 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Progress in bio-based plastics and plasticizing modifications
Tizazu Mekonnena, Paolo Mussonea, Hamdy Khalilb and David Bressler*a
aBiorefining Conversions and Fermentations Laboratory, Department of Agricultural, Food and Nutritional Sciences, University of Alberta, Edmonton, AB T6G 2P5, Canada. E-mail: david.bressler@ales.ualberta.ca; Fax: +1 780-492-4265; Tel: +1 780-492-4986
bThe Woodbridge Group, 8214 Kipling Avenue, Woodbridge, ON, Canada L4L 2A4
First published on 9th August 2013
Abstract
Over the coming few decades bioplastic materials are expected to complement and gradually replace some of the fossil oil based materials. Multidisciplinary research efforts have generated a significant level of technical and commercial success towards these bio-based materials. However, extensive application of these bio-based plastics is still challenged by one or more of their possible inherent limitations, such as poor processability, brittleness, hydrophilicity, poor moisture and gas barrier, inferior compatibility, poor electrical, thermal and physical properties. The incorporation of additives such as plasticizers into the biopolymers is a common practice to improve these inherent limitations. Generally, plasticizers are added to both synthetic and bio-based polymeric materials to impart flexibility, improve toughness, and lower the glass transition temperature. This review introduces the most common bio-based plastics and provides an overview of recent advances in the selection and use of plasticizers, and their effect on the performance of these materials. In addition to plasticizers, we also present a perspective of other emerging techniques of improving the overall performance of bio-based plastics. Although a wide variety of bio-based plastics are under development, this review focuses on plasticizers utilized for the most extensively studied bioplastics including poly(lactic acid), polyhydroxyalkanoates, thermoplastic starch, proteinaceous plastics and cellulose acetates. The ongoing challenge and future potentials of plasticizers for bio-based plastics are also discussed.
 Tizazu Mekonnen | Tizazu Mekonnen earned his BSc degree in Applied Physics (2004) and MSc degree in Chemical Engineering (2009) from Addis Ababa University, Ethiopia. He is currently finalizing his PhD in Bioresource Engineering under the supervision of Professor David Bressler at the Biorefining Conversions and Fermentations Lab at the University of Alberta, Canada. Mr Mekonnen's PhD research work entails hydrolysis and biorefining of hazardous waste protein biomass to fabricate and study novel biopolymers and biopolymer–synthetic polymer hybrid materials for various industrial applications. |
 Paolo Mussone | Dr Mussone holds a position as Research Associate with the Biorefining Conversions and Fermentations Laboratory at the University of Alberta. Dr Paolo Mussone earned an MSc degree in Process Engineering from the Polytechnic University of Milan in Italy and a PhD in Surface Chemistry and Colloidal Science from the University of Manchester in the United Kingdom. Dr Mussone's general research focus is on the conversion of biomass into value-added chemicals and materials. Of particular interest is the development of renewable polymeric surfactant platforms for heavy petroleum processing and waste water treatment processes. |
 Hamdy Khalil | Dr Hamdy Khalil is the Senior Global Director for Advanced Technologies and Innovation. He pioneered the introduction of chemicals derived from renewable resources into the manufacturing of automotive parts and the application of Phase Transfer Catalysis in the synthesis of heterocyclic compounds. Dr Khalil is a member of the Board of Directors of several industrial organizations. He chaired and participated in many International Conferences related to Biotechnology as well as Polyurethane Technology. Dr Khalil is a member of the American Chemical Society and the Canadian Institute of Chemistry. He has several patents and publications in the areas of Bio-polyols, Sealants, Latex and Polystyrene. |
 David Bressler | Dr Bressler is currently a tenured Professor in the Food and Bioresource Technology Division of the Faculty of Agricultural, Life, and Environmental Sciences at the University of Alberta. He is also Founding Director of the Biorefining Conversions Network, an organization focused on facilitating the development of novel, commercially viable biomass conversion technologies, and value-added products within Alberta, Canada. Dr Bressler's general area of research is the industrial application of chemical, thermal, and biological systems for the conversion of conventional agricultural products to platform chemicals and other value-added commodities. |
1. Introduction
Plastics are amorphous organic solid polymers covering a wide range of polymerization products suitable for the manufacture of diversified products. Worldwide annual plastics production is estimated to surpass 300 million tons by 2015 (ref. 1) representing trillions of dollars in terms of global economic returns.2 Plastics are highly valued materials because of their low cost and extraordinary versatility and they constitute the largest petroleum application second only to energy.3 Among the many applications of plastics, packaging accounts for almost one-third of their use followed by construction and consumer products.4 The materials science community has been striving for decades to generate bio-based plastics to substitute or complement conventional synthetic plastics based on exclusively petroleum feedstock. According to current estimates, the global production of bioplastics is expected to grow at an annual rate of up to 30% in the coming decade to reach 3.5 million tonnes in 2020.5Bioplastics may also be bio-based (i.e. polymer derived from renewable feedstock) and biodegradable (i.e. polymer that can return to nature).6 Biodegradability and compostability depend on the chemical structure rather than the feedstock source. According to the US Department of Agriculture (USDA), bio-based products are defined as commercial or industrial goods (other than feed or food) composed in whole or in significant part of biological products.7 Thus, synonymous use of the terms bio-based plastic and biodegradable plastic is not correct. Some of the most commonly known bio-based plastics in today's marketplace in terms of production and renewability are poly(lactic acid) (PLA), polyhydroxyalkanoates (PHAs), starch plastics, cellulose esters and protein based plastics (Fig. 1). Other bio-based plastics, such as bio enriched polyurethane manufactured using modified vegetable oils, polyethylene monomers derived from the dehydration of bio-ethanol, polypropylene monomers derived from dehydration of bio-butanol and poly(ethylene terephthalate) monomers produced via fermentation, catalytic pyrolysis or gasification of biomass,8 that have at least partial sourcing from plants constitute emerging technologies expected to make a significant market impact.
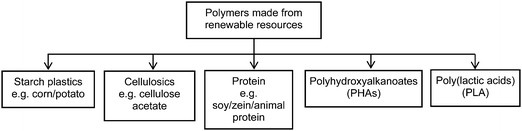 | ||
| Fig. 1 Major bio-based plastics and their production routes.6 | ||
Bio-based plastics could overcome the sustainability issues and environmental challenges posed by the production and disposal of synthetic plastics. However, the large scale commercial deployment of bio-based plastics to replace conventional plastic materials remains challenged by several factors. Some of the challenges are attributed to the relatively poor performance, variability of properties of the feedstock associated with location and the time of harvest, high production cost and lack of infrastructure. Recent development in bacteria synthesized plastics (PHAs) and the utilization of nature's own building blocks such as proteins, fats, carbohydrates, lignin, etc. obtained from agricultural feedstock and agricultural industry wastes constitute a major progress towards bio-based plastics in the last decade.
Plasticizers have long been known for their effectiveness in enhancing the flexibility of synthetic plastics such as polyvinyl chloride (PVC) and epoxy resins. New types of plasticizers compatible with bio-based plastics are being developed. For technical and economic reasons, polymer additives are a large and increasingly significant component of the polymer industry.9 Among the additives, plasticizers constitute about one third of the global additive market,10 with a worldwide consumption of over 4.6 million metric tonnes in 2003 (ref. 11) and over 6.4 metric tonnes in 2011.12 Generally, plasticizers are small, relatively non-volatile, organic molecules that are added to polymers to reduce brittleness, impart flexibility, and improve toughness, reducing crystallinity, lowering glass transition and melting temperatures.13,14 Plasticization reduces the relative number of polymer–polymer contacts thereby decreasing the rigidity of the three-dimensional structure thereby allowing deformation without rupture.15 Consequently, plasticizers improve processability, flexibility, durability and in some cases reduce the cost of polymers.16,17
The use of plasticized polymers in pharmaceutical applications ranging from packaging materials or auxiliary substances in conventional dosage forms to membranes or matrices modifying and controlling the drug release characteristics in therapeutic systems has been reported in the literature.16,18,19 The processing behaviour, such as film formation and coating dispersion, and properties of polymers in various applications are greatly improved by adequate choice of plasticizer type and quantity.16,20 Generally, the choice of these plasticizers to be used as modifiers of plastics is limited by the required safety, environmental favorability, chemical and physical property that dictate their miscibility, processing temperature and required flexibility towards the target application.17
The risk of leaching out of certain plasticizers, such as phthalates during storage or end-user application, constitutes a major safety risk.21–24 This coupled with other shortcomings (e.g. toxicity, poor compatibility) limits some plasticizers from application in the medical, pharmaceutical and food packaging fields. The ideal plasticizer significantly lowers the glass transition temperature (Tg), is biodegradable, nonvolatile, and nontoxic, and exhibits minimal leaching or migration during use or aging.
Recent advances in bio-based plastics are spurred by factors such as public concern over the depletion of petroleum based raw materials, the desire of manufacturing companies to develop more sustainable raw material sources, the improvement in properties as well as cost competitive relationship of bioplastics.25,26 As these bio-based plastic industries continuously grow, the demand for new types of plasticizers with new characteristics, performance and other additives that are compatible with the bioplastics also grows in the same direction.27 In the realm of developing packaging materials from bio-based materials, a high ductility at room temperature is required and thus, there is no tolerance for the polymer film tearing or cracking when subjected to stresses during package manufacturing or use.28 Moreover, increase in the utilization of plasticized polymers for biomedical and pharmaceutical application,16 the search for safer plasticizers for commodity plastics such as poly(vinyl chloride)29–31 and efforts to produce renewable and biodegradable plasticizers29,30 constitute an additional motive for the recent development of new plasticizers. This review briefly reports recent progress in the development of plasticizers utilized for bio-based plastics, and their influence on the performance of bio-based plastics.
2. Plasticization mechanism
Two types of plasticizers are defined in polymer science: internal and external.22,23,32 Internal plasticizers are part of the polymer molecules, co-polymerized into the polymer structure, grafted or reacted with the original polymer thereby making the polymer chains more difficult to fit and compact with each other closely.23 They soften polymers by lowering the glass transition temperature (Tg) and reducing the elastic modulus.22 External plasticizers, on the other hand, are low volatility molecules added to interact with polymers and produce swelling without chemical reaction. Internal molecular forces between plasticizer molecules and between a plasticizer and a polymer such as dispersion forces, induction forces, dipole–dipole interaction, hydrogen bonds are important in external plasticization.23Several theories have been proposed to explain the mechanism and action of plasticizers on polymers. Among those theories, the following plasticizing mechanisms have been widely accepted to describe the effect of plasticizers on polymeric networks:33–36 (a) the lubricity theory: this theory is similar to metal parts lubrication by oil. The plasticizer acts as a lubricant to reduce friction and facilitates polymer chain mobility past one another, consequently lowering deformation; (b) the gel theory: this theory extends the lubricity theory and suggests that a plasticizer disrupts and replaces polymer–polymer interactions (hydrogen bonds, van der Waals or ionic forces, etc.) that hold polymer chains together resulting in reduction of the polymer gel structure and increased flexibility; and (c) the free volume theory: for any polymeric material the free volume is defined as the internal space available in a polymer for the movement of chains. Free volume is usually described as the difference between the observed volume at absolute zero and the volume measured at a selected temperature. Rigid resins are characterized by limited free volume whereas flexible resins have relatively large amounts of free volume. Plasticizers increase the free volume of resins and also maintain the free volume after the polymer–plasticizer mixture post processing is cooled down. The free volume theory explains the effect of plasticizers in lowering the glass transition temperature.
Although these theories are widely accepted and utilized in the selection of plasticizer for polymers, Shtarkman and Razinskaya35 stressed the limitation of the current plasticization theories. According to these authors,35 the plasticization theories are limited and not feasible for plasticizer selection for the following reasons: (1) direct studies of the plasticization mechanism is lacking and (2) the existing plasticization theories have limited predicting capability and are limited to only particular cases. For this purpose, the authors35 suggested the necessity of a compatibility–efficiency–property study that takes into account the structure of the polymeric system to select a specific plasticizer rather than relying on the theories.
The aforementioned plasticization theories/mechanisms were developed for synthetic plastics, particularly PVC. Limited attention has been devoted to developing new theories/mechanisms or improving established theories to explain the plasticization mechanism of the newly developed and emerging bio-based plastics. The complex nature of some of the biological feedstock macromolecules makes bio-based plastics radically different from the common repeating monomer based synthetic polymers. Hence, renewed efforts are required to investigate other more explanatory plasticization possibilities and theories.
3. Plasticization of bio-based plastics
3.1. Poly(lactic acid) plastics
Poly(lactic acid) (PLA) is one of the most promising innovative plastics for various end-use applications. This polymer is thermoplastic, renewable, biodegradable and biocompatible, a set of highly attractive attributes for pharmaceutical, biological and medical applications.37,38 The raw material of PLA, L-lactic acid, can be produced by fermentation of renewable sugar resources such as starch and other polysaccharides.39,40 Moreover, PLA exhibits a remarkable balance of performance properties comparable to traditional thermoplastics39 processed using conventional plastic processing techniques. From a physical property standpoint it is often loosely compared to polystyrene.37 Similar to polystyrene, standard grade PLA has high modulus and strength.37,41 Moreover, the degradation products of polylactides are nontoxic which enhances practical applications in biomedicine.42 PLA is currently being commercialized for a wide spectrum of technologically important fields and applications by companies such as Cargill and Dow Chemicals.38PLA belongs to the family of aliphatic polyesters commonly made from lactic acid (2-hydroxypropionic acid) building block shown in Fig. 3. The synthesis of lactic acid into high-molecular weight PLA can follow two different routes of polymerization,40,43 as depicted in Fig. 4. The monomer lactic acid is condensation polymerized to yield a low-molecular weight, brittle, glassy polymer in the first route, which, for the most part, is unusable unless external coupling agents are used to increase the molecular weight of the polymer.40 The second route of producing PLA is to collect, purify, and ring-open and polymerize lactide to yield high molecular weight (average Mw > 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) PLA.40,43,44
000) PLA.40,43,44
The combination of the chiral lactic acid monomers (Fig. 2) or the depolymerization of low molecular weight PLA (Fig. 3) could give rise to distinct forms of polylactides. These polylactides are poly(L-lactide) (or LL-lactide), poly(D-lactide) (or DD-lactide), poly(LD-lactide) (or meso-lactide) as shown in Fig. 4 or a mixture of L-and D-lactides, called racemic lactide (rac-lactide).38,46,47 While the D- and L- lactides are optically active, meso- is not (Fig. 4).46 Highly crystalline PLA can be obtained with low D content (<2%), fully amorphous PLA on the other hand can be obtained with high D content (>20%).48 Semi-crystalline PLA is obtained with 2 to 20% of D content.2 The amount and stereosequence of these lactides in the polymer backbone give rise to a wide range of molecular weights. These changes as a result impact the melt behavior, thermal, mechanical, optical properties, barrier properties and biological properties of PLA.49,50
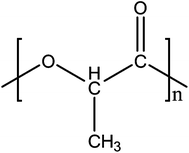 | ||
| Fig. 2 Basic structure of PLA. | ||
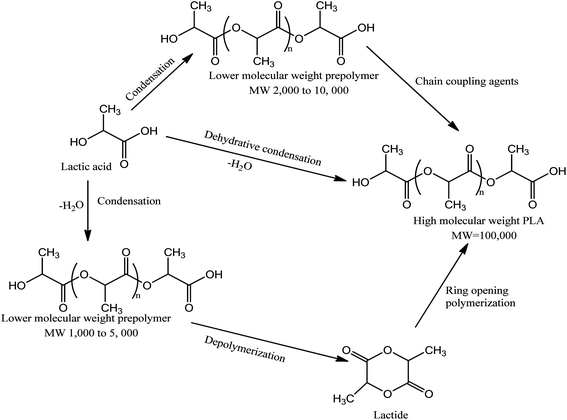 | ||
| Fig. 3 Synthesis methods for high molecular weight PLA.44,45 | ||
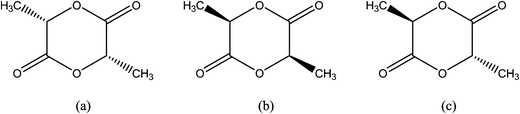 | ||
| Fig. 4 Chemical structures of dimeric (a) D-lactide, (b) L-lactide and (c) meso-lactide. | ||
PLA is brittle, with relatively poor impact strength and low thermal degradation temperature limiting its applicability.39,40 Relatively poor strength, coupled with its hydrophobicity, semi-crystalline properties, limited thermal processability, lack of reactive functional groups along the polymer backbone and high cost constitute the majority of its limitation in wide industrial and medical applications.39,45 Accordingly, to compete with the low-cost and flexible commodity polymers and upgrade the PLA performance, considerable research effort is being carried out. These attempts include modifying PLA with plasticizers, blending with other polymers,51 copolymerization, and incorporation of fillers.37,45,52
| Modifiers | MW (g mol−1) | Conc. (wt%) | Tg (°C) | Ea (MPa) | εa (%) | σa (MPa) |
|---|---|---|---|---|---|---|
| a Tensile modulus (E), tensile stress at yield (σ), and elongation at break (ε). | ||||||
| PLA54,55 | 137000 | 100 | 59 | 1720 | 7 | 51.7 |
| Triethyl citrate54 | 276 | 20 | 32.6 | 382 | 12.6 | |
| Tributyl citrate54 | 360 | 20 | 17.6 | 350 | 7.1 | |
| Acetyl triethyl citrate54 | 318 | 20 | 30 | 320 | 9.6 | |
| Acetyl tributyl citrate54 | 402 | 20 | 17 | 420 | 9.2 | |
| Poly(oxyethylene)55 | 10000 | 21 | 31 | 320 | 7 | 49 |
| Poly(ε-caprolactone)55 | 10000 | 20 | 35 | 961 | 25 | 19 |
| Glycerol51 | 92.09 | 20 | 53 | — | — | |
| PEG monolaurate51 | 400 | 20 | 21 | 142 | — | |
| Plasticized TPS51 | — | 25 | — | 2.9 | 30.2 | |
| PEG62 | 1500 | 10 | 34.3 | 1750 | 150 | 15.1 |
| PEG62 | 1500 | 20 | 23.2 | 1460 | 150 | 14.6 |
The results in Table 1 show that all citrates at 20% concentration reduced the glass transition temperature and improved the flexibility while reducing the tensile strength of the PLA control. A significant improvement of elongation at break was achieved at the expense of tensile strength. Ljungberg and Wesslen63 also investigated the use of triacetin, tributyl citrate, triethyl citrate, acetyl tributyl citrate, acetyl triethyl citrate as potential plasticizers of PLA and reported a drastic lowering of the glass transition temperature of PLA at concentrations as low as 15% resulting in a homogeneous and flexible film. However, it was reported that the migration of citrates onto the film surfaces during aging, especially the low molecular weight citrates, was a major challenge.17 This issue could be addressed by increasing the molecular weight. For instance, Ljungberg and Wesslen17 transesterified tributyl citrate (MW 360 g mol−1) with diethyl glycol that resulted in two oligomeric plasticizers with higher molecular weights (MW 4500 g mol−1 and 63600 g mol−1). The investigation of the effects of these oligomers on thermo-mechanical and aging properties of PLA shows that both oligomers did not lower the Tg as greatly as monomeric citrates. Among the two oligomeric plasticizers, a relatively larger reduction in Tg was achieved by the oligomer with the lower molecular weight.
Similarly, molecular weight variation, concentration and the presence of polar amide groups of plasticizers can positively interact with PLA chains, affecting the compatibility between PLA and the plasticizer and controlling elongation and morphological stability that result in leaching during aging or use.17,63 The plasticizer with lower molecular weight that resulted in lower Tg of PLA may also facilitate the migration of the plasticizer from the bulk of the material compared to the higher molecular weight plasticizer.17 The effect of triacetin (0–30%) and tributyl citrate (0–25%) loading on PLA was studied63 and an almost linear decrease in Tg with the increase of plasticizer content was observed.
Maglio et al.55 studied the copolymerization of PLA with poly(ε-caprolactone) (PCL) and poly(oxyethylene) (PEO) to improve the brittleness and reduce Tg. The copolymers obtained as a result exhibited high elongations at break and as a result much lower tensile moduli than the PLA pure polymer (Table 1). The Tg of the copolymers was also much lower than that of the original PLA. A recent study by Hassouna et al.62 investigated grafting of poly(ethylene glycol) (PEG) onto PLA through reactive extrusion to develop plasticized PLA. It was shown that the Tg and modulus were invariably reduced. In all cases, both Tg and elastic modulus were dependent on the content of PEG grafted onto the PLA. The in situ reactive grafting of PEG onto PLA exhibited a marked Tg reduction than the blending option. Other plasticizers of PLA reported in the literature include epoxidized soybean oil,65 ionic liquids,66 mixed plasticizers,67etc.
In summary, it can be pointed out that several studies have demonstrated that plasticizers play a significant role in determining the performance properties of PLA plastics. Plasticizers can solve most of the problems that occur during processing or in final use. New characteristics of PLA observed during plasticization may also pave the way to novel applications. The limitations of the currently studied PLA plasticizers include leaching during use, lack of thermal stability, need for offering more ductility and more performance, biocompatibility issues, cost, need for high percentage loading to lower price of PLA, need for a bio-based plasticizer that reduces the overall carbon footprint, etc.
3.2. Polyhydroxyalkanoates (PHAs)
Microbial-produced PHAs are fully biodegradable bio-polyesters produced by a wide variety of microorganisms for internal carbon and energy storage as part of their survival mechanism.68,69 PHAs, also known as poly(4-alkan-2-oxelanones) according to IUPAC naming, have attracted much attention recently as alternative polymeric materials that can be produced from renewable and biowaste resources. PHAs, with the general structure shown in Fig. 5, vary widely in their structure and properties (flexibility, crystallinity, melting temperature, etc.), depending on the producing microorganisms, the conditions of biosynthesis and the type of carbon source.70 PHAs are piezoelectric, perfectly isotactic/optically active and biocompatible thermoplastic polyesters amenable to melt-processing into various final forms.71–74 High molecular weight PHAs have attracted considerable attention as potential replacements for non-degradable commodity plastics (e.g. polyethylene and polypropylene), as well as biodegradable and biocompatible biomaterials for implant purposes.72,75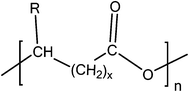 | ||
| Fig. 5 General structure of PHAs. n = 1 (R = hydrogen, poly(3-hydroxypropionate); R = methyl, poly(3-hydroxybutyrate); R = ethyl, poly(3-hydroxyvalerate); R = propyl, poly(3-hydroxyhexanoate); R = pentyl, poly(3-hydroxyoctanoate); R = nonyl, poly(3-hydroxydodecanoate)), n = 2 (R = hydrogen, poly(4-hydroxybutyrate)), and n = 3 (R = hydrogen, poly(5-hydroxyvalerate)). | ||
Poly(3-hydroxybutyrate) (PHB), R = methyl, being the first among the isolated PHAs, is the most extensively studied PHA produced in nature in the presence of excess carbon by bacteria as storage granules providing food, energy and reducing power.76,77 This polymer and its copolymer with polyhydroxyvalerate to make poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) are at present the only known PHAs relevant for practical applications.78 PHB and PHBV are completely biodegradable in the environment and can be extruded, moulded and spun on conventional plastic processing equipment.71,79 These features make these polymers an ideal candidate for the production of biodegradable packaging materials and other disposable articles.79 However, the industrial scale production of PHB/PHBV is hindered by roadblocks. Thermal processing is challenging because of their relative low decomposition temperatures near their melting points, pronounced brittleness, very low deformability and susceptibility to a rapid thermal degradation.78,80 Furthermore, the current cost of production of PHB is high compared to other high-volume synthetic plastics.70,78 Because of its limited thermal stability, the melt flow index changes rapidly with time and its volatile decomposition products need to be handled safely. PHB's slow crystallization rates also lead to tacky products (e.g. fibres, films, etc.).70 Its copolymer with valerate (PHBV) has overall better properties, especially regarding improved toughness with an acceptable loss of strength and modulus.73 However, the present large-scale production cost of PHBV remains higher than that of PHB.78
The toughness and processability of PHB can be improved by incorporation of the hydroxyvalerate (HV) monomers in the bacterial fermentation process.81 While PHBV with a high HV content has high flexibility, low crystallinity, and low crystallization rate, it compromises the yield strength and Young's modulus of PHB, which can result in rubbery materials, meanwhile, it increases the cost of materials.81 Various approaches, such as use of nucleating agents, plasticizers and agents that promote crystallization of the polymer, modification of the polymer structure, blending, etc., have been carried out to overcome the processing and product difficulties and other shortcomings.70,73,78,82
| Sample | Modifier concentration (wt%) | MW (g mol−1) | Tg (°C) | ε (%) | TS (MPa) |
|---|---|---|---|---|---|
| PHBV83 | 100 | 680000 | −6.6 | 6 | 43.1 |
| Soy oil83 | 20 | 814.3 | −3.4 | 3 | 33.7 |
| Epoxidized soy oil83 | 20 | 874.2 | −19.0 | 7.2 | 22.1 |
| Dibutyl phthalate83 | 20 | 278.2 | −28.5 | 10 | 11.7 |
| Triethyl citrate83 | 20 | 276.1 | −30.0 | 10 | 10.9 |
| Epoxy soyate88 | 20 | — | — | 7.51 | 13.8 |
| Soy oil88 | 10 | — | — | 5.09 | 18.7 |
| Epoxidized linseed oil88 | 10 | — | — | 7.46 | 19.6 |
It is observed from Table 2 that the plasticizers induced depression of glass transition temperature and improvement in the elongation at break with all the plasticizing additives used, with the exception of triglyceride soy oil. From Park and Choi's study,83 triethyl citrate was the most effective plasticizer in terms of reduction of the glass transition temperature as well as in terms of improvement of the impact strength and elongation. The difference in the effectiveness of these plasticizers can be attributed to the variation in the combined effect of chemical structure, molecular weight compatibility or solubility of the plasticizer with the polymer.83 On the other hand, studies of impact strength and elongation properties by Seydibeyoglu et al.88 showed that functionalized oils such as epoxy soyate are much more effective than triglycerides of epoxidized soybean or linseed oil. This might be due to the better reactivity of epoxy soyate than the counterpart triglycerides owing to its lower molecular size and simple molecular structure.
The use of low molecular weight, biodegradable and non-toxic compounds as plasticizing additives such as dibutyl sebacate (DBS), dioctyl sebacate (DOS), polyethylene glycol (PEG), Lapro1503 (L503), Lapro15003 (L5003), and a nonpolar polymer polyisobutylene (PIB) with concentration up to 50 wt% was investigated to improve the deformative characteristics of PHB.89 These plasticizers were completely compatible with the polymer and formed a monophase system in mixtures of up to 15–20 wt%. Conversely, when the concentration was beyond 20 wt% the system becomes considerably weak, because of overloading. The majority of the plasticizers examined by the author cause a considerable decrease in crystallization temperature and improvement of mechanical properties. Other plasticizers reported in the literature include dodecanol, lauric acid, tributyrin, and trilaurin.90
Ma et al.81 used a free radical initiator (dicumyl peroxide) to induce compatibilization and partial crosslinking between PHB and PBS. The resulting compatibilized blends were shown to have a smaller particle size, improved interfacial adhesion and consequently resulted in improved tensile strength, impact toughness and elongation at break. Sadi et al.95 evaluated the compatibilization efficiency of polypropylene/PHB blends with copolymers such as poly(propylene-g-maleic anhydride), poly(ethylene-co-methyl acrylate), poly(ethylene-co-glycidyl methacrylate), and poly(ethylene-co-methyl-acrylate-co-glycidyl methacrylate). Their study showed that poly(propylene-g-maleic anhydride), having the strongest adhesion between the phases, was the most efficient in terms of improving the mechanical performance of the blend.
Optimization of the processing conditions, taking the relationship among structure, composition, and polymer properties into account, is of particular importance as well.96 High shearing rates during process operations such as extrusion (10–100000 S−1) and injection molding (1000–100000 S−1)97 in addition to the heating applied during processing are expected to change the molecular weight and as a result its performance. For instance, Yamaguchi et al.98 reported 25–30% molecular weight reduction of PHB through shearing of PHB at 180 °C, at a shear rate of 6.3 s−1 within 5 min interval in addition to an order-of-magnitude decrease in shear viscosity.
3.3. Thermoplastic starch (TPS)
Starch, a polysaccharide of granular structure, is one of the most attractive feedstock for the development of biodegradable polymers because it is relatively inexpensive, abundant and renewable. Starch plays an important role both in the development of the commercialized bio-based plastics99 and in the bio-ethanol industry. The role of starch in the development of bio-based plastics could be in the development of (a) thermoplastic starch (TPS) plastics where TPS is obtained through direct modification of starch, (b) poly(lactic acid) where its feedstock (lactic acid) is originated from starch derived sugars fermentation and (c) PHAs where starch derived sugars are used as a carbon source for the microorganisms producing PHAs.Starch is composed of two homopolymers of D-glucose: the linear (1,4)-linked α-D-glucan amylase, typically constituting about 30% of starch depending on the source of starch, and a highly branched (1,6)-linked α-D-glucan amylopectin (Fig. 6). Commonly, amylopectin takes part in the formation of a crystalline structure and amylose does not.100 Virgin starch is brittle and difficult to process into articles due to its relatively high glass transition and melting temperatures. The Tg of virgin dried starch is estimated to be approximately 240 °C,101 which is above the starting point of its thermal degradation (about 220 °C).102 High Tg and brittleness of starch are mainly caused by the presence of strong inter- and intra-molecular hydrogen bonds between the starch macromolecules.101 Furthermore, TPS polymers based solely on starch are extremely water sensitive103 and can suffer from significant molecular weight change during processing (extrusion or injection molding).104 These drawbacks limit the possible shapes that can be imparted to the materials into films with adequate mechanical properties105 and thus of limited practical value. Therefore, starch must be modified to breakdown the crystalline granules, decrease the Tg and melting temperature (Tm) either by incorporating plasticizers,106 blending with other polymers,51,52 chemical modification or combinations before they can be processed into plastics.107
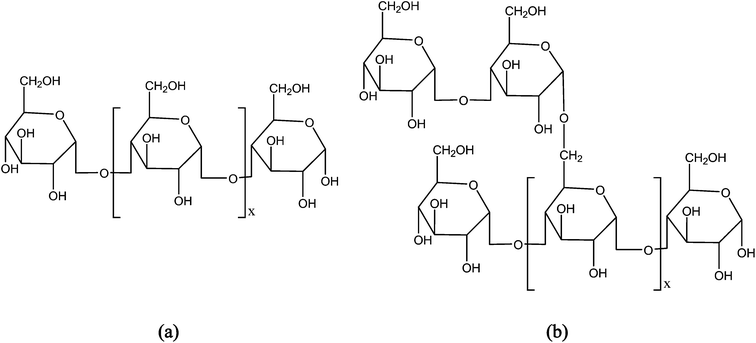 | ||
| Fig. 6 Structure of starch polymers (a) amylose and (b) amylopectin. | ||
There are several substances used as plasticizers for the preparation of thermoplastic starch. Some of the most studied and reported TPS plasticizers in the literature include polyols such as glycerol, glycol, sorbitol, xylitol, maltitol, ethylene glycol, propylene glycol, butanediol;107,110,111 sucrose, fructose, mannose,112 fatty acids (such as myristate or palmitate),113etc. It is also necessary to note that water is a good plasticizer of starch. However, the use of water alone as a plasticizer is not preferable because the resulting product will be brittle when equilibrated with ambient humidity110 and due to volatilization of water. Glycerol, a classical plasticizer of starch, is perhaps the most widely studied and used polyol plasticizer of TPS. This is because of its low cost, nontoxicity (for food and biomedical application) and high boiling point (292 °C). Moreover, the hydrolysis and/or transesterification of lipids (triglycerides) into fatty acids for the biodiesel industry produce glycerol as a by-product. Utilizing such by-products provides glycerol with an additional market driver in addition to the opportunity of improving the economics of both the biodiesel and the bioplastic industries. Nonetheless, glycerol is known to leach out during aging and humidity exposure, a major limitation for large scale applications.
The properties of plasticized starch can be tuned by changing the temperature of processing, water content and the properties and amount of plasticizers. For instance, Yu et al.101 reported that the elongation at break of the thermoplastic starch is significantly improved by plasticization with glycol, glycerol and hexylene glycol. In addition, the thermal properties of plasticized starch are a function of water and plasticizer content.110,114 The source of starch is also important for the property of TPS. This is because starches from various sources have different amylose/amylopectin ratios, molecular weights, molecular weight distributions and granular size crystallinity (Fig. 7). This as a result influences the gelatinization and glass transition temperatures115,116 that are directly correlated with the thermoplasticity of the TPS. The effect of various plasticizers at different concentrations on the gelatinization temperatures, thermal stability, and glass transition temperature has been studied and reported in the literature. Some of the plasticizers and their effects are reviewed and shown in Table 3.
 | ||
| Fig. 7 Scanning electron microscopy micrograph of (a) native corn starch granules, (b) ethylenebisformamide (25%) plasticized TPS, (c) ethylenebisformamide (30%) plasticized TPS (adapted from: ref. 126 John Wiley & Sons, copyright © 2006) and (d) native potato starch. | ||
| Starch source | Plasticizer | Plasticizer concentration (wt%) | Gelatinization onset (°C) | Gelatinization peak/conclusion (°C)/Tg |
|---|---|---|---|---|
| a Gelatinization conclusion.b Gelatinization peak.c Tg. | ||||
| Wheat starch in the presence of water108 | Glycerol | 65 | 74.7 | 91.5a |
| Sorbitol | 65 | 73 | 92a | |
| Diglycerol | 65 | 90 | 115a | |
| Sago starch117 | Starch (control) | 0 | 123.7 | 157.2b |
| Glycerol | 30, 40, 50 | 149, 152, 141 | 169, 175, 164b | |
| Sorbitol | 30, 40, 50 | 124, 126, 122 | 158, 151, 155b | |
| Sorbitol:glycerol (1:1) | 30, 40, 50 | 120, 118, 142 | 150, 147, 176b | |
| Corn starch118,119 | Glycerol,118,119 PLA,120 poly(butylene adipate-co-terephthalate)120 | — | — | — |
| Potato starch111,121 | Glycerol–xylitol | 40 | — | −66.4c |
| Glycerol–sorbitol | 40 | — | −69.3c | |
| Xylitol–sorbitol | 40 | — | −44.1c | |
| Rice starch122–124 | Glycerol | 20, 25, 30, 35 | ||
| Poly(ethylene glycol) | 3, 6, 9 | |||
| Sorbitol | 30, 35, 40, 45 | |||
| Others(formamide,123 urea,124 propylene and triethylene glycol124) | 10–30 | |||
Abdorreza et al.117 showed that the type and concentration of plasticizers govern the heat sealability as well as the seal strength of sago starch based films. The same authors showed that sorbitol-plasticized films exhibited significantly better heat sealability than did the glycerol type. However, the highest seal strength was obtained with a combination of sorbitol and glycerol. The effect of starch gelatinization in the presence of high molecular weight polyol plasticizers and water was also studied under static and dynamic conditions by Taghizadeh and Favis.108 Their investigation showed that glycerol and sorbitol exhibited similar gelatinization temperatures, while an ascending Tg was observed from glycerol to diglycerol and polyglycerol attributed to the viscosity and molecular weight increase and hydroxyl bond density diminution of the latter two plasticizers.
Other plasticizers such as urea, formamide, combinations of urea and formaldehyde,125 used with thermoplastic corn starch at different concentrations were also reported. Property evaluation by Ma et al.125 showed that mixtures of urea (20 wt%) and formamide (10 wt%) plasticized TPS exhibited better thermal stability, water resistance and better mechanical properties than conventional glycerol plasticized TPS. According to Ma et al.,125 the reasons behind such property improvement with the urea–formamide mix plasticizer could be due to the formation of more stable and stronger hydrogen bonds with the hydroxyl groups of starch molecules than with glycerol. Yang et al.126,127 reported ethylenebisformamide, synthesized from methyl formate and ethylenediamine, as a novel and effective plasticizer of corn starch and potato starch. Ethylenebisformamide was shown to be effective in destroying the crystalline morphology of the native starch granule and conversion into a homogeneous phase TPS through plasticization and extrusion under shear and pressure. The morphology of the native crystalline starch and the homogeneous plasticized starch at 25% and 30% ethylenebisformamide loading was studied by scanning electron microscopy (SEM)126,127 and shown in Fig. 7.
The SEM study (Fig. 7) clearly showed that the action of ethylenebisformamide and temperature processing (in this case extrusion) resulted in destruction of the crystalline native starch granules (Fig. 7a and d) morphology to form a continuous phase of TPS having a different crystallinity as further confirmed by X-ray diffraction crystallography.126,127 The effect of plasticizer loading had also an effect on the continuity of the plasticized TPS phase. Higher concentrations resulted in more uniform phases for the studied loading range. Possible hydrogen bond formation between ethylenebisformamide and starch126 during plasticization is shown in Fig. 8 below. The hydrogen bonds formed can be stronger than the intra and intermolecular bonds in starch, and as a result corn and potato starch were effectively plasticized with ethylenebisformamide.126,127 In general, besides the plasticizer type, the quantity of plasticizer used and the processing method applied also affect the physical, thermal and mechanical properties of the resulting starch based bioplastics. For example, Flores et al.128 studied and reported the effect of different gelatinization and drying techniques on the performance of glycerol plasticized starch films. The authors128 finding shows that gelatinization and drying techniques used to obtain TPS films affected network characteristics that as a result determines the changes in physical properties potentially affecting the film performance as well.
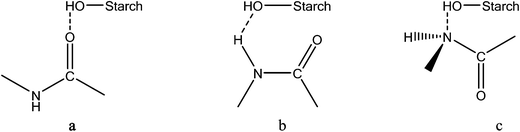 | ||
| Fig. 8 Possible hydrogen bonds between ethylenebisformamide and starch.126 | ||
In recent studies, the use of novel multifunctional ionic liquid plasticizers such as 1-allyl-3-methylimidazolium chloride109,129 and 1-butyl-3-methyl imidazolium chloride as a plasticizer130 and a compatibilizing agent131 of starch has been reported. Ionic liquids, organic salts that are liquid at ambient temperature, are gaining interest because of their unique properties including non-volatility, non-flammability, low viscosity, chemical and electrochemical stability.132 These liquids (examples of structures are shown in Fig. 9) have strong hydrogen bond forming abilities with starch owing to their high concentration of chloride ions. TPS plasticized using 1-butyl-3-methylimidazolium chloride shows less hygroscopicity and a much higher elongation at break in the rubbery state than the control glycerol-plasticized TPS samples.130 The potential application of ionic liquids plasticized starch as solid biopolymer electrolytes was also reported by Wang et al.129 This paves the way for a wide variety of potential applications of TPS bioplastics such as antistatic plastics, electronic shielding, biosensor, and environmentally sensitive membranes.
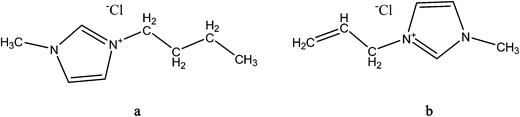 | ||
| Fig. 9 Chemical structure of ionic liquids 1-allyl-3-methylimidazolium chloride and 1-butyl-3-methyl imidazolium chloride. | ||
Other toughening modifications recently reported in the literature to improve performance and overall economics of TPS based polymers include blending of TPS with protein,131 PVA,140 polycaprolactone, polyhydroxybutyrate, polymethacrylate, polystyrene mostly in the presence of urea and polyol plasticizers.103,120 Surface modifications such as polymeric surface coating,109 chemical and photo crosslinking of TPS and blends were also shown to reduce surface hydrophilic characteristics and improve water resistance, increase the tensile strength and Young's modulus while decreasing the elongation at break.140–142 In summary, starch based plastics have grown to represent a major portion of the biodegradable polymer market. The commercial success of TPS polymers is hugely affected by the source, safety, quality, cost and functionality of plasticizers. Selective plasticization/toughening methods of TPS provide an attractive base for developing starch polymers that can be used as biodegradable and renewable packaging materials, environmentally sensitive membranes, and in biomedical and pharmaceutical applications such as drug and protein carriers, tissue engineering applications, etc.
3.4. Protein based plastics
Proteins are renewable, biodegradable and optically active natural143 polymers produced by animals, plants and bacteria. Until recently proteins have been utilized exclusively in the food industries. Recent studies on non-food uses of agricultural feedstock initiated an interest in protein based plastics as well. Due to the continuous and cohesive matrix forming ability of proteins, various proteins of both plant and animal origin have received attention for the production of biodegradable plastics, edible films and sheets.144,145 Furthermore, microencapsulating agents and active compounds in pharmaceutical applications,146,147 adhesives, blend and composite materials,142,148 wound dressing149 and bionanocomposites142,150 can be produced. Protein based biomaterials can also promote tissue regeneration, such as new bone growth,149 integrate into blood clots and stimulate collagen deposition, and stimulate cells to produce new tissue, with no need for expensive growth factors.151 Plant proteins that can be used for bio-based plastics include soy protein,150 corn zein,33 wheat protein,152,153 cottonseed protein,154 sunflower protein,155etc. Animal proteins such as blood meal,156 gelatine and collagen,157 keratin and feather quill,158 egg protein,159 whey protein,155 meat and bone meal160,161 can also be used as a feedstock of such bio-based plastics (Table 4).| Plasticizer | Protein studied | TS (MPa) | E (MPa) | ε (%) | Ref. |
|---|---|---|---|---|---|
| Urea (20%) | Blood meal | 12.3 | 608 | — | 156 |
| Wheat gluten | — | — | — | 165 | |
| Diethyl tartrate (30%) | Feather quill | 19.2 | 1267.9 | 1.6 | 158 |
| Chicken fibers | 19.0 | 907.9 | 3.3 | 166 | |
| Dibutyl tartrate (30%) | Corn zein | 20 | 1325 | — | 167 |
| Glycerol (30%) | Corn zein | 19.3 | 620 | — | 167 |
| Wheat gluten | 6.7 | 51 | 118 | 168 | |
| Soy protein | 13.8 | 250.5 | 177.5 | 169 | |
| Feather quill | 15.2 | 380.5 | 13.6 | 158 | |
| Chicken feathers | 15.7 | 332.3 | 8.5 | 166 | |
| Peanut proteins | 8.0 | 147.0 | 63.0 | 168 | |
| Sunflower protein | 8.5 | — | 140 | 170 | |
| Ethylene glycol (30%) | Chicken feathers | 17.8 | 354.0 | 43.8 | 166 |
| Sunflower protein | 8.7 | — | 23 | 170 | |
| Feather quill | 16.8 | 321 | 64.9 | 158 | |
| Propylene glycol (30%) | Chicken feathers | 22.3 | 811.2 | 7.6 | 166 |
| Sunflower protein | 7.2 | — | 63 | 170 | |
| Feather quill | 20.5 | 529.5 | 11.2 | 158 | |
| Soy protein | 4.5 | 108.4 | 8.5 | 169 | |
| Oleic acid (20%) | Gelatin | 54 | 2500 | 2.9 | 171 |
| Sorbitol (20%) | Gelatin | 52 | 1997 | 4.4 | 171 |
| Mannitol (20%) | Gelatin | 57 | 2250 | 4.5 | 171 |
Proteins are interesting biomaterials based on 20 amino acids which confers a wide range of functional and film-forming properties as a function of various extrinsic or intrinsic conditions such as plasticizer type and concentration.162 The major drawback of protein-based plastics, with the notable exception of keratin, is their sensitivity towards relative humidity.163 For example, Zheng et al.163 reported that soy protein sheets submerged in water for 20 h absorbed up to 180% water. In addition, protein films and coatings are often quite stiff and brittle due to extensive intermolecular interactions between protein chains through hydrogen bonding, electrostatic forces, hydrophobic bonding and disulfide cross-linking.34 Thus, thermoplastic processing of proteins into bio-based plastics is usually accompanied by plasticization and/or other form of modification for the successful development of useful proteinaceous biopolymers. Plasticizers can reduce the aforementioned chain-to-chain interaction and induce flexibility, moisture resistance and ease of processability.
Thermosetting protein plastics processing, on the other hand, occurs through chemical crosslinking that involves the formation of covalent bond bridges between protein chains by using a crosslinking agent. The crosslinkers chiefly target the reaction between themselves and protein functional groups – such as primary amines, carboxyl, hydroxyl, and sulfhydryls – of amino acid residues to provide mechanical strength and moisture resistance.160,161 Protein-polymer grafting is another method of producing a protein based biomaterial usually with complementary advantages of each component. Thermoplastic processing, which involves melting a polymer followed by shaping and cooling, is the most widely adopted method for the production of protein-based bioplastics.
Similar to most other bio-based plastics, the composition, size, and shape of plasticizers influence the mechanical, physical, thermal, moisture permeability and aging behavior of proteinaceous plastics.158,166,170,171 Orliac et al.170 demonstrated that sunflower protein isolate films plasticized with different polyalcohols, such as glycerol, ethylene glycol, propylene glycol, polyethylene glycols, and polypropylene glycols, exhibited high mechanical properties, and good moisture impermeability to the level that it can be used for agricultural mulching. Cao et al.171 compared the plasticizing effect of polyethylene glycol (PEG) with different molecular weights (300, 400, 600, 800, 1500, 4000, 10000, 20000) on gelatin films. The result showed that PEG with lower molecular weight gave better plasticizing effect (higher elongation), lower water vapor permeability and better visual effect. An increase in molecular weight of PEG in contrast induced an increase in the tensile strength, elastic modulus and a decrease in the elongation of gelatin films. Polar groups (–OH) along plasticizer chains are believed to develop polymer–plasticizer hydrogen bonds replacing the polymer–polymer interactions in biopolymer films.172 Thus, hydrogen bonding ability of PEGs was affected by factors such as the number of hydroxyl groups per mole, molecular size, solubility and polarity that will explain the observed variation. Recent studies by Ullah et al.158,166 also demonstrated that the variation in hydrogen bonding interactions between plasticizers (glycerol, diethyl tartrate, propylene glycol and diethyl tartrate) and keratin from poultry feather quills and poultry feather fiber could be responsible for the variation in plasticization efficacy. The best mechanical properties, transparency, flowability, and processability were observed in the case of ethylene glycol plasticized keratin quill and keratin feather, conceivably because of the formation of strong hydrogen bonding between the ethylene glycol and quill keratin.
Proteins are hydrophilic materials and as such they need to be coupled with adequate plasticizers to reduce the water absorbance of the corresponding plastics. Therefore, extensive attempts to improve moisture barrier properties are being conducted.173–175 The introduction of hydrophobic materials such as lipids, long chain fatty acids and waxes incorporated into protein films has shown promising results.173,176,177 For instance, Sohail et al.173 studied and reported the moisture barrier property improvement of protein biopolymers (casein films), as a result of wax incorporation in the film formation and surface wax coating. The wax application on moisture barrier properties was more efficient in wax-coated casein films than wax incorporated biopolymers. While both wax-coating and incorporation improved the flexibility of the films at the expense of tensile strength reduction, the wax incorporated polymers exhibited better flexibility than the coated ones. Pommet et al.176 likewise reported an improvement in the water vapor permeability of gluten protein films with the use of saturated fatty acids with an even number of carbons from 6 to 18 (C6:0:hexanoic acid, C8:0:octanoic acid, C10:0:decanoic acid, C12:0:lauric acid, C14:0:myristic acid, C16:0:palmitic acid, C18:0:stearic acid).
Shellhammer and Krochta178 studied the effect of lipid type and amount on the plasticization of whey protein biopolymer using beeswax, candelilla wax, carnauba wax and a high melting fraction of anhydrous milk fat. According to the authors, an increase in lipid level decreased the strength of the biopolymers. Among the studied lipids, candelilla wax incorporation provided the weakest films, followed by beeswax, milk fat, and carnauba wax. Furthermore, a positive correlation between water vapor permeability of the lipids and the lipid plasticized protein plastics explains the increment in water vapor permeability of some of the biopolymers. Fabra et al.177 reported the formation of bilayer structures by saturated fatty acids in sodium caseinate film forming solution that led to water vapor permeability improvement. The self-association of saturated fatty acid molecules occurs to form bilayers of different sizes in the film forming dispersions, and these laminar structures grow and persist in the dried film. The crystal formations as a result greatly limit water vapor permeability and yield rigid nonflexible films that show opacity and low gloss. According to the same authors,177 unsaturated fatty acids such as oleic acid did not form laminar structures due to the double bond while it provokes a synergic plasticizing effect with water that seriously increased the water vapor permeability and film flexibility at intermediate relative humidity levels.179
The synergetic effects of using mixed glycerol (polar) and oleic acid (amphiphilic) plasticizers on sodium caseinate179 and zein protein biopolymers180 were also reported recently. According to Ibragimo et al.,181 the plasticization obtained by glycerol is structural (inter-packet) and that of oleic acid is molecular (intra-packet). These two different molecules with different plasticization mechanisms provide the possibility for their interaction during film formation. The combination of these two plasticizers in zein films exhibited synergy and as a result a change in tensile strength (highest at 3:1 ratio of oleic acid to glycerol), decrease in glass transition temperature and change in microscopic molecular structure were observed.180
Tummala et al.182 reported the use of glycerol, sorbitol and their blend to plasticize and compatibilize soy-protein and polyester amide, and compared their influence on the performance of the resulting biopolymers. While sorbitol plasticized soy-polyester amide plastics were more rigid, with a higher tensile modulus and tensile strength and thermal stability, glycerol plasticized soy-polyester amide plastics had the highest impact strength. The blend of the two on the other hand provided an intermediate tensile strength and modulus. Other types of protein biopolymer modifications reported in the literature include blending of gelling agents such as agar, agargel, phytagel,183 incorporation of nanoclays,184etc. A recent study by Kim and Netravali183 demonstrated that the blending of gelling agents with soy protein significantly improved the mechanical, thermal stability resistance of the protein biopolymers. This is because of the possible formation of interpenetrating network (IPN) structures between the gelling agents and the protein with a high degree of intermolecular interactions.183
The graft polymerization of styrene on soy protein isolate,191 2-hydroxyethyl methacrylate on soy protein,192 poly(ethylene glycol) on soy protein,193 polycaprolactone on zein,194 waterborne polyurethane on soy protein,195 poly(ethylene oxide) diglycidyl ether on wheat protein,196 methyl methacrylate, ethyl methacrylate and butyl methacrylate on camelina meal197 has been widely reported. Wu et al.194 reported that the grafting of polycaprolactone onto zein protein resulted in a dramatic flexibility improvement, while the strength remained constant. Moreover, the glass transition temperature and melting temperature were also shown to decrease due to the plasticizing effect of polycaprolactone on the protein. Kurniawan et al.196 also showed that the chemical modification of wheat protein based biopolymers with poly(ethylene oxide) diglycidyl ether resulted in the formation of a different network structure of the biopolymer with an improved flexibility, and improved mechanical performance.
Chemical crosslinking modification of protein with various agents to improve the mechanical, thermal and moisture resistance of the resulting biopolymers is another technique that has been widely studied.160,161,196,198,199 Chemical crosslinking of proteins usually depends on the availability of particular chemicals that are capable of reacting with the specific kinds of functional groups that exist in proteins. The most extensively used chemical crosslinking agents of proteins include aldehydes (formaldehyde, glutaraldehyde, glyoxal, benzaldehyde),161,200,201 carbodiimide, maleic anhydride, hydroxysuccinimide, etc.202–204 Most of the studies show that crosslinking improved the tensile strength, tensile modulus, and moisture and solvent resistance, while the flexibility is reduced.160,161 In summary, protein-based plastics can be easily modified through plasticization; grafting or crosslinking due to the presence of several functional groups provides protein-based plastics great promise in a wide range of applications. Further research into plasticizer/modification technique selection that combines the characteristics of the different protein feedstock with performance is necessary if protein based plastics are to achieve their full commercial potential.
3.5. Cellulose acetate
Cellulose is an abundant, renewable, and biodegradable natural polymer that constitutes the skeletal part of plants. It is a homogeneous polysaccharide formed by repeating connection of D-glucose building blocks (Fig. 10), with an average degree of polymerization of 1500 to 3000 depending on the source.205 Industrial materials are being developed from cellulose and its derivative over a broad range of application because of its abundance, environmental and biocompatibility benefits, relatively low cost and ease of modification. Cellulose by itself is poorly soluble in common solvents and is not melt processable as it decomposes before it undergoes melt flow.206 Nonetheless, the chemistry of cellulose opens the way to various chemical modifications, such as esterification and etherification give entry into a broad variety of products including coatings, base for photographic films, filters, pharmaceutics, fragrances, polymer additives, membranes and building materials.207–209 The most industrially relevant and oldest biodegradable cellulose ester derivative is cellulose acetate.208 Cellulose acetate and mixed cellulose esters, such as cellulose diacetate, cellulose triacetate, cellulose acetate propionate, and cellulose acetate butyrate, are all commercially available materials.208 These thermoplastic materials are usually synthesized through esterification of cellulose,210 where other substituent groups replace the hydroxyl groups of cellulose (Fig. 10). For instance, cellulose acetate is the product of esterification reaction between cellulose and acetic anhydride in the presence of sulfuric acid catalyst to form fully acetylated cellulose triacetate, followed by partial hydrolysis to remove acid catalyst and produce a degree of substitution in the polymer that yields the desired working properties.211 Other methods of cellulose ester synthesis, such as transesterification of cellulose with vinyl esters under catalysis, are reported in the literature recently.212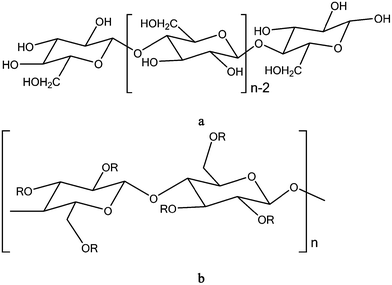 | ||
| Fig. 10 Schematic representation of the molecular structure of (a) cellulose (n-degree of polymerization) and (b) cellulose ester (R-functional group for each type of cellulose ester). | ||
Raw materials such as cotton, recycled paper, wood cellulose, and sugarcane are used in making cellulose ester biopolymers in powder form.210 Cellulose ester powders combined with plasticizers and additives are extruded to produce various grades of commercial cellulosic plastics in pelletized form. Of great interest as potential biodegradable plastics are also long chain aliphatic acid esters of cellulose.206,213 These cellulose esters are characterized by stiffness, moderate heat resistance, high moisture vapor transmission, grease resistance, clarity and appearance, and moderate impact resistance.206 The presence of polar functional groups in the cellulose acetate chain offers an additional advantage of affinity to solvents including plasticizers and lithium ions for the development of polymer electrolytes.214 Nevertheless, owing to the high viscosity and elevated glass transition temperature, cellulose acetate derivatives themselves are not processable as a thermoplastic.209 In an effort to modify its properties and facilitate processing, cellulose acetate is modified through plasticization by various aliphatic and aromatic esters,215,216 chemically modified through grafting onto the polysaccharide backbone, and modification by forming polymer blends.208,216,217
Zepnik et al.221 have recently studied the effect of plasticizer type and concentration on cellulose acetates using benzoate, acetates, phosphate and citrates based plasticizers. An increase in plasticizer concentration resulted in significant broadening of the thermoplastic processing window due to a strong decrease in glass transition temperature. It was thus possible to tune the rheology, melt strength and thermoplastic processing cellulose acetate by changing the plasticizer concentration. On the other hand molecular size, chemical structure, and solubility variation of plasticizers were shown to influence its compatibility, and ultimately the efficacy. It is generally agreed that plasticizers that have higher thermodynamic compatibility with the base polymer cause better plasticization than those with limited compatibility. The selection of an efficient plasticizer for cellulose esters was suggested by Fridman and Sorokina219 who developed a set of criteria for efficient plasticization of cellulose acetate. An efficient plasticizer should take into account the compatibility of components, temperature durability and mechanical properties during processing and service time of the final polymer.
The efficiency of a plasticizer depends also on the loading concentration. Fig. 11 and 12 show the effect of one of the common cellulose acetate plasticizer (diethyl phthalate) concentration on the thermal and mechanical properties of cellulose acetate (drawn from tabulated data reported by Fridman and Sorokina219). As the plasticizer concentration increases, a reduction in glass transition temperature was observed and hence a significantly lower processing temperature is needed, while a substantial thermal stability drop resulted in the cellulose ester plastic (Fig. 11). On the other hand, an increase in plasticizer concentration resulted in an increment of impact strength (Fig. 12) and elongation at break accompanied by a drop in tensile strength. Similar trend of cellulose acetate stiffness and toughness properties was observed by Mohanty et al.210 upon increasing triethyl citrate plasticizer concentration. In summary, the type of the plasticizer and optimum plasticizer concentration are key parameters to reduce the processing temperature without compromising the stability and other performances of the plastic.
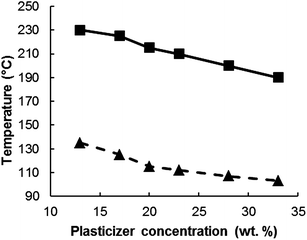 | ||
| Fig. 11 Influence of diethyl phthalate concentration on Tg (▲) and processing temp (■) based on the data from Fridman and Sorokina.219 | ||
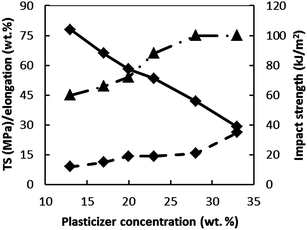 | ||
| Fig. 12 Influence of diethyl phthalate concentration on impact strength (▲), elongation (■) and tensile strength (TS) (♦) based on the data from Fridman and Sorokina.212 | ||
While cellulose acetate or its degradation products are safe, some of its common plasticizers such as phthalates, triacetin, glycerin, polyethylene glycol are associated with high toxicity, relatively high diffusion and water solubility.222,223 For example, deterioration of cellulose acetate as a result of migration or evaporation of plasticizers, reaction of plasticizers with other chemicals in their surroundings to form other products has been documented in the literature.218 As a result of such deterioration, not only unsafe plasticizers and plasticizer reaction products are released to the environment, but materials developed from cellulose ester became prone to cracking, warping, discoloration, exudation, shrinkage and powdering as they age.224
To mitigate these safety issues in addition to awareness of green technology and government legislations, several mitigation efforts are being conducted. These efforts include the development of safe, more stable and more compatible, bio-based, and functional plasticizers. Sugar based plasticizers, such as sorbitan,216,225 polyoxyethylene sorbitan monopalmitate,225 polyoxyethylene sorbitan monostearate,216 are also reported. The use of an ionic liquid plasticizer synthesized from choline chloride and urea, such as deep eutectic solvent (DES), has been recently reported as a safe and novel plasticizer of cellulose acetate.226 These plasticizers have the high solvating potential of crystalline cellulose acetate, and are less expensive, non-toxic and biodegradable in addition to their large electronegativity and delocalization of charge that enables them to positively influence ionic conductivity of cellulose ester.226,227 Other ionic solvent cellulose ester plasticizers reported in the literature include 1-allyl-3-methylimidazolium chloride,228 and other ionic liquids based on methylimidazolium and methylpyridinium cores with allyl-, ethyl-, or butyl-side chains.229
4. Concluding remarks
The rapid technological development of bio-based plastics, such as PLA, polyhydroxyalkanoates, (PHA), bio-based epoxy resin and bio-based PE, has yet to be translated into significant market impact, primarily due to high production cost and performance limitations. Plasticizers are important additives and performance enhancers of polymers. As such they can augment the processability of most of these bio-based plastics, and constitute a significant opportunity and at the same time barrier in the applications of bio-based plastics in various fields of applications. Moreover, the performance, safety, biodegradability, economics and functional utilization of bio-based plastics are strongly dependent on the performance of the incorporated plasticizers. This review highlights that selective use of a plasticizer at an optimum concentration allows controlling the balance between the processability, strength, modulus, toughness and other important mechanical properties of plastics. Such a selection is usually conducted based on reasons for application of plasticizer, mechanism of plasticizer action, interaction and effect of the plasticizer on other additives besides the biopolymer, concentration range depending on the application.In addition to the sanction of some common plasticizers (e.g. phthalates) for various applications, due to environmental and health concern as a result of migration and leaching during aging or use of plastics, newer concerns are emerging with regard to the effect of plasticizers in maintaining the renewability and biodegradability of bio-based plastics. The development and utilization of bio-based plasticizers such as polyols, fatty acids and fatty acid derivatives, epoxidized soy oil, ester amides, citrates and ionic liquids, such as methylimidazolium chloride and deep eutectic solvent are widely reported to tackle such issues. These new forms of plasticizers offer new dimensions of plasticizer selection that provide additional functionality (e.g. electric conductivity by ionic liquids) to the bio-based plastics and some of the others provide high safety to be used even in edible food packaging applications. While most of the fundamental mechanism, physico–chemical interaction or rule of thumb in the selection of a suitable plasticizer were established for the synthetic plastics (mainly for PVC), there are hardly no newer theories/mechanisms for the relatively new bio-based plastics. As a result, most of the current plasticization investigations are conducted under the assumption that the mechanisms developed for PVC would also be valid for the bio-based plastics. A fundamental understanding of the plasticization mechanism of bio-based materials is essential, along with their similarity and difference with PVC, if they are to reach their full potential and success.
Besides the usual purpose of improving flexibility and processability, research is progressing in areas such as the search and modification of plasticizers that impart additional functions of flame retardancy, optical quality, electric conductivity or insulation, thermoxidative stability, chemical and temperature (high and low) resistance in demanding environments; reactive plasticizers that provide chemical integrity, gas and moisture impermeability improvement, provide or improve biodegradability and biocompatibility to the polymers are under investigation. The migration of some of the current bio-based plasticizers through either volatilization or mass transfer to a liquid or solid in contact poses another challenge. These and the other challenges led to the search for newer types of plasticizers and alternative methods of improving the processability and overall performance. Some of the alternative methods to plasticization include molecular orientation, physical and reactive blending, chemical crosslinking and grafting of the bio-based polymers with other polymers to tailor the ultimate product properties.
Acknowledgements
Tizazu H. Mekonnen, Paolo G. Mussone and David C. Bressler are grateful to PrioNet Canada, Alberta Prion Research Institute, Alberta Livestock and Meat Agency, and the Biorefining Conversion Network at the University of Alberta for the financial support they received while they were researching and preparing this work.References
- R. U. Halden, Annu. Rev. Public Health, 2010, 31, 179–194 CrossRef PubMed.
- M. M. Reddy, S. Vivekanandhan, M. Misra, S. K. Bhatia and A. Mohanty, Prog. Polym. Sci., 2013 DOI:10.1016/j.progpolymsci.2013.05.006.
- B. Gervet, The use of crude oil in plastic making contributes to global warming, Lulea University of Technology, Sweden, 2007 Search PubMed.
- M. McCoy, Chem. Eng. News, 1998, 76, 15–18 Search PubMed.
- L. Shen, E. Worrell and M. Patel, Biofuels, Bioprod. Biorefin., 2010, 4, 25–40 CrossRef CAS.
- M. M. Reddy, M. Misra and A. K. Mohanty, Chem. Eng. Prog., 2012, 108, 37–42 CAS.
- USDA Agriculture, ed. USDA, http://www.biocom.iastate.edu/workshop/2012workshop/presentations/pruszko.pdf, Washington, DC., 2008, accessed 21 May 2013.
- R. Mathers, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1–15 CrossRef CAS.
- M. Rahman and C. Brazel, Prog. Polym. Sci., 2004, 29, 1223–1248 CrossRef CAS PubMed.
- I. Lemer, Plasticizers to reach $8 billion by 2004 Look Smart, Ltd, http://www.findarticles.com/cf_dls/m0FVP/9_258/65462810/p1/article.jhtml, accessed January 2010.
- C. E. Wikes, J. W. Summers and C. A. Daniels, PVC handbook, Hanser Gardner Publications, Inc., 2005 Search PubMed.
- S. Cullen, Global Plasticizer Update, http://www.plasticsindustry.org/files/events/Stephen%20Cullen_Tuesday.pdf, accessed 28 April 2013.
- L. Sperling, Introduction To Physical Polymer Science, 4th edn, 2006, pp. 1–845 Search PubMed.
- P. De Groote, J. Devaux and P. Godard, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 2208–2218 CrossRef CAS.
- S. Varughese and D. Tripathy, J. Elastomers Plast., 1993, 25, 343–357 CrossRef CAS PubMed.
- E. Snejdrova and M. Dittrich, Pharmaceutical Applications of Plasticized Polymers, Recent Advances in Plasticizers, ed. Mohammad Luqman, Intech, 2012 Search PubMed.
- N. Ljungberg and B. Wesslen, Polymer, 2003, 44, 7679–7688 CrossRef CAS PubMed.
- D. Klee and H. Hocker, Biomedical Applications: Polymer Blends, 1999, 149, 1–57 CrossRef CAS.
- S. Tomic, M. Micic, S. Dobic, J. Filipovic and E. Suljovrujic, Radiat. Phys. Chem., 2010, 79, 643–649 CrossRef CAS PubMed.
- Y. Zhu, N. Shah, A. Malick, M. Infeld and J. McGinity, Int. J. Pharm., 2002, 241, 301–310 CrossRef CAS.
- Y. Li, C. Wang, G. Wang and Z. Qu, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2008, 23, 100–104 CrossRef CAS PubMed.
- R. Sothornvit and J. M. Krochta, Innovations in Food Packaging, 2005, pp. 403–433 Search PubMed.
- E. H. Immergut and H. F. Mark, Principles of Plasticization, in Plasticization and Plasticizer Processes, American Chemical Society, Washington, DC, 1965 Search PubMed.
- A. S. Wilson, Plasticisers: Principles and Practice, The Institute of Materials, London, UK, 1995 Search PubMed.
- A. Mohanty, M. Misra and L. Drzal, Abstracts of Papers of the American Chemical Society, 2002, vol. 223, pp. D70–D70 Search PubMed.
- P. Sarnacke and W. Stephen, Disposable Bioplastics: Consumer disposables agricultural films, a market opportunity study, United soybean board, Omni Tech International, Ltd., Midland, Michigan, 2008 Search PubMed.
- M. Vieira, M. da Silva, L. dos Santos and M. Beppu, Eur. Polym. J., 2011, 47, 254–263 CrossRef CAS PubMed.
- L. Lim, R. Auras and M. Rubino, Prog. Polym. Sci., 2008, 33, 820–852 CrossRef CAS PubMed.
- N. Gil, M. Saska and I. Negulescu, J. Appl. Polym. Sci., 2006, 102, 1366–1373 CrossRef CAS.
- O. Fenollar, D. Garcia, L. Sanchez, J. Lopez and R. Balart, Eur. Polym. J., 2009, 45, 2674–2684 CrossRef CAS PubMed.
- A. Stuart, M. McCallum, D. Fan, D. LeCaptain, C. Lee and D. Mohanty, Polym. Bull., 2010, 65, 589–598 CrossRef CAS.
- B. Videki, S. Klebert and B. Pukanszky, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 873–883 CrossRef CAS.
- L. Di Gioia, B. Cuq and S. Guilbert, Int. J. Biol. Macromol., 1999, 24, 341–350 CrossRef CAS.
- R. Sothornvit and J. Krochta, J. Food Eng., 2001, 50, 149–155 CrossRef.
- B. P. Shtarkman and I. N. Razinskaya, Acta Polym., 1983, 34, 514–520 CrossRef CAS.
- J. K. Sears and J. R. Darby, The Technology of Plasticizers, John Wiley & Dpmd, New York, NY, 1982 Search PubMed.
- R. Drumright, P. Gruber and D. Henton, Adv. Biomater., 2000, 12, 1841–1846 CAS.
- B. Gupta, N. Revagade and J. Hilborn, Prog. Polym. Sci., 2007, 32, 455–482 CrossRef CAS PubMed.
- X. Hu, X. Jing, S. Sharma and A. Mudhoo, Handbook of Applied Biopolymer Technology: Synthesis, Degradation and Applications, 2011, pp. 291–310 Search PubMed.
- D. Garlotta, J. Polym. Environ., 2001, 9, 63–84 CrossRef CAS.
- A. Singha, V. Thakur, A. Singha and V. Thakur, Green Polymer Materials, 2012, pp. 1–14 Search PubMed.
- J. P. Penning, H. Dijkstra and A. J. Pennings, Polymer, 1993, 34, 942–951 CrossRef CAS.
- J. Lunt, Polym. Degrad. Stab., 1998, 59, 145–152 CrossRef CAS.
- P. Kurcok, A. Matuszowicz, Z. Jedlinski, H. Kricheldorf, P. Dubois and R. Jerome, Macromol. Rapid Commun., 1995, 16, 513–519 CrossRef CAS.
- H. Liu and J. Zhang, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1051–1083 CrossRef CAS.
- L. Averous, M. Belgacem and A. Gandini, Monomers, Polym. Compos. Renewable Resour., 2008, 433–450 CrossRef CAS.
- A. Gupta and V. Kumar, Eur. Polym. J., 2007, 43, 4053–4074 CrossRef CAS PubMed.
- J. Huang, M. Lisowski, J. Runt, E. Hall, R. Kean, N. Buehler and J. Lin, Macromolecules, 1998, 31, 2593–2599 CrossRef CAS.
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS PubMed.
- P. Zhao, Q. F. Wang, Q. Zhong, N. W. Zhang and J. Ren, J. Appl. Polym. Sci., 2010, 115, 2955–2961 CrossRef CAS.
- O. Martin and L. Averous, Polymer, 2001, 42, 6209–6219 CrossRef CAS.
- L. Averous and C. Fringant, Polym. Eng. Sci., 2001, 41, 727–734 CAS.
- S. Jacobsen and H. Fritz, Polym. Eng. Sci., 1999, 39, 1303–1310 CAS.
- L. Labrecque, R. Kumar, V. Dave, R. Gross and S. McCarthy, J. Appl. Polym. Sci., 1997, 66, 1507–1513 CrossRef CAS.
- G. Maglio, A. Migliozzi and R. Palumbo, Polymer, 2003, 44, 369–375 CrossRef CAS.
- G. Maglio, M. Malinconico, A. Migliozzi and G. Groeninckx, Macromol. Chem. Phys., 2004, 205, 946–950 CrossRef CAS.
- G. Maglio, A. Migliozzi, R. Palumbo, B. Immirzi and M. Volpe, Macromol. Rapid Commun., 1999, 20, 236–238 CrossRef CAS.
- A. Gajria, V. Dave, R. Gross and S. McCarthy, Polymer, 1996, 37, 437–444 CrossRef CAS.
- E. Blumm and A. Owen, Polymer, 1995, 36, 4077–4081 CrossRef.
- N. Ogata, T. Tatsushima, K. Nakane, K. Sasaki and T. Ogihara, J. Appl. Polym. Sci., 2002, 85, 1219–1226 CrossRef CAS.
- J. Park and S. Im, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 1931–1939 CrossRef CAS.
- F. Hassouna, J. Raquez, F. Addiego, P. Dubois, V. Toniazzo and D. Ruch, Eur. Polym. J., 2011, 47, 2134–2144 CrossRef CAS PubMed.
- N. Ljungberg and B. Wesslen, J. Appl. Polym. Sci., 2002, 86, 1227–1234 CrossRef CAS.
- F. Hassouna, J. Raquez, F. Addiego, V. Toniazzo, P. Dubois and D. Ruch, Eur. Polym. J., 2012, 48, 404–415 CrossRef CAS PubMed.
- Y. Xu and J. Qu, J. Appl. Polym. Sci., 2009, 112, 3185–3191 CrossRef CAS.
- B. Chen, T. Wu, Y. Chang and A. Chen, Chem. Eng. J., 2013, 215, 886–893 CrossRef PubMed.
- Z. Ren, L. Dong and Y. Yang, J. Appl. Polym. Sci., 2006, 101, 1583–1590 CrossRef CAS.
- P. Suriyamongkol, R. Weselake, S. Narine, M. Moloney and S. Shah, Biotechnol. Adv., 2007, 25, 148–175 CrossRef CAS PubMed.
- S. Park, T. Kim, M. Kim, S. Lee and S. Lim, Biotechnol. Adv., 2012, 30, 1196–1206 CrossRef CAS PubMed.
- T. G. Volova, Polyhydroxyalkanoates plastic material of the 21st century, Nova science publishers, Inc., Hauppauge, New York, 2004.
- K. Snell and O. Peoples, Biofuels, Bioprod. Biorefin., 2009, 3, 456–467 CrossRef CAS.
- G. Chen, Q. Wu, Y. Wang, Z. Zheng, X. Zhang, J. Tanaka, Y. Yu and Y. Tabata, Asbm6: Advanced Biomaterials Vi, 2005, 288–289, 437–440 CAS.
- L. Yu, K. Dean and L. Li, Prog. Polym. Sci., 2006, 31, 576–602 CrossRef CAS PubMed.
- B. Laycock, P. Halley, S. Pratt, A. Werker and P. Lant, Prog. Polym. Sci., 2013, 38, 536–583 CrossRef CAS PubMed.
- G. Chen and Q. Wu, Biomaterials, 2005, 26, 6565–6578 CrossRef CAS PubMed.
- A. Anderson and E. Dawes, Microbiol. Rev., 1990, 54, 450–472 CAS.
- H. Salehizadeh and M. Van Loosdrecht, Biotechnol. Adv., 2004, 22, 261–279 CrossRef CAS PubMed.
- I. Chodak, in Monomers, polymers and composites from renewable resources, Elsevier Ltd., Boston, US, 2008 Search PubMed.
- R. Baltieri, L. Mei and J. Bartoli, Macromol. Symp., 2003, 197, 33–44 CrossRef CAS.
- I. Chodak, in Degradable polymers, Kluwer Academic publishers, The Netherlands, 2002 Search PubMed.
- P. Ma, D. Hristova-Bogaerds, P. Lemstra, Y. Zhang and S. Wang, Macromol. Mater. Eng., 2012, 297, 402–410 CrossRef CAS.
- M. Erceg, T. Kovacic and I. Klaric, Polym. Degrad. Stab., 2005, 90, 313–318 CrossRef CAS PubMed.
- J. Choi and W. Park, Polym. Test., 2004, 23, 455–460 CrossRef CAS PubMed.
- M. Correa, M. Branciforti, E. Pollet, J. Agnelli, P. Nascente and L. Averous, J. Polym. Environ., 2012, 20, 283–290 CrossRef CAS PubMed.
- S. Hong, H. Hsu and M. Ye, J. Therm. Anal. Calorim., 2013, 111, 1243–1250 CrossRef CAS PubMed.
- W. V. Srubar, Z. C. Wright, A. Tsui, A. T. Michel, S. L. Billington and C. W. Frank, Polym. Degrad. Stab., 2012, 97, 1922–1929 CrossRef CAS PubMed.
- M. A. Abdelwahab, A. Flynn, B. S. Chiou, S. Imam, W. Orts and E. Chiellini, Polym. Degrad. Stab., 2012, 97, 1822–1828 CrossRef CAS PubMed.
- M. O. Seydibeyoglu, M. Misra and A. Mohanty, Int. J. Plast. Technol., 2010, 14, 1–16 CrossRef CAS PubMed.
- I. Bibers, V. Tupureina, A. Dzene and M. Kalnins, Mech. Compos. Mater., 1999, 35, 357–364 CrossRef CAS.
- N. Yoshie, K. Nakasato, M. Fujiwara, K. Kasuya, H. Abe, Y. Doi and Y. Inoue, Polymer, 2000, 41, 3227–3234 CrossRef CAS.
- L. Miao, Z. Qiu, W. Yang and T. Ikehara, React. Funct. Polym., 2008, 68, 446–457 CrossRef CAS PubMed.
- T. Sadik, V. Massardier, F. Becquart and M. Taha, J. Appl. Polym. Sci., 2013, 127, 1148–1156 CrossRef CAS.
- C. Del Gaudio, L. Fioravanzo, M. Folin, F. Marchi, E. Ercolani and A. Bianco, J. Biomed. Mater. Res., Part B, 2012, 100, 1883–1898 CrossRef PubMed.
- P. Ma, D. Hristova-Bogaerds, J. Goossens, A. Spoelstra, Y. Zhang and P. Lemstra, Eur. Polym. J., 2012, 48, 146–154 CrossRef CAS PubMed.
- R. Sadi, R. Kurusu, G. Fechine and N. Demarquette, J. Appl. Polym. Sci., 2012, 123, 3511–3519 CrossRef CAS.
- A. Tsui, Z. Wright and C. Frank, Annu. Rev. Chem. Biomol. Eng., 2013, 4, 143–170 CrossRef PubMed.
- D. van Krevelen and K. te Nijenhuis, Thermal Decomposition, Processing Properties, in Properties of Polymers, Elsevier, Amsterdam, 2009 Search PubMed.
- M. Yamaguchi and K. Arakawa, Eur. Polym. J., 2006, 42, 1479–1486 CrossRef CAS PubMed.
- C. Bastioli, Starch/Staerke, 2001, 53, 351–355 CrossRef CAS.
- N. Cheetham and L. Tao, Carbohydr. Polym., 1998, 36, 277–284 CrossRef CAS.
- J. Yu, J. Gao and T. Lin, J. Appl. Polym. Sci., 1996, 62, 1491–1494 CrossRef CAS.
- P. Russell, J. Cereal Sci., 1987, 6, 133–145 CrossRef CAS.
- P. J. Halley, R. W. Truss, M. G. Markotsis, C. Chaleat, M. Russo, A. L. Sargent, I. Tan and P. A. Sopade, Polymer Durability and Radiation Effects, 2008, vol. 978, pp. 287–300 Search PubMed.
- A. D. Sagar and E. W. Merrill, J. Appl. Polym. Sci., 1995, 58, 1647–1656 CrossRef CAS.
- F. Otey and R. Westhoff, Ind. Eng. Chem. Prod. Res. Dev., 1984, 23, 284–287 CrossRef CAS.
- E. Rudnik and E. Rudnik, Compostable Polymer Materials, 2008, pp. 11–36 Search PubMed.
- J. Yu, N. Wang and X. Ma, Starch/Staerke, 2005, 57, 494–504 CrossRef CAS.
- A. Taghizadeh and B. D. Favis, Carbohydr. Polym., 2013, 92, 1799–1808 CrossRef CAS PubMed.
- X. Ma, P. R. Chang and J. Yu, in Starch-Based Polymeric Materials and Nanocomposites, CRC Press, Florida, US, 2012 Search PubMed.
- P. Forssell, J. Mikkila, G. Moates and R. Parker, Carbohydr. Polym., 1997, 34, 275–282 CrossRef CAS.
- R. A. Talja, H. Helen, Y. H. Roos and K. Jouppila, Carbohydr. Polym., 2007, 67, 288–295 CrossRef CAS PubMed.
- Y. Zhang and J. H. Han, J. Food Sci., 2008, 73, E313–E324 CrossRef CAS PubMed.
- S. Raphaelides, G. Dimitreli, S. Exarhopoulos, G. Kokonidis and E. Tzani, Carbohydr. Polym., 2011, 83, 727–736 CrossRef CAS PubMed.
- F. Rodriguez-Gonzalez, B. Ramsay and B. Favis, Carbohydr. Polym., 2004, 58, 139–147 CrossRef CAS PubMed.
- P. Liu, L. Yu, X. Y. Wang, D. Li, L. Chen and X. X. Li, J. Cereal Sci., 2010, 51, 388–391 CrossRef CAS PubMed.
- R. A. de Graaf, A. P. Karman and L. Janssen, Starch/Staerke, 2003, 55, 80–86 CrossRef CAS.
- M. N. Abdorreza, L. H. Cheng and A. A. Karim, Food Hydrocolloids, 2011, 25, 56–60 CrossRef CAS PubMed.
- O. V. Lopez, C. J. Lecot, N. E. Zaritzky and M. A. Garcia, J. Food Eng., 2011, 105, 254–263 CrossRef CAS PubMed.
- Q. Q. Yan, H. X. Hou, P. Guo and H. Z. Dong, Carbohydr. Polym., 2012, 87, 707–712 CrossRef CAS PubMed.
- J. Ren, H. Y. Fu, T. B. Ren and W. Z. Yuan, Carbohydr. Polym., 2009, 77, 576–582 CrossRef CAS PubMed.
- R. A. Talja, H. Helen, Y. H. Roos and K. Jouppila, Carbohydr. Polym., 2008, 71, 269–276 CrossRef CAS PubMed.
- N. Laohakunjit and A. Noomhorm, Starch/Staerke, 2004, 56, 348–356 CrossRef CAS.
- X. Ma and J. Yu, J. Appl. Polym. Sci., 2004, 93, 1769–1773 CrossRef CAS.
- R. Shogren, C. Swanson and A. Thompson, Starch/Staerke, 1992, 44, 335–338 CrossRef CAS.
- X. Ma, J. Yu and F. Jin, Polym. Int., 2004, 53, 1780–1785 CrossRef CAS.
- J. Yang, J. Yu and X. Ma, Chin. Chem. Lett., 2006, 17, 133–136 CAS.
- J. Yang, J. Yu and X. Ma, Carbohydr. Polym., 2006, 63, 218–223 CrossRef CAS PubMed.
- S. Flores, L. Fama, A. Rojas, S. Goyanes and L. Gerschenson, Food Res. Int., 2007, 40, 257–265 CrossRef CAS PubMed.
- N. Wang, X. Zhang, H. Liu and B. He, Carbohydr. Polym., 2009, 76, 482–484 CrossRef CAS PubMed.
- A. Sankri, A. Arhaliass, I. Dez, A. Gaumont, Y. Grohens, D. Lourdin, I. Pillin, A. Rolland-Sabate and E. Leroy, Carbohydr. Polym., 2010, 82, 256–263 CrossRef CAS PubMed.
- E. Leroy, P. Jacquet, G. Coativy, A. Reguerre and D. Lourdin, Carbohydr. Polym., 2012, 89, 955–963 CrossRef CAS PubMed.
- P. Kubisa, Prog. Polym. Sci., 2004, 29, 3–12 CrossRef CAS PubMed.
- A. L. Chaudhary, P. J. Torley, P. J. Halley, N. McCaffery and D. S. Chaudhary, Carbohydr. Polym., 2009, 78, 917–925 CrossRef CAS PubMed.
- A. L. Chaudhary, M. Miler, P. J. Torley, P. A. Sopade and P. J. Halley, Carbohydr. Polym., 2008, 74, 907–913 CrossRef CAS PubMed.
- A. Alissandratos, N. Baudendistel, S. L. Flitsch, B. Hauer and P. J. Halling, BMC Biotechnol., 2010, 10 Search PubMed.
- P. B. Zamudio-Flores, A. V. Torres, R. Salgado-Delgado and L. A. Bello-Perez, J. Appl. Polym. Sci., 2010, 115, 991–998 CrossRef CAS.
- B. Volkert, A. Lehmann, T. Greco and M. H. Nejad, Carbohydr. Polym., 2010, 79, 571–577 CrossRef CAS PubMed.
- N. Reddy and Y. Q. Yang, Biotechnol. Bioeng., 2009, 103, 1016–1022 CrossRef CAS PubMed.
- F. Xie, E. Pollet, P. J. Halley and L. Averous, Prog. Polym. Sci., 2013 DOI:10.1016/j.progpolymsci.2013.05.002.
- Z. Liu, Y. Dong, H. Men, M. Jiang, J. Tong and J. Zhou, Carbohydr. Polym., 2012, 89, 473–477 CrossRef CAS PubMed.
- J. Zhou, J. Zhang, Y. Ma and J. Tong, Carbohydr. Polym., 2008, 74, 405–410 CrossRef CAS PubMed.
- R. Nakamura, A. Netravali, A. Morgan, M. Nyden and J. Gilman, Fire Mater., 2013, 37, 75–90 CrossRef CAS.
- V. Madison and J. Schellma, Biopolymers, 1972, 11, 1041–1076 CrossRef CAS PubMed.
- S. Swain, S. Biswal, P. Nanda and P. Nayak, J. Polym. Environ., 2004, 12, 35–42 CrossRef CAS.
- A. Gennadios, C. L. Weller, M. A. Hanna and G. W. Froning, J. Food Sci., 1996, 61, 585–589 CrossRef CAS.
- K. Dangaran, P. Tomasula, P. Qi, M. Embuscado and K. Huber, Edible Films and Coatings For Food Applications, 2009, pp. 25–56 Search PubMed.
- A. Elzoghby, W. El-Fotoh and N. Elgindy, J. Controlled Release, 2011, 153, 206–216 CrossRef CAS PubMed.
- S. Khosravi, P. Nordqvist, F. Khabbaz and M. Johansson, Ind. Crops Prod., 2011, 34, 1509–1515 CrossRef CAS PubMed.
- Z. Peles and M. Zilberman, Acta Biomater., 2012, 8, 209–217 CrossRef CAS PubMed.
- H. Tian, J. Compos. Mater., 2012, 46, 427–435 CrossRef CAS PubMed.
- M. Santin and L. Ambrosio, Expert Rev. Med. Devices, 2008, 5, 349–358 CrossRef CAS PubMed.
- Y. Song, Q. Zheng and Q. Zhang, J. Cereal Sci., 2009, 50, 376–380 CrossRef CAS PubMed.
- S. Domenek, M. Morel, A. Redl and S. Guilbert, Macromol. Symp., 2003, 197, 181–191 CrossRef CAS.
- H. Yue, Y. Cui, P. Shuttleworth and J. Clark, Green Chem., 2012, 14, 2009–2016 RSC.
- C. Verbeek and L. van den Berg, Macromol. Mater. Eng., 2010, 295, 10–21 CrossRef CAS.
- C. J. R. Verbeek and L. E. van den Berg, J. Polym. Environ., 2011, 19, 1–10 CrossRef CAS.
- A. Denis, N. Brambati, B. Dessauvages, S. Guedj, C. Ridoux, N. Meffre and C. Autier, Food Hydrocolloids, 2008, 22, 989–994 CrossRef CAS PubMed.
- A. Ullah, T. Vasanthan, D. Bressler, A. L. Elias and J. P. Wu, Biomacromolecules, 2011, 12, 3826–3832 CrossRef CAS PubMed.
- A. Jerez, P. Partal, I. Martinez, C. Gallegos and A. Guerrero, J. Food Eng., 2007, 82, 608–617 CrossRef CAS PubMed.
- T. Mekonnen, P. Mussone, N. El-Daher, P. Choi and D. Bressler, Macromol. Mater. Eng., 2013 DOI:10.1002/mame.201200429.
- N. El-Thaher, T. Mekonnen, P. Mussone, D. Bressler and P. Choi, Ind. Eng. Chem. Res., 2013, 52, 4987–4993 CrossRef CAS.
- B. Cuq, C. Aymard, J. Cuq and S. Guilbert, J. Food Sci., 1995, 60, 1369–1374 CrossRef CAS.
- H. Zheng, Z. Tan, Y. Zhan and J. Huang, J. Appl. Polym. Sci., 2003, 90, 3676–3682 CrossRef CAS.
- V. Hernandez-Izquierdo and J. Krochta, J. Food Sci., 2008, 73, R30–R39 CrossRef CAS PubMed.
- M. Pommet, A. Redl, S. Guilbert and M. Morel, J. Cereal Sci., 2005, 42, 81–91 CrossRef CAS PubMed.
- A. Ullah and J. Wu, Macromol. Mater. Eng., 2013, 298, 153–162 CrossRef CAS.
- J. W. Lawton, Cereal Chem., 2004, 81, 1–5 CrossRef CAS.
- N. Reddy, L. H. Chen and Y. Q. Yang, Ind. Crops Prod., 2013, 43, 159–164 CrossRef CAS PubMed.
- X. Q. Mo and X. Z. Sun, J. Am. Oil Chem. Soc., 2002, 79, 197–202 CrossRef CAS.
- O. Orliac, A. Rouilly, F. Silvestre and L. Rigal, Ind. Crops Prod., 2003, 18, 91–100 CrossRef CAS.
- N. Cao, X. Yang and Y. Fu, Food Hydrocolloids, 2009, 23, 729–735 CrossRef CAS PubMed.
- L. Yang and A. T. Paulson, Food Res. Int., 2000, 33, 563–570 CrossRef CAS.
- S. S. Sohail, B. W. Wang, M. A. S. Biswas and J. H. Oh, J. Food Sci., 2006, 71, C255–C259 CrossRef CAS.
- M. Wihodo and C. I. Moraru, J. Food Eng., 2013, 114, 292–302 CrossRef CAS PubMed.
- W. Wu, N. Hettiarachchy and M. Qi, J. Am. Oil Chem. Soc., 1998, 75, 845–850 CrossRef CAS.
- M. Pommet, A. Redl, M. Morel and S. Guilbert, Polymer, 2003, 44, 115–122 CrossRef CAS.
- M. J. Fabra, A. Jimenez, L. Atares, P. Talens and A. Chiralt, Biomacromolecules, 2009, 10, 1500–1507 CrossRef CAS PubMed.
- T. H. Shellhammer and J. M. Krochta, J. Food Sci., 1997, 62, 390–394 CrossRef CAS.
- M. J. Fabra, P. Talens and A. Chiralt, Food Hydrocolloids, 2010, 24, 384–391 CrossRef CAS PubMed.
- H. Xu, Y. W. Chai and G. Y. Zhang, J. Agric. Food Chem., 2012, 60, 10075–10081 CrossRef CAS PubMed.
- Z. Ibragimo, A. Yulchiba, Y. V. Zelenev and K. U. Usmanov, Vysokomol. Soedin., Ser. A, 1973, 15, 1831–1838 Search PubMed.
- P. Tummala, W. J. Liu, L. T. Drzal, A. K. Mohanty and M. Misra, Ind. Eng. Chem. Res., 2006, 45, 7491–7496 CrossRef CAS.
- J. T. Kim and A. N. Netravali, Macromol. Mater. Eng., 2012, 297, 176–183 CrossRef CAS.
- X. S. Huang and A. N. Netravali, Biomacromolecules, 2006, 7, 2783–2789 CrossRef CAS PubMed.
- A. Bhattacharya and B. N. Misra, Prog. Polym. Sci., 2004, 29, 767–814 CrossRef CAS PubMed.
- A. Bhattacharya and P. Ray, in Polymer grafting and crosslinking, John Wiley & Sons, Inc., Hoboken, New Jersey, 2009 Search PubMed.
- R. M. Broyer, G. N. Grover and H. D. Maynard, Chem. Commun., 2011, 47, 2212–2226 RSC.
- Y. X. Hu, D. Samanta, S. S. Parelkar, S. W. Hong, Q. A. Wang, T. P. Russell and T. Emrick, Adv. Funct. Mater., 2010, 20, 3603–3612 CrossRef CAS.
- J. Shu, B. Panganiban and T. Xu, Annu. Rev. Phys. Chem., 2013, 64, 631–651 CrossRef CAS PubMed.
- N. Dube, A. D. Presley, J. Y. Shu and T. Xu, Macromol. Rapid Commun., 2011, 32, 344–353 CrossRef CAS PubMed.
- D. L. Xi, C. Yang, X. Y. Liu, M. Q. Chen, C. Sun and Y. L. Xu, J. Appl. Polym. Sci., 2005, 98, 1457–1461 CrossRef CAS.
- H. Zhou, P. Jiang, C. Yang, J. Q. Jiang, H. Y. Bai, M. Q. Chen and X. Y. Liu, Acta Polym. Sin., 2008, 424–429 CrossRef CAS.
- R. Snyders, K. I. Shingel, O. Zabeida, C. Roberge, M. P. Faure, L. Martinu and J. E. Klemberg-Sapieha, J. Biomed. Mater. Res., Part A, 2007, 83, 88–97 CrossRef PubMed.
- Q. X. Wu, T. Yoshino, H. Sakabe, H. K. Zhang and S. Isobe, Polymer, 2003, 44, 3909–3919 CrossRef CAS.
- H. F. Tian, Y. X. Wang, L. N. Zhang, C. Y. Quan and X. Z. Zhang, Ind. Crops Prod., 2010, 32, 13–20 CrossRef CAS PubMed.
- L. Kurniawan, G. G. Qiao and X. Q. Zhang, Biomacromolecules, 2007, 8, 2909–2915 CrossRef CAS PubMed.
- N. Reddy, E. Q. Jin, L. H. Chen, X. Jiang and Y. Q. Yang, J. Agric. Food Chem., 2012, 60, 4872–4879 CrossRef CAS PubMed.
- N. G. Wang, L. N. Zhang and J. M. Gu, J. Appl. Polym. Sci., 2005, 95, 465–473 CrossRef CAS.
- C. M. Vaz, L. A. de Graaf, R. L. Reis and A. M. Cunha, Polym. Degrad. Stab., 2003, 81, 65–74 CrossRef CAS.
- S. K. Park, D. H. Bae and N. S. Hettiarachchy, J. Am. Oil Chem. Soc., 2000, 77, 1223–1227 CrossRef CAS PubMed.
- C. Marquie, J. Agric. Food Chem., 2001, 49, 4676–4681 CrossRef CAS PubMed.
- V. Tropini, J. P. Lens, W. Mulder and F. Silvestre, Ind. Crops Prod., 2004, 20, 281–289 CrossRef CAS PubMed.
- V. Tropini, J. P. Lens, W. J. Mulder and F. Silvestre, Cereal Chem., 2000, 77, 333–338 CrossRef CAS.
- S. Kim, D. J. Sessa and J. W. Lawton, Ind. Crops Prod., 2004, 20, 291–300 CrossRef CAS PubMed.
- E. Malmstrom and A. Carlmark, Polym. Chem., 2012, 3, 1702–1713 RSC.
- K. Edgar, C. Buchanan, J. Debenham, P. Rundquist, B. Seiler, M. Shelton and D. Tindall, Prog. Polym. Sci., 2001, 26, 1605–1688 CrossRef CAS.
- T. Heinze and K. Petzold, Monomers, Polym. Compos. Renewable Resour., 2008, 343–368 CrossRef CAS.
- R. Quintana, O. Persenaire, L. Bonnaud and P. Dubois, Polym. Chem., 2012, 3, 591–595 RSC.
- P. Zugenmaier, Macromol. Symp., 2004, 208, 81–166 CrossRef CAS.
- A. K. Mohanty, A. Wibowo, M. Misra and L. T. Drzal, Polym. Eng. Sci., 2003, 43, 1151–1161 CAS.
- H. L. La Nieve, in Handbook of Fiber Chemistry, 3rd edn, ed. M. Lewin, 2007, vol. 16, pp. 1–1030 Search PubMed.
- X. Cao, S. Sun, X. Peng, L. Zhong, R. Sun and D. Jiang, J. Agric. Food Chem., 2013, 61, 2489–2495 CrossRef CAS PubMed.
- N. Joly, R. Granet, P. Branland, B. Verneuil and P. Krausz, J. Appl. Polym. Sci., 2005, 97, 1266–1278 CrossRef CAS.
- S. Ramesh and S. C. Lu, J. Power Sources, 2008, 185, 1439–1443 CrossRef CAS PubMed.
- M. Yoshioka, N. Hagiwara and N. Shiraishi, Cellulose, 1999, 6, 193–212 CrossRef CAS.
- J. Amim, L. Blachechen and D. Petri, J. Therm. Anal. Calorim., 2012, 107, 1259–1265 CrossRef CAS.
- D. Roy, M. Semsarilar, J. Guthrie and S. Perrier, Chem. Soc. Rev., 2009, 38, 2046–2064 RSC.
- M. Schilling, M. Bouchard, H. Khanjian, T. Learner, A. Phenix and R. Rivenc, Acc. Chem. Res., 2010, 43, 888–896 CrossRef CAS PubMed.
- O. Fridman and A. Sorokina, Polym. Sci., Ser. B, 2006, 48, 233–236 CrossRef.
- G. Wypych, Handbook of Plasticizers, William Andrew Inc., New York, 2004 Search PubMed.
- S. Zepnik, S. Kabasci, H.-J. Radusch and T. Wodke, J. Mater. Sci. Eng. A, 2012, 2, 152–163 CAS.
- A. Ghebremeskel, C. Vernavarapu and M. Lodaya, Int. J. Pharm., 2007, 328, 119–129 CrossRef CAS PubMed.
- A. Ghebremeskel, C. Vemavarapu and M. Lodaya, Pharm. Res., 2006, 23, 1928–1936 CrossRef CAS PubMed.
- J. Ballany, D. Littlejohn, R. A. Pethrick and A. Quye, Probing the factors that control degradation in museum collections of cellulose acetate artefacts. in Historic textiles, papers, and polymers in museums, American Chemical Society, Washington, DC, 2001 Search PubMed.
- J. Amim, Y. Kawano and D. F. S. Petri, Mater. Sci. Eng., C, 2009, 29, 420–425 CrossRef PubMed.
- S. Ramesh, R. Shanti and E. Morris, Carbohydr. Polym., 2013, 91, 14–21 CrossRef CAS PubMed.
- H. R. Jhong, D. S. H. Wong, C. C. Wan, Y. Y. Wang and T. C. Wei, Electrochem. Commun., 2009, 11, 209–211 CrossRef CAS PubMed.
- S. Ramesh, R. Shanti and E. Morris, Carbohydr. Polym., 2012, 87, 2624–2629 CrossRef CAS PubMed.
- A. Pinkert, K. N. Marsh, S. S. Pang and M. P. Staiger, Chem. Rev., 2009, 109, 6712–6728 CrossRef CAS PubMed.
- G. Stiubianu, A. Nicolescu, A. Nistor, M. Cazacu, C. Varganici and B. Simionescu, Polym. Int., 2012, 61, 1115–1126 CrossRef CAS.
- E. Bianchi, A. Bonazza, E. Marsano and S. Russo, Carbohydr. Polym., 2000, 41, 47–53 CrossRef CAS.
- C. Yan, J. Zhang, Y. Lv, J. Yu, J. Wu, J. Zhang and J. He, Biomacromolecules, 2009, 10, 2013–2018 CrossRef CAS PubMed.
- Y. Teramoto and Y. Nishio, Biomacromolecules, 2004, 5, 407–414 CrossRef CAS PubMed.
- J. Zhong, X. Chai and S. Fu, Carbohydr. Polym., 2012, 87, 1869–1873 CrossRef CAS PubMed.
- C. Buchanan, S. Gedon, A. White and M. Wood, Macromolecules, 1992, 25, 7373–7381 CrossRef CAS.
- C. Biermann, J. Chung and R. Narayan, Macromolecules, 1987, 20, 954–957 CrossRef CAS.
- G. Cheng, T. Wang, Q. Zhao, X. Ma and L. Zhang, J. Appl. Polym. Sci., 2006, 100, 1471–1478 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |