Evidence by infrared spectroscopy of the presence of two types of β-sheets in major ampullate spider silk and silkworm silk
François
Paquet-Mercier
,
Thierry
Lefèvre
,
Michèle
Auger
and
Michel
Pézolet
*
Département de chimie – Centre de recherche sur les matériaux avancés (CERMA), Regroupement québécois de recherche sur la fonction, la structure et l'ingénierie des protéines (PROTEO) Pavillon Alexandre-Vachon, Université Laval, Québec, Québec G1V 0A6, Canada
First published on 16th October 2012
Abstract
The major ampullate (MA) silk of spider is known to be composed of oriented β-sheet nanocrystals dispersed within an amorphous matrix. The presence of an interphase has also been proposed, but it has not been reported for the fibroin of the silkworm Bombyx mori (B. mori). To obtain quantitative information regarding this third phase, the deuteration of B. mori silk and Nephila clavipes MA silk has been probed by attenuated total reflection infrared spectroscopy. The spectral decomposition of the amide II region has allowed determination of the level of orientation and content of the different secondary structures. The data reveal that, in addition to the amorphous domains, part of the β-sheets is deuterated upon immersion in D2O for both silks. The D2O-inaccessible β-sheets are associated with crystallites, while the interphase is composed of D2O-accessible ones. It is found that the former β-sheets are slightly more oriented along the fiber axis than the latter ones, which suggests that the interphase β-sheets are located at both ends of the crystals. The total β-sheet content is similar for B. mori silk (50 ± 4%) and MA silk (46 ± 4%). However, 27 ± 3% of the β-sheets of MA silk are D2O-accessible compared to 8 ± 3% for B. mori silk. These data suggest that around 5 amino acids for B. mori silk and 9 amino acids for MA silk would be involved in the interphase β-sheets. The higher amount of interphase β-sheets for MA silk is believed to contribute to its higher toughness.
Introduction
The major ampullate (MA) silk of spiders is a remarkably hierarchically organized biopolymer that is very resistant to mechanical stretching.1 To optimize the properties of silkworm silk or to produce synthetic spider silk, a better knowledge of the structure of silk is crucial. The generally accepted model of silk consists in highly oriented crystalline β-sheets embedded in a matrix of disordered amorphous structures. Many authors have also proposed the presence of a third structure, named the interphase, although there is no consensus on the nature of this phase.2–18Pioneering theoretical calculations of Termonia have shown that a thin layer of oriented constrained chains surrounding the crystalline β-sheets is necessary to model mechanical properties.2 This model properly reproduces the combination of high initial modulus, strength and toughness of the fiber. Fossey and Tripathy have shown that a layer of constrained chains surrounding a β-sheet crystal gives rise to greater tensile and shear modulus.3 Moreover, they have shown that mechanical properties are optimized when the crystallites are surrounded by a 2 nm layer. These theoretical considerations are consistent with NMR studies that have shown that one third of the Gly residues and half of non-Gly, non-Ala residues of MA silk are assigned neither to crystalline β-sheets nor to amorphous structures, with these amino acids forming an intermediate phase.4 Moreover, other NMR data have indicated that part of the alanine residues are located in highly oriented crystalline β-sheets and that the other part belongs to weakly oriented β-sheets that may allow the coupling between the highly oriented crystalline domains and the amorphous regions.5,613C-D REDOR NMR experiments performed on silk deuterated with liquid D2O have revealed that 35% of glycine and 24% of alanine amide groups exchange rapidly when the fibers are immersed in deuterium oxide and that they are located in the amorphous structures. Another 25% of the glycine and alanine amide protons deuterate within hours or days and reside in the interphase.7 The rest belong to crystalline β-sheets that do not deuterate.
Infrared experiments have suggested that there is a pre-strained amorphous phase whose distribution is linked to the mechanical properties of MA silk.8–10 A recent study has proposed that amorphous chains bypassing crystals in MA silk can transfer the stress applied to the fiber.11 In this model, the interphase interlinks the crystals and is made of glycine-rich chains with no defined structure. Transmission electron microscopy has also brought evidence of the presence of an intermediate phase. Indeed, the data have revealed crystals that are larger than those typically observed by X-ray diffraction experiments, called non-periodic lattice crystals. These crystals would incorporate polyalanine runs and GGX motifs. The authors suggest that the β-strands form crystals without discrete boundaries.12,13 X-ray diffraction indicates the presence of three types of structure: amorphous, oriented amorphous and oriented crystalline material.14–16 Consistently, scanning transmission X-ray microscopy has shown that the oriented domains are larger than individual crystallites for spider and silkworm silk, the orientation distribution of silkworm silk being narrower than that of MA silk.17
Despite these studies, there is still a lack of information on the nature of the third phase, and quantitative measurements of the orientation and the fraction of each structure are required. Currently, the interphase has been evidenced only for MA silk so that a comparison with other types of silk would be useful to gain insight into structure–function relationships. In this study, we have used hydrogen–deuterium (H–D) exchange to differentiate the three structures, namely amorphous, crystalline β-sheets and interphase, taking advantage of the fact that deuteration occurs only in structures permeable to water (i.e. not in crystalline β-sheets), and that the kinetics of H–D exchange depends on the type of secondary structure, accessibility, hydrogen bond strength, etc. Recent studies have shown the possibility of deuterating amorphous structures and the interphase.7,18 Comparison between B. mori silk and MA silk of Nephila clavipes (N. clavipes) spider is also useful to ensure that the effects attributed to deuteration are not due to reorientation of the proteins since MA silk is known to shrink in the presence of water. Infrared spectroscopy is used to make the measurements since deuteration has a strong effect on protein vibrational spectra.19 The ATR sampling method is used as it is an efficient technique to record polarized spectra of oriented fibers without spectral artifacts.20 Peak fitting of the amide II region has allowed calculation of the orientation and the amount of amorphous structures, crystalline β-sheets and interphase in silkworm and spider silk.
Experimental section
B. mori sample preparation
B. mori cocoons provided by the Feeder Factory (Mississauga, Canada) were heated at 90 °C in 0.05% NaHCO3 solution for 45 minutes to remove sericin. They were then rinsed three times with demineralized water at 90 °C to avoid contamination by salt and residual sericin, and then air-dried. Fibers were pulled with tweezers from the degummed cocoon. Care was taken to avoid stretching of the fibers. They were mounted side-by-side on the sample holder to obtain a “bundle” of 30 fibers.N. clavipes sample preparation
Major ampullate (MA) silk from adult N. clavipes spiders was forcibly reeled directly around the sample holder at 1 cm s−1. Care was taken to avoid contamination with other silks. Each sample consisted of a “bundle” of approximately 330 fibers. They were attached to the sample holder with cyanoacrylate glue to avoid contraction when exposed to water.Spectral acquisition and treatment
Using a method previously described, the sample was rotated with respect to the polarization of the electric field of the infrared beam to investigate molecular orientation perpendicular (90°-spectrum) and parallel (0°-spectrum) to the fiber axis, using a mechanical setup described elsewhere that allows the rotation of the sample with a reproducible contact pressure on the ATR crystal.20 A germanium crystal was chosen over a diamond one even if the depth of penetration is lower, to avoid anomalous dispersion effects encountered when the crystal refractive index is too close to the index of the sample.21 As a matter of fact, due to the high level of molecular orientation in silk, the anomalous dispersion effects are different for the 0°- and 90°-spectra, which prevents a correct band decomposition of the amide II band for diamond ATR.Spectra were recorded using a Nicolet Magna 850 Fourier transform infrared spectrometer (Thermo Scientific, Madison, WI) with a liquid nitrogen cooled narrow-band MCT detector using a germanium ATR accessory (Silver Gate, Specac Ltd, London, UK). The electric field of the infrared beam was s-polarized (perpendicular to the plane of incidence) using a ZnSe wire-grid polarizer (Specac Ltd.). The sample compartment was equipped with a small chamber that allowed control of the sample environment.
As MA silk undergoes the well-known phenomenon of supercontraction when subjected to a wet or humid environment, deuterated and non-deuterated samples have to be studied in the same state in order to make relevant comparisons. Thus, the samples were first exposed to a high humidity environment for three hours before recording the polarized spectra. They will be referred to as “before deuteration” or “non-deuterated fiber”. The fibers were subsequently soaked in D2O for an hour and placed under a stream of dry air to remove the excess deuterium oxide until the D2O bands disappeared from the ATR spectra. The sample chamber was saturated with D2O humidity for three hours before the acquisition of the spectra. These samples will be referred to as “after deuteration” or “deuterated fiber”.
Spectral corrections were achieved using the GRAMS/AI 8.0 software (Thermo Galactic, Salem, NH). The spectra were first corrected for wavelength dependency of the penetration depth of the infrared radiation. A linear baseline was subtracted to account for the background drift. The parallel and perpendicular spectra of silk before deuteration were normalized using the intensity of the orientation-insensitive region between 1320 and 1330 cm−1 as described elsewhere.20 Because of the presence of the amide II′ band, normalization of the polarized spectra of deuterated silk cannot be achieved using the same criterion. Therefore, the parallel (0°) and perpendicular (90°) spectra of deuterated silk were normalized assuming that the area of their amide I bands is the same as the area of the corresponding amide I bands of the non-deuterated sample.
Spectral decomposition
The amide I band is generally used to obtain information on the secondary structure of proteins. We have thus performed a spectral decomposition of this region. However, a rational curve-fitting including the bands of two different types of β-sheets for the amide I region was not achievable in practice because of the strong overlapping between bands (see below). The amide I was then fitted with one component for both ν(π,0) and ν(0,π) β-sheet amide I mode. Fortunately, the NH in-plane bending component makes the amide II mode more sensitive to deuteration than the amide I mode22 so that it is expected to give more information on the structure of silk. It has in particular a higher potential to discriminate crystalline β-sheets, intermediate structures and the amorphous phase than the amide I band. Consequently, the spectral decomposition has also been performed in the amide II region. The comparison between both regions has allowed validating the results. The spectral decomposition was performed between 1725 and 1290 cm−1. The minimum number of components was used. Several non-amide bands arising from side-chains and backbone vibrations were required. They are not involved in the calculation of structure content but are necessary to obtain a correct decomposition of the amide bands. As far as possible, each band fitting parameter value (position, width and intensity) was based on experimental results. Indeed, widths and positions were determined by second derivative of the spectra where the bands are more obvious (0°- or 90°-spectra, before or after deuteration). Also, as will be shown below, the amide II band of the 0°-spectrum of the deuterated fiber can be used to determine the band parameters of the water-inaccessible β-sheets. These parameters were then taken to fit the spectra of the non-deuterated fiber, allowing a variation of ±1% for the intensity during the calculation. Pure Lorentzian bands were used for β-sheet and pure Gaussian ones for the other structures as these shapes represent the best choice to fit the spectra. To ensure reliable and robust results, the curve-fitting calculations were based on the criterion stating that the same set of parameters should be used to fit the 0°- and 90°-spectra of a given type of silk, allowing only variations of intensities (except that of the water-accessible β-sheets). To this end, the position and width were constrained as much as possible during the fitting calculation (typically ±1 cm−1 for the position and width). The uncertainty of the values of secondary structures is an estimation that encompasses the experimental error on different fibers and the peak fitting procedure variability.For the determination of the secondary structures and orientation, we assumed that the absorption coefficient does not vary as a function of secondary structure, a common and reasonable assumption in the field.23,24 We have used the intensity A0 that is independent of orientation and that can be calculated using eqn (1).20A∥ and A⊥ are the absorbance measured when the fiber is parallel and perpendicular to the electric field, respectively.
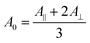 | (1) |
The proportion of secondary structures was calculated by dividing the area of each component of the amide I or amide II band by the total area of the corresponding amide band. The order parameter 〈P2〉 of the transition moments was calculated from eqn (3) for each band using the dichroic ratio defined in eqn (2).
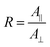 | (2) |
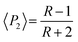 | (3) |
For an isotropic sample, 〈P2〉 = 0. For a perfect parallel orientation 〈P2〉 = 1 and for a perfect perpendicular orientation 〈P2〉 = −0.5.25
Results and discussion
Fig. 1 shows the spectra of the B. mori silk fiber before and after deuteration, parallel (0°-spectrum) and perpendicular (90°-spectrum) to the electric field. The strong dichroism of the amide I and amide II bands arises from the high level of orientation of the β-sheets along the fiber axis. The antiparallel β-sheets give rise to two well-known infrared amide I modes named ν(π,0) and ν(0,π) arising from the coupling between adjacent amide subunits.26,27 The most intense mode is the ν(π,0) mode whose transition moment is perpendicular to the protein chain axis. This band is found at 1624 cm−1 in the 90°-spectra of B. mori silk, in good agreement with the literature.28 The ν(0,π) mode is polarized parallel to the chain axis.28,29 It is observed at 1698 cm−1 in the 0°-spectra of B. mori silk, also in good agreement with the literature.20 This dichroism and normal mode calculations20 undoubtedly demonstrate that this band results from a mode due to β-sheets, as opposed to the erroneous assumption recently made in the literature.30 The band at 1646 cm−1, a position characteristic of disordered chains, is due to the amorphous domains. This band is more apparent in the 0°-spectra because of the low intensity of the β-sheet ν(π,0) band in this orientation. In the amide II region, the band at 1516 cm−1 is due to β-sheets, in agreement with the literature.22,31 It is more apparent in the 0°-spectrum since the amide II mode is dominated by the NH in-plane bending and CN stretching vibrations (both approximately parallel to the protein chain of the β-strands).22 The amide II band of the amorphous structures is located at 1539 cm−1 and is more readily observed in the 90°-spectrum of the non-deuterated fiber.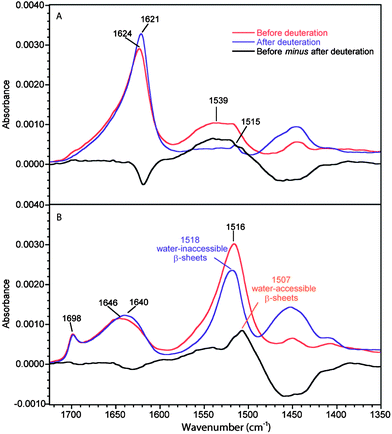 | ||
| Fig. 1 ATR infrared spectra of B. mori silk before and after deuteration. (A) 90°-spectra. (B) 0°-spectra. Real absorbance for the 0°-spectrum before deuteration while the other spectra are normalized following the procedure described in the Experimental section. The black trace represents the difference between the spectra of the non-deuterated fiber and the deuterated one. | ||
For the deuterated B. mori silk, the amide I′ band of amorphous structures (1640 cm−1) is shifted by 6 cm−1 with respect to the amide I band (1646 cm−1). The ν(π,0) amide I′ component of β-sheets is located at 1621 cm−1, i.e. 3 cm−1 lower than that of the non-deuterated fiber. No significant change is observed for the ν(0,π) band of the deuterated fiber. The amide II band at 1539 cm−1 due to amorphous structures disappears completely after deuteration in the 90°-spectrum (Fig. 1a) and the corresponding amide II′ band arises at ∼1453 cm−1, which is consistent with the literature.28 The small remaining band at 1515 cm−1 after deuteration is due to the ν(CC) and δ(CH) of tyrosine residues.32 On the other hand, a strong band at 1518 cm−1 is still present after deuteration in the 0°-spectrum. This band can obviously be assigned to water-inaccessible β-sheets and is most probably related to crystalline β-sheets.
