Imaging deep in tissues requires NIR light in a window where absorption is minimized (between 650 and 900 nm).22 With the conjugation of surface groups to metal nanoparticles, imaging can be performed using the surface plasmon band in the near-infrared region.33 While most imaging with infrared light uses dyes (typically bound to a nanoparticle allowing for targeting or multimodal imaging), it can also be achieved with the absorption of the particle itself using the SPR effect without a fluorophore. This avoids problems such as photo-instability (bleaching) caused by intense illuminating light, and quenching when the dye is too close to the particle,162 both of which are often limiting factors when using dyes. Dickerson et al.172 focussed on demonstrating the feasibility of plasmonic photothermal therapy (PPTT) with gold nanorods. In their work, they presented in vivo imaging with near infrared light after injection of a solution of PEGylated gold nanorods in a mouse. The absorption peak was around 800 nm. Results are presented in Fig. 15, where the accumulation of nanorods after 2 min can be seen for in situ injection or after 24 h for intravenous injection, along with the increase of absorption. This method is sufficient to localize tumours but relies on passive accumulation of AuNR in the tumour, also known as the enhanced permeation and retention or the EPR effect. Choi et al.173 also used gold nanorods functionalized with cyclic RGD peptides for NIR absorption imaging in brain tumours showing the viability of near infrared imaging with gold nanorods.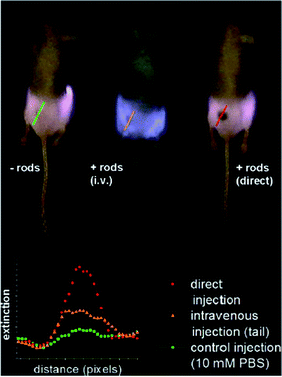 | ||
| Fig. 15 NIR transmission images of mice prior to PPTT treatments. The inset shows intensity line-scans of NIR extinction at tumor sites for control, intravenous, and direct administration of PEGylated gold nanorods. Control mice were interstitially injected with 15 μL 10 mM PBS alone, while directly administered mice received interstitial injections of 15 μL PEGylated gold nanorods (ODλ=800 = 40, 2 min accumulation), and intravenously administered mice received 100 μL PEGylated gold nanorod (ODλ=800 = 120, 24 h accumulation) injections. Reproduced with permission from ref. 172. | ||
Optical coherence tomography (OCT)
OCT is an emerging medical imaging modality that provides cross-sectional subsurface imaging of biological tissue with micrometer-scale resolution. This technique can provide information on the vasculature of tissues up to a depth of 2 mm. It is based on the interferometric detection of backscattered light in the near infrared region. It is also a real time imaging technique and may yield potential for diagnosis of subcutaneous cancers. The source of contrast in OCT is the change in the index of refraction. In turbid tissues, there are many microscopic refractive index mismatches within a resolution element of the OCT system. Limitations occur when adjacent tissues with similar indices exhibit poor contrast, emphasizing the need for contrast agents.174 Gold nanoshells can serve as the contrast agents since they provide a strong backscattering signal on mismatched indices of refraction. Using the passive diffusion of PEGylated AuNSh in tumours, Gobin et al.79 imaged a subcutaneous tumour in live mice after intravenous injection. Fig. 16 shows the difference between normal and tumourous tissue with and without nanoshells. The enhanced brightness suggests that AuNSh provide a contrast for OCT imaging in vivo.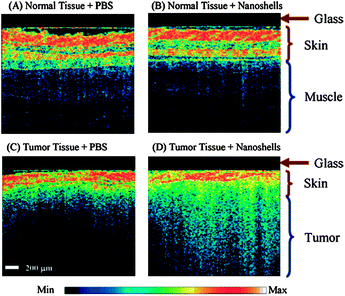 | ||
| Fig. 16 Representative OCT images from normal skin and muscle tissue areas of mice systemically injected with nanoshells (A) or with PBS (B). Representative OCT images from tumors of mice systemically injected with nanoshells (C) or with PBS (D). Analysis of all images shows a significant increase in contrast intensity after nanoshell injection in the tumors of mice treated with nanoshells while no increase in intensity is observed in the normal tissue. The glass of the probe is 200 μm thick and shows as a dark nonscattering layer. Reproduced with permission from ref. 79. | ||
To our knowledge, only the gold nanoshells have been used with OCT in vivo imaging to date. Nevertheless, this modality presents a potential with the combination of gold nanoparticles, but there is a need for more data79,174 in this field to improve the technique.
Photoacoustic tomography (PAT)
Photoacoustic tomography is an imaging modality that combines the high sensitivity of optical imaging and the high resolution of ultrasound. Pulsed laser light is sent into the sample where absorption leads to transient variations of temperature resulting in the generation of acoustics signals due to surface plasmon resonance. Through an ultrasonic probe, these signals are measured and converted into an image of the object. Resolution in PAT depends on the frequency of the ultrasound transducer, for example with a 30 MHz ultrasound transducer the axial, lateral and elevational resolutions were 25, 70, 200 μm at 7 mm depth according to Song et al.175 Due to the high absorption of gold nanoparticles, they are particularly efficient contrast agents for PAT. In fact, all of the described forms of gold nanomaterials reviewed in this article have been tested in vivo using photoacoustic tomography: nanospheres,176 nanoshells,89,177,178 nanorods,60,179–181 nanocages102,103 and nanostars.131Two studies using different nanomaterials for researching sentinel lymph nodes, commonly used for diagnosis of auxiliary staging in breast cancer patients performed by biopsy, are representative of PAT applications. Fig. 17 shows the results from Song et al. of in vivo imaging in the sentinel lymph node of rats.103 An intradermal injection of AuNC of 0.1 mL of 2 nM was performed on the left forepaw pad of rats. After the injection, PA signals increased with time, which was interpreted as a gradual accumulation of nanocages. Peak accumulation occurred at ∼140 min after injection. Significant contrast was obtained up to 33 mm depth with good spatial resolution.
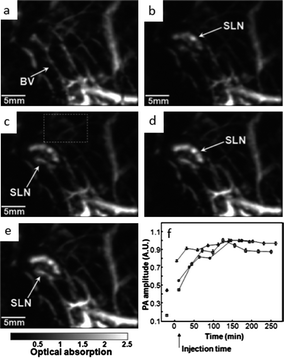 | ||
| Fig. 17 In vivo noninvasive photoacoustic time-course sagittal MAP image. Photoacoustic images acquired before (a) and after (c–f) the nanocage injection: (b) 5 min (SLN started to appear), (c) 59 min, (d) 140 min, (e) 194 min. All images were acquired without signal averaging. (f) Accumulations of nanocages in a SLN over time, in terms of the amplitude changes of PA signals. PA signals from the SLN were normalized by those from adjacent blood vessels (the dotted box in Fig. 2E) to minimize the ultrasonic focal effect, and normalized by maximum. Error bar is standard error. BV, blood vessels; SLN, sentinel lymph node. Color bar represents optical absorptions. Modified and reproduced with permission from ref. 103. | ||
Kim et al. used gold nanostars (AuNSt) to image sentinel lymph nodes in rats after footpad injection.122 In Fig. 18, differential images demonstrate that NSTs can be used for non-invasive in vivo mapping of lymphatic systems.
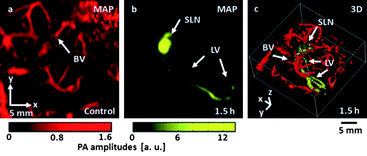 | ||
| Fig. 18 In vivo photoacoustic (PA) mapping of rat lymphatic systems, enhanced using AuNSt. (a) Control PA image acquired before AuNSt injection, displaying only blood vessels (BV). (b) PA difference image acquired post-injection (t¼ 1.5 h), revealing the sentinel lymph node (SLN) and lymphatic vessels (LV). A separate intensity scale (yellow) was applied to the lymphatic system after subtraction of image (a). (c) 3D rendering of (b). Reproduced from ref. 122. | ||
Both groups have validated the benefit of gold nanostructures using PAT for non-invasive, real time, in vivo imaging. Due to these reasons, photoacoustic imaging is currently the technique that has garnered the most attention for in vitro and in vivo imaging using gold. Considering real time imaging, high spatial resolution and the achievable penetration depth makes it an attractive method in the field of biomedical imaging.182
Toxicity in vivo
Along with the increasing interest in gold nanoparticles, in vivo toxicity has always been a concern. Many large and well referenced studies have been done on the subject for the various geometries that exist.63,183–185 Even though gold is touted to be nontoxic, and thus “safe” due to its stability and inertness, some studies show contradictory results.186,187 Comparability and general conclusions become difficult to develop as many individual studies vary significantly over nanomaterial properties such as shapes,188 sizes,189 constituent components,63 concentrations,190 surface charges,191 tendencies for aggregation192 and surface modifications193 of the particular nanomaterial.124 These are critical factors to consider, as toxicity can be induced by any one of them under particular circumstances. For example, when nanostructures are smaller than a set threshold size, they become toxic due to their ability to pervade throughout the body and then penetrate and accumulate inside cells and other biological organelles.189,194 On the other hand, toxicity can be reduced by surface modifiers such as the aforementioned PEG, known as the “stealth” coating that can replace toxic stabilizers and lengthen the circulation of the nanomaterial in vivo.193 However, the optimization of PEG modification must also be moderated as excessive coverage may also encourage non-specific cellular uptake and thus induce toxicity to healthy cells.195 Other common concerns are the presence of toxic chemicals used in the synthetic process, such as the Si core of Au–Si nanoshells and free or bound CTAB stabilizing bilayers, which have been shown to be harmful to cells.19,63,87 Further considerations must be given to experimental parameters which can also vary significantly in targeted cell lines, exposure times, determination methods of toxicity and non-standard benchmarks for toxicity. Jiang et al. present an excellent table that compiles this information in a recent review. Even with their comprehensive compilation, the authors could not provide consistent conclusions for short or long term effects, and urged further work in establishing standard evaluation systems to enable predictability of toxicity.185While toxicity is typically addressed by in vitro tests showing cell survival post exposure to the nanomaterial, translation of these studies to in vivo are often yet to be done.53,196,197 Optimistically, the FDA has already approved phase-I clinical trials for Au-based nanospheres as drug carriers or therapeutic agents.131,136 However, there still remains a need for more research on other gold nanoshapes to successfully attain FDA approval and then move towards the next step: human imaging. Before any of this, long term pharmacokinetics of these systems must also be elucidated. For example, bioavailability, circulation times and clearance of these nanostructures from the body must be comprehensively understood, either bare, or with surface modifiers. In some cases it is more of a matter of exposure time to the material in question rather than particle characteristics.198 While headway on toxicology has been made with gold nanospheres and nanorods, there is a notable lack of specific research on other gold nanomaterial shapes.
The importance of toxicity in this field has commanded appreciable attention in the gold nanomaterials community in recent years.184 General findings seem to indicate increased cell death when particles are very small (exact threshold values differ for different cell types), anionically charged, and possess surface modifiers that are toxic in nature or are in excessive concentrations.184,185,189 It is important to note that the surface functionalization plays an important role in potential toxicity as it forms the primary interface with the biological environment, and as a result can easily alter the expected behaviour of the system.184 As such, selecting synthetic routes for appropriate nanomaterial sizes, and utilizing known biocompatibility enhancing agents can decrease the likelihood of toxic effects.193 Unfortunately, the overall complexities of interactions that exist in vivo make more specific conclusions and guidelines difficult to establish with certainty at this stage.
Conclusions
The unique shapes which range from nano-spheres, -shells, -rods, -cages and -stars impart their characteristic properties to gold nanomaterials based on their symmetry or asymmetry, as well as the presence of internal voids or sharp points. In particular, the surface plasmon resonance peak can be fine tuned by manipulation of these characteristics during synthesis. Thus, the utility of nanogold in terms of these optical properties as well as gold's intrinsic properties, such as photothermal conversion efficiency and biocompatibility, have made it a suitable candidate for a number of imaging modalities for in vivo studies. While researchers have taken advantage of the multimodality199,200 offered by gold nanomaterials as a platform for functionalization, many groups still focus on using the intrinsic optical properties of gold nanomaterials for imaging.Due to the difficulty of obtaining quality images of a system amidst the biological complexity of a living subject, in vivo imaging has been more challenging than in a closed-system i.e. in vitro. As such, in vivo imaging using gold nanomaterials as contrast agents to improve differentiation capability has become an expanding field. Initial studies were performed using shallow techniques such as CT and OCT, and more recently, TPL. With the advent of different geometries possessing optimizable absorbance peaks into the NIR biological window, several groups are now utilizing these properties for deeper internal imaging modalities such as NIR and PAT.
This review has attempted an overview of five different imaging modalities involving gold nanomaterials as contrast agents. Each of these has been significantly improved by the use of nanoparticles, though the number of studies among them are far from equal. Based on reported data, it would seem that while most techniques have shown some progress, the most popular and pursued methodology is photoacoustic imaging. It is clear that gold nanomaterials hold promise for the future in imaging, and with the recent FDA approval of gold nanospheres for clinical studies131 notwithstanding, there still exists uncertainty in short and long term toxicological safety when using gold nanomaterials in the human body. Despite these cautions, imaging using gold nanomaterials has been successfully demonstrated by many groups to be a viable tool in the current field of medicinal nanotechnology.
Notes and references
- D. J. Barber and I. C. Freestone, Archaeometry, 1990, 32, 33–45 CrossRef
.
- M. Faraday, Philos. Trans. R. Soc. London, 1857, 147, 145–181 CrossRef
- G. Mie, Ann. Phys., 1908, 330, 377–445 CrossRef
- S. L. Westcott, S. J. Oldenburg, T. R. Lee and N. J. Halas, Chem. Phys. Lett., 1999, 300, 651–655 CrossRef CAS
- A. M. Schwartzberg, T. Y. Olson, C. E. Talley and J. Z. Zhang, J. Phys. Chem. B, 2006, 110, 19935–19944 CrossRef CAS
- J. Ye, P. Van Dorpe, W. Van Roy, K. Lodewijks, I. De Vlaminck, G. Maes and G. Borghs, J. Phys. Chem. C, 2009, 113, 3110–3115 CAS
- J. Pérez-Juste, I. Pastoriza-Santos, L. M. Liz-Marzán and P. Mulvaney, Coord. Chem. Rev., 2005, 249, 1870–1901 CrossRef
- I. Mannelli and M.-P. Marco, Anal. Bioanal. Chem., 2010, 398, 2451–2469 CrossRef CAS
- H. Wang, D. W. Brandl, F. Le, P. Nordlander and N. J. Halas, Nano Lett., 2006, 6, 827–832 CrossRef CAS
- M. Liu and P. Guyot-Sionnest, J. Phys. Chem. B, 2005, 109, 22192–22200 CrossRef CAS
- F. Kim, S. Connor, H. Song, T. Kuykendall and P. Yang, Angew. Chem., Int. Ed., 2004, 43, 3673–3677 CrossRef CAS
- C. Li, K. L. Shuford, M. Chen, E. J. Lee and S. O. Cho, ACS Nano, 2008, 2, 1760–1769 CrossRef CAS
- D. Seo, J. C. Park and H. Song, J. Am. Chem. Soc., 2006, 128, 14863–14870 CrossRef CAS
- X. Wu, T. Ming, X. Wang, P. Wang, J. Wang and J. Chen, ACS Nano, 2009, 4, 113–120 CrossRef
- Y. N. Xia, W. Y. Li, C. M. Cobley, J. Y. Chen, X. H. Xia, Q. Zhang, M. X. Yang, E. C. Cho and P. K. Brown, Acc. Chem. Res., 2011, 44, 914–924 CrossRef CAS
- A. Guerrero-Martínez, S. Barbosa, I. Pastoriza-Santos and L. M. Liz-Marzán, Curr. Opin. Colloid Interface Sci., 2011, 16, 118–127 CrossRef
- R. Bukasov and J. S. Shumaker-Parry, Nano Lett., 2007, 7, 1113–1118 CrossRef CAS
- L. Dykman and N. Khlebtsov, Chem. Soc. Rev., 2012, 41, 2256 RSC
- E. E. Connor, J. Mwamuka, A. Gole, C. J. Murphy and M. D. Wyatt, Small, 2005, 1, 325–327 CrossRef CAS
- X. J. Xue, F. Wang and X. G. Liu, J. Mater. Chem., 2011, 21, 13107–13127 RSC
- E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759–1782 RSC
- R. Weissleder, Nat. Biotechnol., 2001, 19, 316 CrossRef CAS
- L. Rouleau, R. Berti, V. W. K. Ng, C. Matteau-Pelletier, T. Lam, P. Saboural, A. Kakkar, F. Lesage, E. Rhéaume and J.-C. Tardif, Contrast Media Mol. Imaging, 2012 Search PubMed
- X. Huang and M. A. El-Sayed, Alexandria Journal of Medicine, 2011, 47, 1–9 CrossRef
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC
-
E. A. Hauser and J. E. Lynn, Experiments in Colloid Chemistry, McGraw-Hill Book Company, Inc., New York and London, 1st edn, 1940 Search PubMed
- G. Schmid, Chem. Rev., 1992, 92, 1709–1727 CrossRef CAS
- G. Frens, Nature, 1973, 241, 20–22 CAS
- J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot and A. Plech, J. Phys. Chem. B, 2006, 110, 15700–15707 CrossRef CAS
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC
- P. K. Jain, K. S. Lee, I. H. El-Sayed and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 7238–7248 CrossRef CAS
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 8410–8426 CrossRef CAS
- Y. Yu, S. Chang, C. Lee and C. Wang, J. Phys. Chem. B, 1997, 101, 6661–6664 CrossRef CAS
- S.-S. Chang, C.-W. Shih, C.-D. Chen, W.-C. Lai and C. R. C. Wang, Langmuir, 1998, 15, 701–709 CrossRef
- J. Wiesner and A. Wokaun, Chem. Phys. Lett., 1989, 157, 569–575 CrossRef CAS
- N. R. Jana, L. Gearheart, C. J. Murphy, N. R. Jana, L. Gearheart and C. J. Murphy, Adv. Mater., 2001, 13, 1389–1393 CrossRef CAS
- C. J. Murphy, T. K. Sau, A. M. Gole, C. J. Orendorff, J. Gao, L. Gou, S. E. Hunyadi and T. Li, J. Phys. Chem. B, 2005, 109, 13857–13870 CrossRef CAS
- C. J. Johnson, E. Dujardin, S. A. Davis, C. J. Murphy and S. Mann, J. Mater. Chem., 2002, 12, 1765–1770 RSC
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957–1962 CrossRef CAS
- J. Pérez-Juste, L. M. Liz-Marzán, S. Carnie, D. Y. C. Chan, P. Mulvaney, J. Pérez-Juste, L. M. Liz-Marzán, S. Carnie, D. Y. C. Chan and P. Mulvaney, Adv. Funct. Mater., 2004, 14, 571–579 CrossRef
- C. J. Orendorff and C. J. Murphy, J. Phys. Chem. B, 2006, 110, 3990–3994 CrossRef CAS
- W. M. Park, Y. S. Huh and W. H. Hong, Curr. Appl. Phys., 2009, 9, e140–e143 CrossRef
- D. A. Zweifel and A. Wei, Chem. Mater., 2005, 17, 4256–4261 CrossRef CAS
- C. R. Martin, Chem. Mater., 1996, 8, 1739–1746 CrossRef CAS
- F. Kim, J. H. Song and P. Yang, J. Am. Chem. Soc., 2002, 124, 14316–14317 CrossRef CAS
- E. J. Smythe, E. Cubukcu and F. Capasso, Opt. Express, 2007, 15, 7439–7447 CrossRef
- P. Zijlstra, C. Bullen, J. W. M. Chon and M. Gu, J. Phys. Chem. B, 2006, 110, 19315–19318 CrossRef CAS
- V. Sebastián, S.-K. Lee, C. Zhou, M. F. Kraus, J. G. Fujimoto and K. F. Jensen, Chem. Commun., 2012, 48, 6654–6656 RSC
- X. C. Jiang, A. Brioude and M. P. Pileni, Colloids Surf., A, 2006, 277, 201–206 CrossRef CAS
- B. Nikoobakht, J. Wang and M. A. El-Sayed, Chem. Phys. Lett., 2002, 366, 17–23 CrossRef CAS
- X.-Z. Shi, C.-M. Shen, D.-K. Wang, C. Li, Y. Tian, Z.-C. Xu, C.-M. Wang and H.-J. Gao, Chin. Phys. B, 2011, 20, 076103 CrossRef
- W. Choi, A. Sahu, Y. Kim and G. Tae, Ann. Biomed. Eng., 2012, 40, 534–546 CrossRef
- A. M. Alkilany, L. B. Thompson, S. P. Boulos, P. N. Sisco and C. J. Murphy, Adv. Drug Delivery Rev., 2012, 64, 190–199 CrossRef CAS
- C. Yu and J. Irudayaraj, Biophys. J., 2007, 93, 3684–3692 CrossRef CAS
- C. Wang, Y. Chen, T. Wang, Z. Ma and Z. Su, Chem. Mater., 2007, 19, 5809–5811 CrossRef CAS
- C.-C. Chen, Y.-P. Lin, C.-W. Wang, H.-C. Tzeng, C.-H. Wu, Y.-C. Chen, C.-P. Chen, L.-C. Chen and Y.-C. Wu, J. Am. Chem. Soc., 2006, 128, 3709–3715 CrossRef CAS
- X. Huang, I. H. El-Sayed, W. Qian and M. A. El-Sayed, J. Am. Chem. Soc., 2006, 128, 2115–2120 CrossRef CAS
- A. L. Oldenburg, M. N. Hansen, D. A. Zweifel, A. Wei and S. A. Boppart, Opt. Express, 2006, 14, 6724–6738 CrossRef CAS
- H. Wang, T. Huff, D. Zweifel, W. He, P. Low, A. Wei and J. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15752–15756 CrossRef CAS
- P.-C. Li, C.-R. C. Wang, D.-B. Shieh, C.-W. Wei, C.-K. Liao, C. Poe, S. Jhan, A.-A. Ding and Y.-N. Wu, Opt. Express, 2008, 16, 18605–18615 CrossRef CAS
- X. Huang, S. Neretina, M. A. El-Sayed, X. Huang, S. Neretina and M. A. El-Sayed, Adv. Mater., 2009, 21, 4880–4910 CrossRef CAS
- K.-S. Lee and M. A. El-Sayed, J. Phys. Chem. B, 2005, 109, 20331–20338 CrossRef CAS
- Y. Qiu, Y. Liu, L. Wang, L. Xu, R. Bai, Y. Ji, X. Wu, Y. Zhao, Y. Li and C. Chen, Biomaterials, 2010, 31, 7606–7619 CrossRef CAS
- R. D. Averitt, D. Sarkar and N. J. Halas, Phys. Rev. Lett., 1997, 78, 4217–4220 CrossRef CAS
- S. J. Oldenburg, R. D. Averitt, S. L. Westcott and N. J. Halas, Chem. Phys. Lett., 1998, 288, 243–247 CrossRef CAS
- S. L. Westcott, S. J. Oldenburg, T. R. Lee and N. J. Halas, Langmuir, 1998, 14, 5396–5401 CrossRef CAS
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef
- S. J. Oldenburg, J. B. Jackson, S. L. Westcott and N. J. Halas, Appl. Phys. Lett., 1999, 75, 2897–2899 CrossRef CAS
- L. Hirsch, A. Gobin, A. Lowery, F. Tam, R. Drezek, N. Halas and J. West, Ann. Biomed. Eng., 2006, 34, 15–22 CrossRef
- G. D. Hale, J. B. Jackson, O. E. Shmakova, T. R. Lee and N. J. Halas, Appl. Phys. Lett., 2001, 78, 1502–1504 CrossRef CAS
- L. R. Hirsch, J. B. Jackson, A. Lee, N. J. Halas and J. West, Anal. Chem., 2003, 75, 2377–2381 CrossRef CAS
- S. J. Oldenburg, S. L. Westcott, R. D. Averitt and N. J. Halas, J. Chem. Phys., 1999, 111, 4729–4735 CrossRef CAS
- L. R. Hirsch, R. J. Stafford, J. A. Bankson, S. R. Sershen, B. Rivera, R. E. Price, J. D. Hazle, N. J. Halas and J. L. West, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13549–13554 CrossRef CAS
- S. R. Sershen, S. L. Westcott, J. L. West and N. J. Halas, Appl. Phys. B: Lasers Opt., 2001, 73, 379–381 CrossRef CAS
- D. P. O'Neal, L. R. Hirsch, N. J. Halas, J. D. Payne and J. L. West, Cancer Lett., 2004, 209, 171–176 CrossRef CAS
- R. Bernardi, A. Lowery, P. Thompson, S. Blaney and J. West, J. Neuro-Oncol., 2008, 86, 165–172 CrossRef
- C. Loo, L. Hirsch, M.-H. Lee, E. Chang, J. West, N. Halas and R. Drezek, Opt. Lett., 2005, 30, 1012–1014 CrossRef CAS
- J. R. Cole, N. A. Mirin, M. W. Knight, G. P. Goodrich and N. J. Halas, J. Phys. Chem. C, 2009, 113, 12090–12094 CAS
- A. M. Gobin, M. H. Lee, N. J. Halas, W. D. James, R. A. Drezek and J. L. West, Nano Lett., 2007, 7, 1929–1934 CrossRef CAS
- Y. Sun, B. T. Mayers and Y. Xia, Nano Lett., 2002, 2, 481–485 CrossRef CAS
- Y. Kobayashi, M. Horie, M. Konno, B. Rodríguez-González and L. M. Liz-Marzán, J. Phys. Chem. B, 2003, 107, 7420–7425 CrossRef CAS
- H. Liang, H. Zhang, J. Hu, Y. Guo, L. Wan, C. Bai, H. Liang, H. Zhang, J. Hu, Y. Guo, L. Wan and C. Bai, Angew. Chem., Int. Ed., 2004, 43, 1540–1543 CrossRef CAS
- H.-P. Liang, L.-J. Wan, C.-L. Bai and L. Jiang, J. Phys. Chem. B, 2005, 109, 7795–7800 CrossRef CAS
- Y. Sun and Y. Xia, Anal. Chem., 2002, 74, 5297–5305 CrossRef CAS
- W. Lu, C. Xiong, G. Zhang, Q. Huang, R. Zhang, J. Z. Zhang and C. Li, Clin. Cancer Res., 2009, 15, 876–886 CrossRef CAS
- J. Z. Zhang, J. Phys. Chem. Lett., 2010, 1, 686–695 CrossRef CAS
- M. P. Melancon, W. Lu, Z. Yang, R. Zhang, Z. Cheng, A. M. Elliot, J. Stafford, T. Olson, J. Z. Zhang and C. Li, Mol. Cancer Ther., 2008, 7, 1730–1739 CrossRef CAS
- N. Forbes and J. A. Zasadzinski, Proc. SPIE–Int. Soc. Opt. Eng., 2011, 7911, 79110P CrossRef
- W. Lu, Q. Huang, G. Ku, X. Wen, M. Zhou, D. Guzatov, P. Brecht, R. Su, A. Oraevsky, L. V. Wang and C. Li, Biomaterials, 2010, 31, 2617–2626 CrossRef CAS
- Y. Sun and Y. Xia, Science, 2002, 298, 2176–2179 CrossRef CAS
- Y. Sun and Y. Xia, J. Am. Chem. Soc., 2004, 126, 3892–3901 CrossRef CAS
- X. Lu, L. Au, J. McLellan, Z.-Y. Li, M. Marquez and Y. Xia, Nano Lett., 2007, 7, 1764–1769 CrossRef CAS
- Q. Zhang, C. M. Cobley, J. Zeng, L.-P. Wen, J. Chen and Y. Xia, J. Phys. Chem. C Nanomater Interfaces, 2010, 114, 6396–6400 CAS
- M. A. Mahmoud and M. A. El-Sayed, Langmuir, 2012, 28, 4051–4059 CrossRef CAS
- J. Chen, F. Saeki, B. J. Wiley, H. Cang, M. J. Cobb, Z. Y. Li, L. Au, H. Zhang, M. B. Kimmey, X. D. Li and Y. N. Xia, Nano Lett., 2005, 5, 473–477 CrossRef CAS
- C. Kim, E. C. Cho, J. Chen, K. H. Song, L. Au, C. Favazza, Q. Zhang, C. M. Cobley, F. Gao, Y. Xia and L. V. Wang, ACS Nano, 2010, 4, 4559–4564 CrossRef CAS
- G. D. Moon, S.-W. Choi, X. Cai, W. Li, E. C. Cho, U. Jeong, L. V. Wang and Y. Xia, J. Am. Chem. Soc., 2011, 133, 4762–4765 CrossRef CAS
- M. S. Yavuz, Y. Cheng, J. Chen, C. M. Cobley, Q. Zhang, M. Rycenga, J. Xie, C. Kim, K. H. Song, A. G. Schwartz, L. V. Wang and Y. Xia, Nat. Mater., 2009, 8, 935–939 CrossRef CAS
- J. Y. Chen, D. L. Wang, J. F. Xi, L. Au, A. Siekkinen, A. Warsen, Z. Y. Li, H. Zhang, Y. N. Xia and X. D. Li, Nano Lett., 2007, 7, 1318–1322 CrossRef CAS
- L. Au, Q. Zhang, C. M. Cobley, M. Gidding, A. G. Schwartz, J. Chen and Y. Xia, ACS Nano, 2010, 4, 35–42 CrossRef CAS
- L. Tong, C. M. Cobley, J. Chen, Y. Xia and J.-X. Cheng, Angew. Chem., Int. Ed., 2010, 49, 3485–3488 CrossRef CAS
- X. Yang, S. E. Skrabalak, Z.-Y. Li, Y. Xia and L. V. Wang, Nano Lett., 2007, 7, 3798–3802 CrossRef CAS
- K. H. Song, C. Kim, C. M. Cobley, Y. Xia and L. V. Wang, Nano Lett., 2009, 9, 183–188 CrossRef CAS
- X. Cai, W. Li, C.-H. Kim, Y. Yuan, L. V. Wang and Y. Xia, ACS Nano, 2011, 5, 9658–9667 CrossRef CAS
- C. L. Nehl, H. Liao and J. H. Hafner, Nano Lett., 2006, 6, 683–688 CrossRef CAS
- H. Yuan, C. G. Khoury, H. Hwang, C. M. Wilson, G. A. Grant and T. Vo-Dinh, Nanotechnology, 2012, 23, 075102 CrossRef
- E. Hao, R. C. Bailey, G. C. Schatz, J. T. Hupp and S. Li, Nano Lett., 2004, 4, 327–330 CrossRef CAS
- O. M. Bakr, B. H. Wunsch and F. Stellacci, Chem. Mater., 2006, 18, 3297–3301 CrossRef CAS
- H. Liao, Y. Jiang, Z. Zhou, S. Chen and S. Sun, Angew. Chem., Int. Ed., 2008, 47, 9100–9103 CrossRef CAS
- H. Yuan, W. Ma, C. Chen, J. Zhao, J. Liu, H. Zhu and X. Gao, Chem. Mater., 2007, 19, 1592–1600 CrossRef CAS
- T. K. Sau and C. J. Murphy, J. Am. Chem. Soc., 2004, 126, 8648–8649 CrossRef CAS
- H.-L. Wu, C.-H. Chen and M. H. Huang, Chem. Mater., 2008, 21, 110–114 CrossRef
- G. Kawamura, Y. Yang, K. Fukuda and M. Nogami, Mater. Chem. Phys., 2009, 115, 229–234 CrossRef CAS
- P. Senthil Kumar, I. Pastoriza-Santos, B. Rodríguez-González, F. Javier García de Abajo and L. M. Liz-Marzán, Nanotechnology, 2008, 19, 015606 CrossRef
- C. G. Khoury and T. Vo-Dinh, J. Phys. Chem. C, 2008, 112, 18849–18859 CAS
- J. L. Burt, J. L. Elechiguerra, J. Reyes-Gasga, J. Martin Montejano-Carrizales and M. Jose-Yacaman, J. Cryst. Growth, 2005, 285, 681–691 CrossRef CAS
- J. Xie, J. Y. Lee and D. I. C. Wang, Chem. Mater., 2007, 19, 2823–2830 CrossRef CAS
- B. Van de Broek, F. Frederix, K. Bonroy, H. Jans, K. Jans, G. Borghs and G. Maes, Nanotechnology, 2011, 22, 015601 CrossRef CAS
- P. K. Jain and M. A. Ei-Sayed, J. Phys. Chem. C, 2007, 111, 17451–17454 CAS
- P. Sajanlal and T. Pradeep, Nano Res., 2009, 2, 306–320 CrossRef CAS
- P. Aldeanueva-Potel, E. Carbó-Argibay, N. Pazos-Pérez, S. Barbosa, I. Pastoriza-Santos, R. A. Alvarez-Puebla, L. M. Liz-Marzán, P. Aldeanueva-Potel, E. Carbó-Argibay, N. Pazos-Pérez, S. Barbosa, I. Pastoriza-Santos, R. A. Alvarez-Puebla and L. M. Liz-Marzán, ChemPhysChem, 2012, 13, 2561–2565 CrossRef CAS
- C. Kim, H.-M. Song, X. Cai, J. Yao, A. Wei and L. V. Wang, J. Mater. Chem., 2011, 21, 2841 RSC
- T. Niidome, M. Yamagata, Y. Okamoto, Y. Akiyama, H. Takahashi, T. Kawano, Y. Katayama and Y. Niidome, J. Controlled Release, 2006, 114, 343–347 CrossRef CAS
- W. H. De Jong, W. I. Hagens, P. Krystek, M. C. Burger, A. J. A. M. Sips and R. E. Geertsma, Biomaterials, 2008, 29, 1912–1919 CrossRef CAS
- T. Kawano, M. Yamagata, H. Takahashi, Y. Niidome, S. Yamada, Y. Katayama and T. Niidome, J. Controlled Release, 2006, 111, 382–389 CrossRef CAS
- G. Zhang, Z. Yang, W. Lu, R. Zhang, Q. Huang, M. Tian, L. Li, D. Liang and C. Li, Biomaterials, 2009, 30, 1928–1936 CrossRef CAS
- M. Ogris, P. Steinlein, S. Carotta, S. Brunner and E. Wagner, AAPS J., 2001, 3, 43–53 Search PubMed
- T. Merdan, K. Kunath, H. Petersen, U. Bakowsky, K. H. Voigt, J. Kopecek and T. Kissel, Bioconjugate Chem., 2005, 16, 785–792 CrossRef CAS
- P. Milla, F. Dosio and L. Cattel, Curr. Drug Metab., 2012, 13, 105–119 CrossRef CAS
- F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS
- X. Sun, R. Rossin, J. L. Turner, M. L. Becker, M. J. Joralemon, M. J. Welch and K. L. Wooley, Biomacromolecules, 2005, 6, 2541–2554 CrossRef CAS
- R. Duncan and L. Izzo, Adv. Drug Delivery Rev., 2005, 57, 2215–2237 CrossRef CAS
- C. Kojima, B. Turkbey, M. Ogawa, M. Bernardo, C. A. S. Regino, L. H. Bryant Jr, P. L. Choyke, K. Kono and H. Kobayashi, Nanomed.: Nanotechnol., Biol. Med., 2011, 7, 1001–1008 CrossRef CAS
- T. R. Tshikhudo, Z. Wang and M. Brust, Mater. Sci. Technol., 2004, 20, 980–984 CrossRef CAS
- R. Duncan and L. Izzo, Adv. Drug Delivery Rev., 2005, 57, 2215–2237 CrossRef CAS
- G. Prencipe, S. M. Tabakman, K. Welsher, Z. Liu, A. P. Goodwin, L. Zhang, J. Henry and H. Dai, J. Am. Chem. Soc., 2009, 131, 4783–4787 CrossRef CAS
- D. Bhumkar, H. Joshi, M. Sastry and V. Pokharkar, Pharm. Res., 2007, 24, 1415–1426 CrossRef CAS
- J.-J. Yuan, A. Schmid, S. P. Armes and A. L. Lewis, Langmuir, 2006, 22, 11022–11027 CrossRef CAS
- K. Katti, N. Chanda, R. Shukla, A. Zambre, T. Suibramanian, R. R. Kulkarni, R. Kannan and K. V. Katti, Int. J. Green Nanotechnol.: Biomed., 2009, 1, B39–B52 CrossRef
- R. Shukla, S. K. Nune, N. Chanda, K. Katti, S. Mekapothula, R. R. Kulkarni, W. V. Welshons, R. Kannan and K. V. Katti, Small, 2008, 4, 1425–1436 CrossRef CAS
- R. K. Das, B. B. Borthakur and U. Bora, Mater. Lett., 2010, 64, 1445–1447 CrossRef CAS
- S. P. Dubey, M. Lahtinen and M. Sillanpää, Colloids Surf., A, 2010, 364, 34–41 CrossRef CAS
- P. Babu, P. Sharma, M. Kalita and U. Bora, Front. Mater. Sci., 2011, 5, 379–387 CrossRef
- H. Ding, K.-T. Yong, I. Roy, H. E. Pudavar, W. C. Law, E. J. Bergey and P. N. Prasad, J. Phys. Chem. C, 2007, 111, 12552–12557 CAS
- J. Chen, M. Yang, Q. Zhang, E. C. Cho, C. M. Cobley, C. Kim, C. Glaus, L. V. Wang, M. J. Welch and Y. Xia, Adv. Funct. Mater., 2010, 20, 3684–3694 CrossRef CAS
- S.-W. Tsai, J.-W. Liaw, F.-Y. Hsu, Y.-Y. Chen, M.-J. Lyu and M.-H. Yeh, Sensors, 2008, 8, 6660–6673 CrossRef CAS
- G. Li, D. Li, L. Zhang, J. Zhai and E. Wang, Chem.–Eur. J., 2009, 15, 9868–9873 CrossRef CAS
- B. D. Chithrani and W. C. W. Chan, Nano Lett., 2007, 7, 1542–1550 CrossRef CAS
- P. Nativo, I. A. Prior and M. Brust, ACS Nano, 2008, 2, 1639–1644 CrossRef CAS
- E. Oh, J. B. Delehanty, K. E. Sapsford, K. Susumu, R. Goswami, J. B. Blanco-Canosa, P. E. Dawson, J. Granek, M. Shoff, Q. Zhang, P. L. Goering, A. Huston and I. L. Medintz, ACS Nano, 2011, 5, 6434–6448 CrossRef CAS
- Z. Liang, Y. Liu, X. Li, Q. Wu, J. Yu, S. Luo, L. Lai and S. Liu, J. Biomed. Mater. Res., Part A, 2011, 98A, 479–487 CrossRef CAS
- J. Homola, S. S. Yee and G. Gauglitz, Sens. Actuators, B, 1999, 54, 3–15 CrossRef
- A. K. Sharma, R. Jha and B. D. Gupta, IEEE Sens. J., 2007, 7, 1118–1129 CrossRef
- S. Link and M. El-Sayed, J. Phys. Chem. B, 1999, 103, 4212–4217 CrossRef CAS
- P. Jain, X. Huang, I. El-Sayed and M. El-Sayed, Plasmonics, 2007, 2, 107–118 CrossRef CAS
- X. Huang, P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Nanomedicine, 2007, 2, 681–693 CrossRef CAS
- J. Park, A. Estrada, J. A. Schwartz, P. Diagaradjane, S. Krishnan, A. K. Dunn and J. W. Tunnell, Lasers Surg. Med., 2010, 42, 630–639 CrossRef
- S. Motamedi, T. Shilagard, K. Edward, L. Koong, S. Qui and G. Vargas, Biomed. Opt. Express, 2011, 2, 1194–1203 CrossRef CAS
-
N. K. Balla, C. J. R. Sheppard and P. T. C. So, in Reporters, Markers, Dyes, Nanoparticles, and Molecular Probes for Biomedical Applications Iii, ed. S. Achilefu and R. Raghavachari, Spie-Int Soc Optical Engineering, Bellingham, 2011, vol. 7910 Search PubMed
- S. Motamedi, T. Shilagard, L. Koong and G. Vargas, Proc. SPIE–Int. Soc. Opt. Eng., 2010, 7576, 75760Z CrossRef
- N. J. Durr, T. Larson, D. K. Smith, B. A. Korgel, K. Sokolov and A. Ben-Yakar, Nano Lett., 2007, 7, 941–945 CrossRef CAS
- B. Jang, J.-Y. Park, C.-H. Tung, I.-H. Kim and Y. Choi, ACS Nano, 2011, 5, 1086–1094 CrossRef CAS
- J. Park, A. Estrada, K. Sharp, K. Sang, J. A. Schwartz, D. K. Smith, C. Coleman, J. D. Payne, B. A. Korgel, A. K. Dunn and J. W. Tunnell, Opt. Express, 2008, 16, 1590–1599 CrossRef CAS
- C. Peng, L. Zheng, Q. Chen, M. Shen, R. Guo, H. Wang, X. Cao, G. Zhang and X. Shi, Biomaterials, 2012, 33, 1107–1119 CrossRef CAS
- T. Reuveni, M. Motiei, Z. Romman, A. Popovtzer and R. Popovtzer, Int. J. Nanomed., 2011, 6, 2859–2864 CAS
- J. Hainfeld, D. Slatkin, T. Focella and H. Smilowitz, Br. J. Radiol., 2006, 79, 248–253 CrossRef CAS
- Y. Hwu, W. L. Tsai, J. H. Je, S. K. Seol, B. Kim, A. Groso, G. Margaritondo, K. H. Lee and J. K. Seong, Phys. Med. Biol., 2004, 49, 501–508 CrossRef CAS
- C.-C. Chien, H.-H. Chen, S.-F. Lai, K.-C. Wu, X. Cai, Y. Hwu, C. Petibois, Y. Chu and G. Margaritondo, J. Nanobiotechnol., 2012, 10, 10 CrossRef CAS
- P. Huang, L. Bao, C. Zhang, J. Lin, T. Luo, D. Yang, M. He, Z. Li, G. Gao, B. Gao, S. Fu and D. Cui, Biomaterials, 2011, 32, 9796–9809 CrossRef CAS
- W. Eck, A. I. Nicholson, H. Zentgraf, W. Semmler and S. Bartling, Nano Lett., 2010, 10, 2318–2322 CrossRef CAS
- D. P. Cormode, E. Roessl, A. Thran, T. Skajaa, R. E. Gordon, J.-P. Schlomka, V. Fuster, E. A. Fisher, W. J. M. Mulder, R. Proksa and Z. A. Fayad, Radiology, 2010, 256, 774–782 CrossRef
- E. B. Dickerson, E. C. Dreaden, X. Huang, I. H. El-Sayed, H. Chu, S. Pushpanketh, J. F. McDonald and M. A. El-Sayed, Cancer Lett., 2008, 269, 57–66 CrossRef CAS
- J. Choi, J. Yang, J. Park, E. Kim, J.-S. Suh, Y.-M. Huh and S. Haam, Adv. Funct. Mater., 2011, 21, 1082–1088 CrossRef CAS
- J. C. Y. Kah, M. Olivo, T. H. Chow, K. S. Song, K. Z. Y. Koh, S. Mhaisalkar and C. J. R. Sheppard, J. Biomed. Opt., 2009, 14, 054015 CrossRef
- L. Song, K. Maslov, R. Bitton, K. K. Shung and L. V. Wang, J. Biomed. Opt., 2008, 13, 054028 Search PubMed
- Q. Zhang, N. Iwakuma, P. Sharma, B. M. Moudgil, C. Wu, J. McNeill, H. Jiang and S. R. Grobmyer, Nanotechnology, 2009, 20, 395102 CrossRef CAS
- A. M. Schwartzberg, T. Y. Olson, C. E. Talley and J. Z. Zhang, J. Phys. Chem. B, 2006, 110, 19935–19944 CrossRef CAS
- M.-L. Li, J. C. Wang, J. A. Schwartz, K. L. Gill-Sharp, G. Stoica and L. V. Wang, J. Biomed. Opt., 2009, 14, 010507 CrossRef
- A. Agarwal, S. W. Huang, M. O'Donnell, K. C. Day, M. Day, N. Kotov and S. Ashkenazi, J. Appl. Phys., 2007, 102, 023911 CrossRef
- M. Eghtedari, A. Oraevsky, J. A. Copland, N. A. Kotov, A. Conjusteau and M. Motamedi, Nano Lett., 2007, 7, 1914–1918 CrossRef CAS
- A. Taruttis, E. Herzog, D. Razansky and V. Ntziachristos, Opt. Express, 2010, 18, 19592–19602 CrossRef CAS
- C. Kim, C. Favazza and L. V. Wang, Chem. Rev., 2010, 110, 2756–2782 CrossRef CAS
- A. M. Alkilany and C. J. Murphy, J. Nanopart. Res., 2010, 12, 2313–2333 CrossRef CAS
- N. Khlebtsov and L. Dykman, Chem. Soc. Rev., 2011, 40, 1647–1671 RSC
- X.-M. Jiang, L.-M. Wang, J. Wang and C.-Y. Chen, Appl. Biochem. Biotechnol., 2012, 166, 1533–1551 CrossRef CAS
- Q. Zhang, V. M. Hitchins, A. M. Schrand, S. M. Hussain and P. L. Goering, Nanotoxicology, 2011, 5, 284–295 CrossRef CAS
- H. Yen, S. Hsu and C. Tsai, Small, 2009, 5, 1553–1561 CrossRef CAS
- M. Tarantola, A. Pietuch, D. Schneider, J. Rother, E. Sunnick, C. Rosman, S. Pierrat, C. Sönnichsen, J. Wegener and A. Janshoff, Nanotoxicology, 2011, 5, 254–268 CrossRef CAS
- Y. Pan, S. Neuss, A. Leifert, M. Fischler, F. Wen, U. Simon, G. Schmid, W. Brandau and W. Jahnen-Dechent, Small, 2007, 3, 1941–1949 CrossRef CAS
- L. Wang, Y. Liu, W. Li, X. Jiang, Y. Ji, X. Wu, L. Xu, Y. Qiu, K. Zhao, T. Wei, Y. Li, Y. Zhao and C. Chen, Nano Lett., 2010, 11, 772–780 CrossRef
- J. Lin, H. Zhang, Z. Chen and Y. Zheng, ACS Nano, 2010, 4, 5421–5429 CrossRef CAS
- S. Vesaratchanon, A. Nikolov and D. T. Wasan, Adv. Colloid Interface Sci., 2007, 134–135, 268–278 CrossRef CAS
- T. Niidome, M. Yamagata, Y. Okamoto, Y. Akiyama, H. Takahashi, T. Kawano, Y. Katayama and Y. Niidome, J. Controlled Release, 2006, 114, 343–347 CrossRef CAS
- B. M. Rothen-Rutishauser, S. Schürch, B. Haenni, N. Kapp and P. Gehr, Environ. Sci. Technol., 2006, 40, 4353–4359 CrossRef CAS
- Y. Akiyama, T. Mori, Y. Katayama and T. Niidome, J. Controlled Release, 2009, 139, 81–84 CrossRef CAS
- H. C. Fischer and W. C. Chan, Curr. Opin. Biotechnol., 2007, 18, 565–571 CrossRef CAS
- N. Lewinski, V. Colvin and R. Drezek, Small, 2007, 4, 26–49 CrossRef
- D. B. Warheit, C. M. Sayes, K. L. Reed and K. A. Swain, Pharmacol. Ther., 2008, 120, 35–42 CrossRef CAS
- D.-E. Lee, H. Koo, I.-C. Sun, J. H. Ryu, K. Kim and I. C. Kwon, Chem. Soc. Rev., 2012, 41, 2656 RSC
- B. Li, M. Abran, C. Matteau-Pelletier, L. Rouleau, T. Lam, R. Sharma, E. Rhéaume, A. Kakkar, J.-C. Tardif and F. Lesage, J. Biomed. Opt., 2011, 16, 126010 CrossRef
| This journal is © The Royal Society of Chemistry 2013 |