 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The reaction between Nafion sulfonyl fluoride precursor membrane and 1,4-dimethylpiperazine does not yield reliable anion-exchange membranes†
Donna M.
Hillman
,
Susan H.
Stephens
,
Simon D.
Poynton
,
Sam
Murphy
,
Ai Lien
Ong
and
John R.
Varcoe
*
Department of Chemistry, Faculty of Engineering and Physical Sciences, University of Surrey, Guildford GU2 7XH, UK. E-mail: j.varcoe@surrey.ac.uk; Tel: +44 (0)1483 686838
First published on 23rd November 2012
Abstract
Recent reports that the reaction between Nafion sulfonyl fluoride precursor and the cyclic diamine 1,4-dimethylpiperazine yields stable anion-exchange membranes appear to be premature. On aqueous work up, membranes with high cation-exchange capacities and zero anion-exchange capacities are produced.
There is currently a significant level of research involving the development of alkaline polymer electrolyte fuel cells (APEFC) containing alkaline anion-exchange membranes (AAEM).1 Prior AAEM studies involve either the investigation of different polymer backbones2 or different functional anion-exchange head-groups.3 Studies into soluble alkaline ionomers for membrane electrode assembly fabrication are also intensifying.4
There have been two recent reports of the development of anion-exchange membranes (AEM) based on Nafion perfluorinated membranes.5,6 Both of these prior studies involve the reaction of Nafion® sulfonyl fluoride (–SO2F) precursor membrane (Nafion–SO2F) with the diamine 1,4-dimethylpiperazine (14DMP) (Scheme 1). The first communication on this system was in 2011 by Ramani et al.5 where Nafion®-111 sulfonyl fluoride membrane (Dupont, ca. 30 µm thickness) was reacted with 14DMP at 60 °C for up to 24 h with a subsequent OH− anion-exchange process; they compared the Nafion-based AEMs [Nafion–DMP(OH−)] to polysulfone AEMs. A more recent 2012 report was published by Elabd et al.6 who reacted Nafion–SO2F membranes (111P, Ion Power, 25 µm thickness) with 14DMP at room temperature for 3 h followed by a subsequent OH− anion-exchange process. Both studies used infrared spectroscopic, water uptake and conductivity measurements to characterise the membranes; measurements such as ion-exchange capacity determinations (IEC), thermogravimetry, small angle X-ray scattering and fuel cell testing were also conducted. Note: Elabd et al. have also published a follow up paper on Nafion-based AEMs with different head-group chemistries.7
![The proposed reaction of Nafion® sulfonyl precursor [Nafion–SO2F] with 1,4-dimethylpiperazine [14DMP] to form an AEM in X− anion form [Nafion–DMP(X)]. Also shown is the process to generate the standard PEM form of Nafion® [Nafion–SO3H]. X = anion being studied.](/image/article/2013/TA/c2ta00955b/c2ta00955b-s1.gif) | ||
| Scheme 1 The proposed reaction of Nafion® sulfonyl precursor [Nafion–SO2F] with 1,4-dimethylpiperazine [14DMP] to form an AEM in X− anion form [Nafion–DMP(X)]. Also shown is the process to generate the standard PEM form of Nafion® [Nafion–SO3H]. X = anion being studied. | ||
The Nafion–DMP(OH−) produced by Ramani et al.5 yielded ion conductivities of over 45 mS cm−1 (40 mS cm−1 for Nafion–DMP(CO33332−2−2−2−)) at 70 °C, but they only referred to the theoretical cation-exchange capacity (CEC) of Nafion–SO3H (0.91 meq. g−1 = 1100 equivalent weight) and they did not determine the IEC directly. However, some of the data presented in these prior studies did not correlate well with the proposed synthesis of the AEM. For example, the Nafion–DMP(OH−) produced by Elabd et al.6 yielded conductivities up to 5 mS cm−1 at 50 °C. Unexpectedly, the conductivities at 50 °C for Nafion–DMP(HCO333−−−), Nafion–DMP(CO33332−2−2−2−), and Nafion–DMP(Cl−−) were higher at 7, 7, and 12 mS cm−1 respectively. Could this be evidence of AEM degradation at high pHs in these prior studies? Additionally, the prior determined IECs [measured using the Warder back-titration technique, which yields the anion-exchange capacities (AEC)] were ≤0.1 meq. g−1; these values are <13% of the theoretical AEC value of 0.81 meq. g−1 (calculated from the mono-reaction of 14DMP with Nafion–SO2F as in Scheme 1). These very low AEC values then translated to the unexpectedly high reported λ values (mol H2O molecules per mol anion X−).
The results reported in this communication detail experiments that investigate this intriguing system further. To complement the prior studies, we focused on characterising the membranes using vibrational spectroscopy, solid state NMR, and IEC methods.
All Nafion–DMP(F−) syntheses followed the exemplar procedure detailed in the ESI† but with variations in Nafion–SO2F + 14DMP reaction times (3–503 h) and temperatures (room temperature, 60 °C and 80 °C). Work up involved removal of excess 14DMP, washing with water, and then storage in water until required. Despite washing with water, a continuous smell of 14DMP could still be detected from the vessels containing the reacted transparent brown membranes in their storage water over long periods of time (even with multiple replacement of the storage water). The membranes' colour also faded on prolonged exposure to water.
Ion-exchange of the as-synthesised Nafion–DMP(F−) membranes, into to the various other anion forms, were conducted by immersion of the membranes in excess aqueous solutions (at least 1 mol dm−3 concentrations) for at least 1 day followed by soaking over at least 1 day in 18.2 MΩ cm water. Despite this, the continuous presence of traces of F− anions (even after multiple changes of water) was observed using ion-chromatography of the post-exchange water washings; this tentative observation may suggest either the slow decomposition of the Nafion–DMP(X) AEM or the slow reaction of residual –SO2F groups in the aqueous environment (potentially slightly alkaline considering the presence of amine groups).
Before the Raman spectra of select Nafion–DMP(X) AEMs can be investigated, the Raman spectrum of the starting material (and the potential cation-exchange byproduct) needs to be studied. Fig. S1 in the ESI† gives the FT-Raman spectra of Nafion–SO2F and the commercially available Nafion®-117 proton-exchange membrane (Nafion–SO3H in Scheme 1, EW = 1100 g eq.−1, 0.007 inch in thickness, Alfa Aesar UK). The FT-Raman spectrum of Nafion–SO2F has bands at 1470, 1250, and 648 cm−1, that are well demarcated and not present in the spectrum of Nafion–SO3H; these are therefore diagnostic of the –SO2F functional group. A band at 1470 cm−1 in the FTIR spectrum of Nafion–SO2F was tentatively assigned to the S–F bond motion in a recent prior study8 and to the asymmetric –SO2– stretch (of the –SO2F group) in an earlier prior study.9 The Raman spectrum of Nafion–SO3H is well assigned (Table 2 in ref. 10). For example, the medium intensity band at 1060 cm−1 is due to the symmetrical –SO3− stretch, while the medium intensity band at 973 cm−1 is due to the symmetrical C–O–C stretch.
Fig. 1 below shows how the Raman spectra develop starting with Nafion–SO2F and then Nafion–DMP(F−) membranes synthesised using increasing room temperature reaction times. First consider the disappearance of the –SO2F related band at 1250 cm−1 and note that Elabd et al. used 3 h reaction time with 25 µm thick Nafion–SO2F precursor membranes to synthesise their membranes. With the [thicker] 50 µm Nafion–SO2F precursor membranes used in this study, 3 h is not adequate for reaction at room temperature as this band remains visible until 100+ h of reaction time. This is backed up by the gradual appearance of bands at 1057, 972 and 774 cm−1, all of which plateau in intensity after ca. 100 h of room temperature reaction time; the SO2F-related band at 1470 cm−1 in Nafion–SO2F also appears to increase in intensity on increasing reaction time. A C–H stretch band at 2980 cm−1 (not shown) also develops and plateaus in intensity around 100 h reaction time (the most intense C–H stretch for in the spectrum of 14DMP is located at 2950 cm−1 – see ESI†). Solid state NMR Data (see ESI†) yields further evidence of reaction of the –SO2F functional group of the Nafion–SO2F precursor (this time with a Nafion–DMP(F−) synthesised at 80 °C and 240 h).
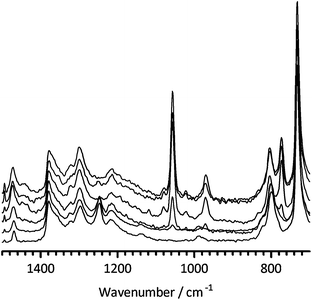 | ||
| Fig. 1 The FT-Raman spectra (diagnostic region 1500–700 cm−1) of the Nafion–DMP(F−) membranes formed on the reaction of Nafion–SO2F and 14DMP at room temperature for (from bottom to top): 0 h (i.e. pristine Nafion–SO2F), 3 h, 24 h, 100 h, 144 h, and 196 h. Spectra were normalised to the band at 1380 cm−1 and then stacked for clarity of presentation. | ||
The Raman spectra of the different Nafion–DMP(F−) membranes are complex. The bands at 1057 and 972 cm−1 that develop on reaction are also present in the spectrum of Nafion–SO3H, but the new band at 774 cm−1 is absent from both the spectra of Nafion–SO3H and Nafion–SO2F; however, there is a strong band at 779 cm−1 in the spectrum of pristine 14DMP [there is also a medium intensity band at 1470 cm−1 in the Raman spectrum of 14DMP]. The bands located in the range 1051–1059 cm−1 in the FTIR spectrum of the Nafion–DMP(F−) and Nafion–DMP(OH−) produced in the prior study by Elabd et al.6 were assigned to the –SO2–N+R3 group. However, considering the spectroscopic data discussed above, this assignment is risky.
An alternative hypothesis is that there is a reaction [or partial reaction] between the 14DMP and Nafion–SO2F but the product membranes [or residual –SO2F groups in the membranes] react with the water [in the aqueous work up and in the presence of the alkaline piperazine-related groups] and form the proton-exchange form Nafion–SO3H membranes. The infrared spectroscopic data presented in the ESI† provides evidence of degradation of a Nafion–DMP(F−) membrane when exposed to alkaline conditions.
Fig. 2 shows an AEC curve (Cl− method)11 of an example Nafion–DMP(Cl−−) AEM. It is immediately obvious that the Cl− AEC cannot be determined as the release of Cl− is below the detection limit of the technique (and well below expected from the theoretical AEC of 0.81 meq. g−1). This was consistently the case for Nafion–DMP(Cl−−) membranes synthesised with different reaction times and temperatures [Nafion–DMP(Cl−−) membranes made from reactions at room temperature for 144–503 h, at 80 °C for 240 h, and 60 °C for 25 days were tested for AEC]. Zero ion-exchange capacities were also measured when the pre-titration KCl exchange solution were replaced with aqueous HCl (1 mol dm−3) [theoretically this modified titration method would enable the Cl− titrations to probe for the presence of non-ionic tertiary amine groups in addition to the ionic quaternary ammonium groups].
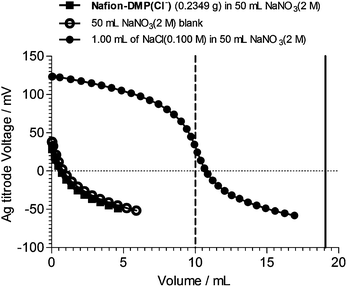 | ||
| Fig. 2 The anion-exchange capacity (AEC) titration curves of a Nafion–DMP(Cl−−) AEM synthesized from Nafion–SO2F and 14DMP at room temperature and 503 h. The method used determines the amount of Cl− anions released from the AEM on immersion in excess NaNO3 (2 mol dm−3). The solid vertical line indicates the expected end point for 0.2349 g of AEM with the theoretical AEC of 0.81 meq. g−1). The dashed vertical line gives the theoretical endpoint for an aqueous solution consisting of 1.00 cm3 of NaCl (0.1000 mol dm−3) added to 50 cm3 NaNO3 (2.0 mol dm−3) which matches the actual control experiment conducted. | ||
AEMs containing the –SO2–N(R′)–R–N+R3″ functional group chemistry (where the N connected to the –SO2– group is not charged) are known,12 but there is little precedence for the functional group –SO2–N+R3 (1-methyl-1-[(4-methylphenyl)sulfonyl]pyrrolidinium is a rare example on ChemSpider13). The question really must be – How stable is the S–N bond in the proposed –SO2–N+ functional group in Scheme 1? To test this, the Nafion–DMP(F−) AEM [synthesised at room temperature and 144 h] was tested for CEC (see ESI† for the standard method used): CEC = 0.79 ± 0.08 meq. g−1 (sd, n = 3). As a control experiment, Nafion–SO2F was hydrolyzed in a solution of 15% mass KOH, 35% mass DMSO, and 50% mass de-ionized water at 80 °C for several hours, followed by thorough washing in water and final conversion to the H+ form Nafion–SO3H membrane by immersion in nitric acid; the CEC of this control membrane = 0.92 ± 0.05 meq. g−1 (sd, n = 3), which compares well to the theoretical 0.91 meq. g−1 for Nafion–SO3H membranes produced from Nafion® PFSA R-1100 sulfonyl fluoride precursor. This is very strong evidence that the end result of the synthesis procedure reported is membranes that are predominantly in the cation-exchange form. These membranes will therefore not be suitable for application in electrochemical devices requiring anion-exchange polymer electrolytes (i.e. APEFCs and alkaline electrolysers).
The authors thank the UK's Engineering & Physical Sciences Research Council (EPSRC grant EP/H025340/1 and Dr John Varcoe's EPSRC Leadership Fellowship funded with grant EP/I004882/1), the University of Surrey (for funds for Susan Stephen's final year undergraduate project) and the EPSRC Solid State NMR service at the University of Durham.
References
- E. H. Yu, X. Wang, U. Krewer, L. Li and K. Scott, Energy Environ. Sci., 2012, 5, 5668 Search PubMed; J. Pan, C. Chen, L. Zhuang and J. Lu, Acc. Chem. Res., 2012, 45, 473 CrossRef CAS; G. Merle, M. Wessling and K. Nijmeijer, J. Membr. Sci., 2011, 377, 1 CrossRef; R. Zeng and J. R. Varcoe, Recent Pat. Chem. Eng., 2011, 4, 93 CrossRef; G. Couture, A. Alaaeddine, F. Boschet and B. Ameduri, Prog. Polym. Sci., 2011, 36, 1521 CrossRef; B. Pivovar, Alkaline Membrane Fuel Cell Workshop Final Report NREL/BK-5600-54297, National Renewable Energy Laboratory, Golden CO, USA, 2011, http://www.osti.gov/bridge CrossRef; E. Antolini and E. R. Gonzalez, J. Power Sources, 2010, 195, 3431 CrossRef; S. Lu, J. Pan, A. Huang, L. Zhuang and J. Lu, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20611 CrossRef; L. A. Adams, S. D. Poynton, C. Tamain, R. C. T. Slade and J. R. Varcoe, ChemSusChem, 2008, 1, 79 CrossRef.
- N. J. Robertson, H. A. Kostalik IV, T. J. Clark, P. F. Mutolo, H. D. Abruña and G. W. Coates, J. Am. Chem. Soc., 2010, 132, 3400 CrossRef CAS; M. R. Hibbs, C. H. Fujimoto and C. J. Cornelius, Macromolecules, 2009, 42, 8316 CrossRef; J. R. Varcoe, R. C. T. Slade, E. Lam How Yee, S. D. Poynton, D. J. Driscoll and D. C. Apperley, Chem. Mater., 2007, 19, 2686 CrossRef.
- O. D. Thomas, K. J. W. Soo, T. J. Peckham, M. P. Kulkarni and S. Holdcroft, J. Am. Chem. Soc., 2012, 134, 10753 CrossRef CAS; Y. Zha, M. L. Disabb-Miller, Z. D. Johnson, M. A. Hickner and G. N. Tew, J. Am. Chem. Soc., 2012, 134, 4493 CrossRef; S. Gu, J. Skovgard and Y. S. Yan, ChemSusChem, 2012, 5, 843 CrossRef; X. Lin, L. Wu, Y. Liu, A. L. Ong, S. D. Poynton, J. R. Varcoe and T. Xu, J. Power Sources, 2012, 217, 373 CrossRef; X. Wang, M. Li, B. T. Golding, M. Sadeghi, Y. Cao, E. H. Yu and K. Scott, Int. J. Hydrogen Energy, 2011, 36, 1002 Search PubMed; Y. Ye and Y. A. Elabd, Macromolecules, 2011, 44, 8494 CrossRef; S. Gu, R. Cai and Y. Yan, Chem. Commun., 2011, 47, 2856 RSC; Q. Zhang, S. Li and S. Zhang, Chem. Commun., 2010, 46, 7495 RSC; X. Kong, K. Wadhwa, J. G. Verkade and K. Schmidt-Rohr, Macromolecules, 2009, 42, 1659 CrossRef; R. Schwesinger, R. Link, P. Wenzl, S. Kossek and M. Keller, Chem.–Eur. J., 2006, 12, 429 CrossRef; M. Tomoi, K. Yamaguchi, R. Ando, Y. Kantake, Y. Aosaki and H. Kubota, J. Appl. Polym. Sci., 1997, 64, 1161 CrossRef; K. J. T. Noonan, K. M. Hugar, H. A. Kostalik IV, E. B. Lobkovsky, H. D. Abruña and G. W. Coates, J. Am. Chem. Soc., 2012, 134, 18161 CrossRef.
- R. Zeng, J. Handsel, S. D. Poynton, A. J. Roberts, R. C. T. Slade, H. Herman, D. C. Apperley and J. R. Varcoe, Energy Environ. Sci., 2011, 4, 4925 Search PubMed; M. Tanaka, M. Koike, K. Miyatake and M. Watanabe, Polym. Chem., 2011, 2, 99 RSC; W. Li, J. Fang, M. Lv, C. Chen, X. Chi, Y. Yang and Y. Zhang, J. Mater. Chem., 2011, 21, 11340 RSC.
- M.-S. J. Jung, C. G. Arges and V. Ramani, J. Mater. Chem., 2011, 21, 6158 RSC.
- H. L. S. Salerno, F. L. Beyer and Y. A. Elabd, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 552 CrossRef CAS.
- H. L. S. Salerno and Y. A. Elabd, J. Appl. Polym. Sci., 2013, 127, 298 CrossRef CAS.
- S. A. Perusich, J. Appl. Polym. Sci., 2011, 120, 165 CrossRef CAS.
- A. J. Greso, R. B. Moore, K. M. Cable, W. L. Jarrett and K. A. Mauritz, Polymer, 1997, 38, 1345 CrossRef CAS.
- A. Gruger, A. Régis, T. Schmatko and P. Colomban, Vib. Spectrosc., 2001, 26, 215 CrossRef CAS.
- O. I. Deavin, S. Murphy, A. Ong, S. D. Poynton, R. Zeng, H. Herman and J. R. Varcoe, Energy Environ. Sci., 2012, 5, 8584 CAS.
- R. Ei Moussaoui and R. Martin, Eur. Pat. EP1612874, 2004; R. Ei Moussaoui and R. Martin, PCT Pat. WO2006003182, 2006.
- http://www.chemspider.com/Chemical-Structure.3526107.html, (accessed 15:39, 14 September 2012) [CSID: 3526107].
Footnote |
| † Electronic supplementary information (ESI) available: Additional details on the syntheses and titrations used and additional spectroscopic results (Raman, FTIR and solid state NMR). See DOI: 10.1039/c2ta00955b |
| This journal is © The Royal Society of Chemistry 2013 |