To highlight the effect of deuteration, the subtractions between the spectra of B. mori silk before and after deuteration are also shown in Fig. 1. In the 0°-difference spectrum, a positive band is seen at 1507 cm−1 that corresponds to the part of the β-sheets that can be deuterated. It is only observed in the 0°-spectra, which means that these β-sheets are preferentially oriented along the fiber axis. It also arises 9 cm−1 lower than the water-inaccessible β-sheets (1516 cm−1). Such a difference is expected for a bending mode in the case of weaker hydrogen bonded β-sheets,33,34 which indicates that the water-accessible β-sheets are less ordered. These results thus show that two types of β-sheets exist in B. mori silk: those that are not permeable to water (crystalline) and those that are accessible to water. The latter β-sheets are less ordered and appear to form the interphase observed previously. The positions of the bands due to the different structures obtained from this analysis are summarized in Table 1 and compared with those of MA silk.
| Position (cm−1) | Assignment | Preferential orientationa | References for the assignment | ||
|---|---|---|---|---|---|
| B. mori silk | MA silk | ||||
| a The symbols ∥, ⊥ and – refer to parallel, perpendicular or no preferential orientation of the vibrational mode with respect to the fiber axis, respectively. | |||||
| 1698 | 1697 | Amide I | β-sheets ν(0,π) | ∥ | 22,28 and 34 |
| 1686 | Amide I′ | ||||
| 1646 | 1653 | Amide I | Amorphous | — | 21 and 28 |
| 1640 | 1638 | Amide I′ | |||
| 1624 | 1627 | Amide I | β-sheets ν(π,0) | ⊥ | 22,28 and 34 |
| 1621 | 1624 | Amide I′ | |||
| 1539 | 1540 | Amide II | Amorphous | — | 21 and 28 |
| ∼1454 | ∼1453 | Amide II′ | |||
| 1518 | 1517 | Amide II | Water-inaccessible β-sheets | ∥ | |
| 1507 | 1506 | Amide II | Water-accessible β-sheets | ∥ | |
| ∼1438 | 1434 | Amide II′ | |||
Fig. 2 shows the 0°- and 90°-spectra of MA silk before and after deuteration. The amide I band due to amorphous structures is located at 1653 cm−1 compared to 1646 cm−1 for B. mori silk. The deuteration-induced shift of this band is larger for MA silk than for B. mori silk (6 and 15 cm−1 for B. mori silk and MA silk, respectively). As the two fibers have different amino acid sequences, one could expect some differences in the local structures of the amorphous phase and in the potential energy distribution of the amide I vibration mode that would lead to different positions and deuteration-induced shift. Upon deuteration, the ν(π,0) amide I mode of β-sheets is shifted from 1627 to 1624 cm−1 while the ν(0,π) mode at 1697 cm−1 is shifted to 1686 cm−1, typical positions for these β-sheet amide I and amide I′ bands, respectively.22,28,34
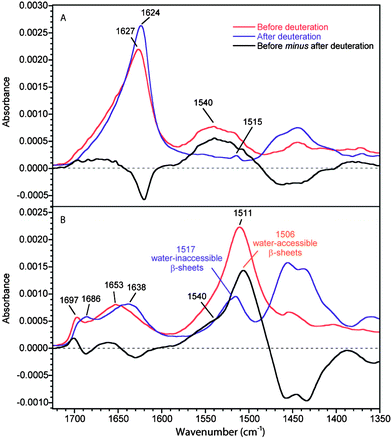 | ||
| Fig. 2 ATR infrared spectra of MA silk before and after deuteration. (A) 0°-spectra. (B) 90°-spectra. Real absorbance for the 0°-spectrum before deuteration while the other spectra are normalized following the procedure described in the Experimental section. The black trace represents the difference between the spectra of the non-deuterated fiber and the deuterated one. | ||
As for B. mori silk, the amide II region in the 90°-spectra shows that the amorphous structures in MA silk are easily deuterated. The 0°-spectra reveal that the loss of intensity of the amide II band due to deuteration is approximately 60% for MA silk compared to 22% for B. mori silk. Since the amide II region of the 0°-spectrum is dominated by the β-sheet conformation, this result clearly indicates that the proportion of water-accessible β-sheets is much higher for MA silk than for B. mori silk. This observation is in agreement with the fact that the β-sheet ν(0,π) amide I band shifts significantly upon deuteration for MA silk and very slightly for B. mori silk. The higher content of water-accessible β-sheets in MA silk is consistent with the lower crystallinity of MA silk.16,35,36
The spectral decomposition of the experimental spectra of B. mori silk and MA silk before deuteration is shown in Fig. 3 and 4, respectively. The position and assignment of the bands due to the different secondary structures obtained from this spectral decomposition are given in Table 2. Small differences between the position of the water-accessible bands obtained from the fit and the positions obtained in the difference spectra (Table 1) are mainly due to the fact that the latter values are affected by the superposition of the contributions of the different bands in this spectral region. Moreover, decompositions performed with a water-accessible component fixed at the position given by the difference spectrum provide unsatisfying fit results, showing that this component is definitely located at a different position. The amide II band of the water-accessible β-sheets is at 1504 cm−1 for MA silk compared to 1510 cm−1 for B. mori silk. This result suggests that hydrogen bonds in water-accessible β-sheets are slightly weaker for MA silk.
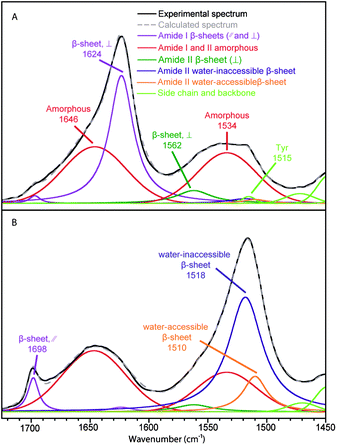 | ||
| Fig. 3 Spectral decomposition of the ATR infrared spectra of B. mori silk. (A) Fiber perpendicular to the fiber axis. (B) Fiber parallel to the fiber axis. | ||
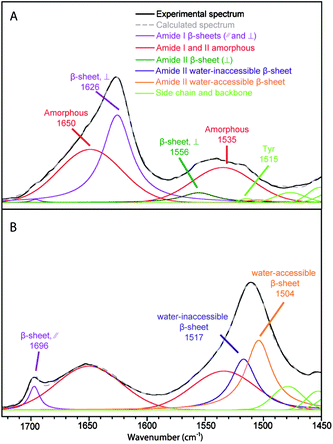 | ||
| Fig. 4 Spectral decomposition of the ATR infrared spectra of MA silk. (A) Fiber perpendicular to the fiber axis. (B) Fiber parallel to the fiber axis. | ||
| Silk | Vibration | Wavenumber (cm−1) | Structure | 〈P2〉 | β-Sheet content (%) |
|---|---|---|---|---|---|
| B. mori silk | Amide I | 1698 | β-Sheet, ∥ | 0.54 ± 0.07 | 48 ± 3 |
| 1646 | Amorphous | 0.04 ± 0.05 | |||
| 1624 | β-Sheet, ⊥ | −0.48 ± 0.02 | |||
| Amide II | 1562 | β-Sheet, ⊥ | −0.20 ± 0.07 | 50 ± 4 | |
| 1534 | Amorphous | −0.06 ± 0.02 | |||
| 1518 | β-Sheet, water-inaccessible | 0.87 ± 0.02 | |||
| 1510 | β-Sheet, water-accessible | 0.76 ± 0.02 | |||
| N. clavipes MA silk | Amide I | 1697 | β-Sheet, ∥ | 0.71 ± 0.07 | 45 ± 3 |
| 1650 | Amorphous | −0.05 ± 0.02 | |||
| 1626 | β-Sheet, ⊥ | −0.50 ± 0.02 | |||
| Amide II | 1556 | β-Sheet, ⊥ | −0.50 ± 0.02 | 46 ± 4 | |
| 1535 | Amorphous | 0.05 ± 0.02 | |||
| 1517 | β-Sheet, water-inaccessible | 0.95 ± 0.06 | |||
| 1504 | β-Sheet, water-accessible | 0.90 ± 0.06 |
Table 2 shows the proportion and the value of the order parameter 〈P2〉 of water-accessible β-sheets, water-inaccessible β-sheets and amorphous structures for B. mori and MA silk obtained from the decomposed spectra of Fig. 3 and 4. The amount of amorphous structures represents 50 ± 3% and 54 ± 3% for B. mori silk and MA silk, respectively. The 〈P2〉 values are near zero for both silks, suggesting that the amorphous phase has almost no orientation. This result differs from Raman37 and solid-state NMR38 studies, which reported a certain degree of orientation along the fiber axis for amorphous structures. The total proportions of β-sheets calculated from the amide I and amide II bands are very close, suggesting that the curve-fitting procedure of the two regions is reliable. In agreement with previous investigations,15,17,37,39Table 2 shows that the crystalline β-sheets of both B. mori and MA silk are highly oriented along the fiber axis with 〈P2〉 close to 1 for the amide II band at 1517–1518 cm−1 and −0.5 for the 1624–1626 cm−1 component of the amide I band. Table 2 further indicates that the interphase β-sheets are also oriented, although slightly less than the water-inaccessible ones.
The total β-sheet content of B. mori silk calculated from the amide II band (50 ± 4%) is close to the previously reported value from Raman analyses of the amide I band by Lefèvre et al. (50 ± 3%)37 and Gillespie et al. (56 ± 5%)40 and from ATR measurements by Boulet-Audet et al. (49 ± 3%).20 The value of the water-inaccessible β-sheet content is 42 ± 3%, which is consistent with the reported values of B. mori silk crystallinity (40–55%).35,36,41,42 The water-accessible β-sheets represent 8 ± 3% of the secondary structure content. It is likely that both the crystalline and interphase β-sheets are formed by the GAGAGS regions as they represent 53% of the amino acid sequence.37 The fact that the orientation of the interphase β-sheets is similar to that of the β-sheet crystallites suggests that the former β-sheets should be formed by small segments located at the beginning and the end of the crystallites. Considering that the β-sheet crystal lengths are correlated with those of the (GAGAGS)n segments,42 it can be estimated from the structure content that about 5 amino acids would form the interphase of B. mori silk (2–3 amino acids on each side of the crystal).
The total β-sheet content of MA silk (45–46 ± 4%) is somewhat higher than the previously reported values from Raman measurements by Lefèvre et al. (37 ± 3%)37 and from solid-state NMR measurements by Jenkins et al. (36%).43 This discrepancy may suggest that the decomposition model used in the amide II region could be too simplistic to represent the actual complexity of the secondary structure of MA silk. In particular, it cannot be totally excluded that another structure, in particular the left-handed 31 helix (PPII), contributes to the band of the water-accessible β-sheets. The values of the crystalline and interphase β-sheet content are 18 ± 3% and 27 ± 3%, respectively. The crystallinity of MA silk determined by wide angle X-ray diffraction ranges between 12 and 29%.16,44,45 The calculation of different indexes of crystallinity on the same sets of wide angle X-ray diffraction data by Plaza et al. has led to two fairly different values (29 ± 4% and 15 ± 1%).44 Also, the groups of Jelinski and Yarger have obtained different values of crystallinity using the same method of calculation (12 ± 3% and 28%).16,45 The present data thus provide an alternative estimation of crystallinity using a different technique. Overall, it may be concluded from the results of the literature as a whole that the MA silk crystallinity is around 20%.
Our results are also basically consistent with the results of Grubb and Jelinski on MA silk from N. clavipes using wide-angle X-ray diffraction that have suggested that 12 ± 3% of silk is crystalline, 34% is non-crystalline oriented material and the remaining 54% is non-crystalline isotropic material.16 The non-crystalline oriented fraction most probably corresponds to the present water-inaccessible β-sheets found in the interphase. This would account for the discrepancy between crystallinity measurements made by X-ray diffraction and the β-sheet content measurements made by vibrational spectroscopy since all β-sheets are not in crystals, as suggested by the groups of Jelinski and Michal.5–7
For MA silk, the polyalanine runs represent 18% of the sequence,37 which is in excellent agreement with the crystalline β-sheet content. The presence of AG and GGA in β-sheets has already been suggested by solid-state NMR.43,46 The amount of AG and GGA blocks flanking the polyalanine motifs (13%) alone is not enough to account for the interphase β-sheets (27 ± 3%) that should also include about one third of GXG motifs (where X = Q, Y, L, R).37 The presence of GXG motifs within the interphase has also been suggested by the non-periodic lattice model12,13 and Raman results.37 The presence of some glycine residues in the interphase is consistent with the fact that they have some orientation along the fiber axis.38 Moreover, the fact that the orientation of the β-sheet interphase of MA silk is similar to that of the β-sheet crystallites suggests that the former structure is made of chain segments flanking the polyalanine blocks. If one considers that a β-sheet crystal is made of an average of 6 alanine residues and based on the proportions of the different types of β-sheets,37 about 9 amino acids would form the interphase (4–5 amino acids on both sides of the crystals).
One might infer that the presence of water-accessible β-sheets observed by IR spectroscopy simply reflects a deuteration phenomenon of the surface of the crystallites, the interior being inaccessible. Indeed, in this case, the fact that the amount of water-accessible β-sheets of MA silk is (3.4 times) higher than that of B. mori silk would be explained by the smaller size of the crystals of MA silk compared to B. mori silk16,42 since the accessible area would be larger. However, the surface-to-volume ratio of a crystal of B. mori silk is “only” 1.3 times that of MA silk, whereas the crystallinity is about three times larger (40–55 and 15%, respectively).16,35,36,41,42 Thus, the total surface of the crystals of B. mori silk appears to be larger than that of MA silk, which rules out a sole “surface” effect. In addition, the water-accessible β-sheets give rise to a distinctive band in the IR spectra that is representative of an independent structure, different from the crystallite β-sheets, characterized in particular by its hydrogen bond strength.
In silk, where there are crystalline β-sheets well oriented along the fiber axis and amorphous structures, it is reasonable to hypothesize that polypeptide segments make the transition between rigid oriented chain and disordered chain. This role seems to be assumed by the interphase. The similar level of molecular orientation and the proportions of crystalline and interphase β-sheets are consistent with this interpretation. In this respect, other studies using X-ray, transmission electron microscopy or scanning transmission X-ray microscopy have shown that the sizes of the oriented fractions (crystals and interphase) are similar for MA silk and B. mori silk although MA silk crystals are smaller.12,13,17,42,47 Thus, these results suggest that the β-sheets of the interphase are on both sides of the crystallites as depicted in Fig. 5. This model is also used in atomistic models of spider silks.48
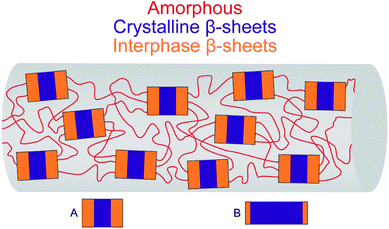 | ||
| Fig. 5 Three-phase model for the MA and B. mori silk. For MA silk (A) and B. mori silk (B), the areas of the blue (crystals) and orange (interphase) rectangles are scaled with respect to the proportion of crystalline and interphase β-sheets, respectively. The length and width of the crystals are scaled with respect to the crystal sizes determined by X-ray scattering.16,42 | ||
The interphase plays an important role in the mechanical properties of the fiber as it has a modulus intermediate between those of the amorphous phase and crystallites.3 Thus, the present results showing that MA silk has a larger interphase than B. mori silk probably account for the better mechanical properties of MA silk. Indeed, the higher proportion of interphase β-sheets for MA silk should lead to a better combination of initial modulus, strength and toughness as the interphase links the stiff crystallites with the extensible amorphous structures. As a matter of fact, due to its reduced interphase, B. mori silk could rather be almost approximated to a two-phase material.
The comparison between the structure of MA silk of different spiders and their mechanical properties would be useful to confirm this hypothesis. As the interphase is important for mechanical properties, the proportion of the three phases should be a criterion when developing protocols for the preparation of regenerated or recombinant silk to obtain a material mimicking MA silk. Moreover, as the permeability and degradation rate of silk films are related to their β-sheet content,49,50 a better knowledge of silk film structure would lead to better silk-based biomaterials which have properties sensitive to the conformation of the silk protein.
Conclusion
The use of ATR infrared spectroscopy to follow hydrogen–deuterium exchange in silk fibers has allowed the differentiation between two types of β-sheets for MA silk and B. mori silk. The water-inaccessible β-sheets are related to the crystalline phase, while the water-accessible β-sheets are associated with the interphase. The presence of an interphase in B. mori silk is demonstrated for the first time. Our data clearly show that although crystals are made of β-sheets, the crystallinity is not directly related to the β-sheet content. This assumption is however commonly made in many studies on silk fibers and films. The total β-sheet contents of B. mori silk and MA silk are close (50 ± 4% and 46 ± 4%, respectively), but the fraction of interphase is significantly higher for MA silk (27 ± 3%) than for B. mori silk (8 ± 3%). This difference probably has a significant effect on the mechanical properties. Our results support the three-phase model, with the third phase assuming the transition between amorphous structures and crystalline β-sheets. Our method could be applied to the study of the structure of other amide based materials like other protein biomaterials and nylons. The decomposition of the amide II region used in this study should be useful for future infrared studies as this region is sensitive to conformation, orientation and H-bond strength. Our method would also allow studying the effect of hydration and supercontraction on the structure of silk.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council (NSERC) of Canada and the Fonds de recherche du Québec – Nature et technologies (FRQ-NT). F.P.-M. gratefully acknowledges NSERC and FRQ-NT for the award of graduate scholarships. The authors thank Jean-François Rioux-Dubé for his technical support.Notes and references
- M. Heim, L. Romer and T. Scheibel, Chem. Soc. Rev., 2010, 39, 156–164 RSC.
- Y. Termonia, Macromolecules, 1994, 27, 7378–7381 CrossRef CAS.
- S. A. Fossey and S. Tripathy, Int. J. Biol. Macromol., 1999, 24, 119–125 CrossRef CAS.
- Z. T. Yang, O. Liivak, A. Seidel, G. LaVerde, D. B. Zax and L. W. Jelinski, J. Am. Chem. Soc., 2000, 122, 9019–9025 CrossRef CAS.
- A. Simmons, E. Ray and L. W. Jelinski, Macromolecules, 1994, 27, 5235–5237 CrossRef CAS.
- A. H. Simmons, C. A. Michal and L. W. Jelinski, Science, 1996, 271, 84–87 CrossRef CAS.
- X. Li, P. T. Eles and C. A. Michal, Biomacromolecules, 2009, 10, 1270–1275 Search PubMed.
- P. Papadopoulos, R. Ene, I. Weidner and F. Kremer, Macromol. Rapid Commun., 2009, 30, 851–857 CrossRef CAS.
- P. Papadopoulos, J. Solter and F. Kremer, Colloid Polym. Sci., 2009, 287, 231–236 CrossRef CAS.
- J. Guan, F. Vollrath and D. Porter, Biomacromolecules, 2011, 12, 4030–4035 Search PubMed.
- R. Ene, P. Papadopoulos and F. Kremer, Soft Matter, 2009, 5, 4568–4574 RSC.
- B. L. Thiel, D. D. Kunkel and C. Viney, Biopolymers, 1994, 34, 1089–1097 Search PubMed.
- B. L. Thiel, K. B. Guess and C. Viney, Biopolymers, 1997, 41, 703–719 CrossRef CAS.
- C. Riekel and F. Vollrath, Int. J. Biol. Macromol., 2001, 29, 203–210 CrossRef CAS.
- C. Riekel, C. Branden, C. Craig, C. Ferrero, F. Heidelbach and M. Muller, Int. J. Biol. Macromol., 1999, 24, 179–186 CrossRef CAS.
- D. T. Grubb and L. W. Jelinski, Macromolecules, 1997, 30, 2860–2867 CrossRef CAS.
- M. E. Rousseau, D. H. Cruz, M. M. West, A. P. Hitchcock and M. Pézolet, J. Am. Chem. Soc., 2007, 129, 3897–3905 CrossRef CAS.
- R. Ene, P. Papadopoulos and F. Kremer, Polymer, 2010, 51, 4784–4789 Search PubMed.
- E. Goormaghtigh, V. Cabiaux and J. M. Ruysschaert, in Physicochemical Methods in the Study of Biomembranes, ed. H. J. Hilderson and G. R. Ralston, Plenum Press, New York, 1994, vol. 23, pp. 363–403 Search PubMed.
- M. Boulet-Audet, T. Lefèvre, T. Buffeteau and M. Pézolet, Appl. Spectrosc., 2008, 62, 956–962 CrossRef CAS.
- M. Boulet-Audet, T. Buffeteau, S. Boudreault, N. Daugey and M. Pézolet, J. Phys. Chem. B, 2010, 114, 8255–8261 CrossRef CAS.
- W. H. Moore and S. Krimm, Biopolymers, 1976, 15, 2465–2483 CrossRef CAS.
- T. Buffeteau, E. Le Calvez, S. Castano, B. Desbat, D. Blaudez and J. Dufourcq, J. Phys. Chem. B, 2000, 104, 4537–4544 CrossRef CAS.
- F. Dousseau and M. Pézolet, Biochemistry, 1990, 29, 8771–8779 CrossRef CAS.
- T. Buffeteau and M. Pézolet, in Handbook of Vibrational Spectroscopy, ed. J. M. Chalmers and P. R. Griffiths, John Wiley & Sons Ltd, London, 2002, vol. 1, pp. 693–710 Search PubMed.
- T. Miyazawa and E. R. Blout, J. Am. Chem. Soc., 1961, 83, 712–719 CrossRef CAS.
- S. Krimm and Y. Abe, Proc. Natl. Acad. Sci. U. S. A., 1972, 69, 2788–2792 CAS.
- E. Goormaghtigh, V. Cabiaux and J. M. Ruysschaert, in Physicochemical Methods in the Study of Biomembranes, ed. H. J. Hilderson and G. R. Ralston, Plenum Press, New-York, 1994, vol. 23, pp. 404–450 Search PubMed.
- M. G. Paterlini, T. B. Freedman and L. A. Nafie, Biopolymers, 1986, 25, 1751–1765 CAS.
- X. Chen, D. P. Knight and Z. Z. Shao, Soft Matter, 2009, 5, 2777–2781 RSC.
- Q. Lu, X. Hu, X. Wang, J. A. Kluge, S. Lu, P. Cebe and D. L. Kaplan, Acta Biomater., 2010, 6, 1380–1387 Search PubMed.
- A. Barth, Prog. Biophys. Mol. Biol., 2000, 74, 141–173 CrossRef CAS.
- J. Kubelka and T. A. Keiderling, J. Am. Chem. Soc., 2001, 123, 12048–12058 CrossRef CAS.
- A. M. Dwivedi and S. Krimm, Macromolecules, 1982, 15, 186–193 CrossRef CAS.
- E. Iizuka, Biorheology, 1965, 3, 1–8 Search PubMed.
- S.-i. Inoue, N. Kawasaki, J. Magoshi and Y. Amemiya, Photon Factory Activity Report, 2003, vol. 20, p. 188 Search PubMed.
- T. Lefèvre, M. E. Rousseau and M. Pézolet, Biophys. J., 2007, 92, 2885–2895 CrossRef CAS.
- J. D. van Beek, S. Hess, F. Vollrath and B. H. Meier, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 10266–10271 CrossRef.
- M. E. Rousseau, T. Lefèvre, L. Beaulieu, T. Asakura and M. Pézolet, Biomacromolecules, 2004, 5, 2247–2257 CrossRef CAS.
- D. B. Gillespie, C. Viney and P. Yager, in Silk Polymers – Materials Science and Biotechnology, ed. D. Kaplan, W. W. Adams, B. Farmer and C. Viney, 1994, vol. 544, pp. 155–167 Search PubMed.
- F. Lucas, J. T. B. Shaw and S. G. Smith, Nature, 1956, 178, 861 Search PubMed.
- L. F. Drummy, B. L. Farmer and R. R. Naik, Soft Matter, 2007, 3, 877–882 RSC.
- J. E. Jenkins, M. S. Creager, R. V. Lewis, G. P. Holland and J. L. Yarger, Biomacromolecules, 2010, 11, 192–200 CrossRef CAS.
- G. R. Plaza, J. Perez-Rigueiro, C. Riekel, G. B. Perea, F. Agullo-Rueda, M. Burghammer, G. V. Guinea and M. Elices, Soft Matter, 2012, 8, 6015–6026 RSC.
- S. Sampath, T. Isdebski, J. E. Jenkins, J. V. Ayon, R. W. Henning, J. P. R. O. Orgel, O. Antipoa and J. L. Yarger, Soft Matter, 2012, 8, 6713–6722 RSC.
- G. P. Holland, J. E. Jenkins, M. S. Creager, R. V. Lewis and J. L. Yarger, Chem. Commun., 2008, 5568 RSC.
- A. Glisovic, T. Vehoff, R. J. Davies and T. Salditt, Macromolecules, 2008, 41, 390–398 CrossRef CAS.
- A. Nova, S. Keten, N. M. Pugno, A. Redaelli and M. J. Buehler, Nano Lett., 2010, 10, 2626–2634 CrossRef CAS.
- K. A. Karve, E. S. Gil, S. P. McCarthy and D. L. Kaplan, J. Membr. Sci., 2011, 383, 44–49 CrossRef.
- C. Vepari and D. L. Kaplan, Prog. Polym. Sci., 2007, 32, 991–1007 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |